
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA DNA-modified hydrogel for simultaneous purification, concentration and detection of targeted cfDNA in human serum†
Xinglu Jiang‡
a,
Chenggui Zhao‡b,
Xiaobo Fana,
Wei Xua,
Rui Zhanga,
Hongbo Xua and
Guoqiu Wu *ab
*ab
aMedical School of Southeast University, Nanjing 210009, People's Republic of China. E-mail: nationball@163.com
bCenter of Clinical Laboratory Medicine, Zhongda Hospital, Southeast University, Nanjing 210009, People's Republic of China
First published on 25th January 2019
Abstract
Cell-free DNA (cfDNA) in blood, which stems from the fetus of pregnant women and tumor in cancer patients, has gained attention in molecular diagnosis. However, cfDNA is less stable, and its amount in the serum is extremely low; these are critical barriers for the utilization of this resource. In this study, a DNA-modified polyacrylamide hydrogel (DNA-Gel) was prepared, and a specialized device was designed to simultaneously catch, purify, concentrate, and detect targeted cfDNA by electrophoresis. We demonstrated that 20–1000 bp ssDNA and dsDNA could be caught and released by the DNA-Gel-based device with high specificity and sensitivity. Upon increasing the number of cycles and electrophoresis time, higher DNA purity and density were achieved, and the separation of serum proteins, untargeted cfDNA, and other charged molecules was promoted. As low as 10 pg μL−1 of DNA could be detected using the DNA-Gel after four cycles of concentration. We also detected 1 fg μL−1 of DNA in the serum with 16 cycles of concentration, followed by 25 PCR cycles. We also designed a device to obtain DNA from the DNA-Gel. We found that the DNA loss rate was around 50%, and A260/A280 was close to 1.7. Thus, we have designed a cost-effective and highly economical device to purify DNA at low concentrations with high specificity and selectivity.
Introduction
Non-invasive diagnosis has been gaining attention since the discovery of circulating cell-free DNA (cfDNA) in blood, which stems from the fetus of pregnant women and tumor in cancer patients. Using this cfDNA for diagnostic testing, the harmful side effects of invasive tissue sampling can be evaded.1–3 Non-invasive prenatal testing (NIPT) for common chromosomal aneuploidies is widely used for clinical diagnosis in over 90 countries, and millions of pregnant women have adopted it for prenatal screening in the past few years.4,5 Recent progress in the analysis of blood samples for cfDNA in cancer patients has identified diagnostic markers for rapid and non-invasive diagnosis of various cancers.6–9 Circulating cfDNA is less stable, with a variable half-life in blood ranging from 15 min to several hours, which gives rise to insufficiently robust and inconsistent results.1,3,10 Classical diagnosis methods include quantitative real-time polymerase chain reaction (PCR)-based, fluorescence-based, and spectrophotometric approaches; next-generation sequencing technologies are currently being applied to plasma DNA analysis and will play a pivotal role in clinical applications.11,12 However, all these methods are limited by the need for specialized equipment and stringent laboratory conditions, resulting in exorbitant costs.13,14 Therefore, significant interest is being focused on the development of a simple, economical, and effective strategy for the purification, concentration, and detection of cfDNA.The amount of cfDNA in serum was reported to be 2–4 times higher than that in plasma; however, it is only 14–45 ng mL−1.15 A high isolation efficiency is vital for a successful cfDNA analysis, particularly in early cancers, as the level of cfDNA has been reported to be correlated with the cancer burden.16,17 Similarly, the fragment size, which is dependent on the cellular release mechanisms, should be considered while analyzing the serum cfDNA.2,18 Detection of the specific DNA changes, such as in MYCN sequences, MSI, and LOH in cfDNA, associated with cancer has been reported to be an effective method for cancer diagnosis.19,20 There are many cfDNA isolation kits such as QIAamp circulating nucleic acid kit (QIA, Qiagen, Antwerp, Belgium), PME free-circulating DNA Extraction Kit (PME, Westburg, Leusden, The Netherlands), Maxwell RSC ccfDNA Plasma Kit (RSC, Promega, Leiden, The Netherlands), and EpiQuik Circulating Cell-Free DNA Isolation Kit (EQ, EpiGentek, Farmingdale, NY).21,22 The commercial kits are extremely expensive and ineffective in specifically separating the targeted DNA. Therefore, we aimed to construct a device for cfDNA isolation, with high sensitivity for low-concentration isolations and high specificity for fragment size-specific isolations.
Gel electrophoresis is the most popular technique to separate proteins and DNA according to their size and charge. It can achieve a limit of detection (LOD) of 10 pg μL−1 for a single fragment of DNA.23 Some high sensitivity electrophoresis systems have been developed to detect pathogens and drug levels and analyze genes.24–26 A commercial capillary electrophoresis instrument using electro-hydrodynamic actuation has been used for the sizing and concentration analysis of cfDNA.27 Although its sensitivity and operational process are satisfactory, the necessary equipment is not available in most laboratories.
In this study, a DNA-modified polyacrylamide hydrogel was designed that could specifically and simultaneously isolate, concentrate, and detect target cfDNA. Moreover, the entire process is relatively easy to implement in a molecular biology laboratory.
Experimental
Chemical reagents
The acrylamide/bisacrylamide solution (30% w/v; 29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), ammonium persulfate (APS), N,N,N′,N′-tetramethylenediamine (TEMED), phosphate buffered saline (PBS), and sodium hydroxide were obtained from Aladdin (Shanghai, China). Nucleic acid oligonucleotides were produced by Sangon Biotech (Shanghai, China). All buffer solutions were prepared using ultrapure water (Millipore, Billerica, MA, USA).
:1), ammonium persulfate (APS), N,N,N′,N′-tetramethylenediamine (TEMED), phosphate buffered saline (PBS), and sodium hydroxide were obtained from Aladdin (Shanghai, China). Nucleic acid oligonucleotides were produced by Sangon Biotech (Shanghai, China). All buffer solutions were prepared using ultrapure water (Millipore, Billerica, MA, USA).
Preparation of the DNA-modified polyacrylamide hydrogel
An aqueous solution containing 1 mL of a 10% acrylamide solution, DNA (100 mM, Acrydite-gaggagccgaaaaggttcca) and APS (0.1 mL, 10% w/v) was prepared. TEMED (0.01 mL, 10% v/v) was added to this mixture, and the mixture was then vortexed to ensure uniform mixing at room temperature. The DNA-modified polyacrylamide hydrogel was obtained after a 1 minute polymerization. The chemical equation is shown in Fig. 1A.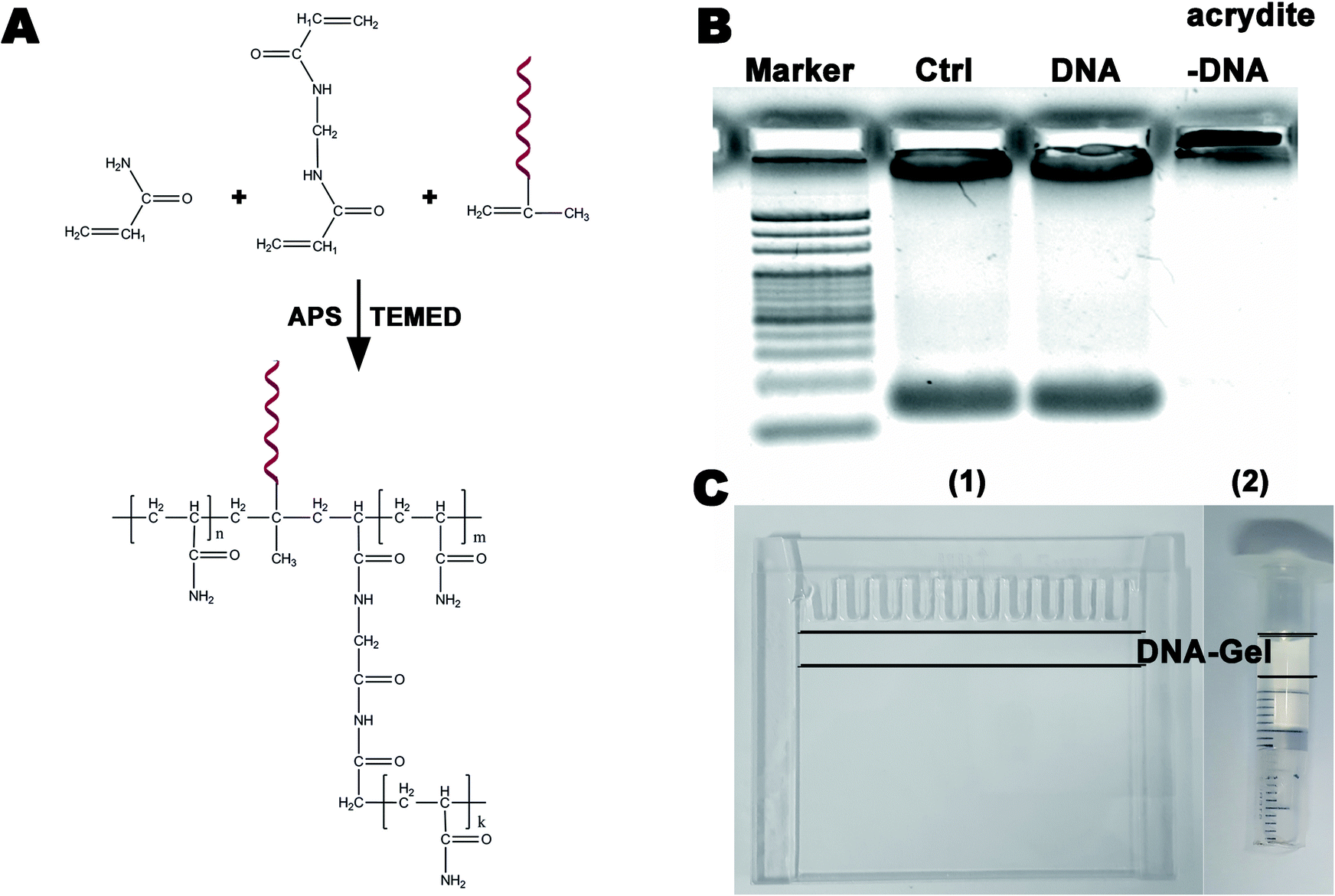 | ||
| Fig. 1 (A) A schematic of the synthesis of the polyacrylamide hydrogel modified by DNA (DNA-Gel); (B) representative results obtained from the electrophoretic assays to verify whether polyacrylamide gel could bind DNA (three groups: control, DNA without acrydite and DNA with acrydite); (C) the device used for specific purification, concentration, and detection of free DNA between two glass sheets (1) or in a plastic injector (2) DNA-Gel was inserted between the two black lines. | ||
Gel electrophoresis
Purification, concentration, and detection of free DNA were performed by gel electrophoresis using an electrophoresis system obtained from Bio-Rad (California, USA). The amount of DNA was estimated using the ethidium bromide staining method.Highly specific DNA catch and release
The sequences of DNA that were caught and released are shown in Table S1.† Herein, 20–70 bp single-stranded DNA (ssDNA) migrated into the DNA-modified polyacrylamide hydrogel in 10 min under a 30 V electric field. Then, 1 min denaturation (95 °C) and 1 min annealing (65 °C) were sequentially performed using constant temperature baths to catch DNA by hybridization. To release DNA, double-stranded DNA (dsDNA) was melted using a 95 °C constant temperature bath, and the resulting ssDNA was maintained in an ice bath. After 1 min denaturation (95 °C), the hydrogel was rapidly placed in an ice bath for 2 min.The target DNA (tggaaccttttcggctcctc) was inserted into the EcoRI/HindIII of pUC57 (2730 bp, Fig. S2A†). Different lengths of dsDNA were then obtained by PCR using the pUC57 template. The primers used are shown in Table S2.† The method to catch and release dsDNA in the DNA-Gel was the same as that used for ssDNA.
Concentration and collection of DNA
DNA (10 pg μL−1 20 bp + 40 bp) was prepared, and 1 ng μL−1 DNA-modified polyacrylamide hydrogel was synthesized and polymerized in a plastic injector to make a device to concentrate DNA. The process of concentration is shown in Fig. 3A. The DNA sample (100 μL 20 bp + 40 bp) was added dropwise to the device. The solution migrated into the DN-modified polyacrylamide hydrogel under the effect of a 30 V electric field and was caught after 1 min denaturation (95 °C) and 1 min annealing (65 °C) in constant temperature baths. This constituted one concentration cycle. Further DNA concentration was achieved by adding multiple cycles.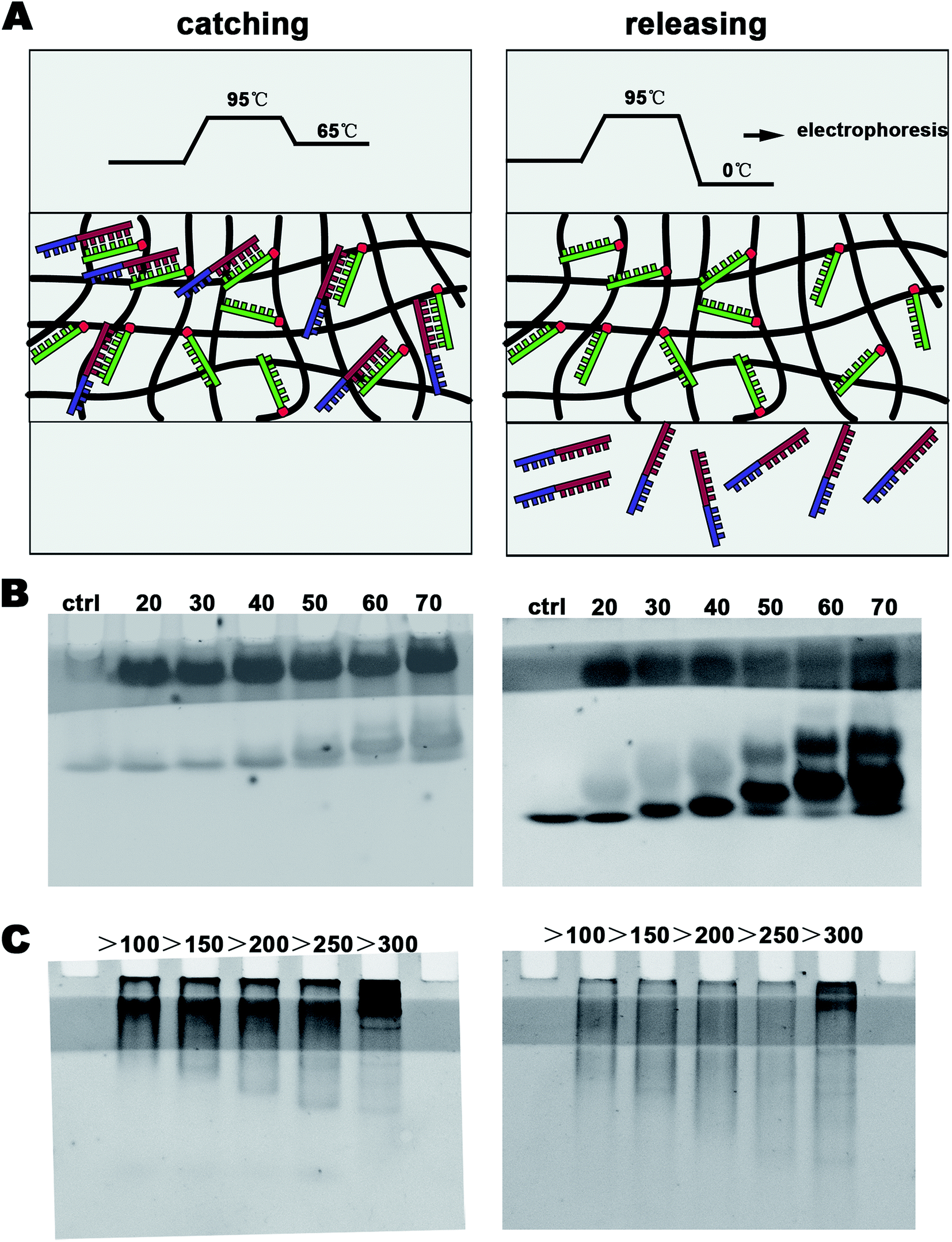 | ||
| Fig. 2 (A) The process diagram to specifically catch and release DNA; (B) 20 bp, 30 bp, 40 bp, 59 bp, 60 bp, and 70 bp single-stranded DNA were caught and released by DNA-Gel; and (C) double-stranded DNA of different lengths were caught and released by the DNA-Gel. | ||
 | ||
| Fig. 3 (A) One cycle of DNA concentration in the DNA-Gel device and (B) the amount of DNA caught by the DNA-Gel in different cycles. | ||
The enriched DNA was caught and trapped in the DNA-modified polyacrylamide hydrogel. Often, DNA analyses, including concentration determination and sequencing, are carried out in solution. Hence, it is necessary to dissolve the trapped DNA in water. Nucleic acids are insoluble in 75% ethyl alcohol. Hence, a filter paper containing 75% ethyl alcohol was inserted between the polyacrylamide hydrogel and the DNA-modified polyacrylamide hydrogel to obtain the enriched DNA (Fig. 4A). At first, the enriched DNA was released from the DNA-modified polyacrylamide hydrogel by rapidly decreasing the temperature from 95 °C to 0 °C. Then, the enriched DNA was allowed to migrate into the filter paper containing 75% ethyl alcohol under an electric field, and it remained there as it was precipitated in ethyl alcohol. After electrophoresis, the filter paper was taken out and placed at room temperature to remove the alcohol. The alcohol-free dry filter paper was then soaked in 50 μL of ultrapure water and vortexed for 2 min to dissolve the enriched DNA. Finally, the filter paper was removed, and the DNA solutions were obtained by centrifugation. The solution mass was measured using a balance, and the DNA concentration was determined using an ultra-micro spectrophotometer (Norllu Berge Colibri, Germany).
 | ||
| Fig. 4 (A) The purge process of free DNA in the serum; (B) the DNA-Gel device stained with ethidium bromide after different cycles of DNA concentration; and (C) the DNA-Gel device stained with Coomassie brilliant blue after different cycles of DNA concentration. | ||
Purification of DNA in serum
A serum containing 1 ng μL−1 20 bp + 40 bp DNA was prepared. It was then subjected to 2, 4, 8, or 16 concentration cycles using the abovementioned system. Serum DNA and proteins would migrate into the polyacrylamide hydrogel under an electric field and get separated along with the target DNA. The amount of DNA was estimated using the ethidium bromide staining method. The amount of protein was estimated using the Coomassie brilliant blue staining method. After the enrichment and collection, the purity of DNA was estimated by A260/280 using the ultra-micro spectrophotometer (Norllu Berge Colibri, Germany).Polymerase chain reaction
DNA (10 fg μL−1 20 bp + 40 bp) was subjected to 2, 4, 8, or 16 concentration cycles. After collection, the enriched DNA was amplified by PCR. The primers for 20 bp + 40 bp DNA were as follows: forward primer: 5′ tggaaccttttcggctcctc 3′ and the reverse primer: 5′ tccgcagtatggatcggcat 3′ (Sangon, Shanghai, China). PCR conditions were as follows: 94 °C, 5 min; (94 °C, 30 s; 57 °C, 30 s; and 72 °C, 30 s) × 20, 25, or 30 cycles; 72 °C, 8 min. The PCR products were assessed by electrophoresis on a 3% agarose gel.Results and discussion
Characterization of the DNA-Gel
Electrophoresis is widely utilized in laboratories to separate macromolecules based on their size and charge. Acrylamide is soluble in water; upon the addition of water, free radicals and a stabilizer, such as ammonium persulfate and TEMED, it polymerizes resulting in the formation of a polyacrylamide gel. The pore diameter of three-dimensional polyacrylamide gels is decided by the total concentration of acrylamides, which can isolate nucleic acids of different sizes via electrophoresis. Functionalized polyacrylamide gels are emerging biomaterials and have recently attracted significant attention in the fields of cell capture and release, biomimetic engineering, and molecular biosensing.28–30 In this study, we used a 20 bp (primer) DNA to modify the polyacrylamide gel to specifically purify, concentrate, and detect DNA of different lengths (>20 bp). Fig. 1A shows the schematic of the synthesis of the DNA-modified polyacrylamide hydrogel (DNA-Gel). The DNA-Gel was prepared by mixing acrylamide, bisacrylamide, and DNA with acrydite, and polymerization was initiated by the addition of APS and TEMED. The detailed procedure is described in the materials and methods section. Agarose gel electrophoresis was used to verify whether the polyacrylamide gel could combine with DNA, and the amount of DNA was estimated using ethidium bromide. The results are shown in Fig. 1B, and it is clear that after electrophoresis, DNA enters the gel in the DNA without the acrydite group (see no. 3 lane), whereas it gets trapped in situ in the experimental group (see no. 3 lane) because of acrylamide polymerization between DNA and the hydrogel. DNA-Gel (between the two black lines in Fig. 1C) was polymerized between two glass sheets (see Fig. 1C(1)) or in a plastic injector (see Fig. 1C(2)) to make a device for purifying specific fragments.Highly specific catch and release of the targeted DNA in the DNA-Gel
In this study, we aimed to design a device that could catch a specific DNA sequence from the cfDNA of serum using a simple electrophoresis configuration. We inserted a DNA-Gel into the polyacrylamide gel (see Fig. 1C(2) and 2) to design the DNA-capture gel. After staining with ethidium bromide, a one-centimeter-wide light stripe of the DNA-Gel appeared in the gel (see Fig. 2B and C). The procedure to catch and release the specific DNA sequence is shown in Fig. 2A as well as provided in the materials and methods section. At first, 20 bp, 30 bp, 40 bp, 59 bp, 60 bp, and 70 bp ssDNA (20 μM) were introduced into the sample wells of the polyacrylamide gel and subjected to a 30 V electric field for 10 min to allow the samples to migrate into the DNA-Gel. Then, 1 min denaturation (95 °C) and 1 min annealing (65 °C) were performed to make 20 bp complementary strands of each group to hybridize them with the DNA-Gel. To prove this, we continued running electrophoresis for 20 min and let the uncombined DNA migrate across the DNA-Gel. In the left panel of Fig. 2B, we could find that there were two bands of DNA. The hybridized DNA was located in the DNA-Gel.It is also very important to release DNA from the DNA-Gel. After denaturation at 95 °C, the double-stranded DNA was melted. At this point, it was cooled down to almost 0 °C in an ice bath to release DNA from the DNA-Gel. In the right panel of Fig. 2B, we could find that there were three bands. The unbound DNA was released and observed as a separate band. Overall, the device could specifically catch the target DNA and release it when needed.
As is known, cfDNA exists as dsDNA in blood. To further study whether the DNA-Gel could catch and release dsDNA, first, the sequence (GAGGAGCCGAAAAGGTTCCA) should bind with the dsDNA. We generated 20 bp ssDNA and inserted it at the EcoRI/HindIII site of pUC57 (2730 bp, Fig. S2A†). Then, dsDNA of different lengths (100–1000 bp) containing the targeted sequence was obtained by PCR (Fig. S3†). Moreover, five kinds of primer sequences are shown in Table S2.† The repeated trials of binding and releasing DNA generated similar results (Fig. 2C). All groups showed the effective capture of dsDNA in the DNA-Gel (left panel of Fig. 2C). After denaturation at 95 °C, all the bound dsDNA could be released from the hydrogel (right panel of Fig. 2C). DNA electrophoresis analyses revealed that the 20 bp fragment was capable of effectively dragging dsDNA of different lengths, and the size ranged up to 1000 bp (Fig. 2C).
Concentration of DNA in the DNA-Gel
Most cfDNA fragments are of 180–200 bp in length, and their levels in the serum are relatively low because of their rapid clearance by the spleen, liver, and kidneys.2,11,16 This makes it challenging to study cfDNA by gel electrophoresis. Therefore, it is necessary to concentrate cfDNA before conducting any studies. Thus, we designed a DNA-Gel-based device to concentrate cfDNA (Fig. 1C and S1†). The methods were verified as follows.The procedure for concentration is shown in Fig. 3A. New samples were added after each cycle of concentration. The DNA increment per concentration cycle was similar. We found that the DNA band in the device became brighter with an increase in the cycle number; this suggested DNA enrichment in the DNA-Gel (Fig. 3B). We detected 10 pg μL−1 of DNA in the DNA-Gel after four concentration cycles.
Purification of DNA in the DNA-Gel
Separation and purification of biological macromolecules, such as proteins, peptides, and nucleic acids, are essential tasks in life science laboratories. The purification of nucleic acids is widely used in genetic engineering, genetic analysis, and genetic therapy. However, the traditional methods based on centrifugal sedimentation and column chromatography are complicated and time-consuming and require toxic chemicals. It is also challenging to implement automation in these processes. Both proteins and nucleic acids carry a net negative charge and migrate towards the positive electrode in a polyacrylamide gel. In our device, the targeted DNA could be specifically caught, and it retained in the DNA-Gel under electric fields. Moreover, proteins and other untargeted DNA were separated under electric fields. A schematic of the process is shown in Fig. 4A. The gels were stained with ethidium bromide and Coomassie brilliant blue to trace the nucleic acids and proteins, respectively, after 2, 4, 8, or 16 cycles. The results are shown in Fig. 4B and C. It was very clear that the targeted DNA stayed in the DNA-Gel (see Fig. 4B), and the serum proteins migrated across the gel under a constant electric field (Fig. 4C). Thus, serum DNA was simultaneously separated and purified from serum proteins.Collection of DNA from the DNA-Gel
Targeted DNA caught by the DNA-Gel should be dissolved in a solution for further analyses such as PCR and gene sequencing. We designed a new device to obtain DNA from the DNA-modified hydrogel. A schematic of the device is shown in Fig. 5A. A filter paper soaked in 75% alcohol was placed between the DNA-Gel and the hydrogel. When the targeted DNA reached the filter paper under the electric field, it precipitated onto the filter paper due to its low solubility in alcohol. To evaluate the DNA precipitation, we stained the device before electrophoresis with ethidium bromide (Fig. 5B). DNA was observed in the upper half of the device (black band in Fig. 5B). We found that most of the DNA was precipitated onto the filter paper with 75% alcohol after electrophoresis, whereas DNA could pass through the filter paper in the absence of 75% alcohol (Fig. 5C). The filter papers with and without alcohol were checked under UV light to evaluate the DNA capture. DNA enrichment was observed in the filter paper with 75% alcohol. Finally, the filter paper was washed with water to dissolve DNA for further analysis. | ||
| Fig. 5 (A) The schematic of a device for collecting DNA from the DNA-Gel device; before (B) and after (C) electrophoresis in two groups – filter paper with and without 75% ethyl alcohol, and the device containing ethidium bromide to trace the amounts of DNA; (D) the filter paper with and without 75% ethyl alcohol stained with ethidium bromide after collecting DNA from the DNA-Gel device. | ||
Efficiency evaluation of the DNA-Gel
To evaluate the DNA loss and the efficiency of the device, DNA at five different initial concentrations (1, 4, 16, 64, and 256 ng μL−1) was subjected to 16 cycles of concentration. The final volumes and the amounts of DNA in the five groups were determined to calculate the concentration multiple and the DNA loss rate (Fig. 6). These two parameters were kept in balance with the increasing concentration of DNA, and a 9-fold concentration efficiency was achieved. Even when the DNA concentration was very low, a significant portion of DNA was caught in the filter paper. We used 200 μL ultrapure water to wash the filter paper and dissolve the DNA, but only about 175 μL water was retrieved; this might have caused a loss of DNA. Although there was a gap between the computed and the measured results, it was within the limits of acceptability. We believe that this gap can be decreased by optimizing the conditions. | ||
| Fig. 6 The concentration multiple and DNA loss rates after 5 different initial concentrations (1, 4, 16, 64, and 256 ng μL−1) of DNA were subjected to 16 cycles of DNA concentration. | ||
After the feasibility and fundamental studies of the device for specific purification, concentration, and detection of cfDNA, we optimized the cycling conditions. At first, it was necessary to understand whether the electrophoresis time would affect the purity of the purified DNA. We found that more proteins would migrate out of the gel with an increase in the electrophoresis time; this would result in an increase in the A260/A280 value, representing an improvement in the purity of DNA. The initial concentration of DNA in the serum was 20 ng μL−1, and eight enrichment cycles were carried out. The final concentration and the value of A260/280 obtained are shown in Fig. 7A. An increase in the electrophoresis time improved the purity of the purified DNA and did not affect the final concentration of DNA (Fig. 7A). Then, to understand the relationship between the concentration cycle and the ultimate density of DNA, we carried out 2, 4, 8, or 16 cycles of concentration, as shown in Fig. 7B. With an increase in the number of cycles, the ultimate density of DNA also significantly increased, and we observed a linear relationship between the cycle number and DNA density (Fig. 7B). Although a decrease in the A260/A280 value was observed with the increasing number of cycles, the value stayed above 1.3. This suggests that DNA enrichment would also lead to protein enrichment.
 | ||
| Fig. 7 (A) The effect of electrophoresis time on concentration multiple and A260/A280 of DNA; (B) the concentration multiple and A260/A280 of DNA after 20 ng μL−1 DNA was subjected to 2, 4, 8, and 16 cycles of DNA concentration. | ||
Allele drop-out, failure of gene amplification, and false-negative results are common challenges for extremely low-concentration DNA PCR. The impurities, such as proteins, polysaccharides, and other organic compounds, would interfere with PCR amplification to generate false-negative results. We used a serum containing 1 fg μL−1 of DNA and subjected it to 2, 4, 8, or 16 concentration cycles on our device. The enriched DNA was obtained, and PCR was performed. After 20, 25, and 30 cycles of amplification, agarose gel electrophoresis was used to analyze the DNA amounts (Fig. 8). We found that the DNA band first appeared in the 25th cycle group after 16 cycles of concentration, and all four lanes showed bright bands after 20 cycles. False-negative detection results would occur if the amplification cycles are insufficient. Evidently, our device could overcome this challenge and improve the diagnostic accuracy.
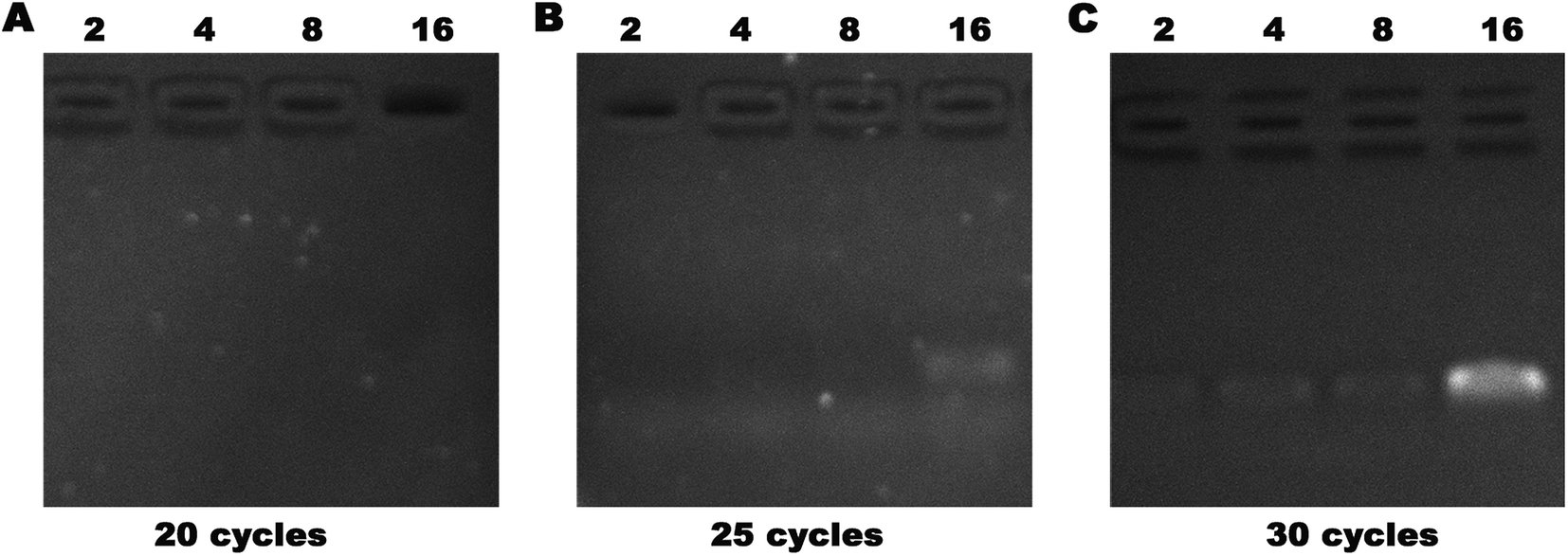 | ||
| Fig. 8 Serum containing 1 fg μL−1 was subjected to 2, 4, 8, or 16 cycles of concentration followed by PCR expansion, and then analyzed by agarose gel electrophoresis. | ||
Conclusions
In this study, a DNA-modified polyacrylamide hydrogel (DNA-Gel) was prepared, and a device was designed that could simultaneously isolate, purify, concentrate, and detect targeted cfDNA via electrophoresis. In our feasibility tests, the device caught 20–1000 bp ssDNA and dsDNA with high specificity and released them upon DNA-Gel denaturation and annealing. The targeted cfDNA was enriched in the DNA-Gel by increasing the number of cycles and electrophoresis time. Serum proteins, untargeted cfDNA, and other charged molecules were separated by electrophoresis. As low as 10 pg μL−1 of DNA could be detected in the DNA-Gel after four cycles of concentration. We could also detect 1 fg μL−1 of DNA with four cycles of concentration followed by 25 PCR cycles. We also designed a device to obtain DNA from the DNA-Gel. We found that the DNA loss rate was around 50%, and A260/A280 was close to 1.7. Thus, we have designed a cost-effective and highly economical device for purifying DNA at low concentrations with high specificity and selectivity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the grants received from the National Science and Technology Major Project (No. 2018ZX09301026-005), the National Natural Science Foundation of China (No. 81773624 & 81603016), the National Natural Science Foundation of Jiangsu Province (No. BE2017746 & BK20160706), and Medical Science and Technology Development Foundation of Nanjing Department of Health (YKK17281).Notes and references
- X. Q. Chen, M. Stroun, J. L. Magnenat, L. P. Nicod, A. M. Kurt, J. Lyautey, C. Lederrey and P. Anker, Nat. Med., 1996, 2, 1033–1035 CrossRef CAS PubMed.
- P. Jiang and Y. M. D. Lo, Trends Genet., 2016, 32, 360–371 CrossRef CAS PubMed.
- R. Deng, K. Zhang and J. Li, Acc. Chem. Res., 2017, 50, 1059–1068 CrossRef CAS PubMed.
- M. Allyse, M. A. Minear, E. Berson, S. Sridhar, M. Rote, A. Hung and S. Chandrasekharan, Int. J. Women's Health, 2015, 7, 113–126 CrossRef CAS PubMed.
- S. Chandrasekharan, M. A. Minear, A. Hung and M. Allyse, Sci. Transl. Med., 2014, 6, 231fs215 Search PubMed.
- N. Sobhani, D. Generali, F. Zanconati, M. Bortul and B. Scaggiante, World J. Clin. Oncol., 2018, 9, 26–32 CrossRef PubMed.
- Z. Kang, S. Stevanovic, C. S. Hinrichs and L. Cao, Clin. Cancer Res., 2017, 23, 6856–6862 CrossRef CAS PubMed.
- W. S. Li, X. Zhang, X. Y. Lu, L. You, Y. Q. Song, Z. G. Luo, J. Zhang, J. Nie, W. W. Zheng, D. N. Xu, Y. P. Wang, Y. Q. Dong, S. L. Yu, J. Hong, J. P. Shi, H. K. Hao, F. Luo, L. C. Hua, P. Wang, X. P. Qian, F. Yuan, L. H. Wei, M. Cui, T. P. Zhang, Q. Liao, M. H. Dai, Z. W. Liu, G. Chen, K. Meckel, S. Adhikari, G. F. Jia, M. B. Bissonnette, X. X. Zhang, Y. P. Zhao, W. Zhang, C. He and J. Liu, Cell Res., 2017, 27, 1243–1257 CrossRef CAS PubMed.
- R. J. Hendriks, S. Dijkstra, F. P. Smit, J. Vandersmissen, H. Van de Voorde, P. F. A. Mulders, I. M. van Oort, W. Van Criekinge and J. A. Schalken, Prostate, 2018, 78, 336–342 CrossRef CAS PubMed.
- S. Salvi, G. Gurioli, U. De Giorgi, V. Conteduca, G. Tedaldi, D. Calistri and V. Casadio, OncoTargets Ther., 2016, 9, 6549–6559 CrossRef CAS PubMed.
- Z. Qin, V. A. Ljubimov, C. Q. Zhou, Y. G. Tong and J. M. Liang, Chin. J. Cancer, 2016, 35 Search PubMed.
- W. Zhang, W. J. Xia, Z. Y. Lv, Y. Xin, C. Ni and L. Yang, Cell. Physiol. Biochem., 2017, 41, 755–768 CrossRef CAS PubMed.
- R. A. van Amerongen, V. P. Retel, V. M. H. Coupe, P. M. Nederlof, M. J. Vogel and W. H. van Harten, Ecancermedicalscienc, 2016, 10 Search PubMed.
- P. Marino, R. Touzani, L. Perrier, E. Rouleau, D. S. Kossi, Z. M. Zou, N. Charrier, N. Goardon, C. Preudhomme, I. Durand-Zaleski, I. Borget, S. Baffert, S. Baffert, E. Barillot, S. Bezieau, I. Borget, L. Coppin, C. Descapentries, I. Durand-Zaleski, S. Forget, T. Frebourd, P. Guardiola, N. Goardon, C. Houdayer, P. Hupe, L. Lacroix, J. Leclerc, A. Lespagnol, S. Longuemare, P. Marino, J. Mosser, M. F. Odou, L. Perrier, C. Preudhomme, F. Revillion, E. Rouleau, N. Sevenet, I. Soubeyran, D. Vaur and N. Grp, Eur. J. Hum. Genet., 2018, 26, 314–323 CAS.
- M. Jung, S. Klotzek, M. Lewandowski, M. Fleischhacker and K. Jung, Clin. Chem., 2003, 49, 1028–1029 CrossRef CAS.
- C. M. Stewart, P. D. Kothari, F. Mouliere, R. Mair, S. Somnay, R. Benayed, A. Zehir, B. Weigelt, S. J. Dawson, M. E. Arcila, M. F. Berger and D. W. Y. Tsui, J. Pathol., 2018, 244, 616–627 CrossRef CAS PubMed.
- M. Murtaza, S. J. Dawson, D. W. Y. Tsui, D. Gale, T. Forshew, A. M. Piskorz, C. Parkinson, S. F. Chin, Z. Kingsbury, A. S. C. Wong, F. Marass, S. Humphray, J. Hadfield, D. Bentley, T. M. Chin, J. D. Brenton, C. Caldas and N. Rosenfeld, Nature, 2013, 497, 108–112 CrossRef CAS PubMed.
- P. Y. Jiang, C. W. M. Chan, K. C. A. Chan, S. H. Cheng, J. Wong, V. W. S. Wong, G. L. H. Wong, S. L. Chan, T. S. K. Mok, H. L. Y. Chan, P. B. S. Lai, R. W. K. Chiu and Y. M. D. Lo, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E1317–E1325 CrossRef CAS PubMed.
- D. M. Marzese, H. Hirose and D. S. B. Hoon, Expert Rev. Mol. Diagn., 2013, 13, 827–844 CrossRef CAS PubMed.
- V. Combaret, C. Audoynaud, I. Iacono, M. C. Favrot, M. Schell, C. Bergeron and A. Puisieux, Cancer Res., 2002, 62, 3646–3648 CAS.
- L. Sorber, K. Zwaenepoel, V. Deschoolmeester, G. Roeyen, F. Lardon, C. Rolfo and P. Pauwels, J. Mol. Diagn., 2017, 19, 162–168 CrossRef CAS PubMed.
- L. Sorber, K. Zwaenepoel, V. Deschoolmeester, G. Roeyen, S. Goethals, C. Rolfo and P. Pauwels, Cancer Res., 2016, 76, 486 Search PubMed.
- A. Valdes, V. Garcia-Canas and A. Cifuentes, Electrophoresis, 2013, 34, 1555–1562 CrossRef CAS PubMed.
- J. W. Lee, D. Lee, Y. T. Kim, E. Y. Lee, D. H. Kim and T. S. Seo, Biosens. Bioelectron., 2017, 91, 388–392 CrossRef CAS PubMed.
- Y. F. Wei, H. Wang, S. J. Sun, L. F. Tang, Y. P. Cao and B. Y. Deng, Biosens. Bioelectron., 2016, 86, 714–719 CrossRef CAS PubMed.
- C. C. Liu, Y. Yamaguchi, S. Sekine, Y. Ni, Z. Q. Li, X. F. Zhu and X. M. Dou, J. Sep. Sci., 2016, 39, 986–992 CrossRef CAS PubMed.
- C. L. Andriamanampisoa, A. Bancaud, A. Boutonnet-Rodat, A. Didelot, J. Fabre, F. Fina, F. Garlan, S. Garrigou, C. Gaudy, F. Ginot, D. Henaff, P. Laurent-Puig, A. Morin, V. Picot, L. Saias, V. Taly, P. Tomasini and A. Zaanan, Anal. Chem., 2018, 90, 3766–3774 CrossRef CAS PubMed.
- J. Li, L. T. Mo, C. H. Lu, T. Fu, H. H. Yang and W. H. Tan, Chem. Soc. Rev., 2016, 45, 1410–1431 RSC.
- Z. Y. Zhang, N. C. Chen, S. H. Li, M. R. Battig and Y. Wang, J. Am. Chem. Soc., 2012, 134, 15716–15719 CrossRef CAS PubMed.
- W. W. Guo, C. H. Lu, R. Orbach, F. A. Wang, X. J. Qi, A. Cecconello, D. Seliktar and I. Willner, Adv. Mater., 2015, 27, 73–78 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra10138h |
| ‡ These authors were equal to this work. |
| This journal is © The Royal Society of Chemistry 2019 |