
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSite-specific labeling of an anti-MUC1 antibody: probing the effects of conjugation and linker chemistry on the internalization process†
Hongjiao
Xu‡
,
Lu
Gan‡
,
Ying
Han
,
Yifan
Da
,
Jiale
Xiong
,
Sihua
Hong
,
Qian
Zhao
,
Nazi
Song
,
Xiaoqing
Cai
 * and
Xianxing
Jiang
* and
Xianxing
Jiang
School of Pharmaceutical Sciences, Sun Yat-sen University, 132 East Outer Ring Road, Guangzhou, 510006, China. E-mail: caixq7@mail.sysu.edu.cn
First published on 15th January 2019
Abstract
Antibody-drug conjugates (ADCs) have recently received enormous attention as an attractive approach for cancer therapy. Although ADC design has been believed to be important for the relative efficacy of ADCs, it remains unexplored how the structural characteristics of ADCs would impact the internalization process and intracellular trafficking of the molecules. Herein, we report our efforts in investigating the cellular endocytosis implications of the conjugation and linker chemistry in designing antibody-based agents. A series of anti-MUC1 single-chain variable fragment (scFv-SM3) conjugates were designed with unique structural characteristics ranging from conjugation methods, sites of attachment and linker chemistry. In vitro confocal imaging showed that both random lysine-conjugation and site-specific conjugation, including C-terminus modification or internal site conjugation, could afford antibody conjugates with similar binding affinity and cellular uptake to target-expressing cells. Time-course internalization studies demonstrated that SM3-conjugates with short polyethylene glycol linkers outcompeted those that lack any hydrophilic linkers for higher cellular uptake and faster internalization rate. The SM3-conjugates with the highest affinity and internalization rate were also tested in mouse xenograft models using MUC1-overexpressing tumor cells. Our results indicate that the linker and conjugation chemistry play an important role in the internalization process of antibody conjugates, and this in turn could impact the therapeutic effects of ADCs.
Introduction
Full-size monoclonal antibodies (mAb) and antibody fragments, such as fragments antigen-binding (Fabs), single-chain variable fragments (scFvs) and single-domain antibodies (sdAb), have been extensively used as excellent targeting and binding molecules in biomedical sciences.1,2 For the past two decades, small molecule-antibody bioconjugates have emerged as a novel class of targeting molecules that rival their individual components in their potential for therapeutic and diagnostic applications.3,4 Bioconjugation methods play a critical role in creating potent antibody-based agents to tackle different medical needs. Traditional conjugation strategies typically target solvent accessible reactive amino acids, such as lysine or cysteine, which are inevitably associated with heterogeneous products with a wide range of conjugation ratios.5 To overcome the limitations of random conjugation, site-specific conjugation strategies have been developed, which generally can be classified into three types: (1) protein engineering with additional cysteine residues;6,7 (2) protein modification at the C or N terminus, especially through enzymatic ligation strategies;8,9 and (3) genetic incorporation of bioorthogonal unnatural amino acids (UAAs) into proteins.10Antibody-drug conjugates (ADCs), which combine the targeting specificity of antibodies with the cytotoxicity of anticancer drugs, have recently received enormous attentions as an attractive approach for cancer therapy.3,11 The mechanism of action of ADCs is thought to be related to the internalization process of the bioconjugates. The continuous internalization moves an ADC from its binding site at the plasma membrane to the lysosome, where the molecule undergoes degradation and releases the toxic drug.5,12,13 In another words, the intracellular behaviors of ADCs, including endocytosis, intracellular trafficking and catabolism, could be very essential for the bioactivity of the payload, which in turn determines the pharmacokinetic profile, toxicity and efficacy of ADCs.14,15 However, it remains unexplored how a particular antibody conjugate functions at the molecular level, especially how the structure of an ADC, including the design and selection of linker and the conjugation chemistry, correlates to its cellular uptake and internalization. As the development of current linker technology, more emphasis is being placed on the characteristics of cleavable and non-cleavable linkers, focusing merely on the drug-release mechanism and other biophysical properties of the linker itself. Very little is known about the correlations between the linker/conjugation chemistry and the internalization process. Therefore, our goal here is to explore the effects of ADC structure, including the selection of linker, the site of attachment and the conjugation chemistry, on the delivery and intracellular trafficking of the molecules.
The model antibody we selected was an anti-MUC1 antibody (scFv-SM3). MUC1 is a transmembrane glycoprotein overexpressed on various cancer cells, including breast cancers and ovarian carcinomas, thus has been recognized as an important molecular target for cancer treatment.16,17 Previous studies showed that antibodies could be internalized upon binding to MUC1-expressing tumor cells, indicating that MUC1 is a suitable target for drug delivery.18 Accordingly, several MUC1-binding ligands have been developed for targeted delivery of toxic compounds to cancer cells in vitro.18 However, to the best of our knowledge, no mechanic studies have been done in elucidating the endocytosis and intracellular trafficking pathways of anti-MUC1 antibodies. Especially, no correlation has been investigated between structure modification and cellular delivery of the MUC1-specific ADCs. Furthermore, there is still a lack of functional in vivo studies in the development of anti-MUC1 bioconjugates.19,20
Herein, we prepared four distinct SM3-conjugates with unique structural characteristics ranging from the conjugation chemistry, the conjugation site and the selection of linker (Fig. 1). Confocal imaging studies were performed to evaluate and compare the binding affinity and cellular uptake of these SM3-conjugates with MUC1-expressing cells. To further investigate the linker chemistry as it relates to cellular uptake and internalization, we also carried out the time-course internalization study to compare the internalization process of SM3-conjugates with short polyethylene glycol linkers and those without any hydrophilic linkers. Lastly, the SM3-conjugates with the highest affinity and internalization rate were tested for their in vivo tumor-targeting activity in mouse xenograft models using MUC1-overexpressing tumor cells.
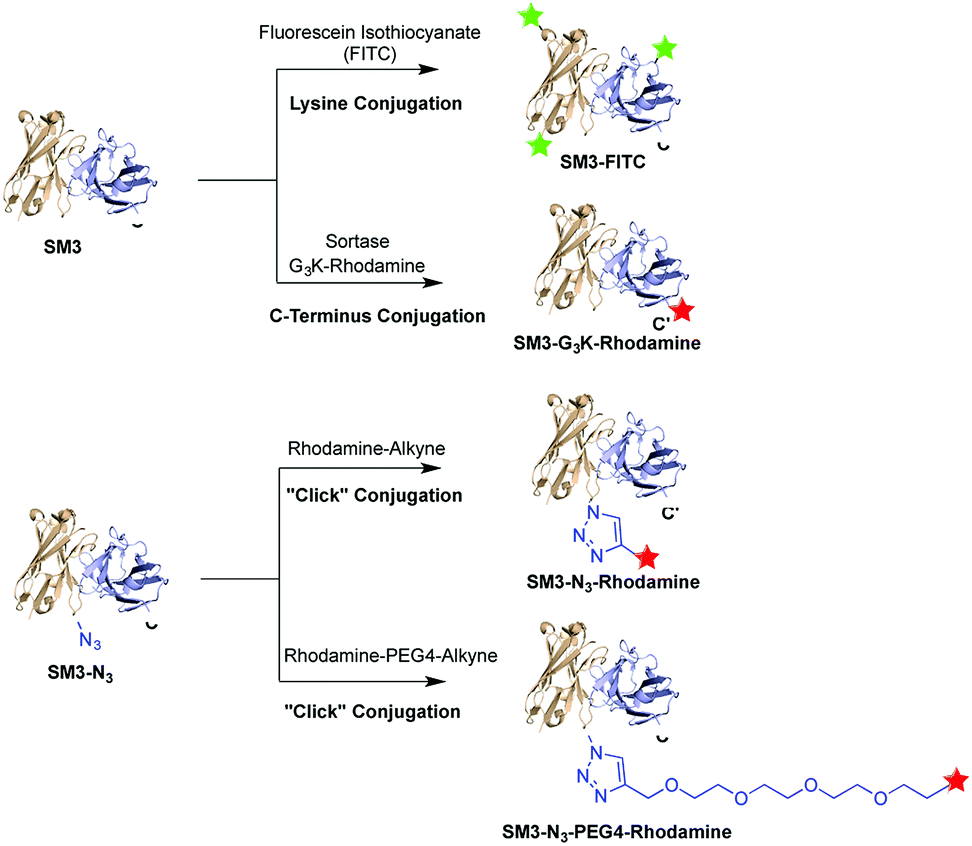 | ||
| Fig. 1 A single-chain variable fragment scFv-SM3 was labeled via three different conjugation methods: random lysine conjugation of FITC (SM3-FITC), sortase-mediated C-terminus labeling of rhodamine (SM3-G3K-rhodamine), “click” chemical conjugation of rhodamine at the internal site of SM3 (SM3-N3-rhodamine or SM3-N3-PEG4-rhodamine). | ||
Results and discussion
Antibody conjugates design
To investigate the biological implication in designing antibody-based conjugates, four anti-MUC1 antibody conjugates, termed SM3-FITC, SM3-G3K-rhodamine, SM3-N3-rhodamine and SM3-N3-PEG4-rhodamine, were designed and prepared (Fig. 1). These bioconjugates were designed based on the following: (1) both random and site-specific conjugation strategies were applied for our ADC design, which was aiming to compare the cellular behavior difference between heterogeneous bioconjugates (SM3-FITC) and homogeneous bioconjugates (SM3-G3K-rhodamine and SM3-N3-rhodamine). (2) To further probe the effect of the conjugation chemistry in terms of conjugation site, two different site-specific conjugation approaches were chosen, i.e., enzyme-mediated ligation strategy to attach the payload at the C-terminus of protein (SM3-G3K-rhodamine), and genetically installation of an azide handle to incorporate the payload into the internal site of the antibody backbone (SM3-N3-rhodamine). (3) A polyethylene glycol (PEG) linker (MW: 192.2) was selected to attach the payload into the internal site of the antibody (SM3-N3-PEG4-rhodamine). Polyethylene glycol moieties have proven useful to decrease the overall hydrophobicity of antibody,21,22 which could be an important feature for our intended studies of ADC endocytosis and intracellular trafficking.Preparation of SM3-G3K-rhodamine through sortase-mediated ligation
Sortases are a family of membrane-anchored transpeptidases found in Gram-positive bacteria, and have been used to modify the C or N terminus of a biological entity with peptides or other functional units.23,24 We first exploited sortase-mediated ligation to attach the payload precisely at the C-terminus of the studied antibody. Our reasoning for such an approach is that this method is site-specific and robust, and most importantly, the reaction site (C-terminus) is well removed from the antigen-binding domain. Accordingly, two essential components are required for this ligation system: (1) a protein substrate engineered with a sortase-recognition motif (LPXTG), and (2) an oligo-glycine molecule as the sortase nucleophile.We began this enzymatic approach through overexpression of the studied antibody, an anti-MUC1 single-chain variable fragment (scFv-SM3, MW: 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 490). The protein SM3 was engineered with a C-terminal LPETG sequence followed by a His6 tag. The His6 tag can be removed during the course of sortase ligation, which should therefore simply the purification process of the protein product.25 The SM3 protein was obtained from the E. coli periplasmic space (∼10 mg l−1), and analyzed by SDS-PAGE (ESI Fig. S1†) and western blot analysis (data not shown). Size-exclusion chromatography was used to observe the monomeric state of the final product under native conditions (ESI Fig. S3†). The in vitro binding activity of SM3 was confirmed through confocal imaging studies using MUC1-expressing cells (ESI Fig. S6A†). Binding of SM3 to MUC1-positive MCF7 cells was measured at various concentrations of SM3 and assessed by flow cytometry, and the results showed a half maximal binding concentration (EC50) of 2.7 ± 0.10 nM (ESI Fig. S6B†).
490). The protein SM3 was engineered with a C-terminal LPETG sequence followed by a His6 tag. The His6 tag can be removed during the course of sortase ligation, which should therefore simply the purification process of the protein product.25 The SM3 protein was obtained from the E. coli periplasmic space (∼10 mg l−1), and analyzed by SDS-PAGE (ESI Fig. S1†) and western blot analysis (data not shown). Size-exclusion chromatography was used to observe the monomeric state of the final product under native conditions (ESI Fig. S3†). The in vitro binding activity of SM3 was confirmed through confocal imaging studies using MUC1-expressing cells (ESI Fig. S6A†). Binding of SM3 to MUC1-positive MCF7 cells was measured at various concentrations of SM3 and assessed by flow cytometry, and the results showed a half maximal binding concentration (EC50) of 2.7 ± 0.10 nM (ESI Fig. S6B†).
SortaseAΔN59 (SrtAΔN59) from Staphylococcus aureus was prepared as reported,26 and the identity and activity of SrtAΔN59 was confirmed by SDS-PAGE (ESI Fig. S2A†) and enzymatic kinetic studies (data not shown). We also synthesized the GGGK-rhodamine, which participates in sortase-mediated ligation to afford SM3-G3K-rhodamine (Fig. 2A). Following conjugation, no His6-tagged SM3 was detected by western blot analysis (data not shown), which indicated efficient cleavage of the tag sequence by this enzymatic ligation. SDS-PAGE demonstrates that SrtAΔN59 was removed completely. Fluorescence scanning (λem = 580 nm) of SDS-PAGE gel indicated that the rhodamine moiety was conjugated successfully with SM3 (Fig. 2B). The identity of SM3-G3K-rhodamine was further confirmed by MALDI-MS (Fig. 2C).
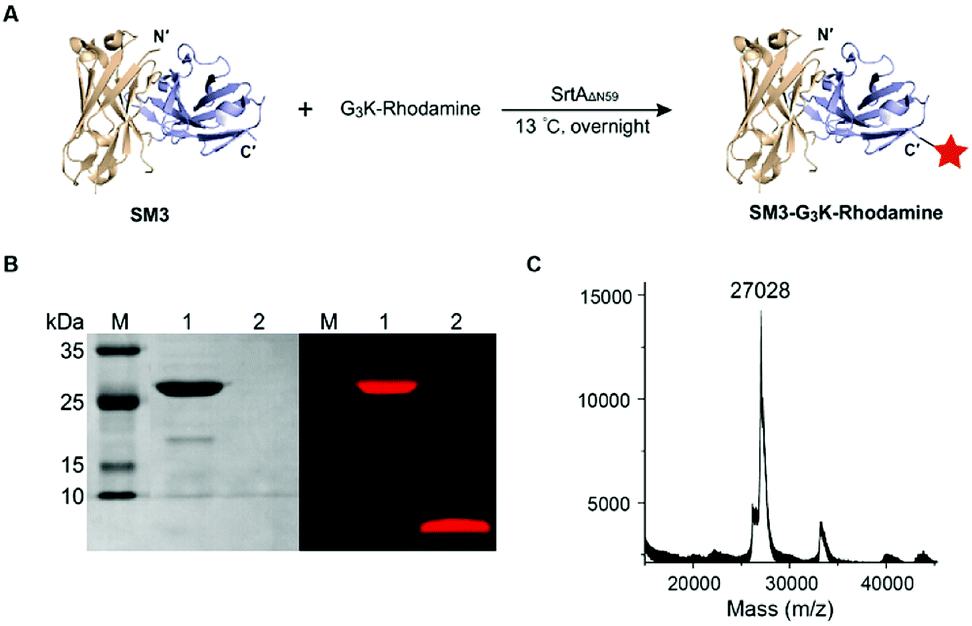 | ||
| Fig. 2 Labeling SM3 with G3K-rhodamine via sortase-mediated ligation. (A) Preparation of SM3-G3K-rhodamine. Condition: 13 °C, overnight. (B) SDS-PAGE analysis of the reaction. Left panel: coomassie-stained gel. Right panel: in-gel fluorescence excited at 580 nm. Lane 1: SM3-G3K-rhodamine; lane 2: unconjugated rhodamine that was removed from the reaction system through a microspin column. (C) MALDI-MS analysis of SM3-G3K-rhodamine. | ||
Preparation of SM3-N3-rhodamine and SM3-N3-PEG4-rhodamine based on expanded genetic codes
Unnatural amino acids (UAA) can be incorporated in proteins to enable unique orthogonal chemical modification, thus presenting a novel platform to create site-specific ADCs.27 As shown in Fig. 3, azidolysine 1 containing a long side chain was chosen as a pyrrolysine derivative to increase the solvent exposure of the azido group. We chose to replace the site K43 of SM3 with azidolysine 1 based on the following principles: (1) it is located in a middle region away from the antigen-binding domain; (2) it is selected from a long loop region, distant from the interface of the heavy and light chains, thus minimizing the possibility to perturb the tertiary structure of the recombinant protein; (3) this site is solvent accessible, and can be further modified through bioorthogonal chemistry. Accordingly, we employed the conventional amber-codon suppression technology to prepare the clickable SM3-N3 containing azidolysine 1 at the site K43. The product was purified by gel filtration chromatography, and then analyzed by SDS-PAGE (ESI Fig. S3†). To further evaluate the specificity of UAA incorporation, trypsin digests of SM3-N3 were analyzed using tandem mass spectrometry (MS/MS), which confirmed the incorporation of azidolysine 1 at the site K43 (ESI Fig. S4 and S5†).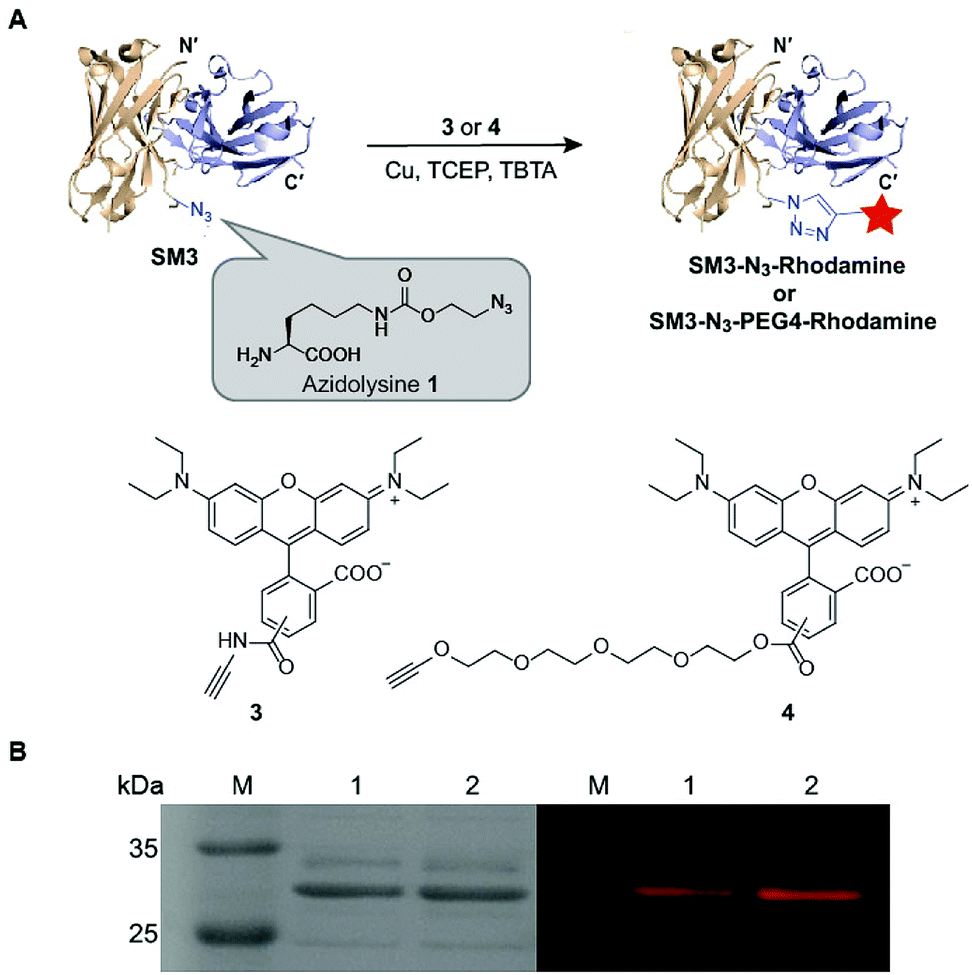 | ||
| Fig. 3 Labeling of SM3-N3 with rhodamine alkyne derivatives via “click” reaction. (A) Preparation of SM3-N3-rhodamine and SM3-N3-PEG4-rhodamine. Condition: 4 °C, overnight. (B) SDS-PAGE analysis of the click reactions. Left panel: coomassie-stained gel. Right panel: in-gel fluorescence excited at 580 nm. Lane 1: SM3-N3-rhodamine; lane 2: SM3-N3-PEG4-rhodamine. | ||
To test the binding specificity and affinity of SM3-N3, we undertook confocal imaging studies and flow cytometry with MUC1-overexpressing MCF7 cells. SM3-N3 revealed a significantly stronger immunofluorescence staining compared with MUC1-negative 293T cells (ESI Fig. S6A†). In addition, a quantitative binding assay was performed using flow cytometry. The results showed a concentration-dependent binding of SM3-N3 with MCF7 cells, exhibiting an EC50 value of 2.7 ± 0.08 nM (ESI Fig. S6C†).
Next, we applied the Cu-catalyzed alkyne–azide cycloaddition to prepare site-specific SM3-N3-rhodamine and SM3-N3-PEG4-rhodamine, respectively (Fig. 3A). As shown in Fig. 3B, SDS-PAGE presented us with a unique fluorescent band, which indicated the successful attachment of the rhodamine-alkyne 3 and rhodamine-PEG4-alkyne 4, respectively, to the clickable SM3-N3.
Preparation of SM3-FITC via random lysine conjugation
Random lysine conjugation was achieved by incubation of scFv-SM3 with fluorescein isothiocyanate (FITC) under the alkaline condition (ESI Fig. S7A†).28 The final product termed SM3-FITC was purified by size-exclusion chromatography, and characterized by SDS-PAGE (ESI Fig. S7B†).In vitro cellular binding and uptake study
With the four SM3-conjugates (SM3-FITC, SM3-G3K-rhodamine, SM3-N3-rhodamine and SM3-N3-PEG4-rhodamine) in hands, confocal imaging study was carried out with MUC1-postive MCF7 cells to determine and compare the cellular activity of the SM3-conjugates. In particular, we are interested in understanding the potential impact of the conjugation methods, and the conjugation site, and the PEG linker on the binding affinity and cellular uptake of the SM3-conjugates. As shown in Fig. 4, we observed significant fluorescent signals for each of the SM3-conjugates after 1 h incubation of the SM3-conjugates with MCF7 cells. First, we compared the cellular activity of those SM3-conjugates that lack PEG4 linkers. As shown in Fig. 4, the SM3-FITC conjugate prepared through lysine conjugation, exhibited similar binding and cellular uptake activity, compared with the other two SM3-conjugates that were prepared through site-specific conjugation, including C-terminus ligation (SM3-G3K-rhodamine) or internal site conjugation (SM3-N3-rhodamine). As noted already, no significant differences were observed between C-terminus ligation and internal site conjugation. Notably, for the treatment with SM3-conjugates that lack the PEG linkers (SM3-FITC, SM3-G3K-rhodamine or SM3-N3-rhodamine), the fluorescent signals were only detected in the cytoplasm, especially in the perinuclear zone of MCF7 cells. However, SM3-N3-PEG4-rhodamine with a PEG linker was found to well distribute in the entire cytoplasm region with a slightly stronger fluorescent signal, and some accumulated fluorescent signals were also observed in the cytoplasmic membrane, which may indicate a possible enhanced surface absorption for SM3-N3-PEG4-rhodamine.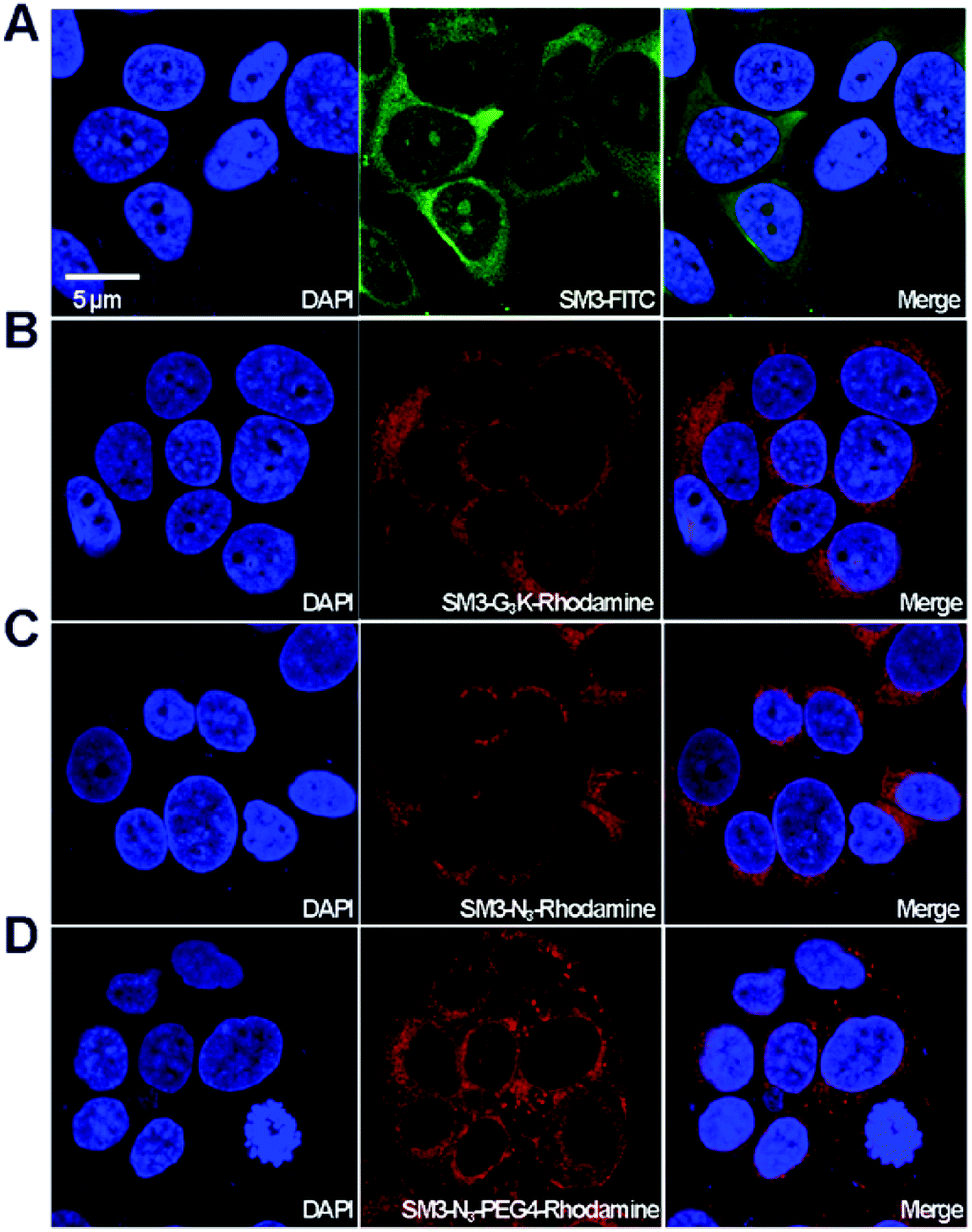 | ||
| Fig. 4 In vitro affinity of SM3 conjugates. MCF7 cells were incubated with (A) SM3-FITC, (B) SM3-G3K-rhodamine, (C) SM3-N3-rhodamine, (D) SM3-N3-PEG4-rhodamine at 37 °C for 1 h. The nucleus was stained with DAPI. The red channel represents as rhodamine (λem = 520–580 nm) and the green channel represents as FITC (λem = 460–530 nm). Scale bar in panel (A) applies to all images. | ||
Taken together, these results suggest that either random or site-specific conjugation, including C-terminus ligation or internal site conjugation, afforded SM3-conjugates with comparable binding affinity and cellular uptake to target-expressing cells. One crucial finding is that a hydrophilic PEG4 linker seems to facilitate the binding of SM3 conjugates to the cell surface, which might in turn promote the cell uptake.
Time-course internalization study
Time-course imaging study was performed to determine and compare the internalization process of SM3-N3-rhodamine and SM3-N3-PEG4-rhodamine. In addition, we focused on measuring the colocalization of these two conjugates with lysosomes. LAMP1 antibody was used as the lysosome marker. Accordingly, MCF7 cells were incubated with SM3-N3-rhodamine and SM3-N3-PEG4-rhodamine for 0.5, 1, 3, 5 and 7 h, and then confocal imaging study was performed immediately to visualize the internalization process. After a 0.5 h incubation, red fluorescent signals were detected in the cytoplasm for both SM3-N3-rhodamine and SM3-N3-PEG4-rhodamine (Fig. 5). Compared to SM3-N3-PEG4-rhodamine, SM3-N3-rhodamine exhibited a lower cellular uptake and slower internalization rate over the course of time. In addition, the red fluorescent intensity of SM3-N3-rhodamine seemed to decrease gradually after 1 h (Fig. 5A). However, we observed significant persistence of the rhodamine signal for SM3-N3-PEG4-rhodamine with the duration of time (Fig. 5B). The results also indicated that both of SM3-N3-rhodamine and SM3-N3-PEG4-rhodamine undertook a lysosome-targeting endocytosis pathway. A faster internalization speed was also shown for SM3-N3-PEG4-rhodamine compared with SM3-N3-rhodamine. Moreover, SM3-N3-PEG4-rhodamine displayed a significantly higher degree of lysosome colocalization over the entire course of time.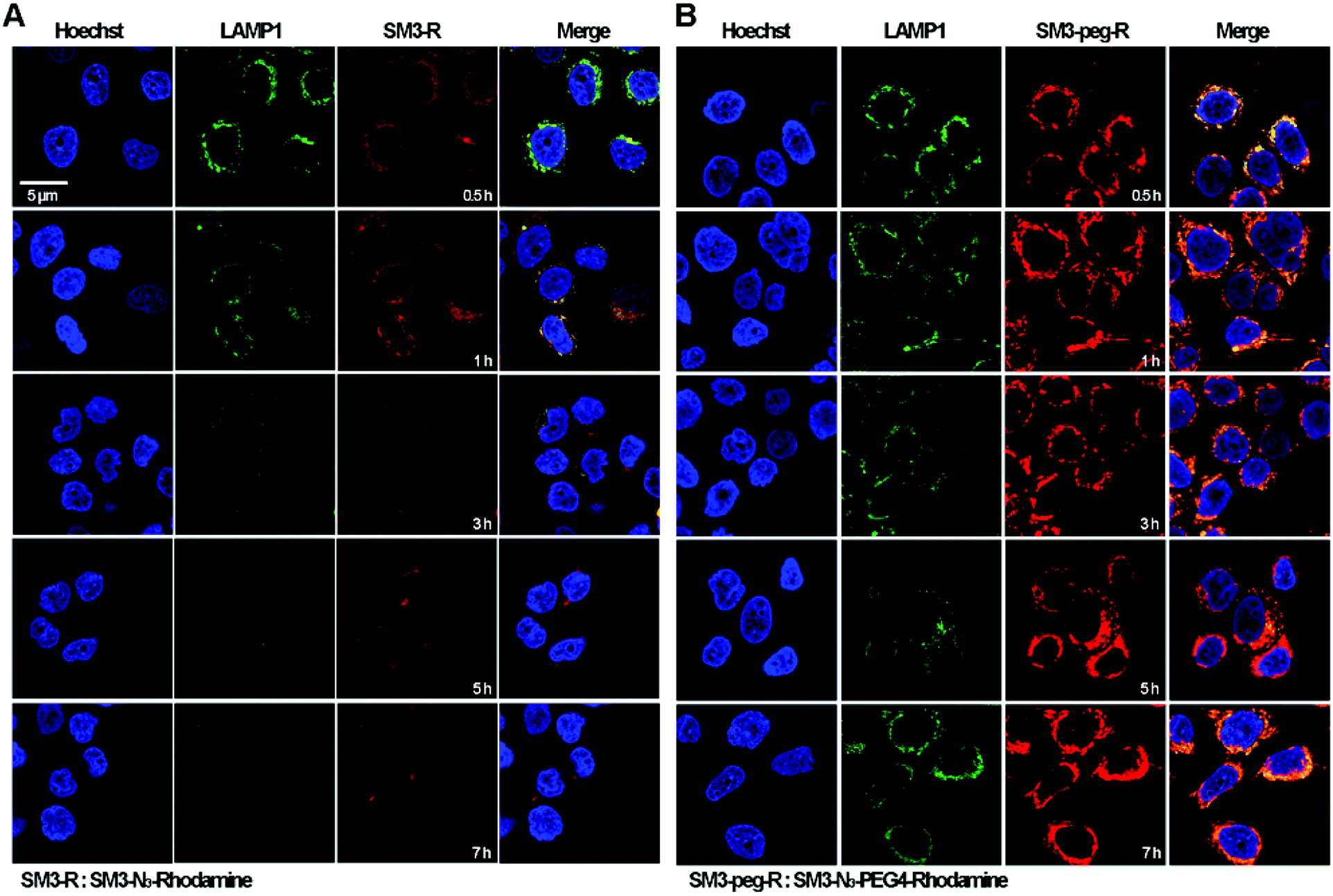 | ||
| Fig. 5 In vitro internalization studies of SM3-N3-rhodamine and SM3-N3-PEG4-rhodamine. MCF7 cell line was incubated with 1 μg ml−1 of LAMP1 at 37 °C for 1 h followed by the treatment of (A) SM3-N3-rhodamine or (B) SM3-N3-PEG4-rhodamine at 37 °C for 0.5, 1, 3, 5 and 7 h. The nucleus was stained with Hoechst 33342. Scale bar in panel (A) applies to all images. | ||
In this study, the attachment of a PEG4 linker at a single internal site of scFv-SM3 was shown to have a direct and on cellular uptake and internalization, suggesting that this short PEG linker was responsible for the internalization of SM3-conjugates. Presumably, the attachment of a short PEG moiety would affect the protein hydrophobicity, which might consequently facilitate the process of receptor-mediated endocytosis. PEGylation is a popular strategy for improving the in vivo pharmacokinetics and stability of proteins or peptides.29 Whether the biophysical property of PEG linkers is associated with the cellular uptake and endocytosis will require additional research to elucidate the endocytosis mechanism of conjugated protein. Future study should also be conducted to determine whether attachment of other hydrophilic linkers or linkers with different biophysical properties would have an impact on the internalization process. However, what is clear is that the unique properties of a chemical linker provide a promising design strategy to increase ADC internalization, which in turn may improve the therapeutic effects of ADCs.
In vivo imaging studies on xenograft models
We used the human breast tumor xenograft model to assess the in vivo tumor-targeting properties of SM3-N3-PEG4-rhodamine in comparison with a commercial monoclonal antibody (mAb-FITC). Animals were inoculated with MUC1-expressing cells, and the subcutaneous tumors were allowed to grow to an average tumor volume of 150 ± 20 mm3 (21 days). Mice with established subcutaneous xenograft tumors were treated with mAb-FITC or SM3-N3-PEG4-rhodamine, and the in vivo bioluminescence was acquired at different time points (2, 4 and 24 h). Meanwhile, a nude mouse without tumor was also treated with SM3-N3-PEG4-rhodamine, and was used as a negative control. To our surprise, neither mAb-FITC nor SM3-N3-PEG4-rhodamine displayed significant tumor-accumulation activity in tumor-bearing mice (Fig. 6). For the nude mouse, a fluorescence signal was detected dominantly in the kidney, and then progressively decreased after 2 h post injection (Fig. 6A), indicating that SM3-N3-PEG4-rhodamine rapidly went through a kidney clearance pathway without signs of MUC1 binding. In contrast, in spite of no specific binding at the tumor site, fluorescence signals could be detected in the whole body of the tumor-bearing mice at 2 h, and the signals persisted over the next 24 h (Fig. 6B and C).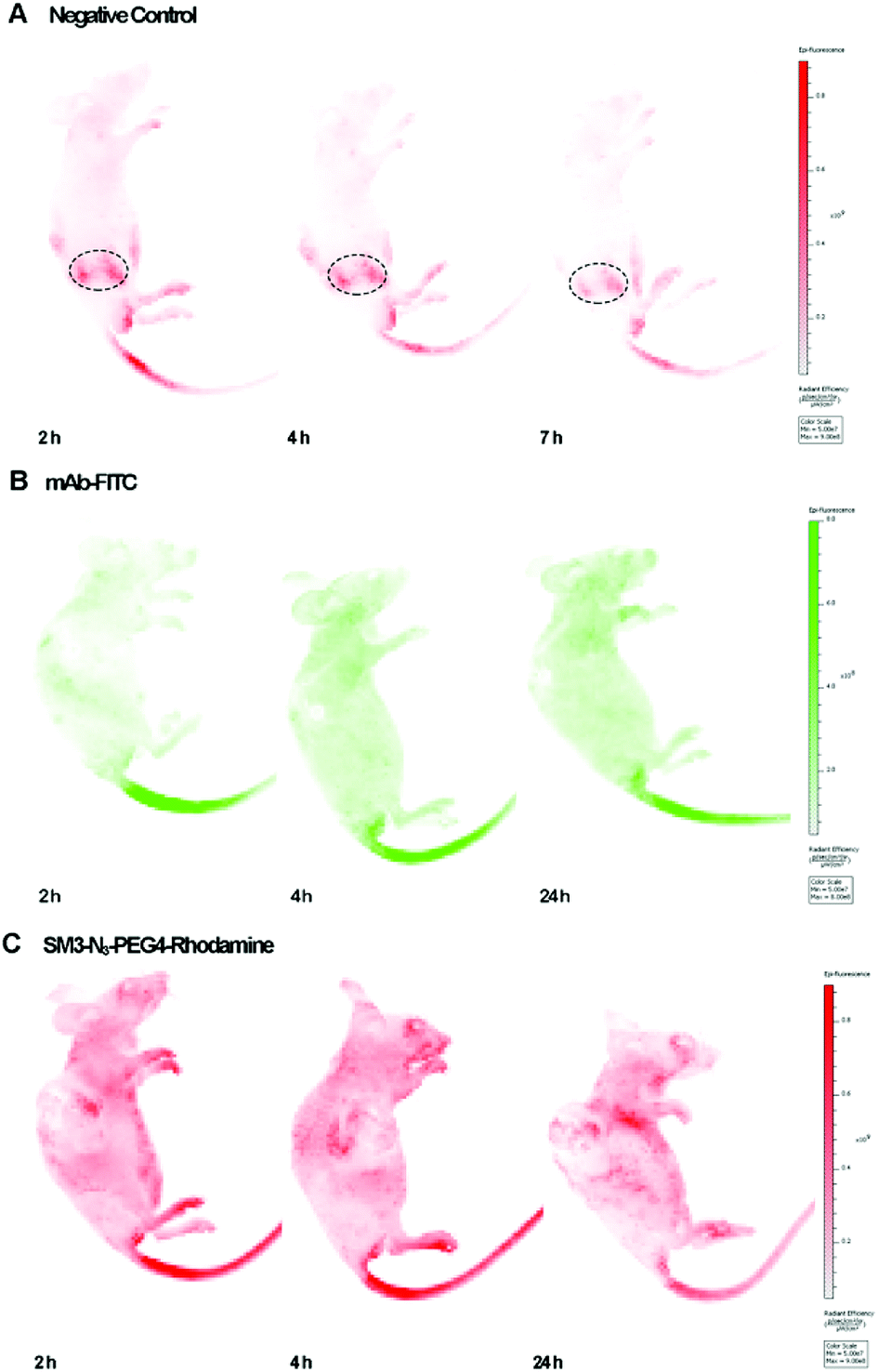 | ||
| Fig. 6 In vivo imaging studies on xenograft models. (A) Representative IVIS imaging of nude mice administrated with SM3-N3-PEG4-rhodamine at 2, 4 and 7 h. λex = 560 nm, λem = 620 nm. (B) Representative IVIS imaging of mice bearing A549 tumor administrated with mAb-FITC at 2, 4 and 24 h. λex = 480 nm, λem = 520 nm. (C) Representative IVIS imaging of mice bearing ZR-75-1 tumor administrated with SM3-N3-PEG4-rhodamine at 2, 4 and 24 h. λex = 560 nm, λem = 620 nm. | ||
Although MUC1 has proven to be a reliable molecular biomarker for cancer diagnosis, previous studies have shown that MUC1 could be cleaved proteolytically into an extracellular α-subunit and a cytoplasmic and transmembrane β-subunit.16,17 Most anti-MUC1 antibodies, including SM3 are actually targeting only the α-subunit located on the cell surface. The α-subunit and β-subunit are generally associated as a stable non-covalent format within the cell membrane; however, during tumor progression, α-subunit could be dissociated from the MUC1 complex, and then released from the cell surface to the circulation system.17
Therefore, it is likely that the persistence of fluorescent signals detected in the whole body of tumor-bearing mice (Fig. 6B and C) were caused by a high level of MUC1 α-subunit in the sera of mice. To test our hypothesis, these three sets of mice were sacrificed followed by the in vivo imaging studies, and the corresponding sera were collected at the time of sacrifice and subjected to a western blot assay, which confirmed the existence of MUC1 α-subunit in the sera of tumor-bearing mice (ESI Fig. S8†). Further in vivo studies of MUC1 from the standpoint of target biology should shed light on the hypothesis.
Conclusions
In summary, four SM3-conjugates were designed and prepared to investigate how the conjugation and linker chemistry would have an impact on ADC endocytosis and intracellular trafficking. Each of these SM3-conjugates represents unique structural characteristics ranging from conjugation methods, sites of attachment and linker chemistry. Both random lysine-conjugated and site-specific SM3-conjugates, including those prepared via C-terminus modification or internal site conjugation, were found to exhibit similar binding affinity and cellular uptake to MUC1-expressing cells. Time-course imaging studies suggested that SM3-conjugates with a short PEG linker (SM3-N3-PEG4-rhodamine) displayed significantly higher cellular uptake and faster internalization rate compared to those that lack any hydrophilic linkers. Surprisingly, in vivo studies showed that neither SM3-N3-PEG4-rhodamine nor commercial mAb possessed any significant tumor-targeting activity. Further antigen analysis suggested that the lack of tumor-targeting activity in SM3-conjugates was probably due to a high level of MUC1 α-subunit in the sera of the tumor-bearing mice. Overall, this present work suggests that the linker and conjugation chemistry is a crucial determinant for the internalization process, and that “linker-structure-tailoring” may represent a unique and powerful approach for developing ADC therapeutics.Experimental procedures
Materials
Fluorescein isothiocyanate (FITC) was purchased from Sangong Biotech, Shanghai, China. The G3K-rhodamine peptide and azidolysine 1 was synthesized according to the reported procedure.9,30 CD227 (MUC1) monoclonal antibody was purchased from eBioscience™, ThermoFisher Scientific, MA, USA. FITC labeled MUC1 (CD227) monoclonal antibody was purchased from BD Pharmingen™, NJ, USA. Anti-6X His-tag antibody (FITC) was purchased from Abcam, Cambridge, UK. The E. coli strain DH5α and BL21 (DE3) were purchased from TransGen Biotech, Beijing, China. The breast cancer cell lines ZR-75-1, MCF7 and A549 were obtained from China Center for Type Culture Collection (CCTCC), China.Construction of SM3 expression plasmids
The expression plasmids of SM3 with a 15-amino-acid,31 flexible glycine-rich linker introduced in between the variable regions VL and VH was synthesized and codon optimized by Genscript (Nanjing, China) for its expression in E. coli. With the purpose of purification and site-specific modification by sortagging, an LPETG pentapeptide recognition motif and a His6-tag and were attached at the C-terminus of the DNA frames. The cloning plasmid (pUC57-SM3) was digested with NdeI and XhoI and cloned into pET29b (Invitrogen) to generate the expression plasmid pET29b-SM3.Expression and purification of SM3
The pET29b-SM3 was transformed to E. coli strain BL21 (TransGen Biotech, China). The pET29b-SM3 transformed cells were grown in LB medium overnight at 37 °C. 1% (v/v) inoculum was transferred to 1 l of Terrific Broth (TB) medium, and incubated at 37 °C until OD600 reached 0.6–0.8. The culture was then induced with 0.2 mM of isopropyl-β-D-thiogalactopyranoside (IPTG, Sigma), and grown for another 16 h at 22 °C for SM3. The cells were harvested by centrifugation (5000g, 25 min at 4 °C), and the cell pellet was suspended in ice-cold Tris–HCl buffer (400 mM NaCl, 50 mM Tris–HCl, 10 μg ml−1 lysozyme, pH 8.0) followed by sonication using an ultrasonic processor. Cell debris was then removed through centrifugation (12000g, 30 min at 4 °C), and the supernatant was gently agitated with Ni-NTA beads (Qiagen, Germany) at 4 °C for 1 h. The protein was eluted with Tris–HCl buffer containing 200 mM imidazole. The eluate containing scFv-SM3 was concentrated, and dialyzed to remove imidazole. Subsequently, gel filtration chromatography was implemented in AKTA explorer 100 chromatography system using Superdex 75 10/300 column (GE Healthcare, USA) with a running buffer of 200 mM NaCl, 20 mM Tris–HCl, pH 8.0 at a flow rate of 1 ml min−1 and 1.0 ml fractions were collected. The concentration of protein was determined by BCA protein assay kit (Pierce, USA).
Expression and purification of SM3-N3
To express SM3 with incorporated UAA (azidolysine 1), residue 43 of scFv-SM3 was mutated to amber codon TAG to generate plasmid pET29b-SM3-43TAG. E. coli BL21 carrying the expression plasmid and the suppressor plasmid (pUltra-ACF) was grown in TB medium containing 50 μg ml−1 kanamycin and 50 μg ml−1 spectinomycin until the OD600 reached 0.6–0.8. Cultures were then induced with 0.2 mM IPTG, and 1 mM of azidolysine 1 was added at the same time. Protein expression was continued for another 16 h at 30 °C. The purification of overexpressed SM3-N3 was performed as described above.Lysine conjugation of SM3 with fluorescein isothiocyanate (FITC)
SM3 was diluted to 10 mg ml−1 in sodium bicarbonate buffer containing Tris since they inhibit the labeling reaction, and the pH of the reaction buffer was then adjusted to 9.0. FITC was slowly added to the SM3 solution as a mass ratio of 80:1 FITC/SM3, and the pH was retained at 9.0. The reaction was carried at 4 °C overnight, followed by centrifugation using an ultrafilter tube with a 10 kDa filter to remove unreacted FITC to provide SM3-FITC. The labeled protein was analyzed by SDS-PAGE. The final concentration of protein was then determined by BCA protein assay kit (Pierce).
Sortase-mediated SM3 labeling
Expression and purification of SrtAΔN59 was performed as previously described.26 G3K-rhodamine peptide was conjugated to SM3 by incubating scFv-SM3 (100 μM) with G3K-rhodamine peptide (500 μM) in the presence 1 mM SrtAΔN59 in a solution containing Tris–HCl (50 mM, pH 7.5), CaCl2 (5 mM), NaCl (150 mM), and 2-mercaptoethanol (5 mM) at 13 °C overnight. Ni-NTA beads were then added to the reaction mixture with gentle shaking at 4 °C for 2 h, followed by configuration to remove SrtAΔN59 and any remaining unreacted His-tagged substrate. The final product was purified by size-exclusion chromatography in ddH2O or Tris–HCl (50 mM, pH 7.5). The labeled protein SM3-G3K-rhodamine was analyzed by SDS-PAGE. The final concentration of protein was then determined by BCA protein assay kit (Pierce, USA).Copper(I)-catalyzed alkyne–azide cycloaddition (CuAAC)
A copper-dependent click-chemistry reaction between SM3-N3 and rhodamine-alkyne 3 or rhodamine-PEG4-alkyne 4 was carried out in a solution containing 40 μM of SM3-N3, 1 mM of rhodamine-alkyne 3 or rhodamine-PEG4-alkyne 4, 1 mM of CuSO4, 1 mM of TCEP and 100 μM of (tris[(1-benzyl-1H-1,2,3-trazol-4-yl)methyl]amine) (TBTA) at 4 °C overnight. Products containing were dialyzed to remove the unreacted molecules to provide the target proteins SM3-N3-rhodamine and SM3-N3-PEG4-rhodamine, respectively. The labeled protein was further analyzed by SDS-PAGE. The final concentration of protein was then determined by BCA protein assay kit (Pierce, USA).Peptide mapping
SM3 and SM3-N3 were reduced in 50 mM NH4HCO3, 5 mM DTT for 30 min at 56 °C.32 Reduced samples were alkylated with 0.1 M iodoacetamide at room temperature in the dark for 30 min. 0.1 μg μl−1 trypsin were added at 1:50 mass ratio (trypsin:protein), and incubated for 3 h at 37 °C. Peptide-mapping samples were purified with 2-layer C18 stage-tips, and then loaded onto a HyPurity C18 1.0 × 150 mm column with 100% mobile phase A (0.1% formic acid in 98% HPLC H2O) and eluted with a gradient of 3–32% phase B (80% acetonitrile) about 65 min. The loading pressure was adjusted to 800 bar and column temperature was set to 25 °C. Mass spectra were collected in positive-ion mode with gas temperatures at 300 °C. The capillary voltage was set to 2200 V. Acquisition was set to auto MS/MS with a collision energy slope of 3.5 V/100 Da and an offset of 2.5 V.
Flow cytometry binding assay
MCF7 or 293T cells were trypsinized and suspended in PBS supplemented with 1% BSA at a concentration of 50000 cells per tube and then incubated with scFv-SM3 or scFv-SM3-N3 with final concentrations of 1 nM, 10 nM, 100 nM, 1 μM, 10 μM, 100 μM at 4 °C for 1 h. Cells were washed three times with PBS + 1% BSA and centrifuged (800g, 3 min) to remove the washing buffer. The cells were then incubated with anti-6X His tag® antibody (FITC) (Abcam, UK) at 4 °C for 1 h. Followed by washing with PBS + 1% BSA and centrifugation for three times, samples were analyzed with a FACS four-color analysis cytometer (Beckman Coulter, USA). FlowJo software (https://www.flowjo.com/) was used to analyze the flow cytometry data. Triplicate was required in this experiment.
Cell confocal imaging
For in vitro binding affinity assay, 50000 cells were seeded on 35 mm glass bottom dish (Cellvis, USA) with 100 μl of complete medium. Adherent cells were fixed with 4% paraformaldehyde on ice for 20 min. The cells were incubated first with 10 μM (274.90 μg ml−1 for SM3, 276.03 μg ml−1 for SM3-N3) SM3 or SM3-N3 followed by anti-6X His tag® antibody (FITC) (Abcam, USA). The nuclei were visible with DAPI staining (Thermo, USA). After washing twice with cold PBS, samples were visualized in a laser confocal microscope (Olympus FV3000, Japan). MUC1 negative cell line 293T was used as negative control.
For intracellular trafficking studies, the adherent cells were treated with FITC-labeled anti-LAMP1 antibody (BD Pharmingen™, USA) at 37 °C for 1 h without fixing. After washing with cold PBS, cells were incubated with 10 μM (281.28 μg ml−1 for SM3-N3-rhodamine, 283.04 μg ml−1 for SM3-N3-PEG4-rhodamine) SM3-N3-rhodamine or SM3-N3-PEG4-rhodamine at 37 °C for variable time phases. The nuclei were visible with Hoechst 33342 staining (Beyotime, China). After washing twice with cold PBS, living cells were visualized in a laser confocal microscope (Olympus FV3000, Japan) and at least 50 images contain 200 cells were collected for each sample. Fluorescence co-localization was calculated with Image J (https://imagej.nih.gov/ij/) and data were analyzed with Graphpad Prism6 (https://www.graphpad.com/). Mucin1 negative cell line 293T was used as negative control.
In vivo bioluminescence imaging
Animal experimental procedure used in this study met the guidelines of the Care and Use of Laboratory Animals as adopted and promulgated by the U.S. National Institutes of Health Animal Care, and was approved by the Laboratory Animals Use Committee of the Sun Yat-sen University. BALB/c nude mice were obtained from the Experimental Animal Center of Sun Yat-sen University. Mice were housed in a 12 hour light/dark cycle and the room temperature was kept at 25 ± 1 °C. These animals had free access to food and water. BALB/c nude mice were inoculated with 2 × 106 ZR-75-1 or A549 cells at the right flank of the animals. The cell preparations were given in a total volume of 200 μl HBSS subcutaneously. Once established, the xenografted tumor was allowed to grow until the average volume up to 150 mm3. Before imaging, mice were anesthetized with 60 mg kg−1 0.6% pentobarbital sodium. Background images of mice were collected followed by the intravenous injection of 100 μg SM3-FITC or SM3-N3-PEG4-rhodamine with a total volume of 0.1 ml. The images were collected on an IVIS spectrum optical imaging system (Berthold Technologies) and analyzed by Living Image 4.0 software. General settings for fluorescence images collecting were as follows: epi-illumination: 10%, λex = 480 nm, λem = 520 nm for SM3-FITC, λex = 560 nm, λem = 620 nm for SM3-G3K-rhodamine, bin: (M) 4, F/stop = 2, acquisition time = 1 s. Images were collected at dorsal view.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the National Natural Science Foundation of China (No. 21708051 to X. C.) for financial support.Notes and references
- R. Etzioni, N. Urban, S. Ramsey, M. McIntosh, S. Schwartz, B. Reid, J. Radich, G. Anderson and L. Hartwell, Nat. Rev. Cancer, 2003, 3, 243–252 CrossRef CAS PubMed.
- T. Hussain and Q. T. Nguyen, Adv. Drug Delivery Rev., 2014, 66, 90–100 CrossRef CAS PubMed.
- J. M. Lambert and A. Berkenblit, Annu. Rev. Med., 2018, 69, 191–207 CrossRef CAS PubMed.
- R. V. J. Chari, M. L. Miller and W. C. Widdison, Angew. Chem., Int. Ed., 2014, 53, 3796–3827 CrossRef CAS PubMed.
- L. Ducry and B. Stump, Bioconjugate Chem., 2010, 21, 5–13 CrossRef CAS.
- N. E. Weisser and J. C. Hall, Biotechnol. Adv., 2009, 27, 502–520 CrossRef CAS PubMed.
- C. Alric, K. Herve-Aubert, N. Aubrey, S. Melouk, L. Lajoie, W. Meme, S. Meme, Y. Courbebaisse, A. A. Ignatova and A. V. Feofanov, et al. , J. Nanobiotechnol., 2018, 16, 18 CrossRef PubMed.
- M. Ritzefeld, Chem.–Eur. J., 2014, 20, 8516–8529 CrossRef CAS.
- T. Fang, J. N. Duarte, J. Ling, Z. Li, J. S. Guzman and H. L. Ploegh, Angew. Chem., Int. Ed., 2016, 55, 2416–2420 CrossRef CAS.
- Q. Y. Hu, F. Berti and R. Adamo, Chem. Soc. Rev., 2016, 45, 1691–1719 RSC.
- P. J. Carter and G. A. Lazar, Nat. Rev. Drug Discovery, 2018, 17, 197–223 CrossRef CAS PubMed.
- S. C. Alley, D. R. Benjamin, S. C. Jeffrey, N. M. Okeley, D. L. Meyer, R. J. Sanderson and P. D. Senter, Bioconjugate Chem., 2008, 19, 759–765 CrossRef CAS PubMed.
- N. Jain, S. W. Smith, S. Ghone and B. Tomczuk, Pharm. Res., 2015, 32, 3526–3540 CrossRef CAS.
- M. Ritchie, L. Tchistiakova and N. Scott, mAbs, 2013, 5, 13–21 CrossRef.
- C. Kulkarni, J. E. Finley, A. J. Bessire, X. Zhong, S. Musto and E. I. Graziani, Bioconjugate Chem., 2017, 28, 1041–1047 CrossRef CAS.
- D. W. Kufe, Cancer Biol. Ther., 2014, 7, 81–84 CrossRef.
- D. W. Kufe, Nat. Rev. Cancer, 2009, 9, 874–885 CrossRef CAS.
- P. R. Hamann, L. M. Hinman, C. F. Beyer, D. Lindh, J. Upeslacis, D. Shochat and A. Mountain, Bioconjugate Chem., 2005, 16, 354–360 CrossRef CAS.
- H. Thie, L. Toleikis, J. Li, R. von Wasielewski, G. Bastert, T. Schirrmann, I. T. Esteves, C. K. Behrens, B. Fournes and N. Fournier, et al. , PLoS One, 2011, 6, e15921 CrossRef CAS.
- E. Sadeqzadeh, F. Rahbarizadeh, D. Ahmadvand, M. J. Rasaee, L. Parhamifar and S. M. Moghimi, J. Controlled Release, 2011, 156, 85–91 CrossRef CAS.
- K. A. Stephenson, R. Chandra, Z. P. Zhuang, C. Hou, S. Oya, M. P. Kung and H. F. Kung, Bioconjugate Chem., 2007, 18, 238–246 CrossRef CAS.
- K. Sano, T. Nakajima, K. Miyazaki, Y. Ohuchi, T. Ikegami, P. L. Choyke and H. Kobayashi, Bioconjugate Chem., 2013, 24, 811–816 CrossRef CAS.
- M. D. Witte, J. J. Cragnolini, S. K. Dougan, N. C. Yoder, M. W. Popp and H. L. Ploegh, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11993–11998 CrossRef CAS.
- L. K. Swee, C. P. Guimaraes, S. Sehrawat, E. Spooner, M. I. Barrasa and H. L. Ploegh, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 1428–1433 CrossRef.
- C. P. Guimaraes, M. D. Witte, C. S. Theile, G. Bozkurt, L. Kundrat, A. E. Blom and H. L. Ploegh, Nat. Protoc., 2013, 8, 1787–1799 CrossRef.
- C. S. Lu, J. Zhu, Y. Wang, A. Umeda, R. B. Cowmeadow, E. Lai, G. N. Moreno, M. D. Person and Z. W. Zhang, Biochemistry, 2007, 46, 9346–9354 CrossRef CAS.
- K. Wals and H. Ovaa, Front. Chem., 2014, 2, 15–26 Search PubMed.
- T. H. The and T. E. Feltkamp, Immunology, 1970, 18, 865–873 CAS.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2004, 2, 214–221 CrossRef.
- D. M. Zhou, C. L. Zhang, S. L. Xiao, T. Z. Yao, F. Yu, L. L. Si and L. H. Zhang, US Pat., US 2016/0297855, 2016.
- N. Martinez-Saez, J. Castro-Lopez, J. Valero-Gonzalez, D. Madariaga, I. Companon, V. J. Somovilla, M. Salvado, J. L. Asensio, J. Jimenez-Barbero and A. Avenoza, et al. , Angew. Chem., Int. Ed., 2015, 54, 9830–9834 CrossRef CAS.
- F. Tian, Y. Lu, A. Manibusan, A. Sellers, H. Tran, Y. Sun, T. Phuong, R. Barnett, B. Hehli and F. Song, et al. , Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1766–1771 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Protein preparation purification and characterization, peptide mapping, additional fluorescence images, schema, and NMR data. See DOI: 10.1039/c8ra09902b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |