
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis and broad-spectrum Bcr-Abl inhibitory activity of novel thiazolamide–benzamide derivatives†
Juan
Liu‡
,
Honglin
Huang‡
,
Xiangping
Deng
,
Runde
Xiong
,
Xuan
Cao
,
Guotao
Tang
 ,
Xin
Wu
,
Shiyu
Xu
and
Junmei
Peng
*
,
Xin
Wu
,
Shiyu
Xu
and
Junmei
Peng
*
Institute of Pharmacy and Pharmacology, Hunan Province Cooperative Innovation Center for Molecular Target New Drug Study, University of South China, Hengyang, China. E-mail: pengmarina@126.com
First published on 15th January 2019
Abstract
Bcr-Abl plays an important role in the pathogenesis and development of chronic myeloid leukemia (CML). But Bcr-Abl is prone to mutation, so it increases the difficulty of clinical treatment. Therefore, it is crucial to design a new class of broad-spectrum Bcr-Abl inhibitors. Herein, forty novel thiazolamide–benzamide derivatives were synthesized and evaluated their broad-spectrum Bcr-Abl inhibitory activities. The newly synthesized compounds were characterized by using spectrum data (1H NMR, APCI-MS and IR) and elemental analysis. The protein kinase results indicated that eight compounds (3a, 3e, 3m, 3n, 3p, 4c, 4f, 4g) showed high activities to wild-type and T315I mutation. The most potent compound 3m exhibited an Abl IC50 value as low as 1.273 μM and showed inhibition to the T315I mutant with IC50 value 39.89 μM. 3m could prove to be a new promising lead compound for the further development of broad-spectrum Bcr-Abl inhibitors to overcome clinical acquired resistance.
1 Introduction
Chronic myeloid leukemia (CML) is a clonal myeloproliferative disorder of a hematopoietic stem cell, marked by the Philadelphia chromosome (Ph).1,2 The appearance of the Ph chromosome is the result of the reciprocal translocation between the Abelson (ABL) gene from the long arm of chromosome 9 and the break-point cluster region (BCR) gene on chromosome 22.3,4 The translocation leads to the creation of abnormal Bcr-Abl fusion protein, which is constitutively active and can increase tyrosine kinase (TK) activity. This dysregulated TK activity is considered to be both a necessary and sufficient starting trigger in CML.5 CML can spread through the bloodstream, causing symptoms such as anemia, bleeding, infection, organ infiltration and reduced immune function. If untreated, it usually progresses to the terminal acute phase within 3 to 5 years and leads to death.6The advent of Bcr-Abl inhibitor therapy has essentially changed the way of treating CML. Imatinib, the first approved ATP-competitive Bcr-Abl inhibitor, has shown great success with a large sum of patients benefiting from it, and now remains as the first-line therapy drug of CML.7 But several studies have reported that imatinib has been associated with a variety of complications, including bleeding quality and thrombosis.8 The most important point is that acquired resistance has become a principal obstacle for treatment of CML, and about 40% of patients have failed treatment.9,10 The main mechanism of imatinib resistance is the mutation of the Bcr-Abl fusion gene, which can affect the binding to imatinib by changing amino acid residues in the Bcr-Abl domain and result in decreased sensitivity and induction of drug resistance.11,12 In order to overcome the resistance of imatinib, a more powerful Bcr-Abl inhibitory-dasatinib has been developed, which can overcome the most of resistance except the resistance caused by T315I mutation.13,14 Ponatinib is the first broad-spectrum Bcr-Abl inhibitor,15 however, published clinical data show that it has a risk of fatal blood clots and severe vascular stenosis.16 Ponatinib was once removed from the market due to its toxic side effects. Now, Ponatinib is only approved for chronic, accelerated or blast-resistant CML and Ph + ALL that are resistant or intolerant to previous TK inhibitor treatments.17
Even though ponatinib is restricted due to the serious side effects, its strategy of design is commendable and encouraged other scientist to develop more CML drugs. Unlikely to other solid tumors, the CML has more mutation types and more mutation risks. It has been reported that more than 90 different amino acid substitutions in Bcr-Abl domain have been identified.18 Among them, the T315I mutation, aptured 15–20%.19 In the progress of CML, there is often a phenomenon that mutants and non-mutants coexist in patients which increases the difficulty of clinical treatment. For example, a combination of imatinib and an imatinib-resistant Bcr-Abl mutants inhibitor was often used to treat this phenomenon, but this may lead to an increased toxicity in normal tissues.20 Hence, it is crucial to design new broad-spectrum Bcr-Abl inhibitors.
In this work, based on the comprehension of the mechanism of imatinib resistance and the high activity of ponatinib on the T315I mutation, a series of novel thiazolamide–benzamide derivatives were designed, synthesized and evaluated for their Bcr-Abl inhibitory activity. The in vitro biochemical assays results showed that some compounds showed good broad-spectrum Bcr-Abl inhibition of both wild-type and T315I mutant.
2 Results and discussion
2.1 Design
The pharmacophore that imatinib binds to the Abl kinase domain is pyrimidine aminobenzoyl.21 This pharmacophore is also present in other Bcr-Abl inhibitors, such as nilotinib and radotinib. Studying the interaction between imatinib and Bcr-AblT315I, it is found that after the 315 threonine mutation to isoleucine, the large volume of isoleucine produces steric hindrance, hindering the interaction with imatinib. The combination result in imatinib being ineffective against Bcr-AblT315I.22,23 Compared to the pyrimidine ring in imatinib, the alkynyl group in ponatinib does not produce steric hindrance to isoleucine due to its small size. This is the main reason why ponatinib is effective to Bcr-AblT315I but imatinib is ineffective.24 So, we speculate that some small volume groups can be used in place of the pyrimidine group in imatinib which reduces its steric hindrance to isoleucine to obtain a CML drug that is effective against both wild-type and T315I mutant.The thiazole skeleton is known as to have pharmacological activities against the chronic myeloid leukemia cell line K562.25,26 Moreover, the result of dasatinib binding to the Abl kinase domain showed that the N3 of thiazole ring was engaged in a vital hydrogen bond interplay with the NH group of Met318.27,28 And the volume of 1,3-thiazole ring is smaller than that of 1,3-pyrimidine ring. So, our team used the 1,3-thiazole ring to instead of 1,3-pyrimidine. At the same time, the structure of the pharmacophore group benzamide was retained. Thus, designed a series of novel thiazolamide–benzamide derivatives were designed (Fig. 1).
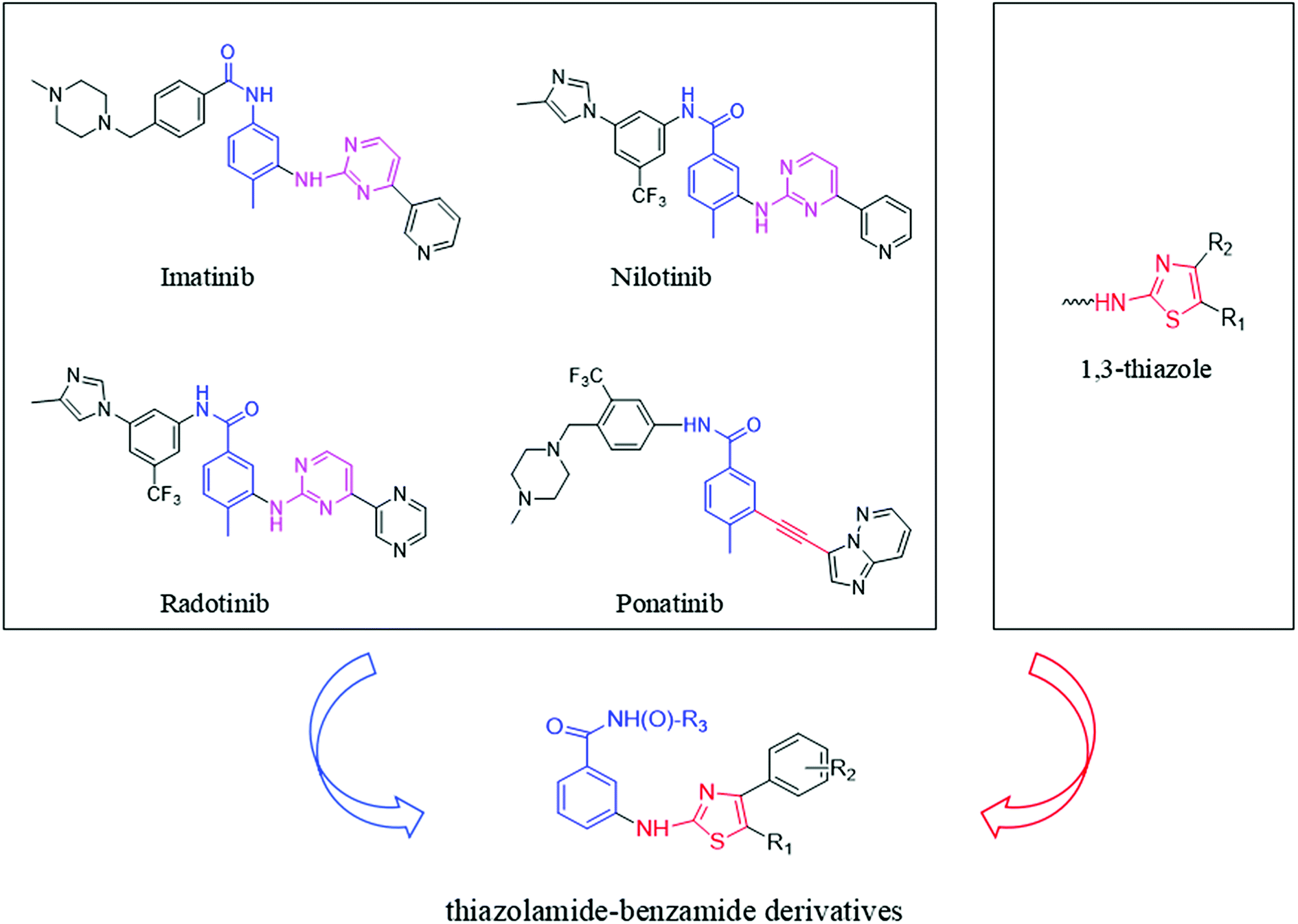 | ||
| Fig. 1 Design of thiazolamide–benzamide derivatives as novel Bcr-Abl inhibitors. | ||
2.2 Molecular
In order to demonstrate our design hypothesis, we used SYBYL-X2.0 to perform molecular docking simulation of the designed compounds with Abl and AblT315I kinases. Wild-type Abl kinase (PDB code: 2GQG27) and its T315I mutant (PDB code: 3IK315) were selected as receptors for docking study. The docking results showed that the designed compounds had a good inhibitory effect on Abl and AblT315I. 3m was taken an example to illustrate the docking mode of the designed compound with Abl and AblT315I. The binding modes of compound 3m with wild-type and AblT315I mutant were illustrated in Fig. 2. As expected, 3m showed a high affinity with Bcr-Abl like imatinib. It displayed perfect binding with the gatekeeper Thr315 residue by two hydrogen bonds. The terminal methoxy group hold the important hydrophobic pocket of Asp381 (part of the DFG-motif of the N-terminal activation ring), which was occupied in an excellent manner by the formation of hydrogen bonds. In addition, the hydroxyl group on the benzene ring of 3m formed an additional hydrogen bond with Thr319 (Fig. 2B).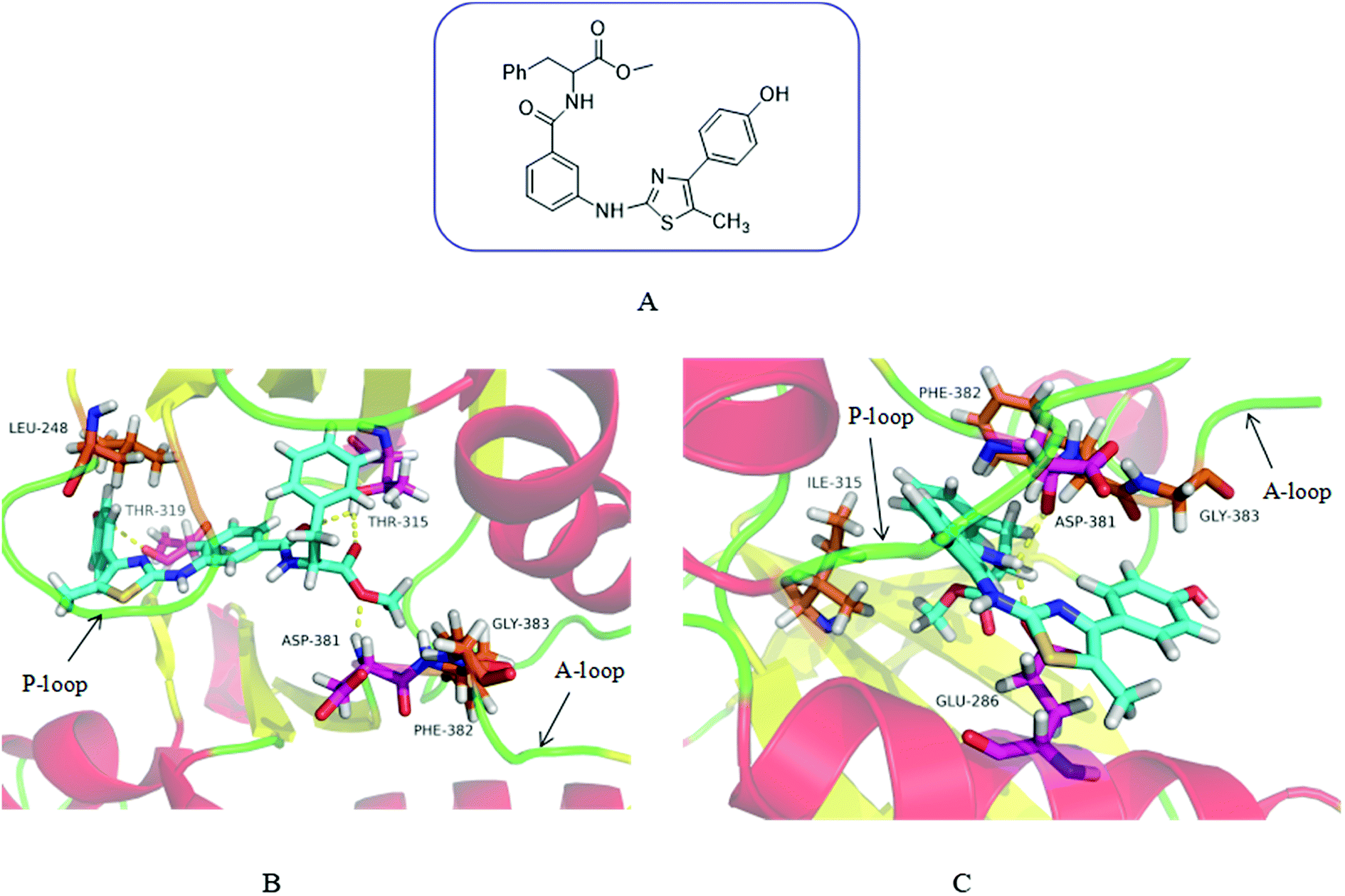 | ||
| Fig. 2 Chemical Structure of 3m and the predicted binding mode of 3m with Abl and AblT315I. (A) Chemical structure of 3m. (B) The binding mode of 3m with the ATP-binding site of Abl. 3m was shown in blue with translucent molecular surface. Hydrogen bonds were highlighted with yellow dashed lines. The residues formed hydrogen bonds with 3m was shown in purple. (C) The binding mode of 3m with the ATP-binding site of AblT315I. The side chain of the mutated gatekeeper residue Ile315 was shown in purple. | ||
3m binded to the ATP pockets formed by the activity of AblT315I mutant, avoiding steric hindrance with the gatekeeper residue Ile315. In addition, NH in the amide group formed two hydrogen bonds with Glu286 and Asp381 which enhanced the affinity of the compound for the ATP binding site (Fig. 2C).
It can be seen from the docking diagram of 3m and Abl and AblT315I that 3m not only has good affinity with the ATP binding site, but also does not produce steric hindrance with Ile315. Therefore, theoretically, this series of compounds may has a good broad-spectrum Bcr-Abl inhibition.
2.3 Chemistry
The synthetic program utilized for the synthesis of titled thiazolamide–benzamide derivatives 2a–2h, 3a–3p, and 4a–4p were outlined in Schemes 1 and 2. The key intermediate 3-thioureidobenzoic acid was synthesized starting from 3-aminobenzoic acid, thiocyanate and benzoyl chloride according to the steps reported in our previous research. A series of bromoaryl ketone (1a–1h) were produced by bromine substitution of different aryl ketones. The vital skeleton of the 1,3-thiazole ring was constructed by a cyclization reaction of the intermediate 3-thioureidobenzoic acid with (1a–1h).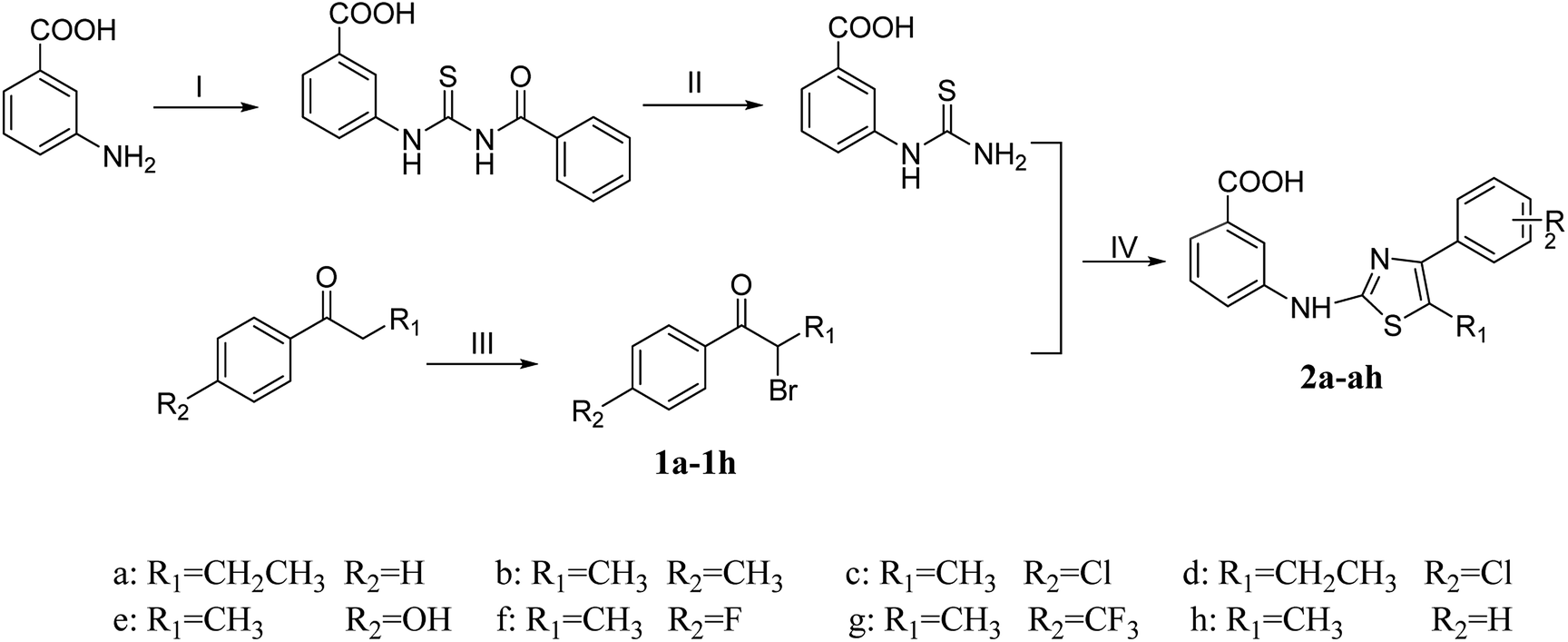 | ||
| Scheme 1 Reagents and conditions: (I) NH4SCN, PhCOCl, CH3COCH3, reflux; (II) 10% NaOH, reflux; (III) CuBr2, EtOH, reflux; (IV) HAc, reflux. | ||
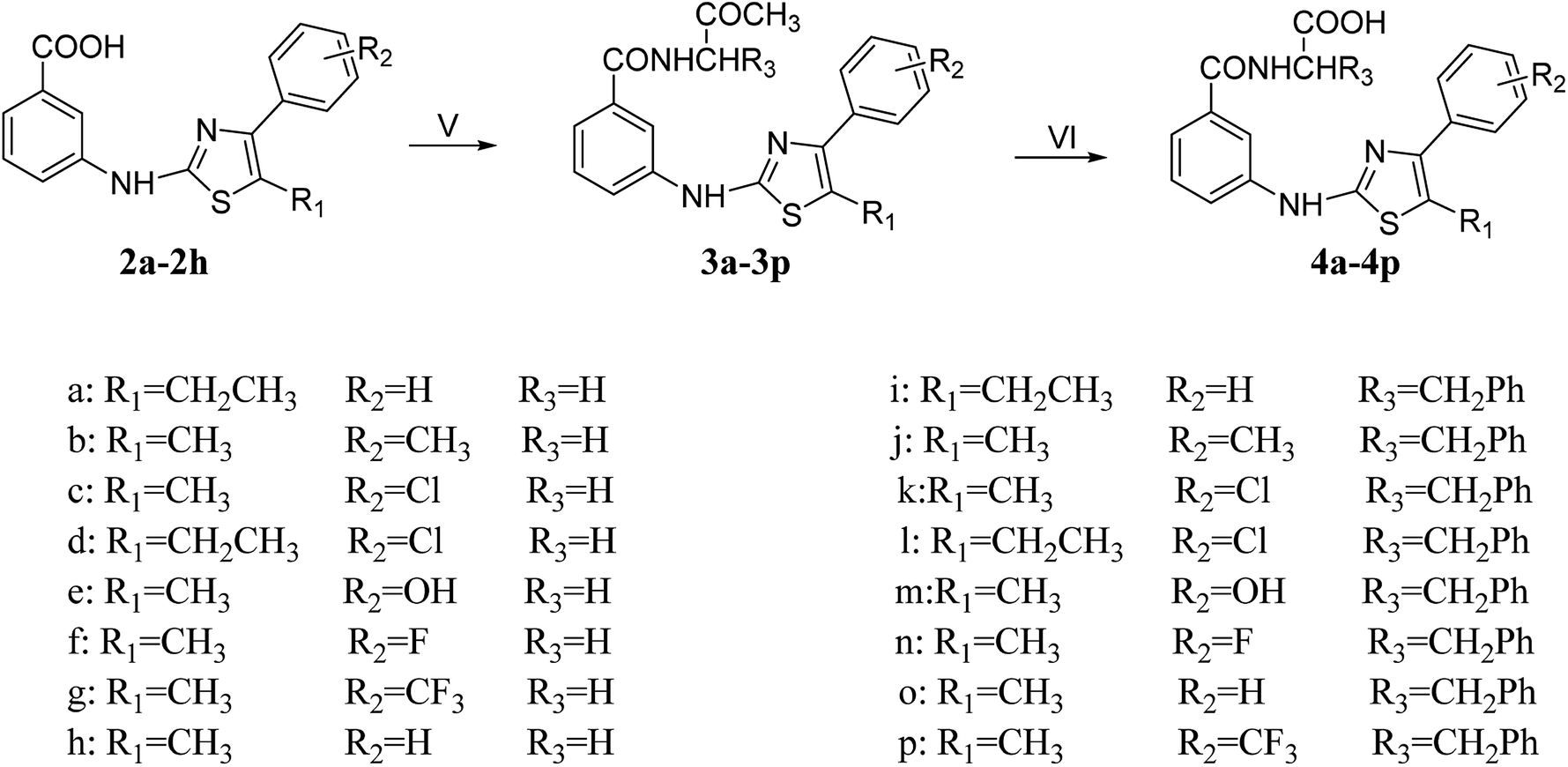 | ||
| Scheme 2 Reagents and conditions: (V) EDCI, HOBT, EtOH, 0 °C; glycine methyl ester hydrochloride/L-phenylalanine methyl ester hydrochloride, DIPEA, DMAP, DMF, 0 °C, 4 h → room temperature; (VI) EtOH, water, reflux. | ||
In the presence of EtOH, HOBt and EDCI were added to compound (2a–2h) under ice bath. After about 4 h, the glycine methyl ester hydrochloride or L-phenylalanine methyl ester hydrochloride, DIPEA, DMAP and DMF were added, and the ice bath was continued for half an hour. Then the resulting mixture was allowed to stir at room temperature for 21–28 h to obtain 3a–3p. The synthesis of 4a–4p was a common hydrolysis reaction, and the yield was also satisfactory.
2.4 Inhibition of Bcr-Abl and Bcr-AblT315I activity
All the target compounds were evaluated for their enzymatic inhibition against both Bcr-Abl and Bcr-AblT315I. The tyrosine kinase inhibitory potency was assayed by using the well-established ADP-Glo assays. The substituents of the tested compounds and their kinase IC50 values were listed in Table 1.| No. | R1 | R2 | R3 | ABL-1 | ABLT315I | No. | R1 | R2 | R3 | ABL-1 | ABLT315I |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a ND = Not determined. | |||||||||||
| 2a | Et | H | — | >100 | ND | 2b | Me | CH3 | — | >100 | >500 |
| 2c | Me | Cl | — | >100 | >500 | 2d | Et | Cl | — | >100 | NDa |
| 2e | Me | OH | — | >100 | >500 | 2f | Me | F | — | >100 | ND |
| 2g | Me | CF3 | — | >100 | ND | 2h | Me | H | — | 70.56 | >500 |
| 3a | Et | H | H | 3.02 | 130.10 | 3b | Me | Me | H | >100 | ND |
| 3c | Me | Cl | H | >100 | ND | 3d | Et | Cl | H | >100 | ND |
| 3e | Me | OH | H | 12.42 | 121.80 | 3f | Me | F | H | >100 | ND |
| 3g | Me | CF3 | H | 35.26 | 268.40 | 3h | Me | H | H | >100 | ND |
| 3i | Et | H | Bz | >100 | ND | 3j | Me | Me | Bz | 42.07 | ND |
| 3k | Me | Cl | Bz | >100 | ND | 3l | Et | Cl | Bz | 39.07 | ND |
| 3m | Me | OH | Bz | 1.27 | 39.89 | 3n | Me | F | Bz | 19.75 | 76.60 |
| 3o | Me | H | Bz | 57.51 | 114.60 | 3p | Me | CF3 | Bz | 12.16 | 59.44 |
| 4a | Et | H | H | 70.56 | >500 | 4b | Me | Me | H | >100 | ND |
| 4c | Me | Cl | H | 36.95 | 167.10 | 4d | Et | Cl | H | 42.07 | >500 |
| 4e | Me | OH | H | >100 | ND | 4f | Me | F | H | 37.18 | 54.98 |
| 4g | Me | CF3 | H | 24.61 | 90.04 | 4h | Me | H | H | 77.37 | 150.70 |
| 4i | Et | H | Bz | >100 | >500 | 4j | Me | Me | Bz | >100 | >500 |
| 4k | Me | Cl | Bz | 73.40 | >500 | 4l | Et | Cl | Bz | >100 | ND |
| 4m | Me | OH | Bz | 77.50 | 104.90 | 4n | Me | F | Bz | 135.30 | 141.50 |
| 4o | Me | H | Bz | >100 | 211.90 | 4p | Me | CF3 | Bz | >100 | 101.90 |
| Imatinib | 0.398 | ND | |||||||||
As shown in Table 1, some compounds showed good inhibitory activities against Bcr-Abl and Bcr-AblT315I. In particular, eight compounds (3a, 3e, 3m, 3n, 3p, 4c, 4f, 4g) showed better activities. The most potent compound, 3m, strongly inhibited Bcr-Abl and Bcr-AblT315I with IC50 values of 1.273 μM and 39.89 μM. Meanwhile, the control drug imatinib displayed excellent Abl potency with IC50 values of 0.398 μM. However, it did not shown an inhibitory effect on the T315I mutant. Compared with imatinib, 3m showed more potent Bcr-AblT315I inhibitory activity.
Eight compounds (2a–2h) without amino acids had no inhibitory effect on Bcr-Abl and Bcr-AblT315I, while compounds with amino acids were significantly increased. The reason may be that the introduction of amino acids can increase the targeting of drugs,29,30 raise solubility.31,32 And the flip-side of using amino acids is the potential accumulation of the drug into the lysosomes as well as exposing the scaffold to potential attack from proteases.33,34 In general, compounds connected with L-phenylalanine were more active than those linked to glycine, which might be that the stronger hydrophobicity of benzyl group than hydrogen atom, resulting in the increased activity of wild-type and T315I mutation.
The enzymatic kinase assays and docking simulation studies have showed that some compounds showed good broad-spectrum Bcr-Abl inhibition of wild-type and T315I mutant. In particular, 3m could prove to be a new promising lead compound for the further development of broad-spectrum Bcr-Abl inhibitors to overcome clinical acquired resistance.
3 Conclusion
In summary, a series of novel thiazolamide–benzamide derivatives were designed, synthesized and evaluated for their Bcr-Abl inhibitory activity. The in vitro biochemical assays results showed that some compounds showed good broad-spectrum Bcr-Abl inhibition of wild-type and T315I mutant. In particular, 3m could prove to be a new promising lead compound for the further development of broad-spectrum Bcr-Abl inhibitors to overcome clinical acquired resistance.4 Experimental
4.1 Chemistry
Chemicals and solvents were purchased from commercial suppliers and used without further purification unless otherwise indicated. The silica powder was 100–200 mesh used to purify the compound by column chromatography. Thin layer chromatography (TLC) was performed to monitor the completion of the reaction and optimize the purity of the reaction, and was observed under ultraviolet light at 254 nm. Melting points were determined on RY-1G melting point apparatus (Tianjin Xintianguang Instrument Company, China) and were uncorrected 1H NMR spectra were recorded (in DMSO-d6 or CDCl3) on a Bruker spectrometer (400 MHz) using TMS as an internal control. 13C NMR spectra were recorded (in DMSO-d6 or CDCl3) on a Bruker spectrometer (101 MHz). IR was acquired on SHIMADZU. Mass spectra were acquired on APCI-MS spectrometer and elemental analysis was achieved on Perkin Elmer 2400 CHN.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :petroleum ether = 4:1). The liquid was cooled, filtered and dried to a pale yellow powder 3-(3-benzoyl thiourea) benzoic acid 28.0041 g, yield 93.33%; mp 186–187 °C.
:petroleum ether = 4:1). The liquid was cooled, filtered and dried to a pale yellow powder 3-(3-benzoyl thiourea) benzoic acid 28.0041 g, yield 93.33%; mp 186–187 °C.
3-(3-Benzoyl thiourea)benzoic acid (0.9913 g, 0.12 mol) and 10% NaOH (33 mL) was added to 100 mL round bottom flask with condensation tube, the mixture was stirred under magnetic pressure, heated to reflux, and the reaction was monitored by TLC (ethyl acetate:petroleum ether = 4:1). The mixture is cooled to room temperature, then adjust pH to 2 with HCl (4 mol L−1). After standing for 24 h, the solid was precipitated, filtered, and dried under vacuum to obtain a 0.6142 g white solid, yield 82%; mp: 186–187 °C. 1H NMR (DMSO-d6, 400 MHz), δ: 2.51 (s, 1H, NH), 3.36 (s, 2H, NH2), 7.43–8.03 (m, 4H, C6H4), 9.89 (s, 1H, COOH).
:petroleum ether = 4:1) detected the reaction process and took about 24 hours. Filter while hot, discarded the filter residue, and concentrate some of the solvents by rotary evaporation. After standing for 24 h, the solid was precipitated, filtered, and dried under vacuum to obtain target compounds.
:petroleum ether = 0.8:1). Ice water was slowly added while stirring, until the solution turned from clarification to turbidity, stirred at room temperature for 30 min, and then put in the refrigerator to separate out the white solid. The crude product was purified by column chromatography on silica gel, eluting with dichloromethane:methanol = 150:1 to obtain target compounds.
:petroleum ether = 1:1). After the reaction is completed, the mixture is filtered, and the residue is discarded. The filtrate was adjusted to pH of 2–3 with dilute HCl, and then put in the refrigerator to separate out the white solid, the solid was precipitated, filtered, and dried under vacuum to obtain target compounds.
4.2 Bcr-Abl and Bcr-AblT315I inhibitory activity assays
The Bcr–Abl inhibitory activity assay was performed using ADP-Glo™ Kinase assay kit (Promega, catalog: V9101), Abl Kinase Enzyme System (Promega, catalog: V1901) and AblT315I Kinase Enzyme System (Promega, catalog: V5320) according to the manufacturer's instructions. The Abl and AblT315I reaction utilized ATP and generate ADP. Then the ADP-Glo™ reagent was added to simultaneously terminate the kinase reaction and deplete the remaining ATP. Finally, the Kinase Detection Reagent was added to convert ADP to ATP and the newly synthesized ATP was converted to light using the luciferase reaction.35,36Abl and AblT315I were incubated with substrates, inhibitors and ATP in a final buffer of 25 mM HEPES (pH 7.4), 10 mM MgCl2, 0.01% Triton X-100, 100 μg mL−1 BSA, 2.5 mM DTT in 384-well plate with the total volume of 10 μL. Then the ADP-Glo™ Kinase Assay was performed in two steps once the kinase reaction was complete. Subsequently, 5 μL ADP-Glo Reagent was added to stop the kinase reaction and deplete the unconsumed ATP. Only ADP and a very low background of ATP were left. Then the mixture was incubated at room temperature for 40 min and added 10 uL of kinase detection reagent to convert ADP to ATP and introduced luciferase and luciferin to detect ATP. At last, the mixture was incubated at room temperature for 30–60 min and measured the luminescence with BioTek Synergy H at 490 nM. The signal was correlated with the amount of ATP present in the reaction and was inversely correlated with the kinase activity. The IC50 value was calculated by Graphpad based on the inhibition rate. All date were expressed as the average of three independent parallel experiments.
4.3 Molecular docking
In order to demonstrate our design hypothesis, we used SYBYL-X2.0 to perform molecular docking simulation of the design compounds with Abl and AblT315I kinases. Wild-type Abl kinase (PDB code: 2GQG) and its T315I mutant (PDB code: 3IK3) were selected as the receptor for docking study. The molecules (3m) were drawn with Chemdraw3D and minimized under Tripos Forcefield with Gasteiger–Huckel charge. The protein structures of wild-type and T315I type were introduced, and the corresponding ligand was used to define the binding pocket and generate the promotional. The inhibitors were docked into the active site using a ligand-based mode. Unwanted water and ligands were removed, and the lost atoms such as hydrogen were added to construct an initial receptor structure for docking. The assigned atom type of the ligand was AMBER7-FF99 and the residues in a radius 5.0 Å around ligands were selected as the active site.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Project is sponsored by Youth Innovative Talents Training Program Cooperative Innovation Center for Molecular Target New Drug Study (No. 0223-0002-0002000-47). The work was also supported by Hengyang Science and Technology Bureau Project (2015KJ18), University of South China Doctoral Start-up Fund (2013XQD20) and Graduate Student Science Foundation of University of South China (2018492), China.References
- A. Sinclair, A. L. Latif and T. L. Holyoake, Br. J. Pharmacol., 2013, 169, 1693–1707 CrossRef CAS PubMed.
- J. Dong, W. Lu and X. Pan, et al. , Bioorg. Med. Chem., 2014, 22, 6876–6884 CrossRef CAS PubMed.
- C. S. Munikrishnappa, S. B. Puranik, G. V. Kumar and Y. R. Prasad, Eur. J. Med. Chem., 2016, 119, 70–82 CrossRef CAS.
- A. K. El-Damasy, N. C. Cho, S. B. Kang, A. N. Pae and G. Keum, Bioorg. Med. Chem. Lett., 2015, 25, 2162–2168 CrossRef CAS PubMed.
- Q. C. Alfonso and C. Jorge, Blood, 2009, 113, 1619–1630 CrossRef.
- J. E. Sokal, M. Baccarani, D. Russo and S. Tura, Semin. Hematol., 1988, 25, 49–61 CAS.
- O. H. Thomas, A. E. Christopher and W. N. D. Michael, Blood, 2007, 110, 2242–2249 CrossRef.
- Q. C. Alfonso, H. Xin, K. Hagop and C. Jorge, Blood, 2009, 114, 261–263 CrossRef PubMed.
- W. Ellen, P. W. Manley, S. W. Cowan-Jacob, H. Andreas and J. D. Griffin, Nat. Rev. Cancer, 2007, 7, 345–356 CrossRef PubMed.
- A. Hochhaus, S. G. O'Brien and F. Guilhot, et al. , Leukemia, 2009, 23, 1054–1061 CrossRef CAS.
- F. Zhao, A. Mancuso and T. V. Bui, et al. , Oncogene, 2010, 29, 2962–2972 CrossRef CAS PubMed.
- Q. C. Alfonso, K. Hagop and C. Jorge, Nat. Rev. Drug Discovery, 2007, 6, 834–848 CrossRef.
- M. Copland, A. Hamilton, L. J. Elrick, J. W. Baird, E. K. Allan, N. Jordanides, M. Barow, J. C. Mountford and T. L. Holyoake, Blood, 2006, 107, 4532–4539 CrossRef CAS PubMed.
- F. Belloc, K. M. Airiau and M. Garcia, et al. , Leukemia, 2009, 23, 679–685 CrossRef CAS.
- T. O'hare, W. C. Shakespeare, X. Zhu, C. A. Eide, V. M. Rivera and F. Wang, et al. , Cancer Cell, 2009, 16, 401–412 CrossRef PubMed.
- A. D. Goodrich, Expert Rev. Hematol., 2014, 7, 513–515 CrossRef CAS PubMed.
- M. Senior, Nat. Biotechnol., 2014, 32, 9–11 CrossRef CAS.
- F. E. Nicolini, Blood, 2011, 118, 1208–1215 CrossRef.
- T. O'Hare, C. A. Eide and M. W. Deininger, et al. , Expert Opin. Invest. Drugs, 2008, 17, 865–878 CrossRef.
- T. O'Hare, D. K. Walters and E. P. Stoffregen, Clin. Cancer Res., 2005, 11, 6987–6993 CrossRef.
- B. Nagar, W. G. Bornmann and P. Pellicena, et al. , Cancer Res., 2002, 62, 4236–4243 CAS.
- N. P. Shah, J. M. Nicoll and B. Nagar, Cancer Cell, 2002, 2, 117–125 CrossRef CAS.
- A. Hochhaus and R. P. La, et al. , Leukemia, 2004, 18, 1321–1331 CrossRef CAS.
- X. Y. Lu, Q. Cai and K. Ding, et al. , Curr. Med. Chem., 2011, 18, 2146–2157 CrossRef CAS.
- T. I. D. Santana, M. D. O. Barbosa, A. C. N. D. Cruz, T. G. D. Silva and A. C. L. Leite, Eur. J. Med. Chem., 2017, 144, 874–886 CrossRef.
- H. He, X. Wang and L. Shi, et al. , Bioorg. Med. Chem. Lett., 2016, 26, 3263–3270 CrossRef CAS.
- J. S. Tokarski, J. A. Newitt and C. Y. J. Chang, et al. , Cancer Res., 2006, 66, 5790–5798 CrossRef CAS.
- F. Manetti, G. A. Locatelli and G. Maga, et al. , J. Med. Chem., 2006, 49, 3278–3286 CrossRef CAS.
- R. S. Koff, Aliment. Pharmacol. Ther., 2014, 39, 478–487 CrossRef CAS.
- R. R. J. Arroo, V. Androutsopoulos, A. Patel, S. Surichan, N. Wilsher and G. A. Potter, Phytochem. Rev., 2008, 7, 431–443 CrossRef CAS.
- K. Beaumont, R. Webster, I. Gardner and K. Dack, Curr. Drug Metab., 2003, 4, 461–485 CrossRef CAS.
- A. Dahan, E. M. Zimmermann and S. Ben-Shabat, Molecules, 2014, 19, 16489–16505 CrossRef.
- J. P. Reeves, J. Biol. Chem., 1979, 254, 8914–8921 CAS.
- M. Rabinovitch, Braz. J. Med. Biol. Res., 1987, 20, 665–674 CAS.
- C. Wang, H. Gao, J. Dong, Y. Zhang, P. Su and Y. Shi, et al. , Bioorg. Med. Chem., 2014, 22, 277–284 CrossRef CAS.
- C. Zhang, C. Tan, X. Zu, X. Zhai and F. Liu, et al. , Eur. J. Med. Chem., 2011, 46, 1404–1414 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra10096a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |