
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceObservation of the generation of peroxynitrite in mouse liver after acetaminophen overdose with a boronate-based ratiometric fluorescence probe†
Ying Maa,
Haigang Tiana,
Zhengyu Jina,
Xiaoyong Lib and
Yiping Li *a
*a
aSchool of Pharmacy, Health Science Center, Xi'an Jiaotong University, Xi'an 710061, Shaanxi, China. E-mail: yipingli@mail.xjtu.edu.cn
bSchool of Science, Xi'an Jiaotong University, Xi'an 710061, Shaanxi, China. E-mail: lixy6658@mail.xjtu.edu.cn
First published on 25th February 2019
Abstract
A ratiometric fluorescent probe, BTPB, for the selective monitoring of hepatic peroxynitrite (ONOO−) in situ after acetaminophen (APAP) overdose has been developed. Our study provided direct evidence for supporting the generation of ONOO− in APAP-induced liver injury. This new probe will be a useful tool for studying the roles of ONOO− in vivo.
Acetaminophen (APAP) is a safe and effective antipyretic/analgesic drug when used at recommended doses, and is one of the most commonly used drugs in the US and throughout the West.1 However, APAP overdose can cause severe acute liver injury with the potential to progress to liver failure. It is responsible for almost 80
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 emergency department visits in the US each year,2 as well as 26000 hospitalizations and nearly 500 deaths.3 In fact, APAP hepatotoxicity is one of the most common etiologies of acute liver failure.4
000 emergency department visits in the US each year,2 as well as 26000 hospitalizations and nearly 500 deaths.3 In fact, APAP hepatotoxicity is one of the most common etiologies of acute liver failure.4
Despite substantial progress in understanding the mechanism of APAP-induced liver injury during the last four decades, many details are still unknown. At therapeutic levels, about 50–70% of the administered APAP is glucuronidated, and 25–35% undergoes sulfation, while only 5–10% is converted to the reactive electrophilic intermediate N-acetyl-p-benzoquinone imine (NAPQI) by cytochrome P450 enzymes.5 NAPQI can be detoxified by reaction with the cysteine sulfhydryl group on glutathione (GSH). Once the administration of APAP overdose, NAPQI can deplete GSH stores, and bind to protein sulfhydryl groups, especially mitochondrial proteins, which triggers mitochondrial oxidant stress. The amplification of mitochondrial oxidative stress further leads to the opening of the mitochondrial membrane permeability transition (MPT) pore, then the rupture of the outer mitochondrial membrane. As a result, there is extensive release of proteins from the intermembrane space, including endonuclease G and apoptosis-inducing factor, both of which can translocate to the nucleus because of their nuclear localization sequences. This then results in DNA fragmentation and, finally, oncotic necrosis.6
During resulting in MPT, it was recognized that the strong oxidant and nitrating peroxynitrite (ONOO−), formed from superoxide (O2˙−) and nitric oxide (NO), is being generated in hepatocytes and a critical mediator in the injury mechanism. The generation of ONOO− was only demonstrated by immunohistochemical staining for 3-nitrotyrosine protein adducts.7,8 The well-known nitration mechanism involves the initial one-electron oxidation of tyrosine to yield tyrosyl radical followed by a diffusion-controlled reaction with nitrogen dioxide (˙NO2) to yield 3-nitrotyrosine.9,10 However, in biological systems, ˙NO2 arises from a variety of sources, except most notably the decomposition of ONOO−, but the hemeperoxidase-dependent oxidation of nitrite,10,11 which is a ONOO−-independent mechanism. Actually, protein 3-nitrotyrosine, which was initially conceived as a specific footprint of ONOO−, typically represents a fairly stable end product that is generated by the action of NO-derived oxidants.12 It seems insufficient for the generation of ONOO− after the administration of APAP overdose only according to the observation of the formation of protein 3-nitrotyrosine. Therefore, there is an urgent need to provide the direct evidence for supporting the generation of ONOO− after APAP overdose.
The lifetime of ONOO− in biological systems is limited to only a few milliseconds, which makes it impossible to quantitate ONOO− in processed cell or tissue samples directly.13 Fluorescence based techniques are advantageous for detection of ONOO−. In accordance with the multiplicity of ONOO− biochemistry,14 the trifluorocarbonyl (HKGreen 1-3)15–17 and phenyl boronic esters (CBA)18 based fluorescent probes, which can directly detect OONO−, have been reported, mainly for in vitro applications. However, these probes are not suitable for biological studies in vivo because of their high molecular weight, bearing easily metabolized lactone group, such as HKGreen 1-3, or a low response signal, such as CBA. Chemiluminescence (CL) probes19,20 can distinguish ONOO− from a variety of other ROS owing to the radicals from the decomposition of ONOO−, such as ONOOH, ˙OH and O2˙−, not directly to the reaction with ONOO−. Additionally, CL probes were not used to detect OONO− in vivo.
In this regard, to provide direct and strong evidence for the formation of OONO− in liver after APAP overdose, we developed a novel and low background phenyl boronic acid-containing fluorogenic probe for sensitive and selective detecting of OONO−. This probe was introduced into the mice administrated APAP overdose by heart perfusion to monitor in situ the formation of OONO− in liver.
The new fluorescent probe was designed by the direct conjugation of an boronic acid reaction group with a (phenyl-2-yl)benzothiazol scaffold, serving as a fluorophore. Phenyl boronic acid group was selected because the group can react with OONO− directly, rapidly and stoichiometrically,21 which is more suitable to detect OONO− than other groups that react with those secondary radicals derived from OONO−, including HONOO, ONOOCO2−, or metal–OONO− complexes. Moreover, phenyl boronic acid group was recently reported to be highly reactive to OONO− nearly a million times faster than H2O2 and two hundred times faster than ClO−.22,23 The (phenyl-2-yl)benzothiazol scaffold was selected because the probe designed reacts with OONO− to yield corresponding hydroxyl derivatives 2-(benzothiazol-2-yl)phenol, which possesses the excellent photophysical properties, such as high photostabilities and high fluorescence quantum yields, of members of this family.24,25 The boronic acid group is converted to the hydroxyl by the reaction with OONO−, with the conversions offering ratiometric responses because 2-(benzothiazol-2-yl) phenylboronic acid (BTPB) and 2-(benzothiazol-2-yl) phenol (BTP) fluoresce at different wavelengths, which makes this probe sensitive. More importantly, benzothiazol group is metabolically stable and a usual drug fragments,26,27 and this probe has a low molecular weight and appropriate water solubility, which makes this probe suitable to use in vivo. The design rationale is depicted in Fig. 1, and illustrated as follows.
 | ||
| Fig. 1 Structure of BTPB and detection of peroxynitrite by oxidation of BTPB. | ||
Probe BTPB was prepared using the one-step reaction shown in ESI.†28 2-(Benzothiazol-2-yl) phenol, BTP, the expected product resulting from reaction of BTPB with ONOO−, was also prepared utilizing a previously described method.29 The structures of BTPB and BTP were confirmed by 1H NMR, 13C NMR, and HRMS spectra (Fig. S1–S6†), which match those of literatures.28,29
Firstly, the spectroscopic response of BTPB to ONOO− was investigated in an abiotic system. We examined the spectroscopic properties of BTPB (1.0 μM) towards ONOO− (1 equiv.) in 100.0 mM PBS buffer with 1.0 mM cetyltrimethylammonium bromide (CTAB) at pH 7.4 at 25.0 ± 0.1 °C. 50 μL of BTPB (1.0 mM) was added to ONOO− (1.0 equiv.), then vigorously stirred for 30 seconds. After dilution to 1.0 μM with 100.0 mM PBS buffer 1.0 mM CTAB, the mixture was vigorously stirred for 1 min before measurement. The absorption and fluorescence spectra of BTPB before and after reaction with ONOO− are shown in Fig. 2. As is seen, BTPB shows a major UV-absorption centered at 296 nm, but its reaction with ONOO− produces two absorption peak at 290 nm and 338 nm. Moreover, BTPB itself displays a low fluorescence with the maximum emission peak at 372 nm, whereas the reaction with ONOO− leads to a fluorescence enhancement with the maximum emission peak at 462 nm.
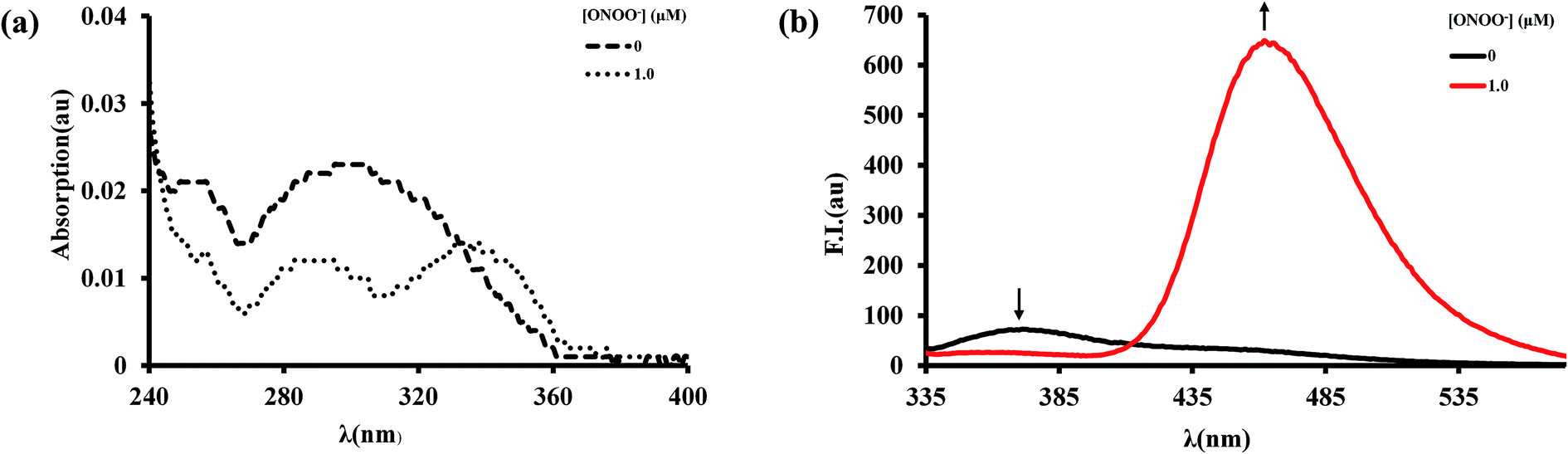 | ||
| Fig. 2 Absorption spectra (a) and fluorescence spectra (b) of BTPB (1.0 μm) before and after reaction with ONOO− (1.0 μm). Spectra were obtained in 100.0 mm phosphate buffer (pH 7.4) with 1.0 mm cetyltrimethylammonium bromide (CTAB) under vigorous stirring for 1 min after incubation of the probe with ONOO− for 30 seconds at 25.0 °C, λex = 296 nm. | ||
The fluorescence response of BTPB to varied concentrations of ONOO− (0–1.0 μM) was examined. When the emission wavelength was set to 372 nm, upon addition of ONOO−, the main excitation peak of BTPB at 296 nm decreases with a concurrent increase in excitation peaks at 296 nm, 332 nm and 386 nm when the emission wavelength was 462 nm (Fig. 3a and b). Thus, the ratiometric feature in the spectra was obtained at an excitation wavelength of 296 nm.
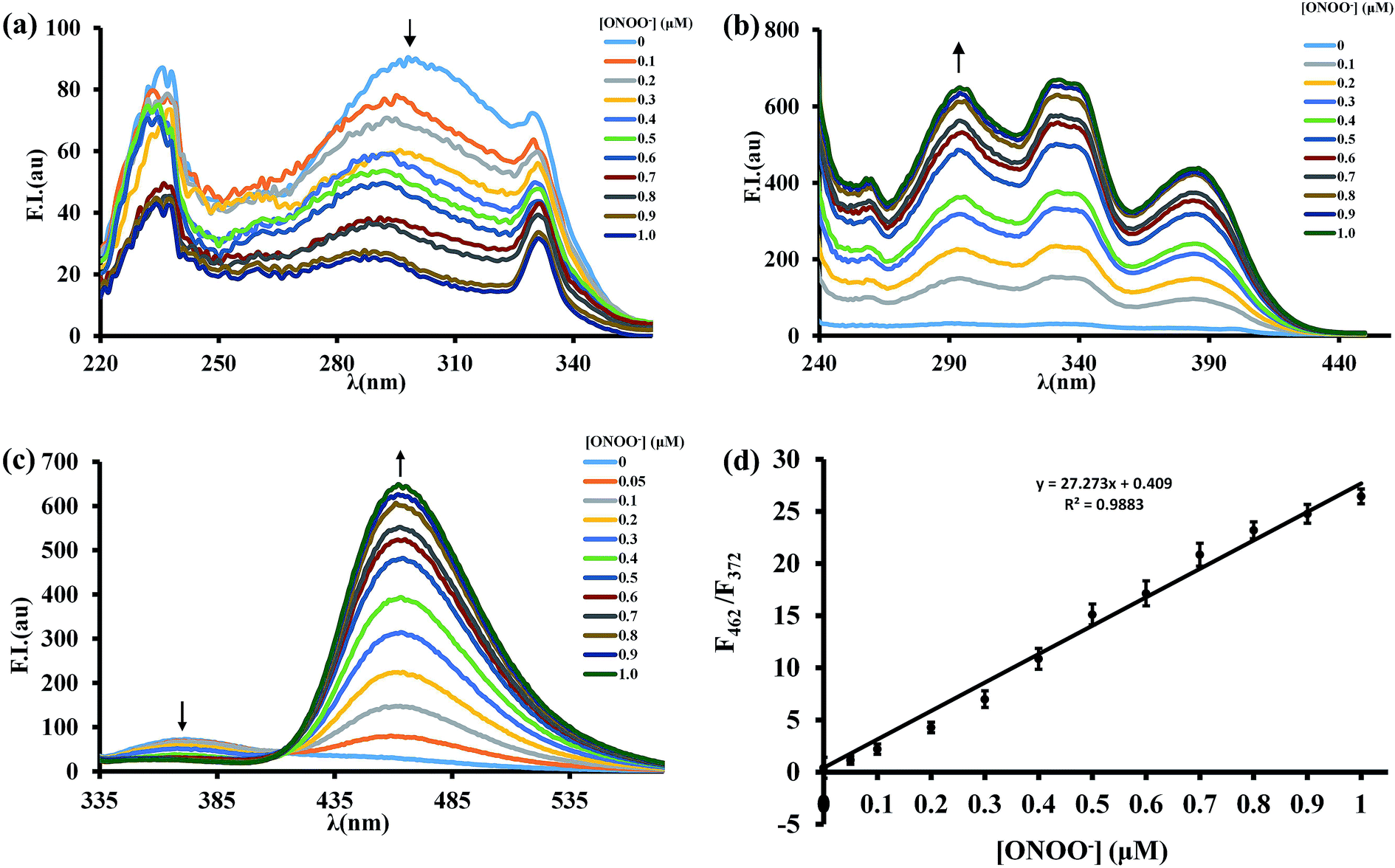 | ||
| Fig. 3 (a) Fluorescence excitation spectra of BTPB (1.0 μm) to OONO− (0–1 equiv.), λem = 372 nm. (b) Fluorescence excitation spectra of BTPB (1.0 μm) to OONO− (0–1 equiv.), λem = 462 nm. (c) Fluorescence emission spectra of BTPB (1.0 μm) to OONO− (0–1 equiv.), λex = 296 nm. (d) Linear relationship of emission intensity ratio (F462/F372) to OONO− (0–1.0 μm). All spectra were obtained in 100.0 mm phosphate buffer (pH 7.4) with 1.0 mm cetyltrimethylammonium bromide (CTAB) under vigorous stirring for 1 min after incubation of the probe with ONOO− for 30 seconds at 25.0 °C. | ||
As expected, in the absence of ONOO−, BTPB (1.0 μM) in 100.0 mM PBS buffer with 1.0 mM CTAB at pH 7.4 showed a very poor fluorescence at 372 nm (ϕF = 0.05); upon addition of ONOO− (1.0 μM), a dramatic fluorescence enhancement was observed immediately at 462 nm (ϕF = 0.33). The ratio of emission intensities (F462/F372) upon excitation at 296 nm varies from 0.41 in the absence of ONOO− to 26.45 after reaction with ONOO−, a ca. 65-fold emission ratio change (Fig. 3c), which is more than the response signal of CBA.18 As shown in Fig. 3d, The ratio of emission intensities (F462/F372) exhibits a good linear relationship in the concentration range of 0–1.0 μM ONOO− with an equation of F = 27.27 × [ONOO−] (μM) + 0.41 (R2 = 0.988), where F represents the ratio of fluorescence intensity of the reaction solution (F462/F372). The detection limit was calculated to be 12.3 nM ONOO−. These results revealed that BTPB could potentially test ONOO− in abiotic systems, which is similar with physiological conditions, both qualitatively and quantitatively.
It is known that CTAB is a cationic surfactant and forms spherical micelles within the concentration range 0.9–100 mM in water. Both BTPB and BTP are low soluble in water; to apply them efficiently in an aqueous environment, CTAB was introduced into the aqueous phase. Thus BTPB molecules could be encapsulated in micelles, which makes more intense emission of BTPB in CTAB-added PBS than that in PBS without CTAB (Fig. S7†). The formation of micelles was also confirmed by means of scanning electron microscopy (Fig. S8†).
The selectivity of BTPB (1.0 μM) toward ONOO− was determined by measuring fluorescence changes that take place in the presence of various ROS and RNS including superoxide (O2˙−), hypochlorite (ClO−), hydroxyl radicals (˙OH), hydrogen peroxide (H2O2), single oxygen (1O2), tert-butoxyl radical (˙OtBu), nitrite (NO2−) and nitric oxide (NO) in phosphate buffer (100 mM, pH 7.4, 1.0 mM CTAB) at 25 °C. As shown in Fig. 4, only ONOO− promotes a dramatic enhancement in fluorescence intensity at 462 nm (F462/F372 = 26.5). In contrast, no changes in emission intensities occur when the probe was incubated with O2˙−, H2O2, ˙OH, 1O2, ˙OtBu, NO2− and NO. ClO− induces a small fluorescence response (F462/F372 = 1.2). Taken together, these results established that BTPB is sensitive enough and also considerably selective toward ONOO− over other ROS or RNS under the conditions of low concentration (1.0 μM) and short reaction time (1.0 min). The observations also corroborate the previous finding that boronate-containing small molecules react with ONOO− much faster than with H2O2 or ClO−.22,23
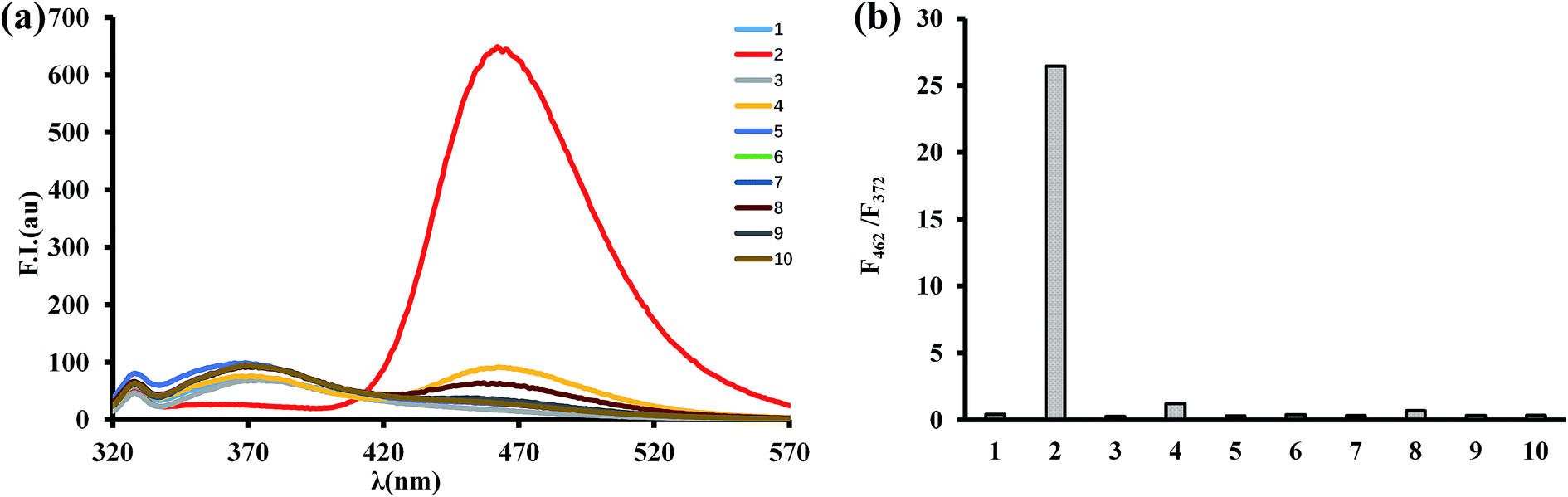 | ||
| Fig. 4 (a) Fluorescence emission spectra of BTPB (1.0 μm) to various ROS and RNS (1 equiv. ONOO− and 10 equiv. for other ROS and RNS). (1) Blank; (2) ONOO−; (3) ˙OH; (4) ClO−; (5) H2O2; (6) NO2−; (7) NO; (8) O2˙−; (9) 1O2; (10) ˙OtBu. (b) Emission intensity ratio (F462/F372) of various ROS and RNS. | ||
Our approach to develop a ONOO− specific probe relies on the use of oxidation of phenyl boronic acid group of BTPB to hydroxyl group of BTP. Subsequently, to confirm this reaction, the process taking place between BTPB and ONOO− was monitored by using HPLC. HPLC analysis of the mixture generated from the reaction of ONOO− with BTPB showed that in the presence of various concentrations of ONOO−, the intensity of the peak of BTPB (eluted at 2.0 min) gradually decreased, in the meanwhile that of the peak of oxidation products BTP eluted at 5.5 min emerged and gradually increased (Fig. S9†). The results confirmed that the fluorescent product was indeed BTP, resulting from the ONOO− induced oxidation reaction of BTPB. The ratiometric fluorescence mechanism is concerned with internal charge transfer (ICT). The boron atom in boronic acid group is sp2 hybridized and possesses fiercely electron-withdrawing character due to the availability of its vacant p orbitals, forming the ICT state with benzothiazol moity as the electron-donating group. After the reaction with ONOO−, the boronic acid group is converted to the electron-donating hydroxyl, which interferes internal charge transfer of BTPB molecule and results in fluorescent emission enhancement. The red shifts in emission maxima could be attributed to the lower-energy emission of the phenolate anion of BTP in PBS at pH 7.4, which matches the peak emission of the deprotonated phenolate anion of benzothiazol derivative in aqueous solution.30,31
MTT assays (Fig. S10†) revealed that BTPB of concentrations below 40 μM does not exhibit obvious cytotoxicity living HepG2 cells. Finally, encouraged by the above promising results, especially fast response rate (within 30 seconds), excellent sensitivity (detection limit = 12.3 nM) and high selectivity, BTPB was employed for confocal imaging of ONOO− in the liver tissue sample. ONOO− formation has long been implicated in APAP-induced liver injury on the basis of indirect detection, i.e., immunostaining of protein nitration, which, however, cannot be used as unique evidence for ONOO− formation. To address this question, the mice administrated APAP overdose (400 mg kg−1) were employed to monitor ONOO− formation in situ. Six hours after administrated APAP overdose, living heart tissues were perfused with BTPB (1.0 μM) in saline for 3 min under anesthesia. Mouse livers were then excised and cryosectioned at 10 μm intervals for confocal imaging and hematoxylin and eosin (HE) staining. The mice without APAP administration were perfused with BTPB as the control.
As shown in Fig. 5, HE staining of liver sections indicate the substantial centrilobular necrosis 6 hours after APAP administration, but no necrosis is observed in the control group without APAP administration, suggesting that APAP overdose induces liver injury, which is consistent with those studies.32,33 Then liver sections were monitored using a confocal microscope over the wavelength range of 417–477 nm with an excitation wavelength at 408 nm. Without APAP administration no fluorescence in the liver sections was observed, while strong blue fluorescence in the centrilobular regions was imaged after treatment with overdose APAP, which correlated well with the areas of necrosis. This result indicates the generation of ONOO− during APAP-induced liver injury, which is the first direct evidence provided by our study. Also, these observations suggest that BTPB has satisfactory cell permeability and is well-retained during histological sample preparation and sensitive enough to visualize endogenous ONOO− in live tissues.
 | ||
| Fig. 5 Hematoxylin and eosin staining of liver sections for hepatic necrosis (a–d) and confocal microscopic fluorescence images of liver sections for detecting of ONOO− generation via BTPB (e–h) in controls and 6 h after 400 mg kg−1 APAP. (a) Control, ×100. (b) Control, ×200. (c) 6 h after 400 mg kg−1 APAP, ×100. (d) 6 h after 400 mg kg−1 APAP, ×200. (e) Control, ×100. (f) 6 h after 400 mg kg−1 APAP, ×100. (g) 6 h after 400 mg kg−1 APAP, ×200. (h) 6 h after 400 mg kg−1 APAP, ×400. | ||
Conclusions
In summary, we present herein a ratiometric fluorescent probe for the highly sensitive and selective detection and hepatic imaging of ONOO− after APAP overdose. This probe was designed based on a ONOO− specific reaction with the boronic acid group. Our study provided the direct evidence for supporting the generation of ONOO− in APAP-induced liver injury. We believe that the probe with excellent spectroscopic properties will find its applications in understanding the actions of ONOO− in vivo.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We acknowledge the National Natural Science Foundation of China (Grant No. 81272448).Notes and references
- D. W. Kaufman, J. P. Kelly, L. Rosenberg, T. E. Anderson and A. A. Mitchell, J. Am. Med. Assoc., 2002, 287, 337–344 CrossRef PubMed.
- D. S. Budnitz, M. C. Lovegrove and A. E. Crosby, Am. J. Prev. Med., 2011, 40, 585–592 CrossRef PubMed.
- P. Nourjah, S. R. Ahmad, C. Karwoski and M. Willy, Pharmacoepidemiol. Drug Saf., 2006, 15, 398–405 CrossRef PubMed.
- W. M. Lee and S. Respir, Crit. Care Med., 2012, 33, 36–45 Search PubMed.
- M. R. McGill and H. Jaeschke, Pharm. Res., 2013, 30, 2174–2187 CrossRef CAS PubMed.
- H. Jaeschke, M. R. McGill and A. Ramachandran, Drug Metab. Rev., 2012, 44, 88–106 CrossRef CAS PubMed.
- J. A. Hinson, S. L. Pike, N. R. Pumford and P. R. Mayeux, Chem. Res. Toxicol., 1998, 11, 604–607 Search PubMed.
- T. R. Knight, A. Kurtz, M. L. Bajt, J. A. Hinson and H. Jaeschke, Toxicol. Sci., 2001, 62, 212–220 CrossRef CAS PubMed.
- S. Bartesaghi, G. Ferrer-Sueta, G. Peluffo, V. Valez, H. Zhang, B. Kalyanaraman and R. Radi, Amino Acids, 2007, 32, 501–515 CrossRef CAS PubMed.
- R. Radi, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4003–4008 CrossRef CAS PubMed.
- R. Radi, Acc. Chem. Res., 2013, 46, 550–559 CrossRef CAS PubMed.
- C. Batthyány, S. Bartesaghi, M. Mastrogiovanni, A. Lima, V. Demicheli and R. Radi, Antioxid. Redox Signaling, 2017, 26, 313–328 CrossRef PubMed.
- R. Radi, J. Biol. Chem., 2013, 288, 26464–26472 CrossRef CAS PubMed.
- Q. Zhang, N. Zhang, Y. T. Long, X. Qian and Y. Yang, Bioconjugate Chem., 2016, 27, 341–353 CrossRef CAS PubMed.
- D. Yang, H. L. Wang, Z. N. Sun, N. W. Chung and J. G. Shen, J. Am. Chem. Soc., 2006, 128, 6004–6005 CrossRef CAS PubMed.
- Z. N. Sun, H. L. Wang, F. Q. Liu, P. K. H. Tam and D. Yang, Org. Lett., 2009, 11, 1887–1890 CrossRef CAS PubMed.
- T. Peng and D. Yang, Org. Lett., 2010, 12, 4932–4935 CrossRef CAS PubMed.
- J. Zielonka, A. Sikora, J. Joseph and B. Kalyanaraman, J. Biol. Chem., 2010, 285, 14210–14216 CrossRef CAS PubMed.
- W. Zhou, Y. Cao, D. Sui and C. Lu, Anal. Chem., 2016, 88, 2659–2665 CrossRef CAS PubMed.
- Z. Wang, X. Teng and C. Lu, Anal. Chem., 2015, 87, 3412–3418 CrossRef CAS PubMed.
- A. Sikora, J. Zielonka, M. Lopez, A. Dybala-Defratyka, J. Joseph, A. Marcinek and B. Kalyanaraman, Chem. Res. Toxicol., 2011, 24, 687–697 Search PubMed.
- A. Sikora, J. Zielonka, M. Lopez, J. Joseph and B. Kalyanaraman, Free Radical Biol. Med., 2009, 47, 1401–1407 CrossRef CAS PubMed.
- J. Zielonka, A. Sikora, M. Hardy, J. Joseph, B. P. Dranka and B. Kalyanaraman, Chem. Res. Toxicol., 2012, 25, 1793–1799 Search PubMed.
- R. Hu, J. Feng, D. Hu, S. Wang, S. Li, Y. Li and G. Yang, Angew. Chem., Int. Ed., 2010, 49, 4915–4918 CrossRef CAS PubMed.
- H. W. Chen, Y. Xing and Y. Pang, Org. Lett., 2011, 13, 1362–1365 CrossRef PubMed.
- P. C. Sharma, A. Sinhmar, A. Sharma, H. Rajak and D. P. Pathak, J. Enzyme Inhib. Med. Chem., 2013, 28, 240–266 CrossRef PubMed.
- R. D. Taylor, M. MacCoss and A. D. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed.
- A. B. Draganov, K. Wang, J. Holmes, K. Damera, D. Wang, C. Dai and B. Wang, Chem. Commun., 2015, 51, 15180–15183 RSC.
- M. A. Chari, D. Shobha and T. Sasaki, Tetrahedron Lett., 2011, 52, 5575–5580 CrossRef.
- M. M. Henary and C. J. Fahrni, J. Phys. Chem. A, 2002, 106, 5210–5220 CrossRef CAS.
- J. E. Kwon and S. Y. Park, Adv. Mater., 2011, 23, 3615–3642 CrossRef CAS PubMed.
- P. James, L. A. MacMillan-Crow and J. A. Hinson, J. Pharmacol. Exp. Ther., 2012, 340, 134–142 CrossRef PubMed.
- C. Saito, C. Zwingmann and H. Jaeschke, Hepatology, 2010, 51, 246–254 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, supplemental spectra, and the 1H-, 13C-NMR, and MS spectra. See DOI: 10.1039/c8ra10053e |
| This journal is © The Royal Society of Chemistry 2019 |