
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVanadyl β-tetrabromoporphyrin: synthesis, crystal structure and its use as an efficient and selective catalyst for olefin epoxidation in aqueous medium†
Tawseef Ahmad Dara,
Reshu Tomara,
Rasel Mohammad Mianb,
Muniappan Sankar *a and
Mannar Ram Maurya*a
*a and
Mannar Ram Maurya*a
aDepartment of Chemistry, Indian Institute of Technology Roorkee, Roorkee, 247667, India
bDepartment of Chemistry, Graduate School of Science, Tohoku University, 6-3 Aza-Aoba, Aramaki, Sendai 980-8578, Japan
First published on 2nd April 2019
Abstract
We hereby report the synthesis, characterization and catalytic applications in the epoxidation of alkenes by a vanadyl porphyrin having bulky bromo substituents at the β-positions viz. vanandyltetrabromotetraphenylporphyrin (1). The synthesized porphyrin was characterized by various spectroscopic techniques like UV-visible, FT-IR, EPR, MALDI-TOF mass spectrometry and single crystal X-ray analysis. Porphyrin 1 has a nonplanar structure as indicated by its X-ray structure, DFT and electrochemical studies. 1 was analyzed for its catalytic application in the epoxidation of various alkenes. The catalytic reactions were carried out in CH3CN/H2O mixture in 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) ratio. 1 displayed good efficiency in terms of mild reaction conditions, lower reaction temperature and minimal catalyst amount consumption. 1 exhibited excellent selectivity, high conversion efficiency and huge TOF (7600–9800 h−1) in a significantly low reaction time of 0.5 h. Catalyst 1 was regenerated at the end of various catalytic cycles making it reusable and industrially important.
:1 (v/v) ratio. 1 displayed good efficiency in terms of mild reaction conditions, lower reaction temperature and minimal catalyst amount consumption. 1 exhibited excellent selectivity, high conversion efficiency and huge TOF (7600–9800 h−1) in a significantly low reaction time of 0.5 h. Catalyst 1 was regenerated at the end of various catalytic cycles making it reusable and industrially important.
Introduction
Despite rapid progress in synthetic organic chemistry over the last several decades, epoxidation of alkenes remains an area of focus owing to the use of its products in further reactions in industry and commercial materials.1,2 Different types of catalysts ranging from pure metals/elements to very complex molecules speed up several useful and important reactions.3,4 Transition metals are more specific catalysts and catalyze many organic reactions like hydrogenation,5 epoxidation,6 polymerization,7 hydrogen transfer,8 hydroxylation,9 halogenation10,11 and some other reactions.12,13 Porphyrins act as multifaceted ligands coordinating with various metals to form metalloporphyrins that have many applications.14–16 Porphyrins have additionally been found to possess remarkable properties in the fields of solar energy,17 medicine,18 sensing,19 catalysis,20 non-linear optics and the like.21 In living systems also, porphyrins perform many vital functions like oxygen transport and storage, photosynthesis, electron transfer and as cofactors to many enzymes.22–24 Unsubstituted porphyrins generally tend to undergo degradation25 in oxidation type reactions and thus β- and meso-functionalization of porphyrins serves well in tunability as well as stability.26–31 Due to this functionalization, porphyrins can be actually tuned to attain very good thermal and chemical stabilities.32,33For epoxidation reaction of alkenes, several catalysts of different structures and natures are already known. The first satisfactory method for asymmetric alkene epoxidation was developed by Sharpless et al. and involved titanium tetraisopropoxide as one of the components.34 Since then a large number of catalysts for epoxidation of alkenes based on chromium and manganese have been developed.35 Other transition metals that have been exploited as good candidates for epoxidation reactions include rhenium,36 iron,37 tungsten,38 titanium39 and molybdenum.40 In earlier times the ‘oxygen provider’ species were mainly molecular oxygen,41 organic hydroperoxides,39 peracids and oxiranes.42 However, in recent times, H2O2, because of its environment friendly nature has emerged as one of the best oxidizing species for epoxidation of alkenes.43–45 Most recently it has been found that presence of bicarbonate ions along with H2O2 speeds up the epoxidation reactions by many folds.46–48 Other than the general coordination complexes,49–51 several metalloporphyrins have also been used as epoxidation catalysts in the presence of H2O2.52–54 We have earlier reported the catalytic efficacy of an β-octachlorovanadylporphyrin complex towards alkene epoxidation.55 In continuity of our efforts to develop versatile catalysts for alkene epoxidation, we hereby report the facile synthesis, characterization, spectral properties, X-ray structure and catalytic applications of vanadyl tetrabromoporphyrin VOTPPBr4 (1). We have also observed that steric (bulky) and electron withdrawing factors play a major role in determining the efficiency of the catalysts for epoxidizing alkenes.
Results and discussion
Synthesis and characterization
Vanadyl tetrabromoporphyrin VOTPPBr4 (1) was synthesized by making certain modifications to existing literature methods. H2TPPBr4 was prepared from H2TPP using NBS.56 Target porphyrin VOTPPBr4 (1) was then prepared from H2TPPBr4 using VOSO4 and DMF by refluxing under argon atmosphere (Fig. 1).55,57 | ||
| Fig. 1 Molecular structure of synthesized vanadyl β-tetrabromoporphyrin 1. | ||
Different spectrometric techniques like UV-visible, FTIR, MALDI-TOF-MS, EPR and single crystal XRD were used to characterize the target compound 1. The UV-visible spectrum of 1 in CH2Cl2 is shown in Fig. 2 and the corresponding data is shown in Table S1 in the ESI.†
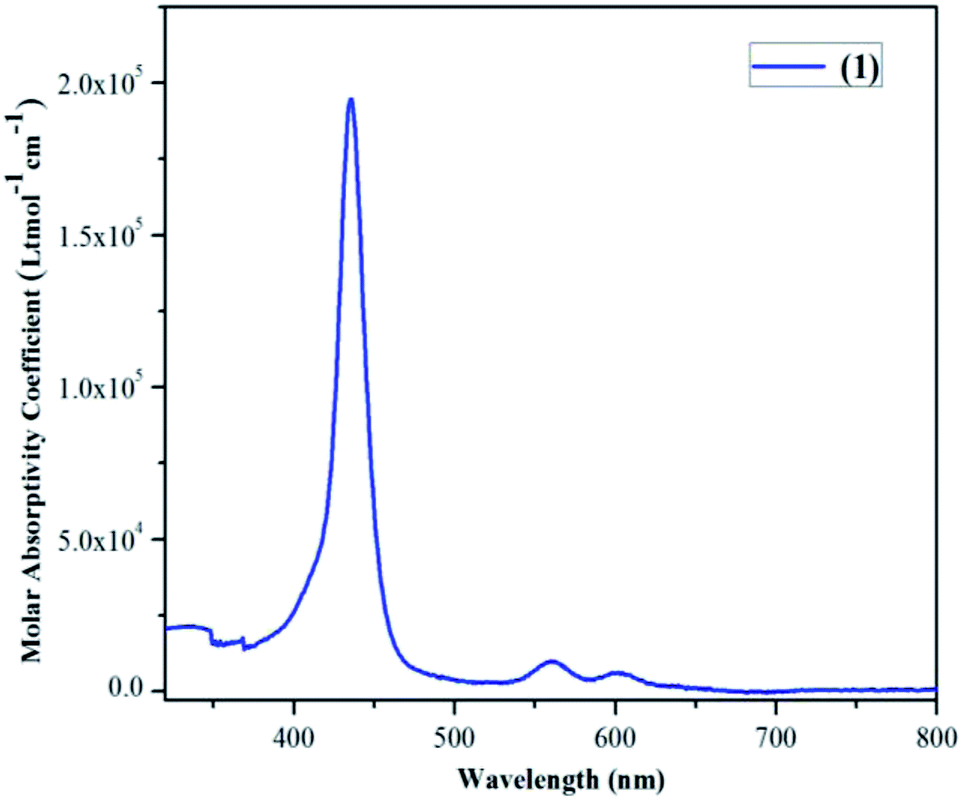 | ||
| Fig. 2 UV-visible spectrum of 1 in CH2Cl2 at 298 K at a conc. of 5.68 × 10−6 M. | ||
The UV-visible spectrum of 1 exhibits one Soret band at 435 nm (ε, 1.94 × 105) which is almost similar to H2TPPBr4 (436 nm) whereas it shows only two Q-bands: 560 nm (ε, 1.94 × 104) and 604 nm (ε, 6.32 × 103) as compared to three in case of H2TPPBr4 (533, 612 and 685 nm). Thus, the first Q-band in case of 1 is 27 nm red shifted and the second Q-band is 8 nm blue shifted relative to H2TPPBr4 due to insertion of vanadium metal as VO2+. The FTIR spectrum of 1 was recorded using KBr pellets (Fig. S1, ESI†). In the FTIR spectrum of 1, we found characteristic peaks corresponding to V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (stretching) at 1010 cm−1 and C–Br (stretching) at 619 cm−1. Notably, the N–H stretching frequency at around 3300 cm−1 was not observed which is generally found in non-metallated (free base) porphyrins. The calculated MALDI-TOF mass value for 1 is in good agreement with the observed value (Fig. S2, ESI†). Single crystals for XRD studies were obtained by diffusing methanol slowly into the saturated toluene solution of 1 (vide infra).
O (stretching) at 1010 cm−1 and C–Br (stretching) at 619 cm−1. Notably, the N–H stretching frequency at around 3300 cm−1 was not observed which is generally found in non-metallated (free base) porphyrins. The calculated MALDI-TOF mass value for 1 is in good agreement with the observed value (Fig. S2, ESI†). Single crystals for XRD studies were obtained by diffusing methanol slowly into the saturated toluene solution of 1 (vide infra).
Electrochemical studies
Cyclic voltammogram of 1 in distilled CH2Cl2 was recorded using Pt working electrode, Ag/AgCl reference electrode and Pt wire as counter electrode at 298 K. TBAPF6 recrystallized twice from ethanol and vacuum dried for 6 hours was used as the supporting electrolyte. 1 exhibited two reversible oxidation peaks at 1.28 V and 1.46 V as shown in Fig. 3 and Table S2 in the ESI.† The first oxidation peak corresponds to the generation of porphyrin cation radical due to loss of a single electron while the second oxidation peak corresponds to the generation of porphyrin dication due to loss of second electron. Similarly the first and second reversible reduction peaks were obtained at −0.84 V and −1.06 V, which correspond to the generation of porphyrin anion radical and porphyrin dianion respectively. Vanadium metal does not undergo any redox changes during cyclic voltammetry. The HOMO–LUMO energy gap (ΔE1/2) on the basis of electrochemical studies was found to be 2.12 V. Cyclic voltammetry supports the non-planar nature of 1 and the electron deficient nature of porphyrin core as compared to VOTPP.58,59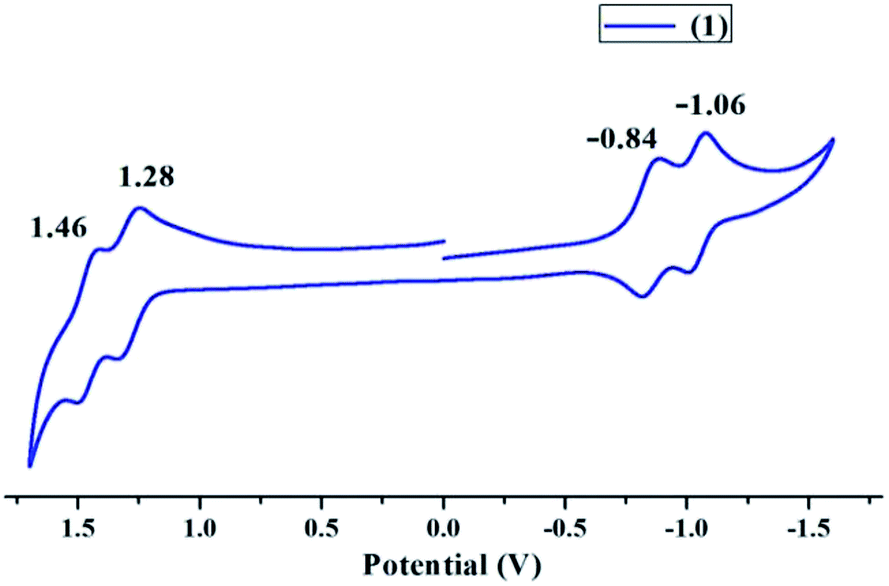 | ||
| Fig. 3 Cyclic voltammogram of 1 in distilled CH2Cl2 at 298 K using TBAPF6 as the supporting electrolyte. | ||
Thermogravimetric analysis
Thermogravimetric analysis of 1 was carried out using alumina as the reference powder with a heating rate of 10 °C min−1 under normal aerial atmosphere. The thermogravimetric profile of 1 is given in Fig. S3 in the ESI.† 1 displayed excellent thermal stability till 637 K despite the labile nature of C–Br bonds having bond energy of 285 kJ mol−1 only. The encouraging thermal stability of 1 can be very useful in terms of industrial applications.EPR spectral studies
X-band EPR spectrum of 1 was recorded in toluene. 1 exhibited a typical EPR spectrum corresponding to the paramagnetic species of vanadium(IV) at 120 K as shown in Fig. 4. The number of lines in the EPR spectrum of 1 were found to be equal to eight corresponding to the I value of vanadium metal (I = 7/2).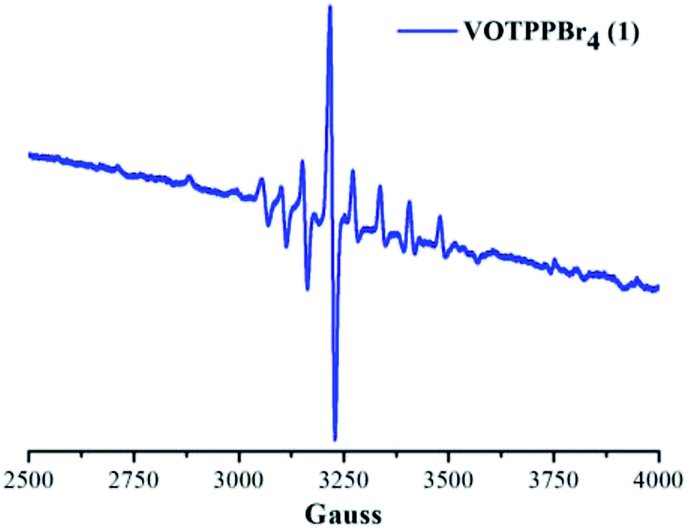 | ||
| Fig. 4 X-band EPR spectrum of 1 in toluene at 120 K. EPR parameters: microwave frequency, 9.10 GHz; incident microwave power, 0.99 mW. | ||
The g∥ value for 1 was found to be 1.96 while the corresponding g⊥ value was observed to be 1.99. Similarly, the A∥ and the A⊥ values obtained for 1 were found to be 158.43 × 10−4 cm−1 and 56.00 × 10−4 cm−1, respectively. The above calculated g and A values fall well within the permissible range for paramagnetic vanadium complexes and highlight that vanadium exists in +4 oxidation state in VOTPPBr4 (1).
DFT optimization studies
DFT optimization studies using B3LYP functional and LANL2DZ basis set was carried out for 1 in gas phase. 1 exhibited non-planar, distorted saddle conformation due to bulkier bromo groups. The optimized geometry of 1 is shown in Fig. 5.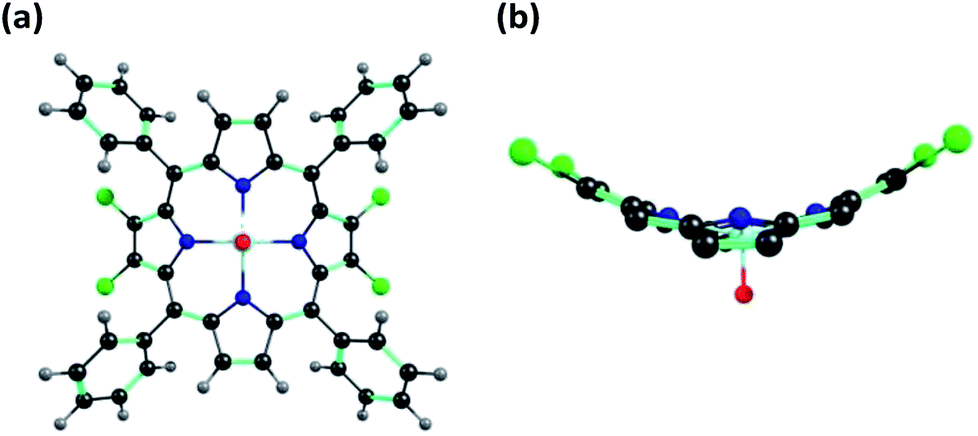 | ||
| Fig. 5 B3LYP/LANL2DZ set optimized geometry of 1 in gaseous phase showing (a) top view and (b) the side view, where β-hydrogens and meso-phenyl substituents have been removed for clarity. | ||
Δ24 in case of 1 was found to be ±0.383 Å. Likewise, ΔCβ (carbon atoms having –Br substituents were taken as β) value was calculated to be ±0.855 Å for 1. Similarly ΔCβ′ (carbon atoms attached to –H atoms were taken as β′) value for 1 was found to be as ±0.725 Å. Likewise, ΔM value was found to be as 0.538 Å. Various bond distance values in 1 were calculated as 1.593 Å (VO), 1.379 Å (Cβ–Cβ) and 1.372 Å (Cβ′–Cβ′) respectively. The full list of various bond lengths and bond angles for 1 is given in Table S3 in the ESI.† It has to be noted that bond length between β-carbon atoms (Cβ–Cβ) having bromo substituents is longer than bond length between β′-carbon atoms (Cβ′–Cβ′) devoid of any substituents, which is a direct consequence of bulkier bromo substituents on these carbon atoms. Similarly, the deviation of carbon atoms having bromine substituents from the mean plane (ΔCβ = ±0.855 Å) is slightly higher than carbon atoms devoid of bromine substituents (ΔCβ′ = ±0.725 Å), which highlights the distortion caused due to bromo substituents.
The frontier molecular orbitals obtained from the optimized geometry of 1 are displayed in Fig. 6. Both the HOMO and the LUMO orbitals are mainly concentrated on the porphyrin core. The energy gap (ΔE) between the HOMO and the LUMO orbitals was calculated to be 2.51 eV on the basis of DFT optimized studies.
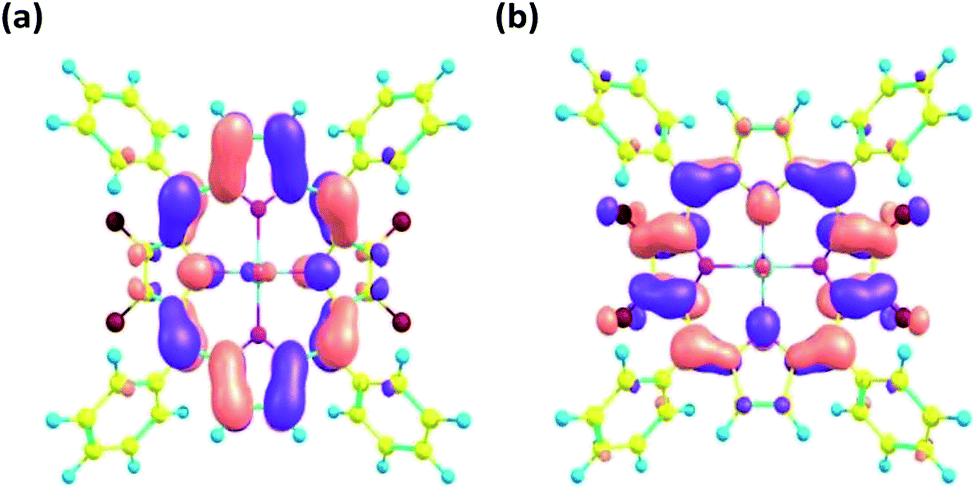 | ||
| Fig. 6 Frontier molecular orbitals (a) HOMO and (b) LUMO as obtained from DFT optimized geometry of 1 using B3LYP/LANL2DZ basis set. | ||
Single crystal XRD studies
X-ray quality single crystals for 1 were obtained by slow diffusion of methanol into a saturated solution of 1 in toluene having a few drops of added chloroform (to aid solubility). 1 crystallizes into a μ-oxo dimeric structure where the two porphyrin units are linked together by a bridging oxygen atom (μ-oxo dimer) resulting into a monoclinic crystal system. Repeated attempts to obtain the monomeric structure of 1 failed. It is possible that the μ-oxo dimer is formed in saturated solutions at low temperatures in presence of methanol which is converted into the monomeric form at room temperature or above. The crystal structure information of the μ-oxo dimer of 1 is given in Table S4 in the ESI† and the corresponding bond lengths and bond angles are listed in Table S5 in the ESI.† The ORTEP images with top and side views for μ-oxo dimer of 1 are shown in Fig. 7.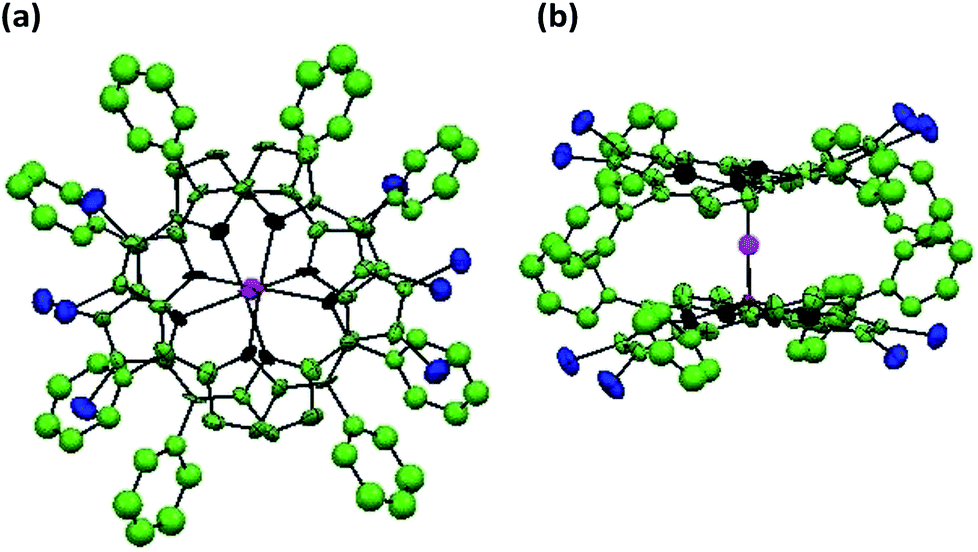 | ||
| Fig. 7 ORTEPs showing (a) top view and (b) side view of μ-oxo dimer of 1. Hydrogen atoms have been removed for clarity. | ||
XRD studies clearly revealed the non-planar, distorted nature of the porphyrin units with Δ24 = ±0.324 Å. The distortion from the mean plane for β-carbon atoms bearing bromo substituents was calculated as 0.663 Å which is much larger than the distortion found in β′-carbon atoms devoid of bromo substituents which was found to be 0.535 Å. Thus bulkier bromo substituents are mainly responsible for the observed distortion in the porphyrin rings. The distance between the mean plane and the metal center in the porphyrin rings was found to be 0.538 Å. Different bond distances in μ-oxo dimer of 1 were calculated as 1.769 Å (V–O), 1.356 Å (Cβ–Cβ) and 1.342 Å (Cβ′–Cβ′) respectively. Finally, the distance between the two metal centers of the porphyrin μ-oxo dimer was calculated as 3.534 Å. It has to be noted that different values of bond lengths and bond angles calculated from DFT studies and XRD structures for monomeric and μ-oxo dimeric form of 1 are in good agreement with each other.
Catalytic epoxidation of olefins
The tendency of vanadium metal to undergo easy changes in oxidation sate from (IV) to (V) during the course of a reaction has made it a first choice catalyst for many reactions. VOTPPBr4 (1) having electron withdrawing and bulky bromo substituents in addition to the vanadium metal (VO2+) center was therefore analyzed for its catalytic efficiency for the epoxidation of alkenes in presence of H2O2 (oxidant) and NaHCO3 (promoter) to generate corresponding epoxides. For performing the catalytic epoxidation of different alkenes, various reaction parameters like catalyst amount, solvent, temperature etc. were first optimized by performing many control experiments using styrene as the model substrate. The details of epoxidation reactions under different conditions are listed in Table 1 and are described in detail in the Experimental section (vide infra).| Entry no. | Catalyst [mg (μmol)] | 30% H2O2 [g (mmol)] | NaHCO3 [g (mmol)] | Temp. [°C] | MeCN/H2O [ml] | Conv. [%] | Selectivity |
|---|---|---|---|---|---|---|---|
| 1 | 1.5 (1.50) | 1.69 (15) | 0.168 (2) | 60 | 2:1 |
93 | 100 |
| 2 | 1.5 (1.50) | 1.69 (15) | 0.168 (2) | 60 | 3:2 |
99 | 100 |
| 3 | 1.5 (1.50) | 1.69 (15) | 0.168 (2) | 60 | 4:3 |
95 | 100 |
| 4 | 1 (1.00) | 1.69 (15) | 0.168 (2) | 60 | 3:2 |
98 | 100 |
| 5 | 2 (2.00) | 1.69 (15) | 0.168 (2) | 60 | 3:2 |
98 | 100 |
| 6 | 1 (1.00) | 0.565 (5) | 0.168 (2) | 60 | 3:2 |
78 | 100 |
| 7 | 1 (1.00) | 1.13 (10) | 0.168 (2) | 60 | 3:2 |
97 | 100 |
| 8 | 1 (1.00) | 1.13 (10) | 0.126 (1.5) | 60 | 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :2 :2 |
96 | 100 |
| 9 | 1 (1.00) | 1.13 (10) | 0.084 (1) | 60 | 3:2 |
93 | 100 |
| 10 | 1 (1.00) | 1.13 (10) | 0.126 (1.5) | 50 | 3:2 |
82 | 100 |
| 11 | 1 (1.00) | 1.13 (10) | 0.126 (1.5) | 40 | 3:2 |
36 | 100 |
| 12 | — | 1.13 (10) | 0.126 (1.5) | 60 | 3:2 |
29 | 100 |
| 13 | — | 1.13 (10) | — | 60 | 3:2 |
9 | 100 |
| 14 | 1 (1.00) | 1.13 (10) | — | 60 | 3:2 |
18 | 100 |
The optimized conditions on the basis of Table 1 were utilized during selective epoxidation of different alkenes with VOTPPBr4 (1) catalyst. The best suited conditions for selective epoxidation of alkenes for efficient conversion rates are: VOTPPBr4 (1) catalyst 1 μmol (1.00 mg), substrate 5 mmol, 30% H2O2 10 mmol (1.13 g), NaHCO3 1.5 mmol (0.126 g), temperature 60 °C, reaction time 0.5 h and solvent mixture CH3CN:H2O in 3:2 (v/v) ratio (entry 8, Table 1). The observed conversion rates and turnover frequency TOF (h−1) for catalyst 1 under the optimized conditions are shown in Table 2. The vanadium porphyrin catalyst, VOTPPBr4 (1) exhibited very good conversion rates, 100% selectivity and high turnover frequency as compared to VOTPP which is devoid of bromo substituents. Under the above optimized conditions VOTPP exhibited a conversion rate of only 54% and a TOF of 3668 h−1 using styrene as the substrate.
:H2O (3:2, v/v) solvent
| S. no. | Substrate | Product | % Conv. | TOF (h−1) |
|---|---|---|---|---|
| 1 | 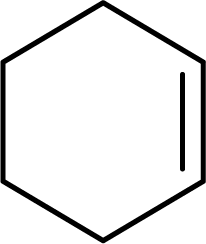 |
 |
96 | 9600 |
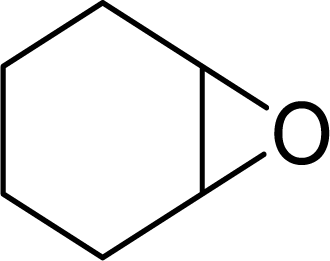
| 2 |  |
 |
98 | 9800 |
| 3 |  |
 |
96 | 9600 |
| 4 |  |
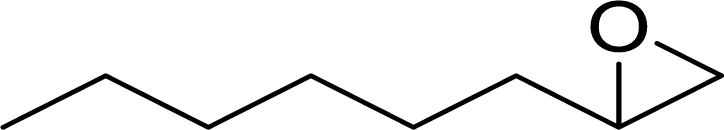 |
97 | 9700 |
| 5 | 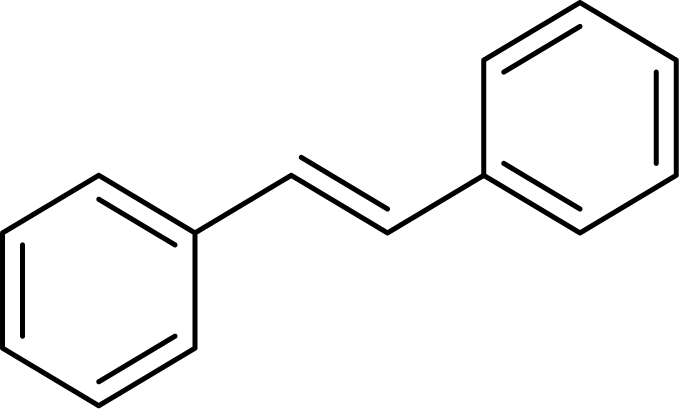 |
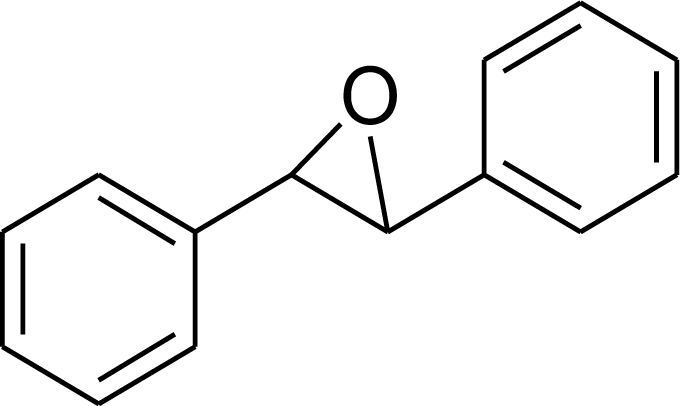 |
94 | 9400 |
| 6 | 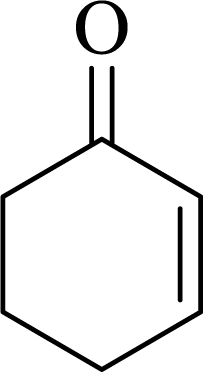 |
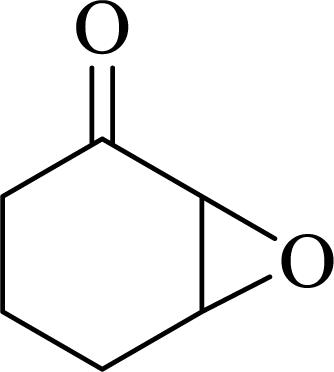 |
95 | 9500 |
| 7 |  |
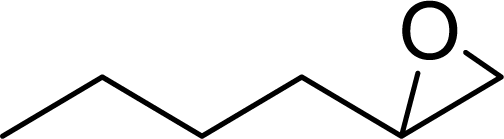 |
96 | 9600 |
| 8 | 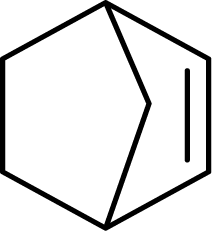 |
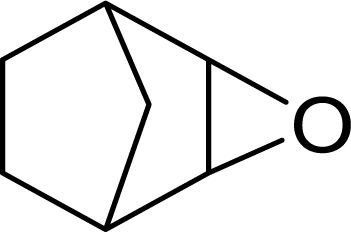 |
93 | 9300 |
| 9 |  |
 |
76 | 7600 |
| 10 |  |
 |
82 | 8200 |
Mechanism of epoxidation
The reaction between H2O2 and NaHCO3 generates peroxymonocarbonate ions by the following reaction;| HCO3− + H2O2 → HCO4− + H2O |
Peroxymonocarbonate ions are very strong oxidizing species in aqueous solutions.60 Peroxymonocarbonate ions transfer oxygen to the vanadium center (VO) in the porphyrins to generate oxidoperoxido species. The formation of such oxidoperoxido/hydroperoxido (oxidoperoxiodo species are more stable) species with different metal centers is well documented in literature.50,51,61–63 The oxidoperoxido species are more efficient and more selective in transferring oxygen to the alkenes which selectively converts the alkenes into epoxides and also regenerates the original catalysts in the process. The generation of oxidoperoxido vanadium(V) species [VO(O2)TPPBr4]− in case of 1 was confirmed by 51V NMR studies in DMSO-d6 showing a clear peak at −1298 ppm as shown in Fig. S5, ESI.† The presence of oxidoperoxido species was alternatively confirmed by adding a small amount of cyclohexene to the above solution which led to decrease in peak intensity. On adding excess amount of cyclohexene the peak from 51V NMR spectrum of the compound disappeared completely because the vanadium(V) center is reduced back to vanadium(IV) which is 51V NMR inactive.
Since the vanadium(V) porphyrin intermediate involved in the epoxidation reaction is an oxidoperoxido negatively charged species, hence electron withdrawing substituents are expected to stabilize it. 1 contains electron withdrawing –Br substituents hence it was expected to be an efficient catalyst which was confirmed by its excellent selectivity, good conversion and high TOF (h−1) as shown in Table 2. Another important factor that determines the efficiency is the size of the substituents which causes distortion or non-planarity in the porphyrin (macrocyclic) core. This can be clearly seen in the efficiency of 1 because the bulkier bromo groups cause more distortion in the porphyrin core making the reactive site easily accessible. The catalyst was recovered at the end of respective catalytic cycles as indicated by its absorption spectrum (Fig. S4, ESI†). Thus the catalyst can be reused again and again for further catalysis of alkenes.
Experimental section
Chemicals and materials
Propionic acid, P2O5, and sodium sulfate were purchased from Thomas Baker, India and used as received. Benzaldehyde, VOSO4, HCl and NaHCO3 were purchased from Himedia, India and used without any further processing. Pyrrole was purchased from SRL Chemicals, India and used without further purification. DMF and CH3CN were purchased from Merck, India and used as such. Various alkenes used in this study were purchased from Alfa Aesar, UK and used as received. H2O2 and silica gel (100–200 mesh) were purchased from Rankem, India and used as such. N-Bromosuccinimide was also purchased from Himedia, India but used after recrystallizing it from hot water and vacuum drying. Various solvents like chloroform, hexane and methanol were purchased from Molychem, India and used after drying over P2O5. Electrochemical studies were carried out in distilled CH2Cl2 using TBAPF6, recrystallized two times from ethanol and vacuum dried for 6 hours, as the electrolyte.Instrumentation
UV-visible spectra were recorded using an Agilent Cary 100 spectrophotometer along with a pair of quartz cells with 3.5 mL volume and 1 cm path length. IR spectra were recorded using a mid IR range 4000–400 cm−1 Perkin-Elmer spectrophotometer by making KBr pellets. For MALDI-TOF spectra we have used Bruker UltrafleXtreme-TN MALDI-TOF-MS spectrometer and HABA matrix. Cyclic voltammetric measurements were performed on a CHI 620E instrument, using a three electrode assembly which consisted of Ag/AgCl as reference electrode, Pt disk working electrode and Pt-wire as counter electrode along with TBAPF6 as the supporting electrolyte. TGA was carried out using a SII EXSTAR 6300 instrument. Single crystal X-ray diffraction data was collected at 100 K on a Bruker APEX-II diffractometer with APEX-II CCD detector and JAPAN thermal Engineering Co. Ltd. Cryo system, DX-CS190LD. The crystal structure was solved by using direct methods (SHELXS-97). The products obtained from the epoxidation reactions were identified using a Shimadzu 2010 plus gas-chromatograph endowed with an Rtx-1 capillary column (30 m × 0.25 mm × 0.25 μm) and an FID detector. The product identities were confirmed using a Perkin-Elmer GC-MS (Clarus 500).Synthetic procedure
ε): 435 (5.28), 560 (4.00), 604 (3.80). MALDI-TOF-MS (m/z): found 995.99 [M]+, calcd 995.24. Anal. calcd for C44H24Br4N4OV: C, 53.10; H, 2.43; N, 5.63. Found: C, 52.99; H, 2.46; N, 5.22.Details of the catalytic studies
The catalytic role of the synthesized vanadyl porphyrin 1 was investigated for the epoxidation of various alkenes by optimizing the reaction conditions initially by taking 1 as the representative catalyst. Styrene was chosen as the representative alkene to optimize the conditions for the best conversion rates and selectivity of the products formed. In a model reaction, styrene (5 mmol, 0.52 g), 30% H2O2 (10 mmol, 1.14 g), NaHCO3 (1.5 mmol, 0.126 g) and catalyst (1) (1.5 μmol, 0.0015 mg) were mixed in aqueous MeCN 5 ml (3:2: v/v) in 50 ml two neck round bottomed flask and refluxed with vigorous stirring at 60 °C for 1 hour. The reaction progress was examined by taking out a drop of sample from the reaction mixture at intervals of 15 minutes. The products formed were extracted with hexane and injecting 0.25 μl of the same into GC/GCMS. The products were identified by using GC-MS (Clarus 500). The reaction was completed in 0.5 h only. The GC traces corresponding to some of the alkene substrates and the reaction mixtures at different stages of the epoxidation reaction are given in Fig. S6–S15 in the ESI.†
A series of reactions were performed to optimize the conditions best for the reactant conversion and product selectivity. Irrespective of the reaction conditions 1 gave only styrene epoxide as the exclusive product as shown in Scheme 1.
 | ||
| Scheme 1 Selective epoxidation of styrene. | ||
Keeping in view our earlier reported results55 and the general solubility of porphyrins, the best solvent for this study was taken to be a mixture of CH3CN and H2O. The solvent (5 ml) ratio (CH3CN:H2O) was fixed by performing three simultaneous reactions at 60 °C for 0.5 h involving 0.52 g (5 mmol) styrene, 1.5 mg (1.50 μmol) catalyst 1, 1.69 g (15 mmol) 30% H2O2 and 0.168 g NaHCO3 (2 mmol) in 2:1, 3:2 and 4:3 solvent mixtures as shown in entries 1, 2 and 3 respectively in Table 1. Substrate conversions corresponding to 93%, 99% and 95% were obtained in case of 2:1, 3:2 and 4:3 (CH3CN:H2O; v/v) respectively. Thus 3:2 (CH3CN:H2O; v/v) was chosen as the ideal solvent for the epoxidation of alkenes in the current study.
The amount of catalyst was optimized by performing three parallel reactions with 1.5 mg (1.50 μmol), 1 mg (1 μmol) and 2 mg (2 μmol) of catalyst 1 in 5 ml CH3CN:H2O (3:2; v/v) solvent at 60 °C keeping all other parameters constant; 0.52 g (5 mmol) styrene, 1.69 g (15 mmol) 30% H2O2 and 0.168 g NaHCO3 (2 mmol): 0.5 h reaction time as shown in entries 2, 4 and 5 in Table 1. Substrate conversions obtained were as 99%, 98% and 98% for catalyst amounts 1.5 mg (1.50 μmol), 1 mg (1 μmol) and 2 mg (2 μmol) respectively. Thus, the ideal amount of catalyst 1 was subsequently taken as 1 mg for the current study.
The amount of oxidant (30% H2O2) was optimized by performing a parallel set of three reactions in 5 ml CH3CN:H2O (3:2; v/v) solvent taking 15 mmol (1.69 g), 5 mmol (0.565 g) and 10 mmol (1.13 g) of 30% H2O2 at 60 °C maintaining other reaction conditions constant; 0.52 g (5 mmol) styrene, catalyst 1 μmol (1 mg) and 0.168 g NaHCO3 (2 mmol): 0.5 h reaction time as depicted in entries 4, 6 and 7 in Table 1. 10 mmol (1.13 g) of 30% H2O2 gave the most feasible conversion (entry 7, Table 1) and was chosen as the ideal amount of oxidant for the current study.
The amount of NaHCO3 (working as promoter by generating peroxymonocarbonate ions)46,47 was also optimized by setting up three parallel reactions taking 2 mmol (0.168 g), 1.5 mmol (0.126 g) and 1 mmol (0.084 g) of NaHCO3 in 5 ml CH3CN:H2O (3:2; v/v) solvent at 60 °C keeping other reaction parameters unchanged: 0.52 g (5 mmol) styrene, catalyst 1 μmol (1 mg) and 1.13 g H2O2 (10 mmol): 0.5 h reaction time as shown in entries 7–9 in Table 1. 1.5 mmol (0.126 g) of NaHCO3 was therefore chosen as the ideal amount of promoter (entry 8, Table 1).
Finally, the effect of temperature on the conversion rate was studied by setting up three parallel reactions at 60 °C, 50 °C and 40 °C in 5 ml CH3CN:H2O (3:2; v/v) solvent maintaining other conditions constant: 0.52 g (5 mmol) styrene, catalyst 1 μmol (1 mg), 1.13 g (10 mmol) H2O2 and 0.126 g (1.5 mmol) NaHCO3: 0.5 h reaction time as shown in entries 8, 10 and 11 respectively in Table 1. Henceforth, the ideal temperature selected for the current study was taken as 60 °C (entry 8, Table 1).
A control reaction in 5 ml CH3CN:H2O (3:2; v/v) solvent at 60 °C without the catalyst was performed to underline the importance of the catalyst keeping other reaction conditions unchanged: 0.52 g (5 mmol) styrene, 1.13 g (10 mmol) H2O2 and 0.126 g (1.5 mmol) NaHCO3: 0.5 h reaction time. The absence of catalyst markedly affected the conversion rate lowering it to just 29% (entry 12, Table 1). A similar reaction without catalyst and without NaHCO3 was performed under identical conditions. A very poor conversion rate amounting to 9% was achieved (entry 13, Table 1). Finally, the reaction in presence of catalyst but without the presence of NaHCO3 gave a conversion rate of only 18% highlighting the role the promoter (entry 14, Table 1). The full details regarding the experimental conditions utilized during the optimization of 1 towards alkene epoxidation are listed in Table 1.
To follow the reaction kinetics better, the epoxidation of one of the alkenes (e.g. cyclohexene) was monitored more closely by taking sample aliquots at different intervals ranging from 0 to 40 minutes. The reaction profile for epoxidation of cyclohexene with 1 as the catalyst is given in Fig. 8.
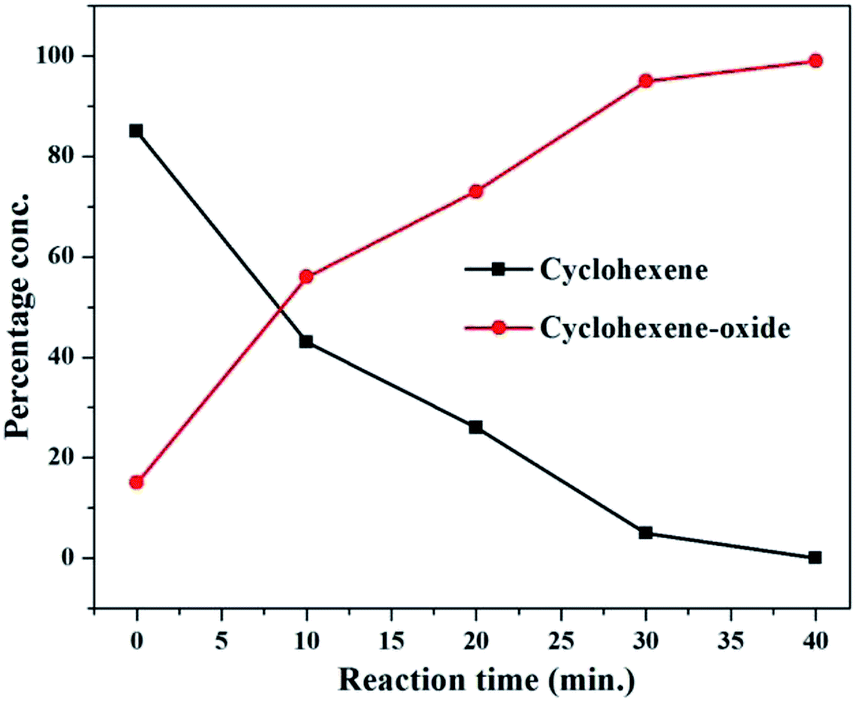 | ||
| Fig. 8 Reaction profile showing progress of epoxidation of cyclohexene with catalyst 1 at different time intervals. | ||
The reaction was initiated by taking cyclohexene (0.410 g; 5 mmol) in 5 ml of the ideal solvent (CH3CN:H2O; 3:2 v/v) in a two neck 50 ml round bottom flask along with the catalyst, H2O2 and promoter under the optimized reaction conditions as shown in entry 8, Table 1. The progress of the reaction was monitored by withdrawing small aliquots from the reaction mixture after 0 min, 10 min, 20 min, 30 min and 40 min. The aliquots from the reaction mixture were injected into and analyzed via gas-chromatography for percentage conversions. Immediately after mixing all the reactants and just shaking, 15% reactant conversion was obtained. As the reaction time increases the amount of the product (cyclohexene epoxide) progressively increases with a simultaneous decrease in the amount of the reactant (cyclohexene) as shown in Fig. 8. The reaction progress was monitored till 40 min (99% reactant conversion), however, no significant changes were observed after 30 min of reaction time (96% reactant conversion). From this we can conclude that the reaction proceeds to completion in only 0.5 h of reaction time.
The conversion rates of the reactant alkenes into epoxides was additionally quantified by using dodecane as the internal standard in one of the key reactions i.e. conversion of cyclohexene into cyclohexene epoxide. Same amount of dodecane (an internal standard) as that of reactant substrate was added to the reaction mixture to confirm the conversion rates, for example, 5 mmol of dodecane was added to the reaction mixture which was started with 5 mmol of cyclohexene. The conversion rates obtained from the two reaction mixtures were very close to each other viz. 93% (with internal standard) and 96% (without internal standard) respectively, signifying the higher selectivity of the porphyrin catalyst 1 (Fig. S16, ESI†).
Conclusions
Vanadyl β-tetrabromoporphyrin VOTPPBr4 (1) having electron withdrawing –Br substituents was synthesized through a facile procedure in quantitative yields and characterized by various spectrometric methods. 1 displayed high thermal stability and encouraging selectivity with high TOF (h−1) during epoxidation of alkenes with H2O2 and NaHCO3. 1 is highly non-planar due to bulkier –Br substituents which makes it a more efficient catalyst as the reactive site is easier to access. The catalyst 1 exhibited excellent selectivity, high conversion efficiency and high TOF (7600–9800 h−1) in significantly less reaction time of 0.5 h in aqueous medium (CH3CN:H2O; 3:2 v/v). The porphyrin catalyst was regenerated at the end of respective catalytic cycles making it reusable and thus industrially important.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
TAD is thankful to MHRD, India for senior research fellowship. We sincerely thank Science and Engineering Research Board (EMR/2016/004016) for financial support.Notes and references
- K. Weissermel and H. J. Arpe, Industrial organic chemistry, VCH, 1997 Search PubMed.
- A. S. Rao, S. K. Paknikar and J. G. Kirtane, Tetrahedron, 1983, 39, 2323–2367 CrossRef CAS.
- (a) M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed; (b) A. Butler, M. J. Clague and G. E. Meister, Chem. Rev., 1994, 94, 625–634 CrossRef CAS; (c) M. M. Abu-Omar, A. Loaiza and N. Hontzeas, Chem. Rev., 2005, 105, 2227–2252 CrossRef CAS PubMed; (d) W. Nam, Y. M. Lee and S. Fukuzumi, Acc. Chem. Res., 2014, 47, 1146–1154 CrossRef CAS PubMed; (e) T. Chaterjee and M. Ravikanth, Inorg. Chem., 2014, 53, 10520–10526 CrossRef PubMed; (f) B. J. Anding and L. K. Woo, Organometallics, 2013, 32, 2599–2607 CrossRef CAS; (g) I. Aviv and Z. Gross, Chem. Commun., 2007, 1987–1999 RSC; (h) M. R. Maurya, A. Kumar and J. Costa Pessoa, Coord. Chem. Rev., 2011, 255, 2315–2344 CrossRef CAS; (i) J. Costa Pessoa, S. Etcheverry and D. Gambino, Coord. Chem. Rev., 2015, 301–302, 24–48 CrossRef; (j) M. Casny and D. Rehder, Dalton Trans., 2004, 839–846 RSC.
- I. Bauer and H. J. Knölker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed.
- T. Ikariya and A. J. Blacker, Acc. Chem. Res., 2007, 40, 1300–1308 CrossRef CAS PubMed.
- K. A. Joergensen, Chem. Rev., 1989, 89, 431–458 CrossRef CAS.
- M. Ouchi, T. Terashima and M. Sawamoto, Chem. Rev., 2009, 109, 4963–5050 CrossRef CAS PubMed.
- G. Zassinovich, G. Mestroni and S. Gladiali, Chem. Rev., 1992, 92, 1051–1069 CrossRef CAS.
- N. Y. Oh, M. S. Seo, M. H. Lim, M. B. Consugar, M. J. Park, J.-U. Rohde, J. Han, K. M. Kim, J. Kim, L. Que Jr and W. Nam, Chem. Commun., 2005, 0, 5644 RSC.
- W. Liu and J. T. Groves, Acc. Chem. Res., 2015, 48, 1727–1735 CrossRef CAS PubMed.
- J.-B. Xia, Y. Ma and C. Chen, Org. Chem. Front., 2014, 1, 468–472 RSC.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC.
- A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkov and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804–6849 RSC.
- S. Yamazaki, Coord. Chem. Rev., 2018, 373, 148–166 CrossRef CAS.
- Z. Sun, J. Li, H. Zheng, X. Liu, S. Ye and P. Du, Int. J. Hydrogen Energy, 2015, 40, 6538–6545 CrossRef CAS.
- Y. Han, H. Fang, H. Jing, H. Sun, H. Lei, W. Lai and R. Cao, Angew. Chem., Int. Ed., 2016, 55, 5457–5462 CrossRef CAS PubMed.
- N. K Subbaiyan and F. D'Souza, Chem. Commun., 2012, 48, 3641–3643 RSC.
- M. Ethirajan, Y. Chen, P. Joshi and R. K. Pandey, Chem. Soc. Rev., 2011, 40, 340–362 RSC.
- Y. Ding, W.-H. Zhu and Y. Xie, Chem. Rev., 2017, 117, 2203–2256 CrossRef CAS PubMed.
- J. Barona-Castaño, C. Carmona-Vargas, T. Brocksom and K. de Oliveira, Molecules, 2016, 21, 310 CrossRef PubMed.
- M. O. Senge, M. Fazekas, E. G. A. Notaras, W. J. Blau, M. Zawadzka, O. B. Locos and E. M. Ni Mhuircheartaigh, Adv. Mater., 2007, 19, 2737–2774 CrossRef CAS.
- R. C. Hardison, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 5675 CrossRef CAS.
- D. Mauzerall, in Photosynthesis I, Springer Berlin Heidelberg, Berlin, Heidelberg, 1977, pp. 117–124 Search PubMed.
- J. Feierabend and S. Dehne, Planta, 1996, 198, 413–422 CrossRef CAS.
- C. K. Chang and M.-S. Kuo, J. Am. Chem. Soc., 1979, 101, 3413–3415 CrossRef CAS.
- M. Vicente and K. Smith, Curr. Org. Synth., 2014, 11, 3–28 CrossRef CAS PubMed.
- P. Bhyrappa and V. Krishnan, Inorg. Chem., 1991, 30, 239–245 CrossRef CAS.
- J. E. Drach and F. R. Longo, J. Org. Chem., 1974, 39, 3282–3284 CrossRef CAS PubMed.
- L. R. Nudy, H. G. Hutchinson, C. Schieber and F. R. Longo, Tetrahedron, 1984, 40, 2359–2363 CrossRef CAS.
- J. E. Baldwin, M. J. Crossley and J. DeBernardis, Tetrahedron, 1982, 38, 685–692 CrossRef CAS.
- S. Hiroto, Y. Miyake and H. Shinokubo, Chem. Rev., 2017, 117, 2910–3043 CrossRef CAS PubMed.
- K. S. Suslick, P. Bhyrappa, J. H. Chou, M. E. Kosal, S. Nakagaki, D. W. Smithenry and S. R. Wilson, Acc. Chem. Res., 2005, 38, 283–291 CrossRef CAS PubMed.
- K. S. Suslick, N. A. Rakow, M. E. Kosal and J. H. Chou, J. Porphyrins Phthalocyanines, 2000, 04, 407–413 CrossRef CAS.
- T. Katsuki and K. B. Sharpless, J. Am. Chem. Soc., 1980, 102, 5974–5976 CrossRef CAS.
- E. M. McGarrigle and D. G. Gilheany, Chem. Rev., 2005, 105, 1563–1602 CrossRef CAS PubMed.
- M. C. A. van Vliet, I. W. C. E. Arends and R. A. Sheldon, Chem. Commun., 1999, 0, 821–822 RSC.
- F. G. Gelalcha, B. Bitterlich, G. Anilkumar, M. K. Tse and M. Beller, Angew. Chem., Int. Ed., 2007, 46, 7293–7296 CrossRef CAS PubMed.
- K. Sato, M. Aoki, M. Ogawa, T. Hashimoto and R. Noyori, J. Org. Chem., 1996, 61, 8310–8311 CrossRef CAS PubMed.
- R. D. Oldroyd, J. M. Thomas, T. Maschmeyer, P. A. MacFaul, D. W. Snelgrove, K. U. Ingold and D. D. M. Wayner, Angew. Chem., Int. Ed. Engl., 1996, 35, 2787–2790 CrossRef CAS.
- A. Corma, A. Fuerte, M. Iglesias and F. Sánchez, J. Mol. Catal. A: Chem., 1996, 107, 225–234 CrossRef CAS.
- R. A. Van Santen and H. P. C. E. Kuipers, Adv. Catal., 1987, 35, 265–321 CAS.
- R. D. Bach, O. Dmitrenko, W. Adam and S. Schambony, J. Am. Chem. Soc., 2003, 125, 924–934 CrossRef CAS PubMed.
- G. Grigoropoulou, J. H. Clark and J. A. Elings, Green Chem., 2003, 5, 1–7 RSC.
- B. S. Lane and K. Burgess, Chem. Rev., 2003, 103, 2457–2474 CrossRef CAS PubMed.
- P. Zhou, X. Wang, B. Yang, F. Hollmann and Y. Wang, RSC Adv., 2017, 7, 12518–12523 RSC.
- D. Quiñonero, D. G. Musaev and K. Morokuma, J. Mol. Struct.: THEOCHEM, 2009, 903, 115–122 CrossRef.
- A. M. Garcia, V. Moreno, S. X. Delgado, A. E. Ramírez, L. A. Vargas, M. Á. Vicente, A. Gil and L. A. Galeano, J. Mol. Catal. A: Chem., 2016, 416, 10–19 CrossRef CAS.
- K. Yuan, T. Song, D. Wang, Y. Zou, J. Li, X. Zhang, Z. Tang and W. Hu, Nanoscale, 2018, 10, 1591–1597 RSC.
- L. Vilas Boas and J. Costa Pessoa, Vanadium, in Comprehensive Coordination Chemistry, ed. G. Wilkinson, R. D. Gillard and J. A. McCleverty, Pergamon, Oxford, 1987, vol. 3, pp. 453–583 Search PubMed.
- N. Gharah, S. Chakraborty, A. K. Mukherjee and R. Bhattacharyya, Chem. Commun., 2004, 0, 2630 RSC.
- S. K. Maiti, K. M. A. Malik, S. Gupta, S. Chakraborty, A. K. Ganguli, A. K. Mukherjee and R. Bhattacharyya, Inorg. Chem., 2006, 45, 9843–9857 CrossRef CAS PubMed.
- W. Nam, H. J. Lee, S. Y. Oh, C. Kim and H. G. Jang, J. Inorg. Biochem., 2000, 80, 219–225 CrossRef CAS PubMed.
- K. Zhang, Y. Yu, S. T. Nguyen, J. T. Hupp, L. J. Broadbelt and O. K. Farha, Ind. Eng. Chem. Res., 2015, 54, 922–927 CrossRef CAS.
- N. A. Stephenson and A. T. Bell, J. Mol. Catal. A: Chem., 2007, 275, 54–62 CrossRef CAS.
- R. Kumar, N. Chaudhary, M. Sankar and M. R. Maurya, Dalton Trans., 2015, 44, 17720–17729 RSC.
- P. K. Kumar, P. Bhyrappa and B. Varghese, Tetrahedron Lett., 2003, 44, 4849–4851 CrossRef CAS.
- W. Chen, T. Suenobu and S. Fukuzumi, Chem.–Asian J., 2011, 6, 1416–1422 CrossRef CAS PubMed.
- T. Takeuchi, H. B. Gray and W. A. Goddard, J. Am. Chem. Soc., 1994, 116, 9730–9732 CrossRef CAS.
- F. D'Souza, M. E. Zandler, P. Tagliatesta, Z. Ou, J. Shao, E. Van Caemelbecke and K. M. Kadish, Inorg. Chem., 1998, 37, 4567–4572 CrossRef.
- H. Yao and D. E. Richardson, J. Am. Chem. Soc., 2000, 122, 3220–3221 CrossRef CAS.
- P. Saisaha, J. W. de Boer and W. R. Browne, Chem. Soc. Rev., 2013, 42, 2059–2074 RSC.
- B. Qi, X.-H. Lu, D. Zhou, Q.-H. Xia, Z.-R. Tang, S.-Y. Fang, T. Pang and Y.-L. Dong, J. Mol. Catal. A: Chem., 2010, 322, 73–79 CrossRef CAS.
- C. J. Schneider, J. E. Penner-Hahn and V. L. Pecoraro, J. Am. Chem. Soc., 2008, 130, 2712–2713 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1880991. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra09825e |
| This journal is © The Royal Society of Chemistry 2019 |