
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew functionalized MIL-53(In) solids: syntheses, characterization, sorption, and structural flexibility†
Lei Wu *ab,
Gérald Chaplaiscd,
Ming Xuee,
Shilun Qiue,
Joël Patarincd,
Angélique Simon-Masseroncd and
Huaxin Chenb
*ab,
Gérald Chaplaiscd,
Ming Xuee,
Shilun Qiue,
Joël Patarincd,
Angélique Simon-Masseroncd and
Huaxin Chenb
aPolymer Materials & Engineering Department, School of Materials Science & Engineering, Chang'an University, Xi'an 710064, China. E-mail: wulei@chd.edu.cn
bEngineering Research Center of Transportation Materials Ministry of Education, Chang'an University, Xi'an 710064, China
cUniversité de Haute-Alsace, CNRS, Institut de Science des Matériaux de Mulhouse (IS2M), Axe Matériaux à Porosité Contrôlée (MPC), UMR 7361, F-68100 Mulhouse, France
dUniversité de Strasbourg, France
eState Key Laboratory of Inorganic Synthesis & Preparative Chemistry, Jilin University, Changchun 130012, China
First published on 15th January 2019
Abstract
The syntheses and characterization of a series of functionalized MIL-53(In) solids have been reported. Chemical groups with variations in steric hindrance and chemical nature (–(OH)2, –Br or –NO2 groups) were introduced through the terephthalate linker to modify the pore surface. Single crystal X-ray diffraction data, N2 adsorption–desorption isotherms, and infrared spectra were systematically investigated to explore the impact of the functional groups grafted onto the organic linker on the dynamic behaviour of these highly flexible hybrid porous frameworks. Owing to the distinctive steric hindrance and chemical nature, the different substituents can influence the interactions between the framework and the trapped molecules, further influencing the flexibility of the materials. Dihydroxyl modified MIL-53(In) exhibits no nitrogen accessible porosity. Notably, functionalization by –Br and –NO2 groups leads to the different capabilities of the corresponding solids to accommodate N2 molecules.
Introduction
The flexible or dynamic MOFs (metal–organic frameworks), which were designed as ‘3rd generation porous coordination compounds’ by Kitagawa,1 have continuously attracted significant attention due to the scientific interest in finding novel phenomena and their potential applications.2–6 Among the large variety, in terms of chemical compositions and structural topologies of MOFs, the flexible MIL-53(M)-type materials (where M = Cr, Al, Fe, Ga, In, Sc)7–16 manifest unprecedented large breathing effects in relation to their flexible diamond-shaped scaffolds (Fig. 1). The one-dimensional channels in the framework may expand or shrink depending on temperature, pressure or trapped molecules with a variation in the unit cell volume up to 40%.17,18 It is noteworthy that the structural transition is fully reversible under appropriate conditions. However, the flexibility, which is also defined as the breathing effect, has been shown to be dependent on the nature of the metal center.10,13,19,20 This phenomenon was highlighted by studies based on Variable Temperature X-ray Diffraction (VT-XRD) and/or N2 adsorption–desorption measurements.8–10,19,21 As represented in Fig. 1, for the Cr- and Al-based MIL-53 materials, the removal of the trapped molecules triggers the direct structural change from either the loaded narrow pore (np_guest) or the loaded large pore (lp_guest) forms to the empty large pore (lp) form. Such a reopening capability is different for MIL-53(Ga), as a closed and empty (cp) form can be encountered before reopening, which is evidenced by a gate opening pressure and the progressive uptake during the N2 sorption measurements.19,21 For MIL-53(Fe), according to the literature,10,11,22 it is impossible to overcome an energy barrier to reopen the shrunken channel by heating or by N2 adsorption processes. The flexibility of MIL-53(Sc) is similar to MIL-53(Fe).15,16 No fully open form can be obtained from the corresponding anhydrous form of this material, but an even more contracted form of the structure is achieved by cooling. As for MIL-53(In), to the best of our knowledge, no studies on the structural flexibility have been reported. | ||
| Fig. 1 Scheme showing the breathing effects of MIL-53(M) with temperature or pressure: lp = large pore, np = narrow pore. | ||
The functionalization of MIL-53-type materials has been reported in a large number of publications, which experimentally and computationally demonstrated that functionalization with the appropriate substituents allows the achievement or improvement of the exceptional abilities of the materials, such as the thermal or chemical stabilities, as well as the selectivity in the gas separation process.20,23–45 Most of the research is devoted to Fe- and Al-based MIL-53 materials. Devic et al. investigated the impact of the functionalization of MIL-53(Fe) with the use of a wide range of organic groups (–Cl, –Br, –CH3, –(CF3)2, –NH2, –(OH)2, or –(CO2H)2).23 A full physicochemical characterization of the related materials showed that only MIL-53(Fe)_(CF3)2 displays N2 accessible porosity, featured by a BET surface area of around 100 m2 g−1, which is, nevertheless, higher than that of the non-porous MIL-53(Fe) parent materials. Besides, the adsorption of CO2 in these materials indicates that less polar functional groups are favourable for pore opening due to a modulation of the CO2–framework interactions, in some cases with a disappearance of the initial intra-framework μ-OH/X hydrogen bonds.24 Stock et al. produced a comparable series of functionalized MIL-53(Al) materials.38,39 The microporosity of these materials was evaluated by N2, CO2, H2O, and methanol sorption measurements. VT-XRD analysis provided evidence that each modification of the chemical composition resulted in significant changes in the flexible behaviour as compared to the parent MIL-53(Al). Nevertheless, reports about the functionalization of In-based MIL-53 materials are scarce. Gascon et al. reported amino modified MIL-53(In), which displayed stepped adsorption of CO2 in good agreement with the breathing behaviour of this type of material.20 It has been proven that the electropositivity of the metal node-associated grafted amino group synergistically influences the flexibility of the framework. Stock et al. synthesized NO2/NH2 modified MIL-53(In) with 2-amino-5-nitro-terephthalate.36 However, because of the low crystalline and centrosymmetric structure, this material shows no NLO activity.
Inspired by the advantages of the functionalization and the interest that MIL-53-type materials have attracted, we incorporated different functional groups with distinctive size and polarity, i.e., –(OH)2, –Br or –NO2 into parent MIL-53(In). In the present work, we report the syntheses, characterization and sorption behaviour of the above-mentioned functionalized MIL-53(In) solids. Single crystal X-ray diffraction measurements, powder X-ray diffraction analysis, IR and 1H NMR spectra, TG analyses and N2 sorption isotherms have been fully investigated to determine the influence of the functionalization on the structural flexibility.
2. Experimental section
2.1 Reagents
All reagents and solvents were used as received from commercial suppliers without further purification. Indium nitrate hydrate, In(NO3)3·xH2O, Alfa Aesar, 99%; terephthalic acid, H2BDC, Alfa Aesar, 98%; 2,5-dihydroxyterephthalic acid, H2BDC-(OH)2, Aldrich, 98%; 2-bromoterephthalic acid, H2BDC-Br, Alfa Aesar, 97%; nitroterephthalic acid, H2BDC-NO2, Alfa Aesar, 99%; N,N-dimethylformamide, DMF, Aldrich, >99%; hydrofluoric acid, HF, Prolabo, 40%; perchloric acid, HClO4, Prolabo, 70%; anhydrous ethanol, EtOH, Prolabo, 99.8%; nitric acid, HNO3, Fisher, 70%; anhydrous methanol, MeOH, Aldrich, 99.9%.2.2 Syntheses of materials
MIL-53(In)_(OH)2_lp_DMF (bulk crystal) was prepared from a mixture of In(NO3)3·xH2O (0.194 g, 0.50 mmol), H2BDC-(OH)2 (0.148 g, 0.75 mmol), DMF (4.00 mL) and H2O (1.00 mL). HF (40%, 0.03 mL) and HClO4 (70%, 0.22 mL) were successively added. Then, the mixture was placed in a Teflon-lined autoclave (15.0 mL) with an occupancy rate of 35%, and then heated for 16 h at 80 °C in an oven. The resulting light yellow lozenge crystals were filtered, washed with DMF and dried in air. The yield was around 75% based on indium.MIL-53(In)_(OH)2_lp_DMF (microcrystalline) was prepared from a mixture of In(NO3)3·xH2O (0.116 g, 0.30 mmol), H2BDC-(OH)2 (0.099 g, 0.50 mmol), DMF (1.50 mL), H2O (1.50 mL) and EtOH (1.50 mL). The mixture was placed in a bottle with a screw cap (8.00 mL) with an occupancy rate of 56% and then heated for 24 h at 100 °C in an oven. The resulting light yellow powder (10–20 μm) was filtered, washed with DMF and dried in air. The yield was 90% based on indium. Elem. and ICP analyses (%): calculated, C 32.02, H 3.64, N 3.40; found, C 32.49, H 3.75, N 3.28.
MIL-53(In)_Br_lp_DMF (bulk crystal) was prepared from a mixture of In(NO3)3·xH2O (0.194 g, 0.50 mmol), H2BDC-Br (0.183 g, 0.75 mmol), DMF (4.00 mL) and H2O (1.00 mL). HF (40%, 0.03 mL) and HNO3 (67%, 0.18 mL) were successively added. The mixture was placed in a Teflon-lined autoclave (15.00 mL) with an occupancy rate of 35% and then heated for 24 h at 80 °C in an oven. The resulting transparent lozenge crystals were filtered, washed with DMF and dried in air. The yield was around 55% based on indium.
MIL-53(In)_Br_lp_DMF (microcrystalline) was prepared from a mixture of In(NO3)3·xH2O (0.149 g, 0.38 mmol), H2BDC-Br (0.28 g, 1.14 mmol) and DMF (1.24 mL). The mixture was placed in a Teflon-lined home-made stainless steel autoclave (2.40 mL) with an occupancy rate of 52% and heated for 5 h at 125 °C in an oven. The resulting white powder (6–50 μm) was filtered and then washed with DMF. The yield was 90% based on Indium. Elem. and ICP analyses (%): calculated, C 28.83, H 2.72, N 2.71; found, C 28.79, H 2.75, N 2.98.
MIL-53(In)_NO2_lp_DMF (bulk crystal) was prepared from a mixture of In(NO3)3·xH2O (0.194 g, 0.50 mmol), H2BDC-NO2 (0.158 g, 0.75 mmol), DMF (4.00 mL) and H2O (1.00 mL). HF (40%, 0.03 mL) and HClO4 (70%, 0.50 mL) were successively added. The mixture was placed in a Teflon-lined autoclave (15.0 mL) (occupancy rate: 37%), and then heated for 16 h at 80 °C in an oven. The resulting transparent lozenge crystals were filtered, washed with DMF and dried in air. The yield was around 60% based on indium.
MIL-53(In)_NO2_lp_DMF (microcrystalline) was prepared by a method similar to that of the microcrystalline MIL-53(In)_Br_lp_DMF sample, except that H2BDC-Br (0.280 g, 1.14 mmol) was replaced by H2BDC-NO2 (0.241 g, 1.14 mmol). The resulting white powder (10–50 μm) was filtered, washed with DMF and dried in air. The yield was 95% based on indium. Elem. and ICP analyses (%): calculated, C 31.24, H 2.95, N 6.39; found, C 31.79, H 2.75, N 6.98.
MIL-53(In)_Br_np_H2O and MIL-53(In)_NO2_np_H2O were prepared from the microcrystalline MIL-53(In)_Br_lp_DMF and MIL-53(In)_NO2_lp_DMF samples by the following procedure. The as-synthesized samples were soaked in anhydrous EtOH (for MIL-53(In)_Br_lp_DMF) or MeOH (for MIL-53(In)_NO2_lp_DMF) for three days and the solvent was changed twice a day. The soaked samples were filtered and then desolvated at 200 °C (for MIL-53(In)_Br_lp_DMF) and 250 °C (for MIL-53(In)_NO2_lp_DMF) for 5 h under N2 flow. At last, the dry samples were rehydrated in air under ambient conditions to obtain the target compounds.
2.3 Activation process
Before gas adsorption measurements, the MIL-53(In)_Br_np_H2O and MIL-53(In)_NO2_np_H2O samples were degassed at 200 °C for 12 h under vacuum. As seen in Section 3.5, because of the chemical instability of the MIL-53(In)_(OH)2_lp_DMF (microcrystalline) sample, the activation stages using organic solvent and calcination were not followed. Instead, tests for activating MIL-53(In)_(OH)2_lp_DMF were undertaken by heating directly at four different temperatures (150, 200, 250 and 300 °C) under vacuum overnight.2.4 Techniques of characterization
Diffraction intensities were obtained on a computer-controlled Bruker SMART CCD diffractometer equipped with graphite-monochromated Mo Kα (λ = 0.71073 Å) radiation at room temperature by using the ω-scan technique. Raw data for all structures were processed using SAINT-5.0.46 All calculations were performed using the SHELXTL-97 program.47 These structures were solved by direct methods. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were fixed at calculated positions and refined by using a riding mode. The powder X-ray diffraction (PXRD) patterns were obtained on a STOE STADI-P diffractometer equipped with a curved germanium (111) primary monochromator and a linear position-sensitive detector using Cu Kα1 radiation λ = 1.5406 Å. The patterns were recorded in the 3–50° 2θ range with a scanning step of 0.12° per sec. The unit–cell parameters were determined using DICVOL4 algorithm.48 The elemental analyses for C, H, N were carried out on a Perkin-Elmer 240C elemental analyzer. The size and the morphology of the crystals were determined by scanning electron microscopy (SEM) using a Philips XL 30 FEG microscope. 1H NMR spectra were obtained on a Bruker 400 UltraShieldTM by using tetramethylsilane as standard. Typically, around 3 mg of sample were digested in a solution of DCl/D2O 35 wt% and then dispersed in DMSO-d6. The Infrared spectra (IR) were recorded (400–4000 cm−1) on a SHIMADZU IRAffinity-1 Fourier transform infrared spectrometer by using the KBr pellet method. The thermogravimetric analysis (TGA) was performed under an air atmosphere with a heating rate of 5 °C min−1 up to 800 °C using a Perkin-Elmer TGA 7 thermogravimetric analyzer. N2 adsorption isotherms were performed on a Micromeritics ASAP 2420 apparatus. Prior to the adsorption measurements, the samples were out-gassed at 200 °C overnight under vacuum. The microporous volumes were evaluated by the t-plot method (Harkins–Jura model). The specific surface areas for MIL-53(In)_NO2_np_H2O sample were calculated according to the criteria given in the literature49 from Brunauer–Emmett–Teller (BET) and Langmuir models in the 0.018 < p/p° < 0.081 range.3. Results and discussion
The crystallographic information of MIL-53(In)_X_lp_DMF (X = –(OH)2, –Br or –NO2) materials were collected by single-crystal X-ray diffraction with the corresponding bulk crystal samples. However, considering the advantages of the short syntheses duration, high production rate, the mild syntheses conditions and the isomorphism of all the corresponding microcrystalline materials, the other characterizations and measurements were performed on the corresponding microcrystalline samples.3.1 Structural description of MIL-53(In)_X_lp_DMF
The main crystallographic data and structure refinement parameters of MIL-53(In)_X_lp_DMF (bulk crystal) are summarized in Table 1. CCDC 1535494 (MIL-53(In)_(OH)2_lp_DMF), CCDC 1535495 (MIL-53(In)_Br_lp_DMF) and CCDC 1535496 (MIL-53(In)_NO2_lp_DMF) contain the supplementary crystallographic data for this paper. The selected bond lengths and bond angles are listed in Table S1–S3 as ESI.†| Compound | MIL-53(In)_(OH)2_lp_DMF | MIL-53(In)_Br_lp_DMF | MIL-53(In)_NO2_lp_DMF |
|---|---|---|---|
| a R = Σ‖Fo| − |Fc‖/Σ|Fo|.b Rw = [Σw(Fo2 – Fc2)/Σw(Fo2)2]1/2. | |||
| Empirical formula | C8H4InO8 | C8BrInO5 | C8InNO7 |
| Formular weight | 342.93 | 370.81 | 336.91 |
| λ (Å) | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | Orthorhombic | Orthorhombic | Orthorhombic |
| Space group | Cmca (no. 64) | Imma (no. 74) | Imma (no. 74) |
| a (Å) | 37.4012 (11) | 17.4254 (9) | 18.6290 (9) |
| b (Å) | 23.0052 (7) | 7.2516 (3) | 7.3019 (4) |
| c (Å) | 7.1553 (2) | 13.3713 (6) | 11.4527 (6) |
| α (°) | 90.00 | 90.00 | 90.00 |
| β (°) | 90.00 | 90.00 | 90.00 |
| γ (°) | 90.00 | 90.00 | 90.00 |
| V (Å3) | 6156.6 (3) | 1689.62 (14) | 1557.88 (14) |
| Z | 16 | 4 | 4 |
| Temperature (K) | 293 (2) | 293 (2) | 293 (2) |
| θ Range (°) | 1.09 to 25.00 | 3.05 to 24.99 | 2.09 to 25.00 |
| ρcalcd (g cm−3) | 1.480 | 1.458 | 1.436 |
| μ (mm−1) | 1.555 | 3.760 | 1.533 |
| GOF on F2 | 1.011 | 1.058 | 1.036 |
| Ra, Rwb [I > 2δ (I)] | 0.0363, 0.1375 | 0.0435, 0.1183 | 0.0472, 0.1550 |
| Ra, Rwb (all data) | 0.0384, 0.1400 | 0.0486, 0.1235 | 0.0538, 0.1682 |
Single-crystal X-ray diffraction analyses reveal that MIL-53(In)_(OH)2_lp_DMF crystallizes in the orthorhombic system (space group Cmca (no. 64)). As shown in Fig. 2a, the asymmetric unit consists of one crystallographically independent In(III) center, one BDC-(OH)2, one μ-OH (O8) and guest water molecules (O10). MIL-53(In)_Br_lp_DMF and MIL-53(In)_NO2_lp_DMF crystallize in the space group Imma (no.74). As shown in Fig. 2b and c, each asymmetric unit consists of one crystallographically independent In(III) center, one linker and one μ-OH (O2). Disorders involving –NO2 groups, as well as Br atoms and the aromatic ring, are notably highlighted for MIL-53(In)_NO2_lp_DMF and MIL-53(In)_Br_lp_DMF, respectively.
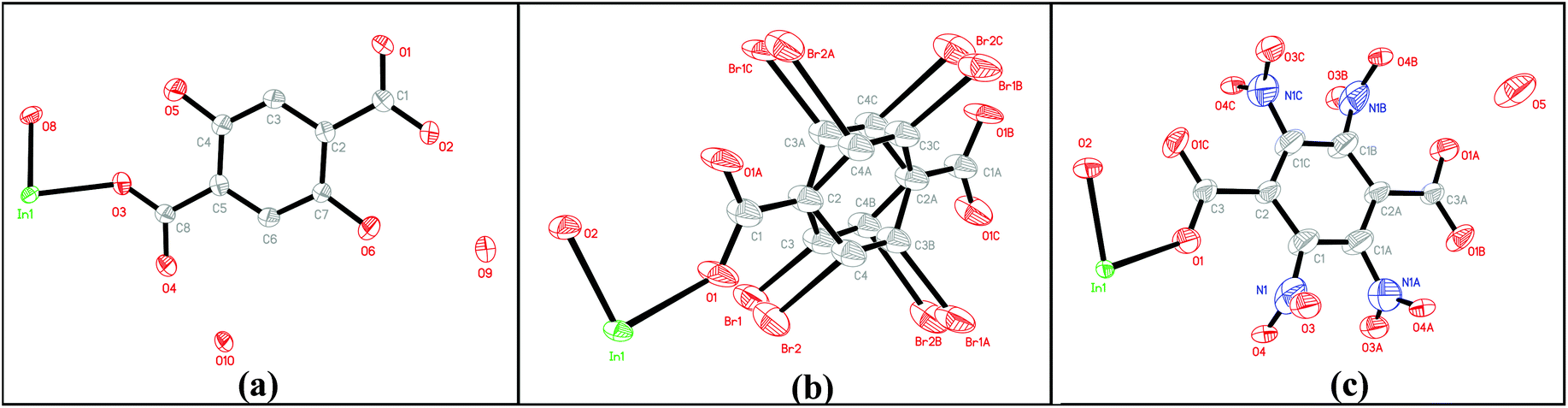 | ||
| Fig. 2 Asymmetric unit of (a) MIL-53(In)_(OH)2_lp_DMF; (b) MIL-53(In)_Br_lp_DMF; (c) MIL-53(In)_NO2_lp_DMF. Thermal ellipsoids for the nonhydrogen atoms were drawn at the 50% probability level. Hydrogen atoms were omitted for clarity. | ||
As shown in Fig. 3, the structures of the functionalized MIL-53(In)_X_lp_DMF materials are isomorphic. The network, like in the prototypical MIL-53(In), is built up from infinite chains of corner-sharing InO4(OH)2 octahedral bridging by μ-OH vertexes and BDC-X linkers, thereby creating one-dimensional diamond-shaped channels. The main difference is characterized by the position and the direction of the substituents. The OH groups turn along the channel axis and are arranged in the pore wall, whereas Br and NO2 groups point into the channel due to the steric hindrance of the involved groups.
 | ||
| Fig. 3 Representations of networks for (a) MIL-53(In)_(OH)2_lp_DMF; (b) MIL-53(In)_Br_lp_DMF; (c) MIL-53(In)_NO2_lp_DMF. | ||
3.2 PXRD of MIL-53(In)_X_lp_DMF and MIL-53(In)_X_np_H2O
The PXRD pattern of the as-synthesized microcrystalline MIL-53(In)_(OH)2_lp_DMF is very similar to the simulated XRD pattern from the single crystal determination of the corresponding bulk crystal sample (Fig. S1a†). This indicates the purity of the microcrystalline sample as well as its isomorphism with bulk crystal. The same correspondence is observed between the bulk crystal and microcrystalline powder of MIL-53(In)_NO2_lp_DMF, as shown in Fig. S1b.† Simultaneously, no significant differences can be observed between the PXRD pattern of MIL-53(In)_Br_lp_DMF and MIL-53(In)_NO2_lp_DMF samples, except for a very weak peak at 12° in the pattern of MIL-53(In)_Br_lp_DMF, which may be attributed to the impurity. Even so, the good fit of the patterns highlights the isomorphism of the two materials.Because of the instability of the MIL-53(In)_(OH)2_lp_DMF material in common organic solvents (such as ethanol, methanol or CH2Cl2) and its low thermal stability, the corresponding hydrated narrow pore form of MIL-53(In)_(OH)2 cannot be obtained by solvent exchange (Fig. S2†) or heating procedures. On the contrary, the MIL-53(In)_Br_lp_DMF and MIL-53(In)_NO2_lp_DMF show good stability in anhydrous methanol and remarkable thermal stability. Thus, their corresponding hydrated narrow pore forms, named MIL-53(In)_Br_np_H2O and MIL-53(In)_NO2_np_H2O, can be obtained with the aforementioned approach. The determinations of the unit–cell parameters of the two hydrated narrow pore forms were performed from their PXRD patterns (Fig. S3†). Both of the patterns were indexed in the monoclinic system (C2/c space group) and the cell parameters are displayed in Table 2. The comparison of the lattice parameters of MIL-53(In)_Br_np_H2O and MIL-53(In)_NO2_np_H2O exhibits minor differences between the two structures, which can be evidenced by the similar PXRD patterns of the two corresponding hydrated phases.
| Compound | MIL-53(In)_Br_lp_DMF | MIL-53(In)_Br_np_H2O | MIL-53(In)_NO2_lp_DMF | MIL-53(In)_NO2_np_H2O |
|---|---|---|---|---|
| Crystal system | Orthorhombic | Monoclinic | Orthorhombic | Monoclinic |
| Space group | Imma | C2/c | Imma | C2/c |
| a (Å) | 17.425 | 19.915 | 18.629 | 19.848 |
| b (Å) | 7.251 | 7.779 | 7.302 | 7.967 |
| c (Å) | 13.371 | 7.215 | 11.453 | 7.224 |
| α (°) | 90.00 | 90.00 | 90.00 | 90.00 |
| β (°) | 90.00 | 93.49 | 90.00 | 94.03 |
| γ (°) | 90.00 | 90.00 | 90.00 | 90.00 |
| V (Å3) | 1689.6 | 1115.8 | 1557.9 | 1138.7 |
3.3 Crystal morphology of MIL-53(In)_X_lp_DMF
The morphologies of MIL-53(In)_X_lp_DMF samples were determined from SEM images shown in Fig. 4. These images indicate the good purity of the samples. The crystallites of MIL-53(In)_(OH)2_lp_DMF display a regular rhombic morphology, with a size of 6 × 20 × 20 μm3. For the two other homologous solids, the crystallites consist of elongated crystals with a similar size of 5 × 10 × 45 μm3. | ||
| Fig. 4 SEM images of microcrystalline samples: (a) MIL-53(In)_(OH)2_lp_DMF; (b) MIL-53(In)_Br_lp_DMF; (c) MIL-53(In)_NO2_lp_DMF. | ||
3.4 1H NMR spectroscopy of MIL-53(In)_X_lp_DMF and MIL-53(In)_X_np_H2O
The 1H NMR spectra of the organic reactants, the corresponding MIL-53(In)_X_lp_DMF and MIL-53(In)_X_np_H2O samples are depicted in Fig. 5. The distinct signals assignable to the aromatic protons of the three linkers appear clearly in the 1H NMR spectra as shown in Fig. 5a–c(i) (1H NMR (400 MHz, DCl/D2O/DMSO-d6) of H2BDC-(OH)2: 7.25 (s, 1H); H2BDC-Br: 7.79 (d, 1H, 3J = 8.06 Hz), 7.95 (d, 1H, 3J = 8.06 Hz), 8.11 (s, 1H); H2BDC-NO2: 8.36 (s, 1H), 8.26 (d, 1H, 3J = 7.81 Hz), 7.92 (d, 1H, 3J = 7.81 Hz)). Three additional signals (denoted by *) corresponding to DMF molecules are distinctly visible on the MIL-53(In)_X_lp_DMF spectra, which evidences the existence of the trapped DMF molecules (Fig. 5a–c(ii)). According to the integration of these signals, the following formulas are proposed: In(OH)(BDC-(OH)2)·(DMF)1.0(H2O)y, In(OH)(BDC-Br)·(DMF)0.85(H2O)y, In(OH)(BDC-NO2)·(DMF)0.85(H2O)y, for MIL-53(In)_X_lp_DMF samples. The disappearance of the DMF signals in the 1H NMR spectra for the MIL-53(In)_Br_np_H2O and MIL-53(In)_NO2_np_H2O samples confirms the effectiveness of the activation method for removing the trapped DMF molecules in the as-synthesized samples (Fig. 5b–c(iii)).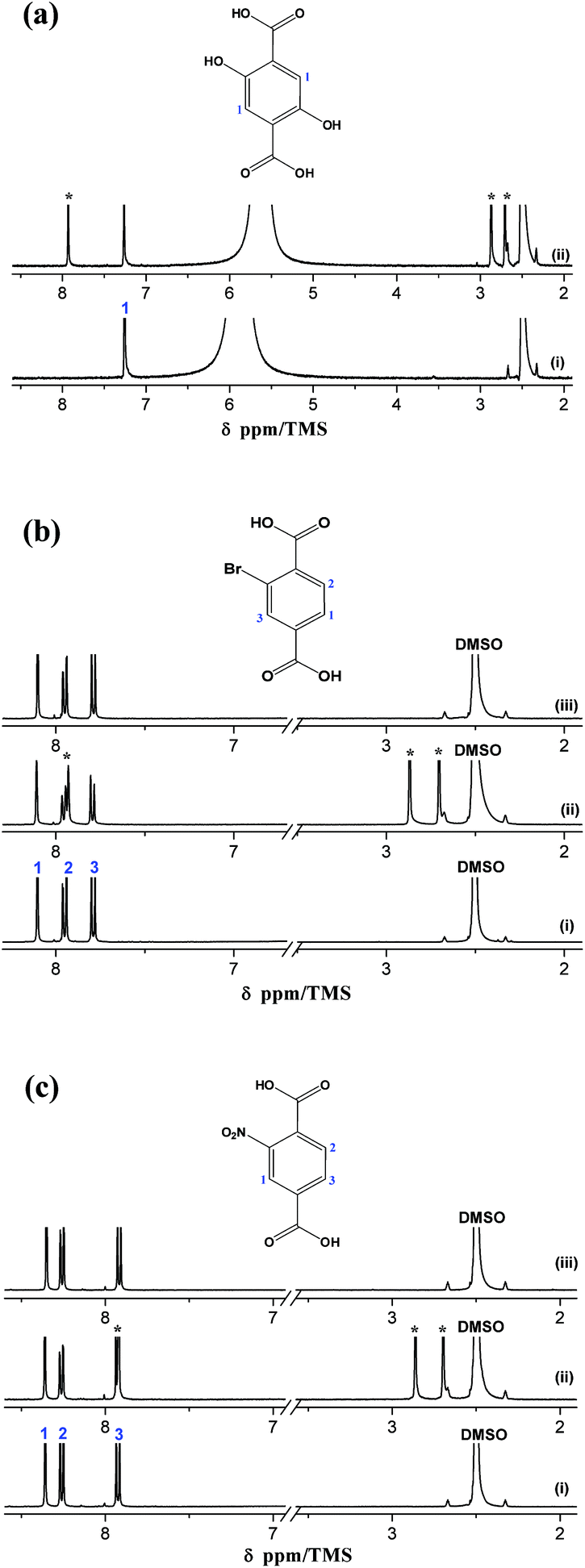 | ||
| Fig. 5 (a) 1H NMR spectra of (i) H2BDC-(OH)2; (ii) MIL-53(In)_(OH)2_lp_DMF; (b) 1H NMR spectra of (i) H2BDC-Br; (ii) MIL-53(In)_Br_lp_DMF; (iii) MIL-53(In)_Br_np_H2O; (c) 1H NMR spectra of (i) H2BDC-NO2; (ii) MIL-53(In)_NO2_lp_DMF; (c) MIL-53(In)_NO2_np_H2O. (* 1H NMR signals of DMF). | ||
3.5 Thermal and chemical stabilities of MIL-53(In)_X
The TG curve of the crude MIL-53(In)_(OH)2_lp_DMF sample is shown in Fig. 6. It indicates a continuous but stepped weight loss up until 500 °C. The first weight loss from room temperature up to 80 °C is assigned to the departure of 0.5 trapped water molecule per metal unit (obs.: 2.2%; calc.: 2.2%). The second event (between 80 and 250 °C) corresponds to the removal of the 1.0 occluded DMF molecule per metal unit (obs.: 17.3%; calc.: 17.6%). Therefore, a combination with the result from 1H NMR enables the deduction of the formula of the compound: In(OH)(BDC-(OH)2)·(DMF)1.0(H2O)0.5. | ||
| Fig. 6 TG curve of MIL-53(In)_(OH)2_lp_DMF. | ||
The absence of any obvious plateau between the loss of the trapped molecules and the transformation into In2O3 implies the difficulty in removing the trapped molecules and keeping the framework intact because the departure of the trapped molecules may be accompanied by its collapse. The similar impossibility of removing all the trapped molecules without the collapse of the framework can be observed in the prototypical as-made MIL-53(In) material.14 According to the TG curve reported in the mentioned publication, although a plateau is formed after the departure of half of the trapped molecules in MIL-53(In), the whole skeleton collapses with the removal of the occluded terephthalic acid molecules.
In order to prepare MIL-53(In)_(OH)2_np_H2O from MIL-53(In)_(OH)2_lp_DMF, we first attempted to remove the trapped molecules by means of solvent exchange using low boiling point solvents (ethanol, methanol and CH2Cl2). The corresponding PXRD patterns are shown in Fig. S2.† It was found that the instability of MIL-53(In)_(OH)2_lp_DMF in these organic solvents leads to phase transitions but not the corresponding hydrated narrow pore form. In conclusion, due to the thermal behaviour and chemical stability of MIL-53(In)_(OH)2_lp_DMF, it was impossible to remove the trapped molecules to obtain the open framework by the usual solvent exchange/heating process.
In contrast, the thermal behaviours of the as-synthesized MIL-53(In)_Br_lp_DMF and MIL-53(In)_NO2_lp_DMF samples are significantly different (Fig. 7). Two similar weight losses were observed in both cases. The first weight loss from room temperature up to around 180 °C is assigned to the departure of 0.85 DMF molecules per indium atom (obs.: 14.5 wt%, calc.: 14.2 wt% for MIL-53(In)_Br_lp_DMF and obs.: 15.6 wt%, calc.: 15.4 wt% for MIL-53(In)_NO2_lp_DMF). The formulas deduced from these thermal analyses combined with the ones from 1H NMR spectroscopy are In(OH)(BDC-Br)·(DMF)0.85 and In(OH)(BDC-NO2)·(DMF)0.85. The plateaus were observed until 400 °C and 350 °C, which indicates that the frameworks can be stable up to these temperatures after the removal of all the trapped molecules. Thus, the functionalization of MIL-53(In) with –Br or –NO2 groups induces a gain in terms of the chemical stability of the network.
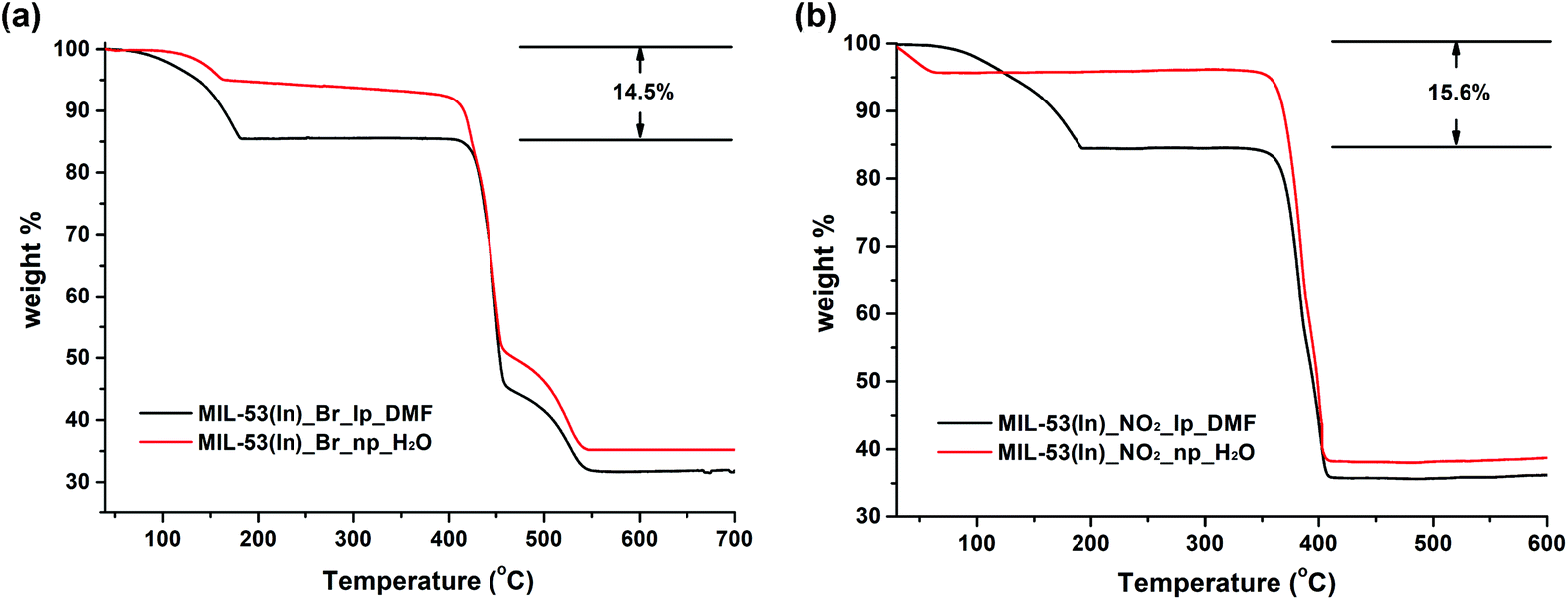 | ||
| Fig. 7 TG curves of (a) MIL-53(In)_Br; (b) MIL-53(In)_NO2 (lp_DMF phase, black; np_H2O phase, red). | ||
3.6 IR spectroscopy of MIL-53(In)_X_lp_DMF and MIL-53(In)_X_np_H2O
In order to better understand the difference related to the stability of the MIL-53(In)_X_lp_DMF materials, IR-based analyses were performed for the as-synthesized MIL-53(In)_X_lp_DMF samples and the hydrated narrow pore forms of MIL-53(In)_X_np_H2O samples. The spectra are displayed in Fig. 8 and for comparison, the spectra of the organic sources are depicted in Fig. S4.† | ||
Fig. 8 IR spectra of materials: (a) MIL-53(In)_(OH)2_lp_DMF; (b) MIL-53(In)_Br_lp_DMF; (c) MIL-53(In)_Br_np_H2O; (d) MIL-53(In)_NO2_lp_DMF; (e) MIL-53(In)_NO2_np_H2O ((♣) ν(μ–OH); (■), νs(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); (*) νs(CO); (**) νas(CO); (♠) ν(C–O); (♦) ν(C–Br); (●) νs(C–N)). O); (*) νs(CO); (**) νas(CO); (♠) ν(C–O); (♦) ν(C–Br); (●) νs(C–N)). | ||
All of the IR spectra of the as-synthesized samples and the hydrated narrow pore forms of samples exhibit the typical vibrational bands of the carboxylic acid function in the region of 1400–1700 cm−1.50 The adsorption bands of the carboxyl groups of the linker coordinated to the metal centers are visible at around 1560 cm−1 and 1390 cm−1, and are attributed to νs(CO) and νas(CO), respectively (denoted by * and ** marks). The band assigned to ν(CO) of DMF (with ■ mark) appears at around 1670 cm−1 in the IR spectra of the as-synthesized MIL-53(In)_X_lp_DMF materials (Fig. 8a, b and d). The absence of this characteristic band of DMF in the spectra of MIL-53(In)_X_np_H2O samples indicates that there are no free DMF molecules in the corresponding hydrated narrow pore forms of these solids (Fig. 8c and e).
Moreover, the peaks sensitive to the –OH group were observed at 1200 cm−1 for MIL-53(In)_(OH)2_lp_DMF in Fig. 8a (represented by the ♠ symbol). The main band involving the C–Br bond vibration, ν(C–Br) (represented by ♦ symbols), is localized at 1039 cm−1 for MIL-53(In)_Br_lp_DMF and MIL-53(In)_Br_np_H2O (Fig. 8b and c) and also for the H2BDC-Br organic source (Fig. S4†). The –NO2 group shows the absorption band at 1302 cm−1 for MIL-53(In)_NO2_lp_DMF and MIL-53(In)_NO2_np_H2O (Fig. 8d and e) assigned to the symmetric stretching vibration, νs(C–N), marked by ● symbols, and also found at the same wavenumber in the spectrum of the H2BDC-NO2 reactant (Fig. S4†).
To probe the different stabilities of the as-synthesized MIL-53(In)_(OH)2_lp_DMF compared to the other two isomorphs, the potential of hydrogen bonds was examined by IR, focusing on the bridging μ-OH group. For MIL-53(In)_(OH)2_lp_DMF, the absence of ν(O–H) bands usually located at approximately 3600 cm−1 and 3700 cm−1 suggest strong hydrogen bond interactions between the bridging μ-OH groups, free –OH groups and trapped molecules (Fig. 8a), which have been previously reported for the MIL-53(Fe)_(OH)2.23 This could be the reason for the framework collapse due to strong host–guest interactions. On the contrary, the sharp absorption peak of ν(O–H) was observed at around 3630 cm−1, indicated by ♣ symbols in the spectra of both the large pore form with DMF and the hydrated narrow pore form of MIL-53(In)_Br and MIL-53(In)_NO2 samples (Fig. 8b–e). In these cases, this implies no strong host–guest interactions. Similar phenomena have been observed in MIL-53(Fe)_X (X = Cl, Br).23 This also explains why the typical solvent exchange/heating process used for these materials enables guest molecules to be released because of weak host–guest interactions.
3.7 N2 adsorption–desorption isotherms of MIL-53(In)_X_np_H2O
The porosity features and the structural flexibility of the functionalized MIL-53(In) materials were evaluated by N2 sorption measurements at 77 K. As mentioned above, the chemical instability and thermal behaviour of the MIL-53(In)_(OH)2_lp_DMF sample did not allow us to obtain the guest free form with the common approaches. However, according to the TG curve, four activation temperatures (150 °C, 200 °C, 250 °C and 300 °C) were tested to treat the crude samples in vacuum overnight before N2 adsorption. Negligible N2 uptake was observed in all cases, as shown in Fig. 9a, revealing a framework collapse and/or the presence of occluded molecules. | ||
| Fig. 9 N2 sorption isotherms of compounds at 77 K: (a) MIL-53(In)_(OH)2_lp_DMF; (b) MIL-53(In)_Br_np_H2O; (c) MIL-53(In)_NO2_np_H2O (adsorption, full symbols; desorption, empty symbols). | ||
For the MIL-53(In)_Br_np_H2O sample, a non-type I adsorption isotherm was obtained as shown in Fig. 9b. Indeed, negligible N2 uptake was observed at low relative pressures (p/p° < 0.07), which can be explained by the predominance of the dehydrated and closed (non-porous) phase. Upon increasing the relative pressure (p/p° ∼0.07), a very sharp and major N2 uptake takes place. At higher relative pressure than this peculiar point (p/p° > 0.07), the adsorbed volume reaches 236 cm3 g−1 (STP) at p/p° = 0.99. The unusual isotherm is similar to and even more pronounced than, the one observed for MIL-53(Ga) (also called IM-19),19 which was explained by the existence of the closed pore form at low relative pressure, and then switched to the final porous polymorph, i.e. the large pore and empty form, with the increase of the pressure. The microporous volume obtained is equal to 0.35 cm3 g−1.
In contrast to the other two homologous compounds, MIL-53(In)_NO2_np_H2O exhibited a classical type Ia isotherm characteristic of microporous solids with a sharp uptake in the low-pressure region (10−5 < p/p° < 10−1),51 and reached the uptake of 266 cm3 g−1(STP) at p/p° = 0.99 (Fig. 9c). The specific surface areas were calculated according to the BET and Langmuir models and reached 964 m2 g−1 and 1076 m2 g−1, respectively. The microporous volume obtained was 0.39 cm3 g−1.
When the microporous volumes of these two materials are based on the weight of the In(OH)(BDC-X) unit, they become 130 cm3 per In(OH)(BDC-Br) unit and 133 cm3 per In(OH)(BDC-NO2) unit for the guest-free and porous form of MIL-53(In)_Br and MIL-53(In)_NO2, respectively. By using the same approach, the microporous volumes of guest-free and large pore MIL-53(M) series materials (based on the weight of the M(OH)(BDC) unit) have been reported to be equal to 118 cm3, 122 cm3 and 119 cm3 per M(OH)(BDC) unit for MIL-53(Al), MIL-53(Cr) and MIL-53(Ga), respectively.19 It is worth noting that both of the unit-based N2 uptakes (thereby ruling out the contributions of the molecular weights of the metal and the functionalized terephthalate linker) for MIL-53(In)_Br_np_H2O and MIL-53(In)_NO2_np_H2O are slightly higher as compared to the nonfunctionalized MIL-53(M) series. This demonstrates in these cases the ameliorative influence of the functionalization on the final microporous volume. Thus, the functional groups –Br and –NO2 effectively improve the porosity of the prototypical MIL-53(In) material.
In conclusion, from N2 adsorption measurements at 77 K, the porosity of dihydroxyl modified MIL-53(In) cannot be assessed. In contrast, both functionalizations by –Br and –NO2 groups lead to the capability of the corresponding solids to accommodate N2 molecules. The S-shaped isotherm of the brominated material can be also underscored, suggesting the presence of a closed form at low pressure.
3.8 Structural flexibility of MIL-53(In)_X
The observations provided by the study concerning the activation process, TG and N2 sorption isotherms clearly indicate three different structural flexibilities of the functionalized MIL-53(In). In particular, the narrow pore phase of the dihydroxyl modified MIL-53(In) in both the hydrated and dry forms cannot be obtained by thermal activation or solvent exchange activation; the activated phase also shows no porosity in the N2 sorption. In contrast, the hydrated narrow pore phase of MIL-53(In)_Br and MIL-53(In)_NO2 can be prepared. It is worth noting that there was a decrease in the unit cell volume by about 34% for MIL-53(In)_Br and 27% for MIL-53(In)_NO2 as compared to the as-synthesized large pore phases with occluded DMF.In order to analyze the structural flexibility, a combination of IR analyses and structural determinations in addition to N2 sorption measurements were conducted. According to the literature,20,23–26,39 there exist strong intra-framework interplays in the MIL-53(M)_X materials, such as bridging μ-OH groups-trapped molecules, bridging μ-OH groups-functionalized groups, functionalized groups-trapped molecules, aromatic rings-aromatic rings (Fig. 10). All of these intra-framework interactions significantly contribute to influencing the scaffold flexibility of MIL-53(M)-type materials. In the present work, the crystal structures of MIL-53(In)_X from single crystal determinations indicate various positions and orientations of the substituent groups. The relatively large substituents –Br and –NO2 point into the pore, as shown in Fig. 3b and c. No additional hydrogen bonds were established between the –Br group and the guest molecules or between the –NO2 group and the guest molecules, which was also attested by IR analyses. The great steric hindrances of –Br and –NO2 groups act as barriers between bridging μ-OH groups and trapped molecules to weaken the interactions between host and guest, which facilitates the structural transition towards the narrow pore form of MIL-53(In)_Br and MIL-53(In)_NO2 from the as-synthesized large pore phase by the activation. In contrast, the structure of the MIL-53(In)_(OH)2 analogue emphasizes that the relatively small hydroxyl groups are arranged along the wall of the channel (Fig. 3a). The existence of –OH groups on the terephthalic moiety even strengthens the interactions between the bridging μ-OH of the framework and the guest molecules, which has been proved by the existence of strong hydrogen bond interactions from the corresponding IR analyses. Therefore, this explains why it is much more difficult to remove guest molecules of MIL-53(In)_(OH)2 to obtain the narrow pore form as compared to the other two homologous structures.
 | ||
| Fig. 10 Scheme of the interactions in the structure of MIL-53(M)_X. | ||
4. Conclusions
The synthesis and the full characterization combined with a description of structural and textural features of a series of functionalized MIL-53(In) materials (by –(OH)2, Br or NO2 groups) have been reported. Single-crystal X-ray diffraction analyses reveal that all functionalized solids display the same diamond-shaped topology as the unmodified parent MIL-53(In) with various positions and orientations of the substituent groups on the terephthalate ligands. Moreover, by modulating the organic component by the presence of additional functional groups, and owing to a subtle combination of steric hindrance, chemical nature and intra-framework interactions, these functionalized MIL-53(In) materials lead to distinctly different structural flexibilities. Indeed, their N2 adsorption behaviour showed that functionalization by both –Br and –NO2 groups led to distinctive nitrogen accessible porosity for the corresponding solids.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by Natural Science Basic Research Plan in Shaanxi Province of China (No. 2017JQ2005), China Postdoctoral Science Foundation (No. 2017M613037), International Science and Special Fund for Basic Scientific Research of Central Colleges, Chang'an University (No. 300102318403).References
- S. Kitagawa and M. Kondo, Bull. Chem. Soc. Jpn., 1998, 71, 1739–1753 CrossRef CAS.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC.
- G. Férey, Dalton Trans., 2016, 45, 4073–4089 RSC.
- M. L. Foo, R. Matsuda, Y. Hijikata, R. Krishna, H. Sato, S. Horike, A. Hori, J. Duan, Y. Sato, Y. Kubota, M. Takata and S. Kitagawa, J. Am. Chem. Soc., 2016, 138, 3022–3030 CrossRef CAS PubMed.
- J. Pang, S. Yuan, D. Du, C. Lollar, L. Zhang, M. Wu, D. Yuan, H.-C. Zhou and M. Hong, Angew. Chem., Int. Ed., 2017, 56, 14622–14626 CrossRef CAS PubMed.
- M. K. Taylor, T. Runcevski, J. Oktawiec, J. E. Bachman, R. L. Siegelman, H. Jiang, J. A. Mason, J. D. Tarver and J. R. Long, J. Am. Chem. Soc., 2018, 140, 10324–10331 CrossRef CAS PubMed.
- F. Millange, C. Serre and G. Férey, Chem. Commun., 2002, 822–823 RSC.
- C. Serre, F. Millange, C. Thouvenot, M. Nogues, G. Marsolier, D. Louër and G. Férey, J. Am. Chem. Soc., 2002, 124, 13519–13526 CrossRef CAS PubMed.
- T. Loiseau, C. Serre, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille and G. Férey, Chem.–Eur. J., 2004, 10, 1373–1382 CrossRef CAS PubMed.
- F. Millange, N. Guillou, R. I. Walton, J. M. Grenèche, I. Margiolaki and G. Férey, Chem. Commun., 2008, 4732–4734 RSC.
- P. Horcajada, C. Serre, G. Maurin, N. A. Ramsahye, F. Balas, M. Vallet-Regi, M. Sebban, F. Taulelle and G. Férey, J. Am. Chem. Soc., 2008, 130, 6774–6780 CrossRef CAS PubMed.
- M. Vougo-Zanda, J. Huang, E. Anokhina, X. Q. Wang and A. J. Jacobson, Inorg. Chem., 2008, 47, 11535–11542 CrossRef CAS PubMed.
- C. Volkringer, T. Loiseau, N. Guillou, G. Férey, E. Elkaim and A. Vimont, Dalton Trans., 2009, 2241–2249 RSC.
- E. V. Anokhina, M. Vougo-Zanda, X. Q. Wang and A. J. Jacobson, J. Am. Chem. Soc., 2005, 127, 15000–15001 CrossRef CAS PubMed.
- J. P. S. Mowat, S. R. Miller, A. M. Z. Slawin, V. R. Seymour, S. E. Ashbrook and P. A. Wright, Microporous Mesoporous Mater., 2011, 142, 322–333 CrossRef CAS.
- J. P. S. Mowat, V. R. Seymour, J. M. Griffin, S. P. Thompson, A. M. Z. Slawin, D. Fairen-Jimenez, T. Duren, S. E. Ashbrook and P. A. Wright, Dalton Trans., 2012, 41, 3937–3941 RSC.
- G. Férey and C. Serre, Chem. Soc. Rev., 2009, 38, 1380–1399 RSC.
- G. Férey, C. Serre, T. Devic, G. Maurin, H. Jobic, P. L. Llewellyn, G. de Weireld, A. Vimont, M. Daturi and J. S. Chang, Chem. Soc. Rev., 2011, 40, 550–562 RSC.
- G. Chaplais, A. Simon-Masseron, F. Porcher, C. Lecomte, D. Bazer-Bachi, N. Bats and J. Patarin, Phys. Chem. Chem. Phys., 2009, 11, 5241–5245 RSC.
- P. Serra-Crespo, E. Gobechiya, E. V. Ramos-Fernandez, J. Juan-Alcaniz, A. Martinez-Joaristi, E. Stavitski, C. E. A. Kirschhock, J. A. Martens, F. Kapteijn and J. Gascon, Langmuir, 2012, 28, 12916–12922 CrossRef CAS PubMed.
- A. Boutin, D. Bousquet, A. U. Ortiz, F.-X. Coudert, A. H. Fuchs, A. Ballandras, G. Weber, I. Bezverkhyy, J.-P. Bellat, G. Ortiz, G. Chaplais, J.-L. Paillaud, C. Marichal, H. Nouali and J. Patarin, J. Phys. Chem. C, 2013, 117, 8180–8188 CrossRef CAS.
- C. Zlotea, D. Phanon, M. Mazaj, D. Heurtaux, V. Guillerm, C. Serre, P. Horcaj-ada, T. Devic, E. Magnier, F. Cuevas, G. Férey, P. L. Llewellyn and M. Latroche, Dalton Trans., 2011, 40, 4879–4881 RSC.
- T. Devic, P. Horcajada, C. Serre, F. Salles, G. Maurin, B. Moulin, D. Heurtaux, G. Clet, A. Vimont, J.-M. Grenèche, B. Le Ouay, F. Moreau, E. Magnier, Y. Filinchuk, J. Marrot, J.-C. Lavalley, M. Daturi and G. Férey, J. Am. Chem. Soc., 2010, 132, 1127–1136 CrossRef CAS PubMed.
- T. Devic, F. Salles, S. Bourrelly, B. Moulin, G. Maurin, P. Horcajada, C. Serre, A. Vimont, J. C. Lavalley, H. Leclerc, G. Clet, M. Daturi, P. L. Llewellyn, Y. Filinchuk and G. Férey, J. Mater. Chem., 2012, 22, 10266–10273 RSC.
- T. Ahnfeldt, D. Gunzelmann, T. Loiseau, D. Hirsemann, J. Senker, G. Ferey and N. Stock, Inorg. Chem., 2009, 48, 3057–3064 CrossRef CAS PubMed.
- E. Stavitski, E. A. Pidko, S. Couck, T. Remy, E. J. M. Hensen, B. M. Weckhuysen, J. Denayer, J. Gascon and F. Kapteijn, Langmuir, 2011, 27, 3970–3976 CrossRef CAS PubMed.
- T. Lescouet, E. Kockrick, G. Bergeret, M. Pera-Titus and D. Farrusseng, Dalton Trans., 2011, 40, 11359–11361 RSC.
- M. Pera-Titus, T. Lescouet, S. Aguado and D. Farrusseng, J. Phys. Chem. C, 2012, 116, 9507–9516 CrossRef CAS.
- L. Bolinois, T. Kundu, X. R. Wang, Y. X. Wang, Z. G. Hu, K. Koh and D. Zhao, Chem. Commun., 2017, 53, 8118–8121 RSC.
- S. Marx, W. Kleist, J. Huang, M. Maciejewski and A. Baiker, Dalton Trans., 2010, 39, 3795–3798 RSC.
- J. Gascon, U. Aktay, M. D. Hernandez-Alonso, G. P. M. van Klink and F. Kapteijn, J. Catal., 2009, 261, 75–87 CrossRef CAS.
- C. Volkringer and S. M. Cohen, Angew. Chem., Int. Ed., 2010, 49, 4644–4648 CrossRef CAS PubMed.
- H. Hintz and S. Wuttke, Chem. Mater., 2014, 26, 6722–6728 CrossRef CAS.
- R. L. Li, Y. Jiang, J. Zhao, D. Ramella, Y. Peng and Y. Luan, RSC Adv., 2017, 7, 34591–34597 RSC.
- S. Bauer, C. Serre, T. Devic, P. Horcajada, J. Marrot, G. Férey and N. Stock, Inorg. Chem., 2008, 47, 7568–7576 CrossRef CAS PubMed.
- K. Markey, M. Krüger, T. Seidler, H. Reinsch, T. Verbiest, D. E. D. Vos, B. Champagne, N. Stock and M. A. van der Veen, J. Phys. Chem. C, 2017, 121, 25509–25519 CrossRef CAS PubMed.
- P. Serra-Crespo, M. A. van der Veen, E. Gobechiya, K. Houthoofd, Y. Filinchuk, C. E. A. Kirschhock, J. A. Martens, B. F. Sels, D. E. De Vos, F. Kapteijn and J. Gascon, J. Am. Chem. Soc., 2012, 134, 8314–8317 CrossRef CAS PubMed.
- A. S. Munn, R. S. Pillai, S. Biswas, N. Stock, G. Maurin and R. I. Walton, Dalton Trans., 2016, 45, 4162–4168 RSC.
- S. Biswas, T. Ahnfeldt and N. Stock, Inorg. Chem., 2011, 50, 9518–9526 CrossRef CAS PubMed.
- S. Biswas, T. Remy, S. Couck, D. Denysenko, G. Rampelberg, J. F. M. Denayer, D. Volkmer, C. Detavernier and P. Van Der Voort, Phys. Chem. Chem. Phys., 2013, 15, 3552–3561 RSC.
- D. Himsl, D. Wallacher and M. Hartmann, Angew. Chem., Int. Ed., 2009, 48, 4639–4642 CrossRef CAS PubMed.
- N. Reimer, B. Gil, B. Marszalek and N. Stock, CrystEngComm, 2012, 14, 4119–4125 RSC.
- A. S. Munn, F. Millange, M. Frigoli, N. Guillou, C. Falaise, V. Stevenson, C. Volkringer, T. Loiseau, G. Cibin and R. I. Walton, CrystEngComm, 2016, 18, 8108–8114 RSC.
- S. Biswas, S. Couck, D. Denysenko, A. Bhunia, M. Grzywa, J. F. M. Denayer, D. Volkmer, C. Janiak and P. Van Der Voort, Microporous Mesoporous Mater., 2013, 181, 175–181 CrossRef CAS.
- P. G. Yot, K. Yang, V. Guillerm, F. Ragon, V. Dmitriev, P. Parisiades, E. Elkaim, T. Devic, P. Horcajada, C. Serre, N. Stock, J. P. S. Mowat, P. A. Wright, G. Férey and G. Maurin, Eur. J. Inorg. Chem., 2016, 4424–4429 CrossRef CAS.
- G. M. Sheldrick, SADABS: Programs for Scaling and Absorption Correction of Area Detector Data, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELXS 97: Program for Crystal Structure Refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- A. Boultif and D. Louër, J. Appl. Crystallogr., 2004, 37, 724–731 CrossRef CAS.
- K. S. Walton and R. Q. Snurr, J. Am. Chem. Soc., 2007, 129, 8552–8556 CrossRef CAS PubMed.
- K. Nakamoto, Infrared and raman spectra of inorganic and coordination compounds, Wiley, New York, USA, 4th edn, 1986 Search PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1535494–1535496. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra08522f |
| This journal is © The Royal Society of Chemistry 2019 |