
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnzymatic synthesis of enantiopure alcohols: current state and perspectives
Bi-Shuang Chen
 ab and
Fayene Zeferino Ribeiro de Souza
*c
ab and
Fayene Zeferino Ribeiro de Souza
*c
aSchool of Marine Sciences, Sun Yat-Sen University, Guangzhou 510275, China
bSouth China Sea Bio-Resource Exploitation and Utilization Collaborative Innovation Center, Sun Yat-Sen University, Guangzhou, 510275, China
cDepartamento de Química, Faculdade de Ciências, Universidade Estadual Paulista, UNESP, Bauru 17033-360, Brazil. E-mail: faylittlefay@yahoo.com.br/; Tel: +55-014-32452580
First published on 15th January 2019
Abstract
Enantiomerically pure alcohols, as key intermediates, play an essential role in the pharmaceutical, agrochemical and chemical industries. Among the methods used for their production, biotechnological approaches are generally considered a green and effective alternative due to their mild reaction conditions and remarkable enantioselectivity. An increasing number of enzymatic strategies for the synthesis of these compounds has been developed over the years, among which seven primary methodologies can be distinguished as follows: (1) enantioselective water addition to alkenes, (2) enantioselective aldol addition, (3) enantioselective coupling of ketones with hydrogen cyanide, (4) asymmetric reduction of carbonyl compounds, (5) (dynamic) kinetic resolution of racemates, (6) enantioselective hydrolysis of epoxides, and (7) stereoselective hydroxylation of unactivated C–H bonds. Some recent reviews have examined these approaches separately; however, to date, no review has included all the above mentioned strategies. The aim of this mini-review is to provide an overview of all seven enzymatic strategies and draw conclusions on the effect of each approach.
 Bi-Shuang Chen | Bi-Shuang Chen received her B.Sc. degree in Chemistry from Sun Yat-Sen University (China) in 2008. After she received her M.Sc. degree in Chemistry from Xiamen University (China), she started her Ph.D. studies at the Delft University of Technology (The Netherlands) under the guidance of Professor Ulf Hanefeld studying the biocatalysis and protein engineering related sciences and received her Ph. D. in 2015. She then returned to Sun Yat-Sen University and started as a post-doctoral researcher with Professor Lan Liu. Since 2018, she was pointed as an associated professor of Sun Yat-Sen University where her research focuses on marine enzymes. |
 Fayene Zeferino Ribeiro de Souza | Fayene Z. R. de Souza obtained her M.Sc degree in Chemistry from São Paulo University (Brazil) in 2008. In 2013, she received her PhD in Chemistry from São Paulo University, having performed her research both in São Paulo University and Delft University of Technology, (the Netherlands). Her research was focused on Xylella fastidiosa bacterium, including a biocatalysis project. After her first post-doctoral position at São Paulo University (2014-206) she joined Universidade Estadual Paulista (UNESP, Brazil) as a post-doctoral researcher and colaborator professor, where currently her research focuses on biotechnology area. |
Introduction
Currently, enantiomerically pure alcohols are important building blocks that are widely used as intermediates in the chemical and pharmaceutical industries for synthesis of fine products with chiral centers such as, flavourings, agrochemicals, drugs, liquid crystals and specialty materials.1,2 Thus, their synthesis is continuously of great interest to researchers. Considering that the traditional synthetic methods predominantly use toxic metals or expensive complex ligands, enzyme-catalysed syntheses offer significant advantages to these methods, such as remarkable chemo-, regio-, and stereoselectivity, environmentally benign processes and energy-efficient operations.3,4 In the traditional chemical methods enantiomerically pure alcohols also lack any handles for stereo- and regio-control during the synthesis and side reactions such as the intermolecular aldol reactions, and follow-up reactions (e.g. dehydration) can easily occur due to the extremely chemical conditions.5 In addition to these features, the natural sources of the biocatalysts, whether from microorganisms, plants, animals, their isolated enzymes or whole cell system, are reproducible and can be easily decomposed in the environment after use, thus making enzymatic methodologies a viable alternative to conventional chemical processing.6 The biocatalytic production of enantiomerically pure alcohols is possible via several pathways. Seven main strategies for the enzyme-catalysed synthesis of enantiomerically pure alcohols can be distinguished as follows:(1) Enantioselective addition of water to α,β-unsaturated ketones, which is a highly attractive reaction because it allows direct access to hydroxyl ketones;7,8 (2) enantioselective aldol addition, which produces hydroxyl ketones via biocatalytic carbon–carbon formation in an asymmetric manner;9–11 (3) enantioselective coupling of ketones or aldehyde with hydrogen cyanide, which leads to nitrile-substituted chiral alcohols (cyanohydrins)12–14 4) asymmetric reduction of carbonyl compounds, which has been widely studied and applied in the asymmetric synthesis of chiral alcohols;15,16 (5) (dynamic) kinetic resolution of racemic mixtures, which play an essential role in the preparation of enantiomerically pure alcohols;17,18 (6) enantioselective hydrolysis of epoxides, which affords the corresponding vicinal diols or analogues;19 and (7) regio- and stereoselective hydroxylation to functionalize C–H bonds, which yields the hydroxylated products.20,21
Recent reviews have been considered these enzymatic approaches separately, e.g., enantioselective addition of water to alkenes,22 asymmetric synthesis of hydroxyl ketones catalysed by aldol addition,23 enzyme-catalysed enantioselective synthesis of cyanohydrins,24 chiral alcohols by biocatalytic reduction of ketones,25 enzymatic chiral resolution using lipase (EC 3.1.1.3),26 and enantioconvergent bioconversions to prepare active alcohol with epoxide hydrolase (EC 3.3.2.-).27 Additionally, although Müller28 gave an overview of the synthesis of tertiary alcohols, no review has included all the enzymatic strategies for enantiopure alcohol synthesis. Thus, it is interesting and essential to review all these strategies to have a comprehensive view of the power that they have in potential applications.
Here, we present the application of all seven enzyme-catalysed strategies for the synthesis of enantiopure alcohols that is accessible for organic chemists. These enzymes are readily available and can be used with standard equipment under standard conditions. The discussion is focused on the product class, and we have restricted ourselves to secondary alcohols, tertiary alcohols, vicinal diols, β-hydroxy carbonyl compounds and cyanohydrins. The emphasis here is on enantioselective enzymatic synthesis, which is likely to extend the use of enantiopure alcohols as building blocks in organic synthesis, and not on the enzymes themselves.
Production of secondary alcohols
Regardless of whether they are primary (1°), secondary (2°), or tertiary (3°), chiral alcohols are predominantly used as optically active intermediates in an area of growing demand in everyday life. At first glance, they seem quite straightforward molecules to chemists. However, it is often very difficult to synthesize enantiopure alcohols. In particular, significant differences exist in the challenges encountered when selectively synthesizing 2° and 3° alcohols compared with 1° alcohols.The huge consumption of enantiopure drugs makes the pharmaceutical industry to have more attention on the production of the chiral compounds and chiral secondary alcohols are the most frequently required as chiral compounds to produce different drugs.29 For instance, fluoxetine, which is used for the treatment of depression among others mental disorders, and (R)-benzyl-4-hydroxyl-2-pentynoate, which is a potential therapeutic for Alzheimer's disease, are synthesized from the chiral intermediate secondary alcohol (R)-4-(trimethylsilyl)-3-butyn-2-ol, respectively.30,31 (R)-2-Octanol was proven to be a precursor compound for the several optically active pharmaceuticals products.32 Isoproterenol, is a pharmaceutical drug which treats bradycardia, and neobenodine, which has antihistaminic and anticholinergic action, and both are obtained from chiral secondary alcohols.33
Chiral secondary alcohols can be efficiently synthesized as follows: (i) via racemic kinetic resolution, often using lipase (EC 3.1.1.3);34,35 (ii) through the asymmetric reduction of ketones, usually using carbonyl reductases (EC 1.1.1.-);36,37 (iii) through the regioselective reductive opening of epoxides, for example, using halohydrin dehalogenase (EC 4.5.1.-);38,39 or (iv) through the regio- and stereoselective oxidation of unactivated C–H bonds, for example, using cytochrome P450 (EC 1.14.13.70).20
Secondary alcohols are the most frequently used molecules in lipase-catalysed kinetic resolutions not only because of their importance in organic synthesis but also because lipases usually show much higher enantioselectivity in resolutions towards secondary alcohols than in those towards primary or tertiary alcohols.31 In enzymatic kinetic resolutions of secondary alcohols, the outcome is normally highly predictable, and it follows Kazlauskas' rule.40 Usually, the (R)-alcohol is converted into (R)-acetate at a higher rate than its antipode with high ee, leaving the (S)-alcohol as an enantiomerically pure unreacted enantiomer. The reverse reaction consists of the lipase-catalysed hydrolysis of the (R)-acetate, affording the (R)-alcohol in high ee and enabling the separation of the two enantiomers. Numerous examples can be found in the available scientific literature (Fig. 1), it should be mentioned that trans-4-phenyl-3-buten-2-ol (1q), a substrate that possesses an allylic strain, has been successfully utilized on the industrial scale via lipase-catalysed resolution to obtain the single enantiomer.41 Notably, Baumann et al.42 reported four substrates, pantolactone (1n), 3-butyn-2-ol (1m), 1-methoxy-2-propanol (1o), and 3-hydroxy-tetrahydrofuran (1p), entitled ‘difficult to resolve’ for the enantioselective resolution using different lipases. The stereoselective ring opening of epoxides catalysed by halohydrin dehalogenase (HHDH) (EC 4.5.1.-) is an attractive way used in the asymmetric synthesis of enantiopure β-substituted alcohols.43–45 A series of chiral secondary alcohols were obtained using different nucleophiles (Br−, Cl−, I−, CN−, NO2−, N3−, OCN−, SCN−, and HCOO−) such as β-azido alcohols, β-nitro alcohols, and β-hydroxyl nitriles (Fig. 2).36 Among the existing nucleophiles, cyanide is the most interesting one because its nitrile group can be converted into amino-, amide-, or carboxyl- groups by specific enzymes.41,46 Detailed studies have reported the use of a recombinant HHDH cloned from the Corynebacterium sp. strain N-1074 for the ring opening of epichlorohydrin by cyanide.47 Elenkov et al.43 further discussed the cyanide-mediated ring-opening reactions of epoxides catalysing by three groups of HHDH.
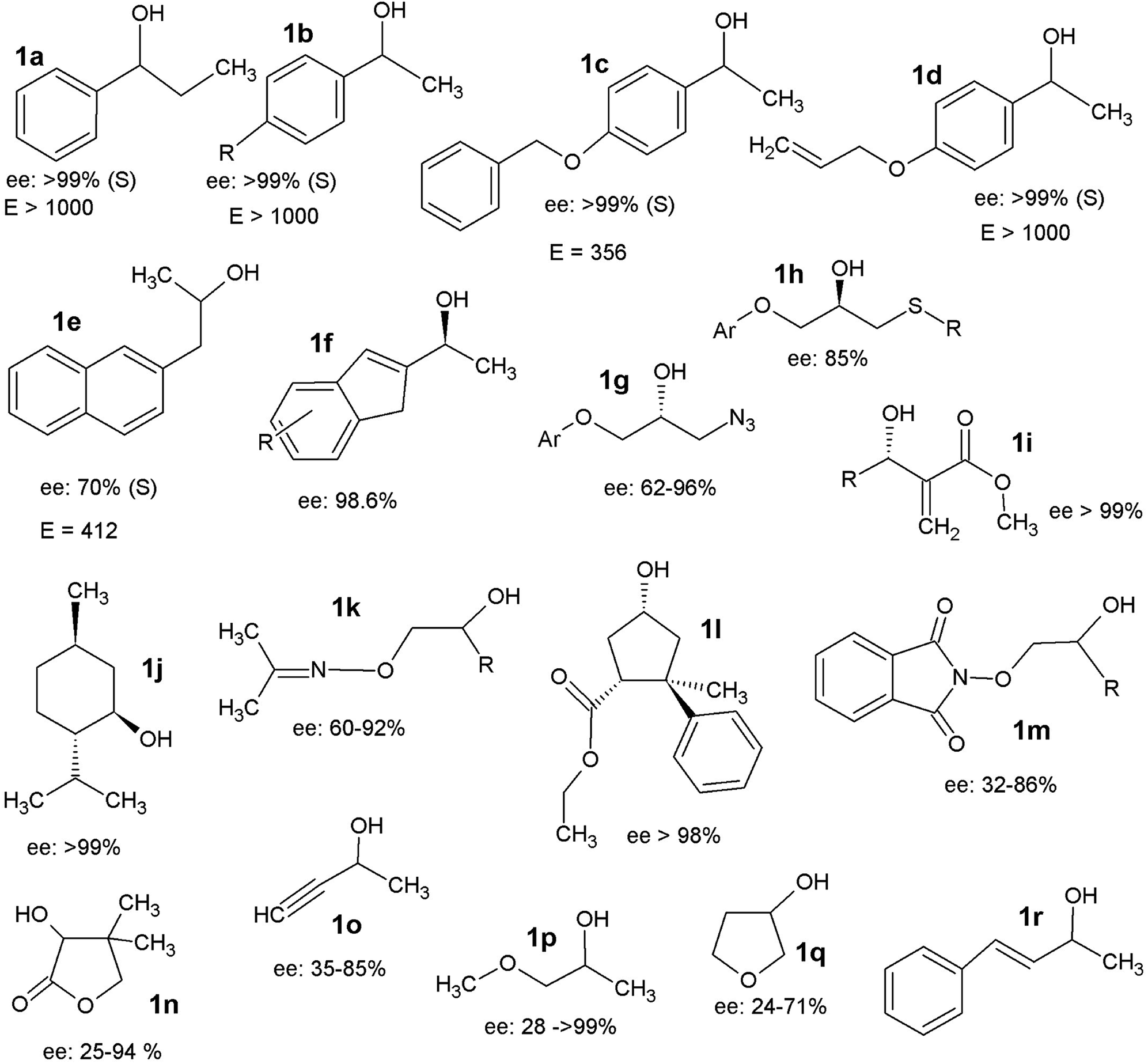 | ||
| Fig. 1 Selected examples showing the kinetic resolution of secondary alcohols.35–38 | ||
 | ||
| Fig. 2 Synthesis of enantiopure β-substituted secondary alcohols by halohydrin dehalogenases.33,37,39 | ||
The enantioselective bioreduction of ketones is a reliable, scalable and straightforward route to produce enantiopure secondary alcohols.35 Ketoreductases (EC 1.1.1.-) are found in several different organisms and catalyse the reduction of ketones to chiral secondary alcohols, with the stoichiometric consumption of the nicotinamide adenine dinucleotide cofactor (NAD+) or its phosphorylated analogue NADP+. The scope of ketoreductases (EC 1.1.1.-) is very vast, and they accept several substrates.48 For example, marine-derived fungi have been successfully used for the asymmetric reduction of a series of aromatic ketones in our laboratory.2 Novel industrially relevant ketoreductases continue to become available, and novel process engineering concepts have been developed. For example, an attractive approach has been developed using an engineered ketoreductase, which allows the bioreduction of the ketoreductases in the synthesis of atorvastatin and atazanavir, two drugs widely used,48 to occur with. Moreover, for NAD(P)H-dependent enzymes, cofactor regeneration can be efficiently achieved by the addition of isopropanol to the buffer, which is then transformed into acetone49–51 Several examples can be found in the scientific literature which highlights that cofactors are no longer cost determinants.48
Another promising route to produce chiral secondary alcohols is regio- and stereoselective monohydroxylation at or near the middle of the carbon chain. The most prominent enzymes for these monohydroxylations are cytochrome P450 monooxygenases (P450, EC 1.14.13.70).48 Notably, many hydroxylations were performed in whole-cell biotransformation without identifying the enzymes involved.28 For example, the Sphingomonas sp. HXN-200 strain is known to catalyse the hydroxylation of pyrrolidines 3a–3c to prepare enantiomerically pure 3-hydroxypyrrolidines 3a′–3c′, which are useful intermediates in the preparation of several pharmaceuticals (Fig. 3).52 The ‘docking/protecting’ (d/p) group concept has been employed to modify a substrate and thereby improve a given biohydroxylation.53 This concept was applied for the first time to increase the stereoselectivity, as exemplified for the hydroxylation of substrate 3d with Beauveria bassiana ATCC 7159 (ref. 54) (Fig. 3). The hydroxylation of confertifolin (3e) to 3-hydroxyconfertifolin (3e′) with Aspergillus niger ATCC 9142 (ref. 55) represented the best example of hydroxylation of natural products (Fig. 3). Hydroxylation of benzylic and allylic carbon atoms have also been reported. For instance, the hydroxylation of propylbenzene (3f and 3g) to (R)-3f′ (and 3g′) was obtained with excellent regioselectivity and good enantioselectivity by Bacillus megaterium56 (Fig. 3). The bacterial strain Rhodococcus opacus PWD4 could catalyse the hydroxylation of D-limonene (3h) exclusively in the 6-position, producing enantiomerically pure (+)-trans-carveol (3h′)57 (Fig. 3).
 | ||
| Fig. 3 Selected examples showing region- and stereoselective hydroxylation to produce chiral secondary alcohols.44,46–49 | ||
Tertiary alcohols
Tertiary alcohols are a desire class of compounds and they are found in many drugs and natural products. For instance, tramadol (analgesic), erythromycin (antibiotic), doxorubicin (antitumour agent), and ethinylestradiol (oestrogen), as well as the prostaglandin analogue, frontalin and the vitamin D3 metabolite,58 are derived from tertiary alcohols. In addition, chiral tertiary alcohols, taking advantage of their steric requirements, are used as auxiliaries in noncatalytic asymmetric syntheses and as ligands in nonenzymatic asymmetric catalysis. Overall, the demand for chiral tertiary alcohols is continuously growing within the fine chemical industry. However, compared with the great progress developed to produce chiral secondary alcohols, the challenges for the preparation of chiral tertiary alcohols are immense.28 Thus, more focus has been placed on developing the process for the asymmetric or enantioselective synthesis of tertiary alcohols.Due to the difficulty associated with the accommodation of tertiary alcohols to the active site of lipases, the kinetic resolution of such substrates is not as well covered in the literature as secondary alcohols are.59,60 Krishna and coworkers used lipase A from Candida antarctica (CAL-A) (CAL-A-C1) (EC 3.1.1.3) as catalysts and vinyl acetate as an acyl donor in organic solvent for the racemic resolution of tertiary alcohol 4a (Fig. 4).61 Ohta and coworkers reported the enantioselective transesterification of tertiary alcohol 4b (Fig. 4) to obtain (R)-(−)-mevalonolactone.62 A selected series of other tertiary alcohols that have been excellently resolved is shown in Fig. 4. Similarly, different enzymatic processes have been developed for the hydrolysis of sterically demanding esters to produce tertiary alcohol, including those utilizing esterases (EC3.1.1.1), lipases (EC 3.1.1.3), and proteases EC 3.4.21.53).63,64 Notably, the best represented examples of the enzymatic hydrolysis of tert-butyl esters,60 partially on an industrial scale, provide further illustration. Bornscheuer, Pleiss, and coworkers described the structure–function relationship for some hydrolases accepting sterically demanding substrates. The sequence motifs commonly found in hydrolases that accept tertiary alcohols as substrates were identified, which were not restrictive.65,66 Bornscheuer and coworkers compared the new enzymatic processes using bioinformatics, structural, and protein biochemical methods.59,67 These articles all attempt to provide new possibilities for the synthesis of chiral tertiary alcohols, which are only discussed in brief here.
 | ||
| Fig. 4 Selected examples of lipase-catalyzed kinetic resolution of tertiary alcohols.52–58 | ||
An attractive route to produce chiral tertiary alcohols is the 2,2-disubstituted epoxide opening with other nucleophiles rather than water, giving the analogous α-functionalized tertiary alcohols. For example, the halohydrin dehalogenases (EC 4.5.1.-) from Agrobacterium radiobacter AD1 (HheC) catalysing nucleophilic ring opening of 2,2-disubstituted epoxide with cyanide and azide for the preparation of enantiopure tertiary alcohols was studied by Elenkov et al.68 Molinaro et al.69 used commercially available Codex halohydrin dehalogenase (HHDH) (EC 4.5.1.-) enzyme purchased from CODEXIS, for the ring opening of 2,2-disubstituted epoxide with azide nucleophiles to afford chiral tertiary alcohols. Due to the limitation of the substrate range (the epoxide and nucleophile), only a few examples of their application exist for the synthesis of tertiary alcohols, but in relation to exploiting perfect atom economy, we can say that this reaction is a suitable method to produce chiral tertiary alcohols.28
Cytochrome P450 (CYP) enzymes catalyse the hydroxylation at position 25 of vitamin D3 (5a), resulted in tertiary alcohol functionality in calcifediol (5a′) (Fig. 5). It has been reported that CYP2R1(EC1.14.14.24) is mainly responsible for this transformation, although there are several human enzymes and bacterial enzymes (such as CYP105A1 from Streptomyces griseolus) associated with this hydroxylation.70–72 Many more CYP enzymes that catalyse the hydroxylation to tertiary alcohols are known, such as the hydroxylation at C9 and C17 of the steroid scaffold.73,74 In addition to cytochrome P450 enzymes, other enzyme families, such as α-keto glutarate (α-KG)-dependent (EC 1.14.11.-) enzymes, are known to catalyse the hydroxylation to tertiary alcohols.75 Another example is that Fuchs and coworkers observed hydroxylation at C25 of the steroid scaffold cholest-4-en-3-one (5b) catalysed by Sterolibacterium denitrificans.76 It is worth noting that many hydroxylations were performed in whole-cell biotransformations without identifying the enzymes involved. For instance, hydroxylation at C14 of 5c in whole-cell biotransformation with Pseudomonas putida was observed by Lister et al.77 for the formation of β-hydroxycodeinone (5c′).
 | ||
| Fig. 5 Preparation of chiral tertiary alcohols through hydroxylation catalyzed by different enzyme families.61–68 5a → 5a′: the hydroxylation at position 25 in 5a results in the 3o alcohol functionality in calcifediol 5a′; 5b → 5b′: hydroxylation at C25 of cholest-4-en-3-one 5b results in the 3o alcohol functionality in 5b′; 5c → 5c′: hydroxylation of 5c results in the formation of 14-hydroxycodeinone 5c′; 5d → 5d′: hydroxylation of 5d results in the formation of the 3o alcohol 5d′. | ||
Vicinal diols
This type of compound requires a special attention due to the two asymmetric centers in its molecular configuration which can be converted into several complex chiral systems.78 Enantiomerically pure diols are exceptionally valuable as a class of important and versatile building blocks or intermediates.79 Many vicinal diols are an essential part or intermediate of several highly consumed pharmaceutical and nutraceutical products.80 For example, (1S,2S)-3-chlorocyclohexa-3,5-diene-1,2-diol is useful to prepare ascorbate;81 (3R,4S,5R)-5-amino-3,4-dihydroxycyclohex-1-enecarboxylic acid is an essential component of the antiviral drug oseltamivir;82 a diol of (1S,2R)-2,3-dihydro-1H-indene-1,2-diol could serve as an intermediate for indinavir sulfate synthesis, which is a protease inhibitor in HIV treatment;83 another diol of (3S,4R)-3,4-dihydroxy-2,2-dimethyl-3,4-dihydro-2H-chromene-6-carbonitrile could be used to synthesize the potassium channel opening vasodilator cromakalim.79The preparation of chiral vicinal diols has been an extensively studied area, and numerous routes and attempts have been reported.79 Some advantages like atom economy, high selectivity and environmental friendliness can be cited when using two chiral centers via a single enzymatic step and diverse enzymes are able to introduce this simultaneous chiral centers. Hydrolases and oxidoreductase, more specific dicarbonyl reductases (EC 1.1.1.-), dioxygenase (EC 1.13.11.-) and epoxide hydrolase (EC 3.3.2.-), are the best examples of enzyme's classes which catalyze the formation of enantiomerically pure diols.79,84–86
Dicarbonyl reductases (EC 1.1.1.-), which are from the class of oxidoreductases, can stereoselectively reduce two carbonyl groups to their corresponding chiral diols. Currently, many dicarbonyl reductases have been discovered from natural sources, such as yeasts, bacteria, fungi, and even plant tissues, and subsequently investigated. Because there are three enzymes in this class and “diol” is a basic concept, we focus on only one particular group of diols, vicinal diol, in which the two hydroxy functional groups are attached to adjacent carbon atoms. For vicinal diol production, the best presented example would be the microbial production of 2,3-butanediol catalysed by 2,3-butanediol dehydrogenases (EC 1.1.1.76).78,80 In terms of the catalytic mechanisms, Wu87 and coworkers have described that dicarbonyl reduction is a consecutive process. During the reduction of ethyl 2-oxo-4-phenylbutanoate 6a, two stable monohydroxy intermediates (6b and 6c), as illustrated in Fig. 6, appeared at the early phase of the reaction and were then further reduced to yield vicinal diol 6d. The detection and isolation of 6b and 6c clearly indicated that the dicarbonyl reduction is a consecutive process. Even with all the different properties, the dicarbonyl reductases exhibit distinct stereoselectivity towards α,β-diketo substrates (Fig. 7), for example, mammalian dicarbonyl reductases catalyse 2-amino-6-(2-oxopropanoyl)-5,6,7,8-tetrahydropteridin-4(3H)-one (PPH4) to form (1R,2S)-vicinal diol (L-erythro-BH4),88 on the other hand, the products of tetrahydrobiopterin and their analogues using dicarbonyl reductases (EC 1.1.1.-) were obtained from different sources (Chlorobium tepidum and Dictyostelium discoideum Ax2).89,90 This example shows us that dicarbonyl reductases can cause notable variations in the product formation and stereoselectivity.
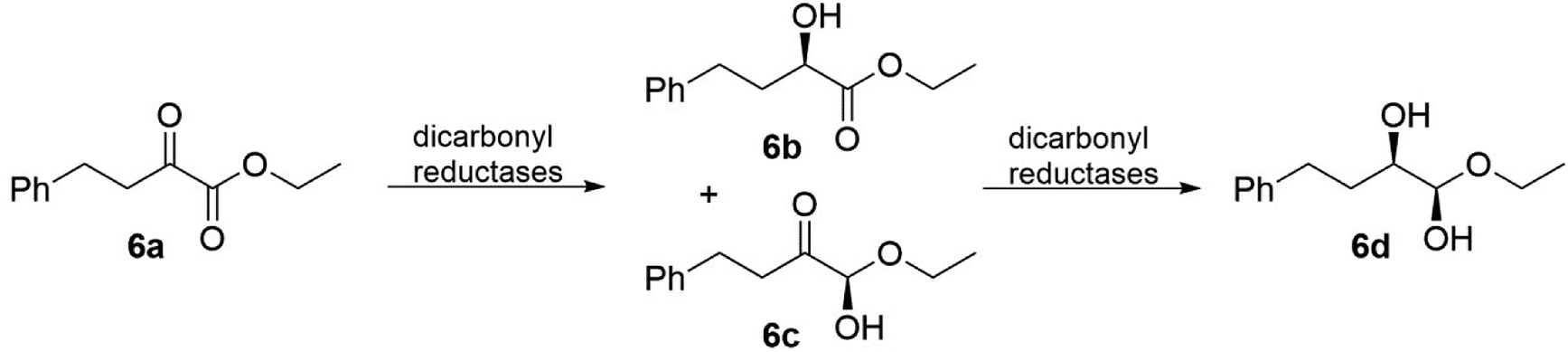 | ||
| Fig. 6 Reactions catalyzed by dicarbonyl reductases from Acinetobacter baylyi.78 | ||
 | ||
| Fig. 7 Bicarbonyl reductases from different sources exhibit different stereoselectivity towards α,β-diketo substrates.79–81 | ||
From the field of the nonenzymatic preparation of vicinal diols, the most extraordinary example is the Nobel-winning methodology, the Sharpless asymmetric dihydroxylation.91,92 The enzymatic variation of this reaction is catalysed by dioxygenases (EC 1.13.11.-). Dioxygenases are widely spread in bacteria to assist the addition of two molecular oxygens into a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, either conjugated with or within an aromatic system, yielding a vicinal cis-diol as the main product.93,94 Aromatic hydrocarbon dioxygenases belong to a multicomponent enzyme systems which consists of several proteins, that in the aromatic nucleus is added oxygen to produce arene vicinal cia-diols,95,96 for example, cis-1,2-dihydroxycyclohexa-3,5-diene-1-carboxylate (8a′) was obtained from benzoate 8a in a high optical purity on a large scale by whole-cell biotransformation (Alcaligenes eutrophus) with benzoate dioxygenase (EC 1.14.12.10) (Fig. 8).97 The underlying benzoate dioxygenase belongs to the known Rieske non-heme iron oxygenases (EC 1.13.11.-) family. These enzymes are referred to as benzene, toluene (EC 1.14.12.11), naphthalene (EC 1.14.12.12), or biphenyl dioxygenase (EC 1.14.12.18), depending on the substrates.96 Thus far, an extensive types of vicinal arene cis-diols have been detected as the major reaction products in the oxidation of aromatic hydrocarbons.98 Notably, Rieske dioxygenases also performed dihydroxylations of conjugated CC bonds,99 as shown in Fig. 8, for the oxidation of α-methylstyrene (8b) to (R)-2-phenylpropane-1,2-diol (8b′) by whole-cell biotransformation (Pseudomonas putida) with naphthalene dioxygenase (EC 1.14.12.12).100
C bond, either conjugated with or within an aromatic system, yielding a vicinal cis-diol as the main product.93,94 Aromatic hydrocarbon dioxygenases belong to a multicomponent enzyme systems which consists of several proteins, that in the aromatic nucleus is added oxygen to produce arene vicinal cia-diols,95,96 for example, cis-1,2-dihydroxycyclohexa-3,5-diene-1-carboxylate (8a′) was obtained from benzoate 8a in a high optical purity on a large scale by whole-cell biotransformation (Alcaligenes eutrophus) with benzoate dioxygenase (EC 1.14.12.10) (Fig. 8).97 The underlying benzoate dioxygenase belongs to the known Rieske non-heme iron oxygenases (EC 1.13.11.-) family. These enzymes are referred to as benzene, toluene (EC 1.14.12.11), naphthalene (EC 1.14.12.12), or biphenyl dioxygenase (EC 1.14.12.18), depending on the substrates.96 Thus far, an extensive types of vicinal arene cis-diols have been detected as the major reaction products in the oxidation of aromatic hydrocarbons.98 Notably, Rieske dioxygenases also performed dihydroxylations of conjugated CC bonds,99 as shown in Fig. 8, for the oxidation of α-methylstyrene (8b) to (R)-2-phenylpropane-1,2-diol (8b′) by whole-cell biotransformation (Pseudomonas putida) with naphthalene dioxygenase (EC 1.14.12.12).100
 | ||
| Fig. 8 Dihydroxylation of (a) benzoate 8a with Alcaligenes eutrophus and (b) α-methylstyrene 8b with Pseudomonas putida.87,90 | ||
Other group of enzymes that furnish vicinal diol are the epoxide hydrolases (EC 3.3.2.-), these enzymes catalyse the hydrolytic ring opening of an epoxide.79 These biocatalysts were isolated from different sources like animals, plants and microorganisms.19 Epoxide hydrolases are independent of cofactors or a prosthetic group or metal ions for their activity and could selectively hydrolyse epoxides of various structural types,101 such as monosubstituted epoxide (type I), styrene oxide-type epoxide (type II), 2,2-disubstituted epoxide (type III), 2,3-disubstituted and trisubstituted epoxides (type IV).19 As shown in Fig. 9, the consequence of this mechanism, where the epoxide opens in a trans-specific fashion, is that only one oxygen from water is transformed into the product. The absolute configuration of the formed vicinal diols may be retained or inverted because the mechanism takes place via nucleophilic attack on the oxirane carbon atom and the stereochemical pathway depends on the structure of the substituent in the carbon atom which is being attacked and the epoxide hydrolase (EC 3.3.2.-) is responsible for the regioselectivity. Consequently, the absolute configuration of both the desired vicinal diol product and the epoxide that was not hydrolysed must be determined independently.19 Several epoxide hydrolases have been applied to the preparative-scale production of chiral epoxides and vicinal diols, indicating that the catalytic power of epoxide hydrolases provides new possibilities for efficient, specific and sustainable technologies to be developed for the production of useful vicinal diols.
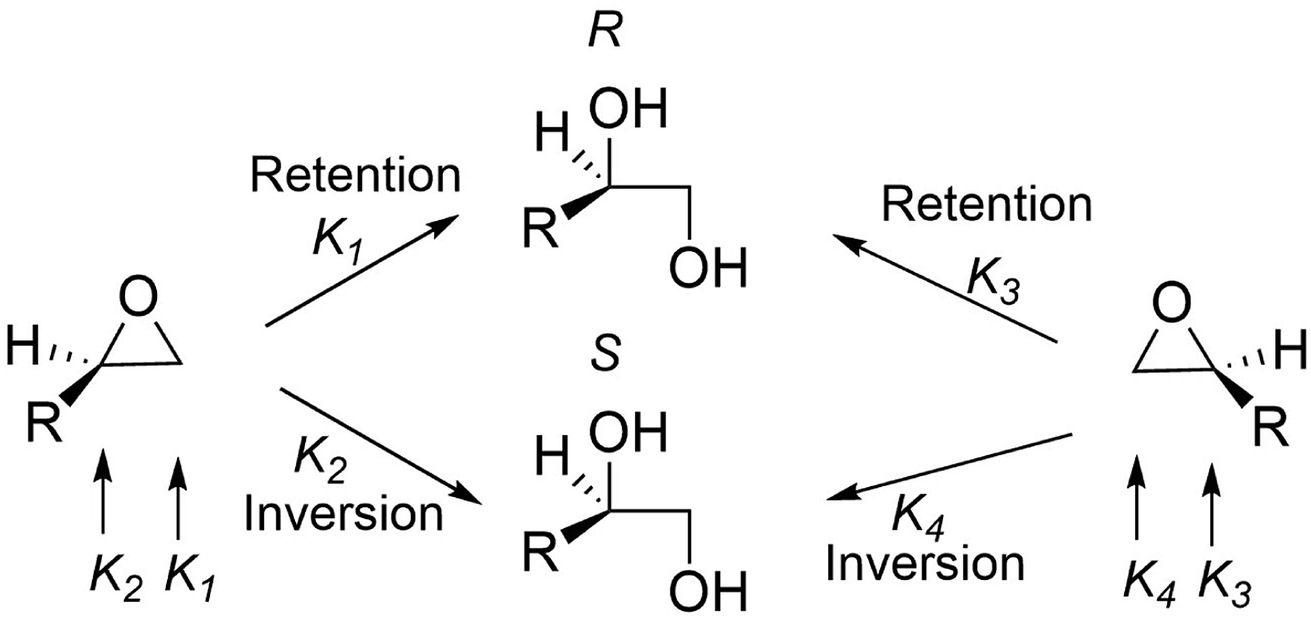 | ||
| Fig. 9 Stereochemical pathways of epoxide hydrolases.16 | ||
β-Hydroxy carbonyl compounds
The cyclic and acyclic β-hydroxy carbonyl compounds are important structures in various biologically active organic compounds because they provide two different functionalities in the same molecule ready for manipulation.102 One of these compounds that are used as potencial building blocks is the chiral β-hydroxy ketones which are capable to synthese amino alcohols, diols, lactones, and natural products.103 For example, (S)-β-hydroxy cyclohexanone is used as a precursor for the synthesis of 25-hydroxy-19-norvitamin D3 analogues.104 Notably, the 25-hydroxy-19-norvitamin D3 analogues are known for their antiproliferative activities towards prostate cancer cells.Although the molecules themselves look rather simple, their synthesis can be challenging. The direct Michael addition of water to the conjugated double bond is a straightforward approach to chiral β-hydroxy carbonyl compounds. The reaction is very interesting with the obvious advantages of cofactor independency, the use of water as a nucleophile and solvent allowing a green route and perfect atom economy, but it has rarely been investigated in organic synthesis. Although very few chemical methods can be used, hydratases can perform this reaction, and many examples have been described.21,105 Prominent and well-studied examples include fumarase (EC 4.2.1.2), malease (EC 4.2.1.31), citraconase (EC 4.2.1.35) and enoyl-CoA hydratase (EC 4.2.1.17), which have been successfully used on an industrial scale, and they show excellent enantioselectivities values. One drawback of the enzymes reported is that most hydratases are involved in metabolic pathways; therefore, they exhibit a very narrow substrate scope. Recently, some remarkable examples of enzymes that catalyse asymmetric hydration have been described and seem to be much broader regarding the substrates of different compound classes. We have reported the use of whole cells from Rhodococcus rhodochrous to catalyse the addition of water to various α,β-unsaturated carbonyl compounds (Fig. 10a). Important β-hydroxy carbonyl compounds were achieved in good yields and excellent enantioselectivities with this method.7,22 Although the enzymes involved in this transformation have not yet been identified, the results are still promising for future investigations on hydratases with a more flexible substrate scope. An artificial metalloenzyme with hydratase activity has been reported to catalyse the addition of water to α,β-unsaturated 2-acyl imidazoles (Fig. 10b) to form the corresponding β-hydroxy carbonyl compounds.106,107
 | ||
| Fig. 10 Preparation of β-hydroxy carbonyl compounds by water addition to (a) α,β-unsaturated carbonyl compounds and (b) α,β-unsaturated 2-acyl imidazoles.19,96 | ||
β-Hydroxy carbonyl compounds can be prepared using aldolases (EC 4.1.2.13), the reaction takes place by the aldol addition of the donor component (i.e., nucleophile, usually ketone) through, typically, an enolate or enamine that was generated at the enzyme's active site, onto an acceptor component (i.e., electrophile, usually aldehyde).108 For example, when α-keto esters (such as pyruvate) were used as acceptors, acetolactate (11a′) (in the case of pyruvate also as donors) or analogous acetohydroxy acids were obtained (Fig. 11a).109,110 Acetolactate is the simplest derivative of this substance class. An example of a more complex β-hydroxy carbonyl compound that can be synthesized with aldol addition was described by Balskus and Walsh111 in their biosynthetic investigations: the asymmetric decarboxylation of indole-3-pyruvate (11b) with p-hydroxyphenylpyruvate (11b′) resulted in the formation of 11b′′ (Fig. 11b). Several other aldolases accept different α-keto acids as donors or acceptors, thereby opening alternative routes to the substances of β-hydroxy carbonyl compounds. Notably, Lehwald et al.112 have described the biosynthesis of β-hydroxy carbonyl compounds via aldol addition, through the decarboxylation of pyruvate (11a), to cyclic and acyclic ketones as substrate, using YerE from Yersinia pseudotuberculosis was possible to produce chiral tertiary alcohol (Fig. 11c). The products (β-hydroxy carbonyl compounds) synthesized so far, many on a semipreparative scale, already represent an impressive variety, especially considering that a single wild-type enzyme was used.
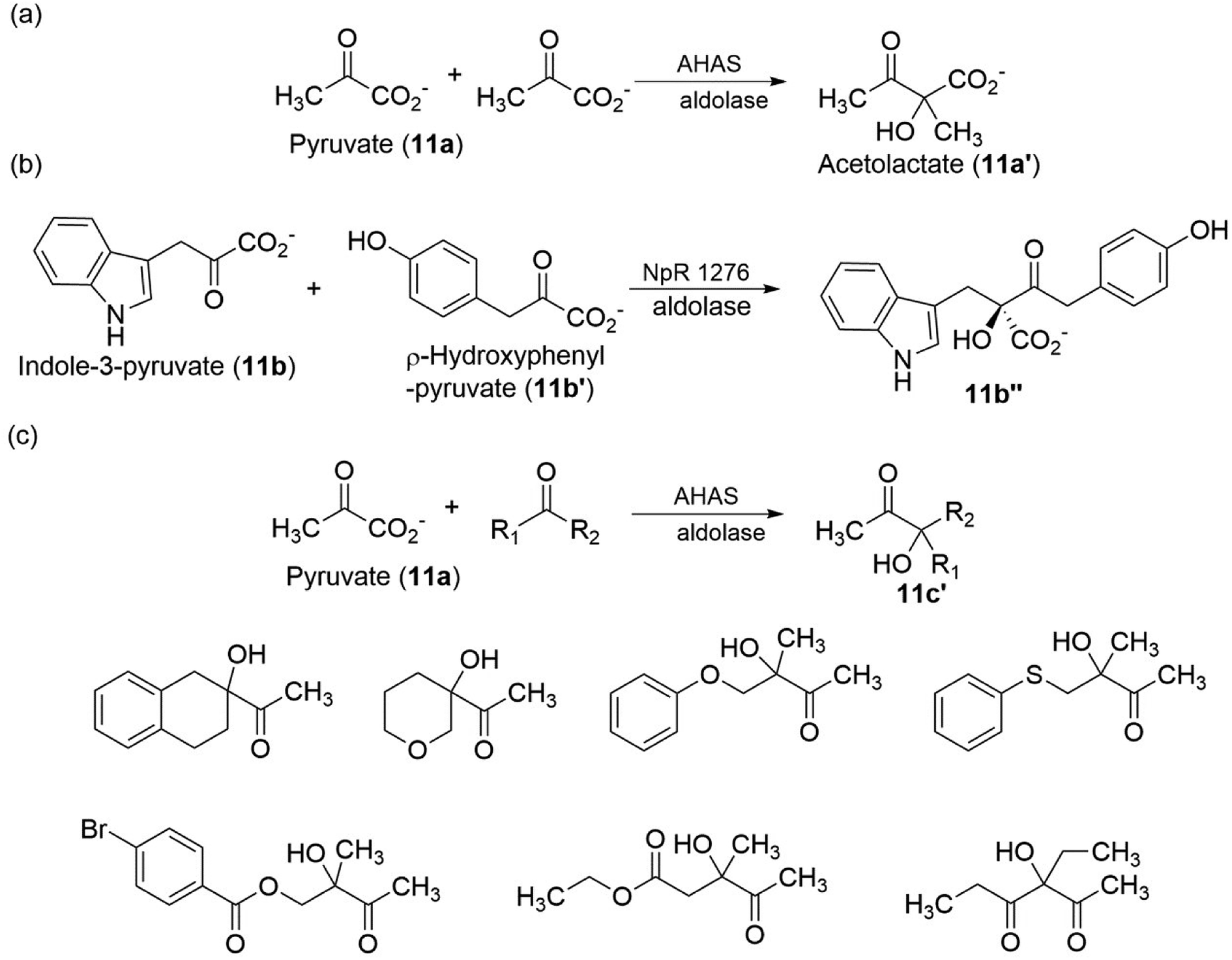 | ||
| Fig. 11 Preparation of β-hydroxy carbonyl compounds by aldol addition.100–102 | ||
Cyanohydrins
Cyanohydrins are valuable building blocks in organic synthesis. They enable an extensive chemistry that can be implemented starting either from the nitrile or hydroxyl functionality. The preparation of cyanohydrins from prochiral aldehydes and ketones by the addition of HCN has therefore always attracted attention and has been the topic of review articles.11,23,57 Hydroxynitrile lyases (HNL) are versatile enzymes for the synthesis of both (R)- and (S)-cyanohydrins, which have been used in industrial applications.113 Here, two examples are given for the possible preparation of both enantiomers of cyanohydrins regarding the use of hydroxynitrile lyases. One uses Arabidopsis thaliana HNL (AtHNL) (EC 4.1.2.47)114 as a catalyst for the addition of HCN to hexan-2-one (12a) and 2-fluoroacetophenone (12b), resulting in the formation of (R)-configured cyanohydrins 12a′ and 12b′, respectively, in high yield and enantioselectivity (Fig. 12). The second example uses Linum usitatissimum HNL (LuHNL) (EC 4.1.2.46) to catalyse the addition of HCN to phenylacetones 12c to obtain the (S)-configured product 12c′ (Fig. 12).115 Regarding the biotechnological potential, unfortunately, the problem of non-enzymatic reaction for the addition of hydrogen cyanide onto carbonyl group must be related for the most HNL-catalysed reactions. As a result, the stereochemical purity of the desired products was reduced.116 In this context, the improvement in yield by the performance of reaction engineering to establish suitable solutions such as, two-phase systems, microreactors, and enzyme membrane reactors, should be mentioned. In this way, the desired cyanohydrins could be obtained in excellent yield and with significantly improved ee values. | ||
| Fig. 12 Enantioselective synthesis of cyanohydrins with hydroxynitrile lyases (HNL).104,105 | ||
Conclusions and outlook
Biocatalysis has become an established technology for the production of predominantly chiral fine chemicals and is increasingly being applied in industry. An established trend is the continued use of biocatalytic methods, which have successfully replaced the well-established chemical methods to produce enantiopure alcohols. The selected examples in this mini-review point to the advantages of biological catalysts in terms of substrate spectrum and enantioselectivity. In this context, it is essential to increase the number and variety of enzyme activities from novel sources. The elucidation of the enzymes' mechanism of action, in combination with the characterization of their structures and biochemical performance, is of paramount importance. Together, this knowledge, along with modern molecular biology methods such as protein engineering, immobilization techniques and metalloenzymes, is essential to push for the development of more versatile and robust (bio)catalysts or catalytic entities for use in the synthesis of enantiomerically pure alcohols. Moreover, the stereospecificity of the reactions which is catalysed by enzymes has been a useful source of information about the mechanisms of enzyme catalysis; the use of stereospecifically labelled substrates enables the study of the reaction's course. To understand the mechanistic and kinetic details, the biocatalysis provides a very promising opportunity even to the most complex enzymatic reactions.The enantioselective water addition to alkenes represents a very efficient and green method to produce enantiopure alcohols. To date, this chemistry has not been highly used, and there are very few examples of it; however, enzyme-catalysed reactions are a mainstay of life. Most hydratase enzymes, however, are involved in metabolic pathways and therefore exhibit a very narrow substrate range. More work remains to be done in broadening the substrate spectrum. Aldolase-catalysed stereoselective aldol addition represents a strategic reaction for the preparation of enantiopure alcohols and analogues. However, aldolases are somewhat limited due to their high selectivity towards the donor substrate, and they do not produce the desired alcohols with a high level of enantioselectivity, particularly when handling non-natural acceptor substrates. Work in this area has been focused on overcoming the above mentioned limitations through directed evolution and reaction engineering. More work still needs to be done in the discovery of novel enzyme activities and broadening the substrate range. The application of enzymes for the enantioselective synthesis of cyanohydrins is a well-established technique that is a key step in many syntheses. Mono-phasic, bi-phasic and pure organic solvent systems are all used with great success. Efficient reaction engineering, enabling a more straightforward application of these versatile enzymes for organic chemistry, is the next stage of development. The biocatalytic reduction of carbonyl compounds to the corresponding enantiopure alcohols represents one of the most common reactions in organic chemistry. The combination of rational and irrational protein design technologies will continue to be applied to improve the catalytic properties of enzymes for their industrial applications in the production of high-value enantiopure alcohols. Chiral kinetic resolution by lipases provides access to a wide range of enantiopure alcohols, remaining a versatile and valuable method in organic synthesis. The epoxide hydrolases (EHs)-mediated enantioconvergent process is also a valuable synthetic tool for the preparation of chiral vicinal diols and is particularly important because no chemical approach for the enantioconvergent hydrolysis of epoxides to yield chiral vicinal diols has been developed. More work is highly anticipated to develop engineered epoxide hydrolases that are less sensitive to diol product inhibition, which is one of the main limitations in EHs-mediated enantioconvergent hydrolysis. Regio- and stereoselective hydroxylation has proven to be a useful tool in the production of enantiopure alcohols, and there have been significant advances in the development of new processes to perform this reaction. The continued search for new methodologies and enzymatic activities, and the optimization of these features, such as the development of new reactor systems, is highly expected in this area.
The challenges for the future in the enzymatic synthesis of enantiopure alcohols lie in philosophical, rather technical aspects. Biocatalysis is a multidisciplinary field, and its development requires a combination of chemical and biological techniques. The strategies given here are used with great successes to produce enantiopure alcohols. It is hopeful that this gives chemists ample room to consider enzymes as tools for envisaged synthesis and, likewise, for biologists to consider the chemical applications of enzymes. With a dedicated biocatalysis group using a combination of chemistry and biology, enzyme-catalysed reactions may be applied in a straightforward manner, with the synthetic strategies designed from the outset. In this way, the potential value of the enzymatic synthesis of enantiopure alcohols for laboratory and industrial use can truly be realized.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.Conflicts of interest
The authors declare that they have no conflict of interest.Acknowledgements
In-depth discussions with Prof. Dr Ulf Hanefeld from Delft University of Technology, helped shaping this review. This study was funded by the National Natural Science Foundation of China (Grant No. 41706148), the Natural Science Foundation of Guangdong Province (Grant No. 2017A030310232), the Basic Research Program of Sun Yat-Sen University (Grant No. 17lgpy58), and the National Science and Technology Major Project of the Ministry of Science and Technology of China (2018ZX09735010).References
- Z. Tan, H. Ma, Q. Li, L. Pu, Y. Cao, X. Qu, C. Zhu and H. Ying, Enzyme Microb. Technol., 2016, 93–94, 191–199 CrossRef CAS PubMed.
- H. Liu, B.-S. Chen, F. Z. Ribeiro de Souza and L. Liu, Mar. Drugs, 2018, 16, 1–15 CrossRef PubMed.
- T. Hiraishi and S. Taguchi, Mini-Rev. Org. Chem., 2009, 6, 44–54 CrossRef CAS.
- R. N. Patel, ACS Catal., 2011, 1, 1056–1074 CrossRef CAS.
- H. Liu, F. Z. R. de Souza, L. Liu and B.-S. Chen, Molecules, 2018, 23, 2144 CrossRef PubMed.
- K. Faber, Biotransformations in organic chemistry, Springer, Berlin, Heidelberg, 7th edn, 2018 Search PubMed.
- B.-S. Chen, V. Resch, L. G. Otten and U. Hanefeld, Chem.–Eur. J., 2015, 21, 3020–3030 CrossRef CAS PubMed.
- V. Resch and U. Hanefeld, Catal. Sci. Technol., 2015, 5, 1385–1399 RSC.
- S. M. Dean, W. A. Greenberg and C. H. Wong, Adv. Synth. Catal., 2007, 349, 1308–1320 CrossRef CAS.
- P. Clapés, W. D. Fessner, G. A. Sprenger and A. K. Samland, Curr. Opin. Chem. Biol., 2010, 14, 154–167 CrossRef PubMed.
- Y. Yamashita, T. Yasukawa, W.-J. Yoo, T. Kitanosono and S. Kobayashi, Chem. Soc. Rev., 2018, 47, 4388–4480 RSC.
- T. Purkarthofer, W. Skranc, C. Schuster and H. Griengl, Appl. Microbiol. Biotechnol., 2007, 76, 309–320 CrossRef CAS PubMed.
- U. Hanefeld, Chem. Soc. Rev., 2013, 42, 6308–6321 RSC.
- P. Bracco, H. Busch, J. von Langermannb and U. Hanefeld, Org. Biomol. Chem., 2016, 14, 6375–6389 RSC.
- C. M. Nealon, M. M. Musa, J. M. Patel and R. S. Phillips, ACS Catal., 2015, 5, 2100–2114 CrossRef CAS.
- R. Zhang, Y. Xu and R. Xiao, Biotechnol. Adv., 2015, 33, 1671–1684 CrossRef CAS PubMed.
- P. Adlercreutz, Chem. Soc. Rev., 2013, 42, 6406–6436 RSC.
- B. Andualema and A. Gessesse, Biotechnology, 2012, 11, 100–118 CrossRef CAS.
- A. Steinreiber and K. Faber, Curr. Opin. Biotechnol., 2001, 12, 552–558 CrossRef CAS PubMed.
- E. O'Reilly, S. J. Aitken, G. Grogan, P. P. Kelly, N. J. Turner and S. L. Flitsch, Beilstein J. Org. Chem., 2012, 8, 496–500 CrossRef PubMed.
- C. Klein and W. Hüttel, Adv. Synth. Catal., 2011, 353, 1375–1383 CrossRef CAS.
- B.-S. Chen, L. G. Otten and U. Hanefeld, Biotechnol. Adv., 2015, 33, 526–546 CrossRef CAS PubMed.
- A. Szekrenyi, A. Soler, X. Garrabou, C. Guérard-Hélaine, T. Parella, J. Joglar, M. Lemaire, J. Bujons and P. Clapés, Chem.–Eur. J., 2014, 20, 12572–12583 CrossRef CAS PubMed.
- P. Bracco, H. Busch, J. von Langermann and U. Hanefeld, Org. Biomol. Chem., 2016, 14, 6375–6389 RSC.
- Y.-G. Zheng, H.-H. Yin, D.-F. Yu, X. Chen, X.-L. Tang, X.-J. Zhang, Y.-P. Xue, Y.-J. Wang and Z.-Q. Liu, Appl. Microbiol. Biotechnol., 2017, 101, 987–1001 CrossRef CAS PubMed.
- A. Belafriekh, F. Secundo, S. Serra and Z. Djeghaba, Tetrahedron: Asymmetry, 2017, 28, 473–478 CrossRef CAS.
- M. Schober and K. Faber, Trends Biotechnol., 2013, 31, 468–478 CrossRef CAS PubMed.
- M. Müller, ChemBioEng Rev., 2014, 1, 14–26 CrossRef.
- D. Alsafadi, S. Alsalman and F. Paradisi, Org. Biomol. Chem., 2017, 15, 9169–9175 RSC.
- N. A. Salvi, L. P. Badheka and S. Chattopadhyay, Biotechnol. Lett., 2003, 25, 1081–1086 CrossRef CAS PubMed.
- X.-H. Chen, X.-T. Wang, W.-Y. Lou, Y. Li, H. Wu, M.-H. Zong, T. J. Smith and X.-D. Chen, Microb. Cell Fact., 2012, 11, 119 CrossRef CAS PubMed.
- P.-X. Du, P. Wei, W.-Y. Lou and M.-H. Zong, Microb. Cell Fact., 2014, 13, 84 CrossRef PubMed.
- E. Şahin and E. Dertli, Chem. Biodiversity, 2017, 14, e1700269 CrossRef PubMed.
- A. Ghanem and H. Y. Aboul-Enein, Chirality, 2005, 17, 1–15 CrossRef CAS PubMed.
- W. S. Qayed, A. S. Aboraia, H. M. Abdel-Rahman and A. F. Youssef, J. Chem. Pharm. Res., 2015, 7, 311–322 CAS.
- W. Kroutil, H. Mang, K. Edegger and K. Faber, Curr. Opin. Chem. Biol., 2004, 8, 120–126 CrossRef CAS PubMed.
- R. Chen, X. Liu, J. Lin and D. Wei, Biosci., Biotechnol., Biochem., 2014, 78, 1350–1356 CrossRef CAS PubMed.
- G. Hasnaoui-Dijoux, M. M. Elenkov, J. H. L. Spelberg, B. Hauer and D. B. Janssen, ChemBioChem, 2008, 9, 1048–1051 CrossRef CAS PubMed.
- J. Koopmeiners, C. Diederich, J. Solarczek, H. Voß, J. Mayer, W. Blankenfeldt and A. Schallmey, ACS Catal., 2017, 7, 6877–6886 CrossRef CAS.
- R. J. Kazlauskas, A. N. E. Weissfloch, A. T. Rappaport and L. A. Cuccia, J. Org. Chem., 1991, 56, 2656–2665 CrossRef CAS.
- A. Ghanem and V. Schurig, Tetrahedron: Asymmetry, 2003, 14, 57–62 CrossRef CAS.
- M. Baumann, B. H. Hauer and U. T. Bornscheuer, Tetrahedron: Asymmetry, 2000, 11, 4781–4790 CrossRef CAS.
- M. M. Elenkov, B. Hauer and D. B. Janssen, Adv. Synth. Catal., 2006, 348, 579–585 CrossRef CAS.
- M. M. Elenkov, L. Tang, B. Hauer and D. B. Janssen, Org. Lett., 2006, 8, 4227–4229 CrossRef PubMed.
- A. Schallmey and M. Schallmey, Appl. Microbiol. Biotechnol., 2016, 100, 7827–7839 CrossRef CAS PubMed.
- J. Chen, R.-C. Zheng, Y.-G. Zheng and Y.-C. Shen, Adv. Biochem. Eng./Biotechnol., 2009, 113, 33–77 CAS.
- T. Nakamura, T. Nagasawa, F. Yu, I. Watanabe and H. Yamada, Tetrahedron, 1994, 50, 11821–11826 CrossRef CAS.
- R. O. M. A. de Souza, L. S. M. Miranda and U. T. Bornscheuer, Chem.–Eur. J., 2017, 23, 12040–12063 CrossRef CAS PubMed.
- W. Hummel and M.-R. Kula, Eur. J. Biochem., 1989, 184, 1–13 CrossRef CAS PubMed.
- U. Kragl, W. Kruse, W. Hummel and C. Wandrey, Biotechnol. Bioeng., 1996, 52, 309–319 CrossRef CAS PubMed.
- C. Wang, F. Xin, X. Kong, J. Zhao, W. Dong, W. Zhang, J. Ma, H. Wu and M. Jiang, Biotechnol. Biofuels, 2018, 11, 12 CrossRef PubMed.
- Z. Li, H.-J. Feiten, D. Chang, W. A. Duetz, J. B. van Beilen and B. Witholt, J. Org. Chem., 2001, 66, 8424–8430 CrossRef CAS PubMed.
- A. de Raadt, H. Griengl and H. Weber, Chem.–Eur. J., 2001, 7, 27–31 CrossRef CAS.
- A. De Raadt, B. Fetz, H. Griengl, F. Klingler, G. Ott, P. Plachota, I. Kopper, B. Krenn, F. Münzer, H. J. Weber, G. Braunegg, W. Mosler and R. Saf, Eur. J. Org. Chem., 2000, 3835–3847 CrossRef CAS.
- G. Aranda, L. Moreno, M. Cortés, T. Prangé, M. Maurs and R. Azerad, Tetrahedron, 2001, 57, 6051–6056 CrossRef CAS.
- W. Adam, Z. Lukacs, C. Kahle, C. R. Saha-Möller and P. Schreier, J. Mol. Catal. B: Enzym., 2001, 11, 377–385 CrossRef CAS.
- W. A. Duetz, A. H. M. Fjällman, S. Ren, C. Jourdat and B. Witholt, Appl. Environ. Microbiol., 2001, 67, 2829–2832 CrossRef CAS PubMed.
- S. T. Chen and J. M. Fang, J. Org. Chem., 1997, 62, 4349–4357 CrossRef CAS PubMed.
- R. Kourist and U. T. Bornscheuer, Appl. Microbiol. Biotechnol., 2011, 91, 505–517 CrossRef CAS PubMed.
- S. Z. Wang, J. P. Wu, G. Xu and L. R. Yang, Biochem. Eng. J., 2012, 65, 57–62 CrossRef CAS.
- S. H. Krishna, M. Persson and U. T. Bornscheuer, Tetrahedron: Asymmetry, 2002, 13, 2693–2696 CrossRef.
- T. Sugai, H. Kakeya and H. Ohta, Tetrahedron, 1990, 46, 3463–3468 CrossRef CAS.
- M. Pogorevc and K. Faber, J. Mol. Catal. B: Enzym., 2000, 10, 357–376 CrossRef CAS.
- Y. Luo, Y. Chen, H. Ma, Z. Tian, Y. Zhang and J. Zhang, Sci. Rep., 2017, 7, 42064 CrossRef CAS PubMed.
- E. Henke, J. Pleiss and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2002, 41, 3211–3213 CrossRef CAS PubMed.
- E. Henke, U. T. Bornscheuer, R. D. Schmid and J. Pleiss, ChemBioChem, 2003, 4, 485–493 CrossRef CAS PubMed.
- R. Kourist, P. D. de Maria and U. T. Bornschuer, ChemBioChem, 2008, 9, 491–498 CrossRef CAS PubMed.
- M. M. Elenkov, H. W. Hoeffken, L. Tang, B. Hauer and D. B. Janssen, Adv. Synth. Catal., 2007, 349, 2279–2285 CrossRef CAS.
- C. Molinaro, A. A. Guilbault and B. Kosjek, Org. Lett., 2010, 12, 3772–3775 CrossRef CAS PubMed.
- J. B. Cheng, M. A. Levine, N. H. Bell, D. J. Mangelsdorf and D. W. Russell, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 7711–7715 CrossRef CAS PubMed.
- J. G. Zhu, J. T. Ochalek, M. Kaufmann, G. Jones and H. F. DeLuca, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15650–15655 CrossRef CAS PubMed.
- H. Sugimoto, R. Shinkyo, K. Hayashi, S. Yoneda, M. Yamada, M. Kamakura, S. I. Ikushiro, Y. Shiro and T. Sakaki, Biochemistry, 2008, 47, 4017–4027 CrossRef CAS PubMed.
- D. Zehentgruber, F. Hannemann, S. Bleif, R. Bernhardt and S. Lütz, ChemBioChem, 2010, 11, 713–721 CrossRef CAS PubMed.
- M. V. Donova and O. V. Egorova, Appl. Microbiol. Biotechnol., 2012, 94, 1423–1447 CrossRef CAS PubMed.
- W. Hüttel, Chem. Ing. Tech., 2013, 85, 809–817 CrossRef.
- Y. R. Chiang, W. Ismail, M. Müller and G. Fuchs, J. Biol. Chem., 2007, 282, 13240–13249 CrossRef CAS PubMed.
- D. L. Lister, G. Kanungo, D. A. Rathbone and N. C. Bruce, FEMS Microbiol. Lett., 1999, 181, 137–144 CrossRef CAS PubMed.
- J. Stankevičiūtė, S. Kutanovas, R. Rutkienė, D. Tauraitė, R. Striela and R. Meškys, PeerJ, 2015, 3, e1387 CrossRef PubMed.
- Y. Chen, C. Chen and X. Wu, Chem. Soc. Rev., 2012, 41, 1742 RSC.
- J. Zhao, W. Hua-lei, J. Zhang, W. Gao, X.-J. Fan, H.-H. Chang, W.-L. Wei and J.-H. Xu, Biotechnol. Lett., 2017, 40, 349–358 CrossRef PubMed.
- M. G. Banwell, B. S. Blakey, B. G. Harfoot and R. W. Longmore, Aust. J. Chem., 1999, 52, 137–142 CrossRef CAS.
- L. Werner, A. Machara and T. Hudlicky, Adv. Synth. Catal., 2010, 352, 195–200 CrossRef CAS.
- B. C. Buckland, Metab. Eng., 1999, 1, 63–74 CrossRef CAS PubMed.
- V. B. Urlacher and R. D. Schmid, Curr. Opin. Chem. Biol., 2006, 10, 156–161 CrossRef CAS PubMed.
- Y. Huang, N. Liu, X. R. Wu and Y. J. Chen, Curr. Org. Chem., 2010, 14, 1447–1460 CrossRef CAS.
- E. Busto, V. Gotor-Fernández and V. Gotor, Chem. Soc. Rev., 2010, 39, 4504 RSC.
- Y. Chen, C. Chen and X. Wu, Chem. Soc. Rev., 2012, 41, 1742–1753 RSC.
- H. Ichinose, S. Katoh, T. Sueoka, K. Titani, K. Fujita and T. Nagatsu, Biochem. Biophys. Res. Commun., 1991, 3, 41–42 Search PubMed.
- S. H. Cho, J. U. Na, H. Youn, C. S. Hwang, C. H. Lee and S. O. Kang, Biochem. J., 1999, 503, 497–503 CrossRef.
- Y. A. Kim, H. J. Chung, Y. J. Kim, Y. K. Choi, Y. K. Hwang, S. W. Lee and Y. S. Park, Mol. Cells, 2000, 10, 405–410 CAS.
- K. B. Sharpless and K. Akashi, J. Am. Chem. Soc., 1976, 98, 1986–1987 CrossRef CAS.
- M. A. Andersson, R. Epple, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 472–475 CrossRef CAS PubMed.
- T. Hudlicky and J. W. Reed, Synlett, 2009, 0685–0703 CrossRef CAS.
- R. Friemann, M. M. Ivkovic-Jensen, D. J. Lessner, C. L. Yu, D. T. Gibson, R. E. Parales, H. Eklund and S. Ramaswamy, J. Mol. Biol., 2005, 348, 1139–1151 CrossRef CAS PubMed.
- D. T. Gibson and R. E. Parales, Curr. Opin. Biotechnol., 2000, 11, 236–243 CrossRef CAS PubMed.
- C. S. Nunes and V. Kumar, Enzymes in human and animal nutrition: principles and perspectives, Academic Press, 2018, p. 315 Search PubMed.
- A. G. Myers, D. R. Siegel, D. J. Buzard and M. G. Charest, Org. Lett., 2001, 3, 2923–2926 CrossRef CAS PubMed.
- S. Y. Sun, X. Zhang, Q. Zhou, J. C. Chen and G. Q. Chen, Appl. Microbiol. Biotechnol., 2008, 80, 977–984 CrossRef CAS PubMed.
- K. Faber, Biotransformations in Organic Chemistry: A Textbook, 7th edn, 2017 Search PubMed.
- D. R. Boyd, N. D. Sharma, N. I. Bowers, I. N. Brannigan, M. R. Groocock, J. F. Malone, G. McConville and C. C. R. Allen, Adv. Synth. Catal., 2005, 347, 1081–1089 CrossRef CAS.
- M. Widersten, A. Gurell and D. Lindberg, Biochim. Biophys. Acta, Gen. Subj., 2010, 1800, 316–326 CrossRef CAS PubMed.
- B.-S. Chen, H. Liu, F. Z. R. de Souza and L. Liu, Mar. Biotechnol., 2017, 19, 351–360 CrossRef CAS PubMed.
- S. K. Karmee, R. Van Oosten and U. Hanefeld, Tetrahedron: Asymmetry, 2011, 22, 1736–1739 CrossRef CAS.
- M. A. Arai, R. Tsutsumi, H. Hara, T. C. Chen, T. Sakaki, N. Urushino, K. Inouye and A. Kittaka, Heterocycles, 2005, 66, 469–479 CrossRef CAS.
- J. Jin and U. Hanefeld, Chem. Commun., 2011, 47, 2502 RSC.
- J. Bos, A. García-Herraiz and G. Roelfes, Chem. Sci., 2013, 4, 3578 RSC.
- A. J. Boersma, D. Coquiére, D. Geerdink, F. Rosati, B. L. Feringa and G. Roelfes, Nat. Chem., 2010, 2, 991–995 CrossRef CAS PubMed.
- P. Clapés and X. Garrabou, Adv. Synth. Catal., 2011, 353, 2263–2283 CrossRef.
- M. Müller, D. Gocke and M. Pohl, FEBS J., 2009, 276, 2894–2904 CrossRef PubMed.
- H. C. Hailes, D. Rother, M. Müller, R. Westphal, J. M. Ward, J. Pleiss, C. Vogel and M. Pohl, FEBS J., 2013, 280, 6374–6394 CrossRef CAS PubMed.
- E. P. Balskus and C. T. Walsh, J. Am. Chem. Soc., 2008, 130, 15260–15261 CrossRef CAS PubMed.
- P. Lehwald, M. Richter, C. Röhr, H. W. Liu and M. Müller, Angew. Chem., Int. Ed., 2010, 49, 2389–2392 CrossRef CAS PubMed.
- G. Torrelo, F. Z. Ribeiro De Souza, E. Carrilho and U. Hanefeld, ChemBioChem, 2015, 16, 625–630 CrossRef CAS PubMed.
- J. Andexer, J. Von Langermann, A. Mell, M. Bocola, U. Kragl, T. Eggert and M. Pohl, Angew. Chem., Int. Ed., 2007, 46, 8679–8681 CrossRef CAS PubMed.
- C. Roberge, F. Fleitz, D. Pollard and P. Devine, Tetrahedron: Asymmetry, 2007, 18, 208–214 CrossRef CAS.
- J. N. Andexer, J. V. Langermann, U. Kragl and M. Pohl, Trends Biotechnol., 2009, 27, 599–607 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |