
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe neutralization of heparan sulfate by heparin-binding copolymer as a potential therapeutic target
Bartlomiej Kalaska a,
Joanna Miklosza,
Kamil Kamińskib,
Bogdan Musielakb,
Shin-Ichi Yusac,
Dariusz Pawlaka,
Maria Nowakowskab,
Krzysztof Szczubiałka*b and
Andrzej Mogielnicki*a
a,
Joanna Miklosza,
Kamil Kamińskib,
Bogdan Musielakb,
Shin-Ichi Yusac,
Dariusz Pawlaka,
Maria Nowakowskab,
Krzysztof Szczubiałka*b and
Andrzej Mogielnicki*a
aDepartment of Pharmacodynamics, Medical University of Bialystok, Mickiewicza 2c, 15-089 Bialystok, Poland. E-mail: amogiel@umb.edu.pl
bFaculty of Chemistry, Jagiellonian University, Gronostajowa 2, 30-387 Krakow, Poland. E-mail: szczubia@chemia.uj.edu.pl
cDepartment of Applied Chemistry, Graduate School of Engineering, University of Hyogo, Himeji, Hyogo, Japan
First published on 23rd January 2019
Abstract
Besides regulating ligand–receptor and cell–cell interactions, heparan sulfate (HS) may participate in the development of many diseases, such as cancer, bacterial or viral infections, and their complications, like bleeding or inflammation. In these cases, the neutralization of HS could be a potential therapeutic target. The heparin-binding copolymer (HBC, PEG41-PMAPTAC53) was previously reported by us as a fully synthetic compound for efficient and safe neutralization of heparins and synthetic anticoagulants. In a search for molecular antagonists of HS, we examined the activity of HBC as an HS inhibitor both in vitro and in vivo and characterized HBC/HS complexes. Using a colorimetric Azure A method, isothermal titration calorimetry and dynamic light scattering techniques we found that HBC binds HS by forming complexes below 200 nm with less than 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry. We confirmed the HBC inhibitory effect in rats by measuring activated partial thromboplastin time, prothrombin time, anti-factor Xa activity, anti-factor IIa activity, and platelet aggregation. HBC reversed the enhancement of all tested parameters caused by HS demonstrating that cationic synthetic block copolymers may have a therapeutic value in various disorders involving overproduction of HS.
:1 stoichiometry. We confirmed the HBC inhibitory effect in rats by measuring activated partial thromboplastin time, prothrombin time, anti-factor Xa activity, anti-factor IIa activity, and platelet aggregation. HBC reversed the enhancement of all tested parameters caused by HS demonstrating that cationic synthetic block copolymers may have a therapeutic value in various disorders involving overproduction of HS.
Introduction
Heparan sulfate (HS)1,2 is a glycosaminoglycan (GAG), a naturally sulfated polysaccharide with a total content of approximately one sulfate per disaccharide unit. The molecular weight of HS is quite disperse (14–45 kDa),3 and its structure varies between tissues.2 It is synthesized in the Golgi compartments of the cells, covalently attached to the core proteins forming heparan sulfate proteoglycans (HSPGs). These complexes are associated with the surface of virtually all cells and with extracellular matrix (ECM) of animal tissues. HSPGs play a vast number of physiological roles. The activity of HSPGs is based on the electrostatic interactions of highly charged HS chains with proteins.4 A degree of selectivity in the binding of HSPGs to various ligands (e.g., growth factors, cytokines and morphogens) has been observed. HSPGs act as receptors for proteases, facilitate cell–ECM attachment and cell–cell interactions, modulate the activity of angiogenic factors,5 and play a prominent role in the immune system by regulating leukocyte development and migration, immune activation, and inflammatory processes.6 Last but not least, HS, being structurally very close to heparin, shows anticoagulative properties, although weaker than those of the latter.1 Like in the case of heparins, the degree of sulfation of HS may vary significantly and both low-7 and highly-sulfated8 forms of HS have been found depending on the tissue of origin or in some pathologies.9–11Unfortunately, HS also participates in many pathological processes. HS may play a crucial role in cancer progression, angiogenesis, and metastasis12,13 showing both tumor progressing14 or tumor inhibiting activity.15 HS was present in the fluid of an ascitic form of a chemically-induced pancreatic ductal adenocarcinoma in the Syrian golden hamster. Moreover, plasma containing the fluid showed markedly prolonged coagulation times, indicating that HS may be largely responsible for the bloody ascites.16 More recently it was found that high serum HS concentration was responsible for the poor treatment outcome with epidermal growth factor receptor-tyrosine kinase inhibitors in patients with non-small cell lung cancer.17 Since in this study HS concentrations were significantly higher in patients with progressive cancer than in those with a non-progressive form of cancer, HS was proposed as a candidate biomarker for treatment resistance.
Pathologically increased plasma levels of endogenous HS may also be responsible for serious bleeding. Palmer and co-authors have identified HS in the blood of a patient with multiple myeloma who had lethal bleeding.18 The authors concluded that recognition of the existence of endogenous anticoagulant inhibitors in bleeding patients could be crucial because of the potential for treatment with proper antidotes. Recently, it has been shown that patients with untreated mucopolysaccharidosis (MPS) had higher levels of HS in blood in comparison to age-matched controls.19 Tolar et al. found that increased levels of GAGs including HS were responsible for the increased risk of life-threatening pulmonary bleeding.20
HSPGs have been found to be the components of the attachment receptors of many viruses.21 It has been shown that the interaction of dengue virus (DV) with human cells is mediated by HSPGs,22 and a highly sulfated type of HS is a cellular receptor utilized by DV envelope protein to bind to target cells.23 Other ligands of this receptor such as heparin or the polysulfonated pharmaceutical suramin prevented DV infection, indicating that the interaction with HS is a critical determinant of infectivity. Furthermore, acute HS levels were significantly higher in dengue patients and decreased at convalescence.24 The authors concluded that developing pharmaceuticals that inhibit target cell binding may be an effective strategy for treating flavivirus infections.23 HS-containing receptors are also involved in the entry of many other viruses causing both severe diseases (human immunodeficiency virus (HIV),25 hepatitis C virus (HCV),26 herpes simplex virus (HSV),27 Japanese encephalitis virus),28 mild pathologies (Sindbis virus29 foot-and-mouth disease virus,30 coxsackievirus A),31 and non-pathogenic ones (human foamy virus).32 Recently, it was also shown that blocking the interaction of HS with Ebola virus leads to significant inhibition of basolateral infection of epithelial cells.33
Myocardial dysfunction in sepsis has been linked to inflammation caused by pathogen-associated and host danger-associated molecular patterns, which include soluble HS.34 Indeed, the administration of endotoxin from a Gram-negative bacterium (Escherichia coli) to pigs resulted in increased serum levels of HS, signalizing a shedding of the glycocalyx.35 The experiments in HL-1 cardiomyocytes showed that compounds antagonizing HS might have the potential for further development as broad-spectrum anti-inflammatory agents in sepsis-induced myocardial inflammation and dysfunction.34
All of these findings indicate that HS could be a novel therapeutic target in the treatment of cancer, viral infections, bleeding, inflammatory processes, MPS and possibly other diseases. Therefore, both low-molecular-weight and macromolecular compounds have been developed as the inhibitors of various activities of HS or its biosynthesis. For example, surfen, a small molecule capable of binding to HS, was found to inhibit HS-dependent binding, and signaling by fibroblast growth factor 2 (FGF2), infection by HSV-1, cell attachment, and endothelial sprouting stimulated by vascular endothelial growth factor and FGF2.36 Its interaction with HS also resulted in strong and general inhibition of differentiation and promotion of pluripotency in mouse embryonic stem cells.37 In a very recent study surfen and its derivative, oxalyl surfen, showed potential in the inhibition of tau hyperphosphorylation, which was found to strongly correlate with the progression and memory loss in Alzheimer's disease.38 It was suggested that preventing tau hyperphosphorylation was due to surfen-induced alterations in HS synthesis, metabolism, structure, and stability. Moreover, compounds that inhibit HS–protein interactions have considerable potential as anti-inflammatory therapeutics.39 Also, glucosamine analogs which inhibit HS biosynthesis have been proposed as possible drugs in the treatment of amyloid diseases.40 However, inhibiting HS synthesis may result in unpredictable adverse effects taking into account the multiple physiological roles of HS.
In this study, we have focused on the possibility of inhibition of the anticoagulant activity of HS, particularly because there are no reports on the reversal of HS-induced bleeding. This action of HS was previously neutralized by protamine.41 However, the inhibition was weaker than that of unfractionated heparin (UFH), especially when measured by anti-factor Xa (anti-fXa) activity. Moreover, taking into account the safety issues related to protamine use, it is not a good therapeutic candidate.42 We have recently developed a heparin-binding copolymer (HBC, PEG41-PMAPTAC53) well-tolerated by rats and mice, which may constitute an alternative to protamine in the efficient neutralization of UFH. Importantly, we found that HBC also reverses anticoagulation caused by low-molecular-weight heparins (LMWHs),43 fondaparinux,43 and sulfonated synthetic polymers.44 Here, our aim was to check whether HBC may bind and neutralize HS in the living organism. We used the sample of HS as a model to show potential of HBC as a new therapeutic for various disorders involving increased concentration of HS.
Materials and methods
Materials
HS (95.95%) from 6C USA Inc was used as received. 4-Cyano-4-(phenylcarbonothioylthio)pentanoic acid was synthesized according to the method reported by Mitsukami et al.45 Poly(ethylene glycol)methyl ether (average molecular weight ∼ 2000 Da) from Aldrich was used as received without further purification. Poly(ethylene glycol)4-cyano-4-(phenylcarbonothioylthio)pentanoate (PEG41-CTA) was synthesized according to the literature with slight modification.46 3-(Methacryloylamino)propyl trimethylammonium chloride (MAPTAC, 50 wt% in water) from Aldrich was passed through an inhibitor removal column. 4,40-Azobis(4-cyanopentanoic acid; V-501, 98%) from Wako Pure Chemical was used as received without further purification. Azure A chloride (Fluka standard) was purchased from Fluka (Switzerland). Water was purified with a Millipore Milli-Q system. Other reagents were used as received.HBC preparation and characterization
The preparation, recovery, and characterization of HBC have been described previously.43 Briefly, MAPTAC (5.53 g, 25.0 mmol), V-501 (69.2 mg, 0.247 mmol), and PEG41-CTA (1.13 g, 0.501 mmol) were dissolved in water (41.0 mL). The mixture was degassed by purging with Ar gas for 30 min. Reversible Addition-Fragmentation chain Transfer (RAFT) polymerization was carried out at 70 °C for 5 h. The polymerization mixture was dialyzed against pure water for two days. HBC was recovered using a freeze-drying technique (5.58 g, 83.8%).In vitro anticoagulant properties of HS
The anticoagulant properties of HS were analyzed by measuring activated partial thromboplastin time (aPTT), prothrombin time (PT), and anti-fXa and anti-factor IIa (anti-fIIa) activity. In brief, 200 μL of pooled rat plasma were mixed with 20 μL of HS (0.005, 0.01, 0.05, 0.1 mg mL−1 of pooled plasma). After 5 min of incubation (37 °C), coagulation parameters were measured according to the manufacturer instructions. aPTT and PT were analyzed with an optical method (Coag Chrom 4000, Bio-Ksel) and routine laboratory reagents (Bio-Ksel). Anti-fXa and anti-fIIa activities were determined by chromogenic assays (Dynex Tech.) and the manufacturer instructions (Sekisui Diagnostics).In vitro binding of HS by HBC
The ability of HBC to bind HS was assessed with a colorimetric method using Azure A, as described previously.43,44,47 Briefly, HS forms a complex with Azure A. Dimers of Azure A absorb at 513 nm while the monomeric forms of Azure A absorb at 630 nm. The addition of HS results in the disruption of Azure A dimers and consequently increases 630 nm and decreases 513 nm absorption. The initial concentration of HS in the mixture was 0.2 g L−1 whereas the concentration of the polycation added was 1.6 g L−1. The volumes were mixed in such a way that phosphate buffer solution (PBS) and a polycation solution was added to 0.5 mL of HS so that the total volume of the sample was 1 mL.In vitro neutralization of HS by HBC
The ability of HBC to neutralize HS was assessed by measuring aPTT, PT, and anti-fXa and anti-fIIa activity, as described previously.43,44,47 Briefly, 10 μL of HS (0.05 mg mL−1 of plasma) was mixed with 200 μL of pooled plasma. After 2 min and 30 s of incubation, 10 μL of a solution containing increasing concentrations of HBC (0.0125, 0.025, 0.0375, 0.05 mg mL−1 of pooled plasma; mass ratios 0.25:1, 0.5:1, 0.75:1, 1:1) was added. After 2 min and 30 s of incubation, the aPTT, PT, and anti-fXa and anti-fIIa activity were automatically determined according to the methods described above.
Dynamic light scattering (DLS) and zeta potential measurements
The size of complexes formed by HBC and HS was studied with the DLS technique in serum and PBS. HS was dissolved in PBS or bovine serum (0.4 mg mL−1). To that mixture, the HBC solutions in PBS buffer were added (12 mg mL−1), and the formation of complexes was monitored by DLS and zeta potential using a Zetasizer Nano-ZS (Malvern Instruments, UK).Isothermal titration calorimetry (ITC) measurements
ITC measurements were made using Malvern MICROCAL PEAQ-ITC. HBC was dissolved in PBS; its concentration was 8 g L−1 (0.001078 mol L−1). HS was also dissolved in PBS in the concentration of 0.5 g L−1. The measurement temperature was 25 °C and the mixing speed of 750 rpm. The measuring chamber contained 270 μL of HS solution to which 19 portions of HBS solution, each 2 μL, were added.Animals
Male Wistar rats obtained from the Centre of Experimental Medicine in the Medical University of Bialystok were bred in a 12 h light/dark cycle in a humidity- and temperature-controlled room in specific pathogen-free conditions. They allowed having ad libitum access to standard chow and sterilized tap water. All the procedures involving animals were approved by Local Ethical Committee on Animal Testing (Permit Number 60/2018) and conducted according to Directive 2010/63/EU of the European Parliament and the Council on the protection of animals, ARRIVE guidelines, and the national laws.In vivo neutralization of HS by HBC
Thirty-three male Wistar rats weighing 182–212 g were randomly assigned to 3 groups, anesthetized by an intraperitoneal injection of pentobarbital (45 mg kg−1), and placed in a supine position on a heated operating table. Rats were intravenously injected into the right femoral vein with HBC (4.5 mg kg−1) 5 min after injection with HS (6 mg kg−1). Twenty minutes after HS administration, the blood samples were collected from the heart and drawn into 3.13% trisodium citrate. Neutralization of HS by HBC was monitored by measuring aPTT, PT, anti-fXa and anti-fIIa activity, and platelet aggregation. Coagulation parameters were determined as described above whereas platelet aggregation was measured after incubation of sodium citrate-anticoagulated blood with 0.9% NaCl solution (volume ratio 1:1) for 20 min at 25 °C and then for 15 min at 37 °C. The changes in impedance registered as aggregation curve were recorded for 6 min after collagen addition (7.5 μg mL−1) using a Chrono-log aggregometer (Chrono-log Corp.). Four parameters were automatically calculated: maximal extension (MaxA), lag phase (Lag), the slope of platelet aggregation (Slp), and area under the curve (AUC). The white blood cell count (WBC) was automatically performed using an animal blood counter (scil Vet abc Plus, Horiba, France).
Statistical analysis
The data is shown as median with lower and upper limits or mean ± SD and analyzed using the Mann–Whitney test or unpaired Student's t-test, depending on the normal distribution results. Results were analyzed and graphically presented using GraphPad Prism 6 software (USA). P values less than 0.05 were considered significant.Results
The anticoagulant properties of HS in rat plasma
HS concentration-dependently and maximally increased aPTT and showed both anti-fXa and anti-fIIa activities. The maximum anticoagulant effect occurred at 0.05 mg mL−1 as measured by aPTT, anti-fXa and anti-fIIa activity tests, while PT was prolonged two-fold for HS at 0.1 mg mL−1 (Fig. 1). We chose 0.05 mg mL−1 concentration of HS for further in vitro studies.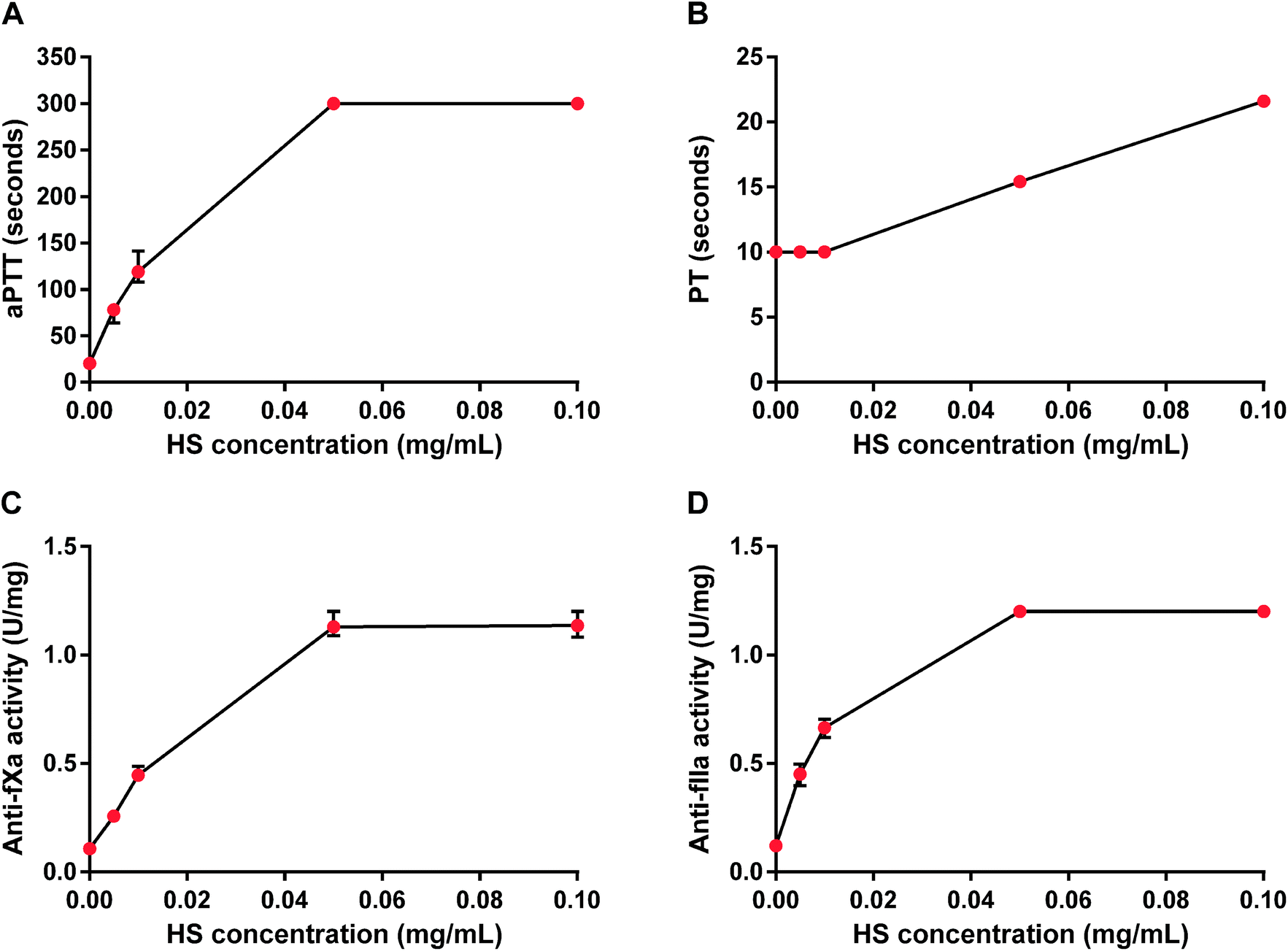 | ||
| Fig. 1 Anticoagulant properties of heparan sulfate (HS) in rat plasma. Effects of HS were monitored by measuring activated partial thromboplastin time (aPTT, A), prothrombin time (PT, B), anti-factor Xa (anti-fXa, C) and anti-factor IIa (anti-fIIa) activity (D). Results are shown as median values with lower and upper limits. | ||
The binding and the neutralization of HS by HBC in phosphate-buffered saline and rat plasma
From among a large panel of polymers we have previously studied as the inhibitors of UFH43,47–51 and synthetic polymeric anticoagulants44 we have selected HBC as a potential HS inhibitor (Fig. 2).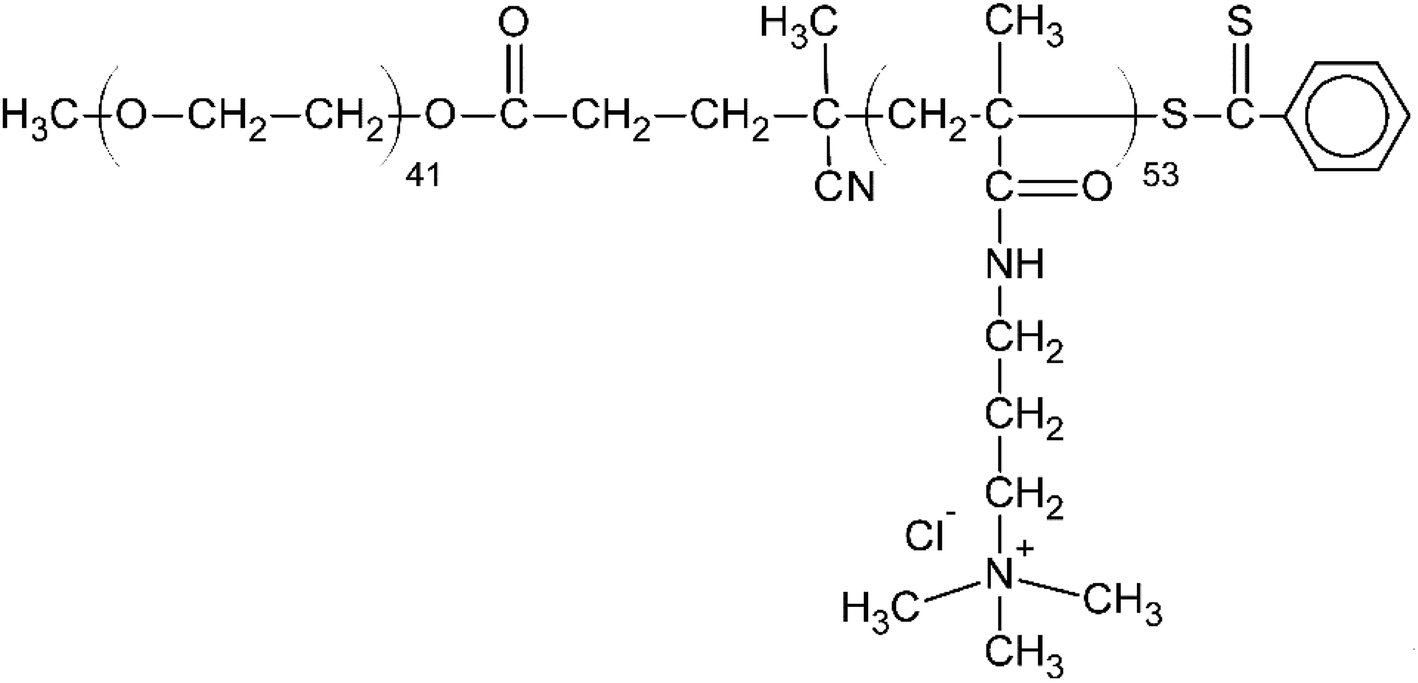 | ||
| Fig. 2 The chemical structure of heparin-binding copolymer (HBC). | ||
HBC is a block copolymer composed of a cationic block of poly(3-(methacryloylamino)propyl trimethylammonium chloride) (PMAPTAC) and a neutral block of poly(ethylene glycol) (PEG). HBC was synthesized using RAFT allowing for the preparation of the block polymers with very low molecular weight dispersity. The molecular weight of HBC and its dispersity index were found to be 14 kDa (nuclear magnetic resonance, NMR) and 1.02 (gel permeation chromatography, GPC), respectively. The molecular weight of HBC was calculated based on the integral intensity ratios of signals of the methylene protons in the PEG block and the pendant methylene protons in the PMAPTAC block and assuming the molecular weight of the PEG block of 2000 Da provided by the manufacturer. HBC showed superior in vitro and in vivo properties as a UFH inhibitor compared to other studied polymers and protamine.43,47–51 To find out if HS is negatively charged enough to form a complex with cationic HBC, we used a colorimetric method using Azure A, a cationic dye. The experiment revealed that HBC completely bound HS in PBS with an optimal ratio of 0.9 mg of the copolymer for 1 mg of HS (Fig. 3).
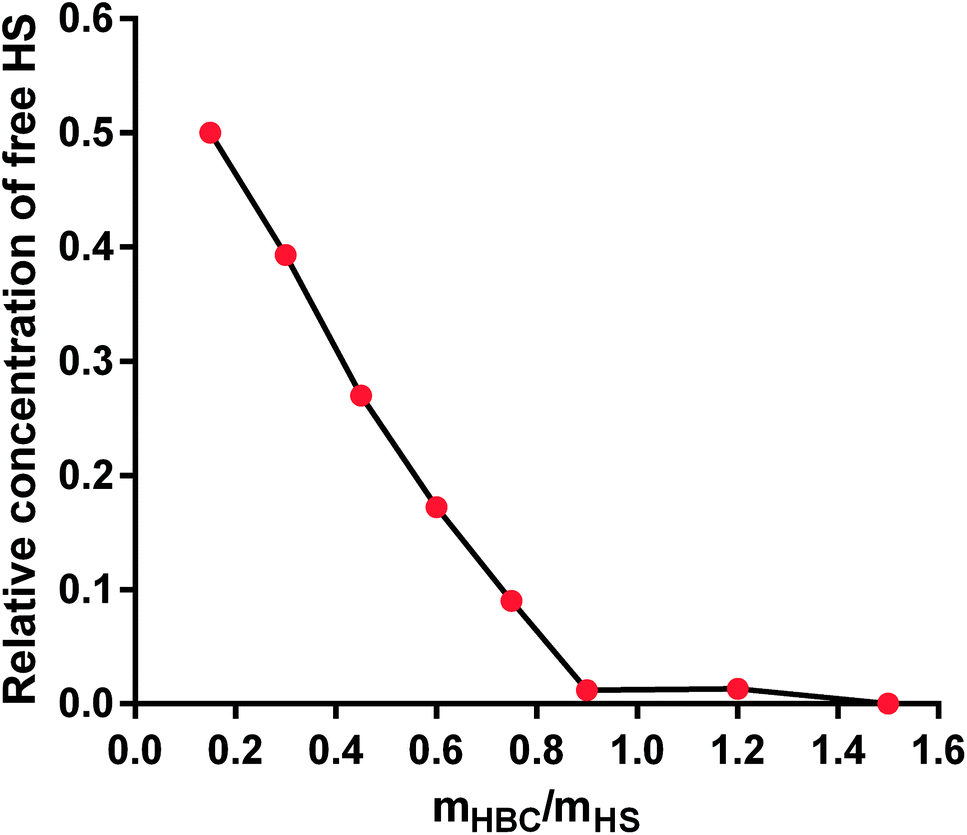 | ||
| Fig. 3 The binding of heparan sulfate (HS) by heparin-binding copolymer (HBC) in phosphate buffer solution (PBS). | ||
For further analysis of the effectiveness of HBC, we studied the in vitro neutralization of HS measured as aPTT, PT, anti-fXa activity, and anti-fIIa activity in rat plasma. The results of the coagulation tests indicated that the full neutralization of HS required an HBC to HS ratio of about 0.75:1. Anti-fIIa activity in the presence of HS was decreased by HBC even below the desired normal value (Fig. 4). We chose 0.75:1 (HBC:HS) mass ratio for further in vivo studies.
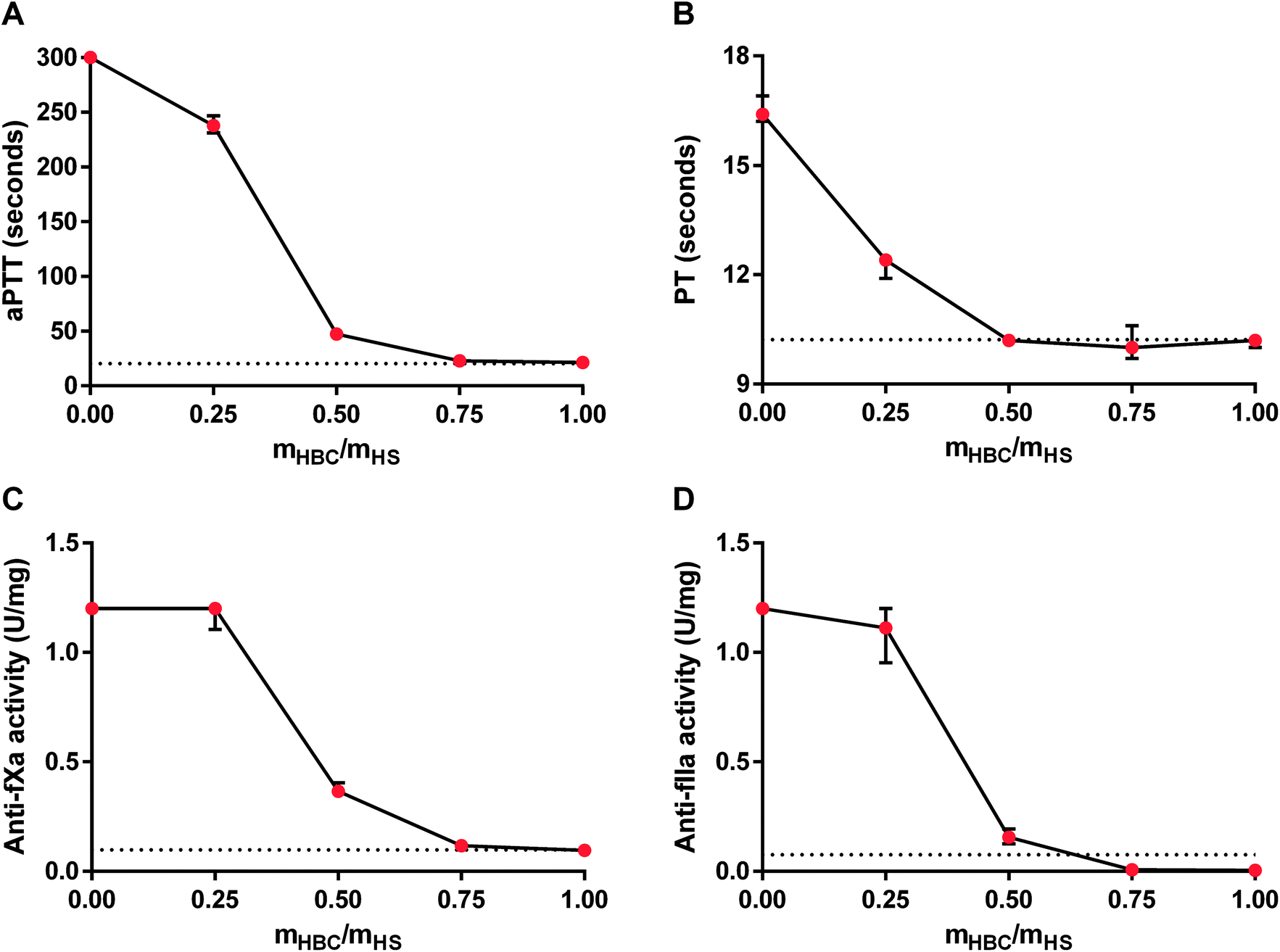 | ||
| Fig. 4 Neutralization of heparan sulfate (HS) by heparin-binding copolymer (HBC) in rat plasma. Neutralization was monitored in vitro by measuring activated partial thromboplastin time (aPTT, A), prothrombin time (PT, B), anti-factor Xa (anti-fXa) activity (C), and anti-factor IIa (anti-fXa) activity (D). Results are shown as median values with lower and upper limits. Dotted lines indicate normal values for coagulation parameters of rat plasma. | ||
The neutralization of HS by HBC in rats
We used the same coagulation parameters to determine the in vivo inhibitory potency of HBC on HS in Wistar rats as in the in vitro studies. HBC completely neutralized HS as assessed by aPTT, PT, and anti-IIa assays (Fig. 5). Anti-fXa assay showed more than 90% neutralization of HS activity by HBC. HBC had no procoagulant properties as observed in the in vitro anti-IIa assay.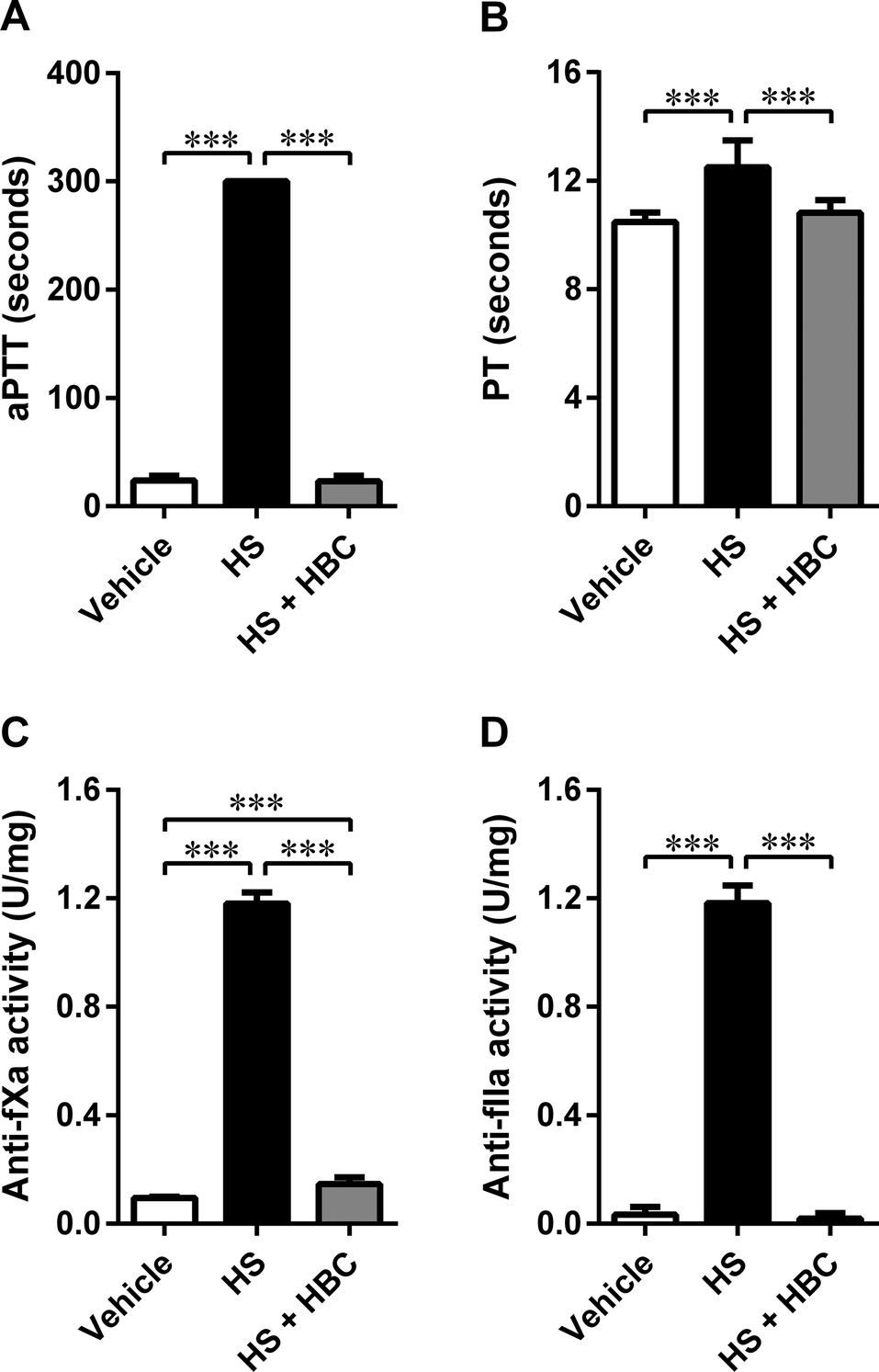 | ||
| Fig. 5 Neutralization of heparan sulfate (HS) (6 mg kg−1, 1 mL kg−1) by heparin-binding copolymer (HBC) (4.5 mg kg−1, 1 mL kg−1 by weight) in Wistar rats. Neutralization was assessed by measuring activated partial thromboplastin time (aPTT, A), prothrombin time (PT, B), anti-factor Xa activity (anti-fXa, C), and anti-factor IIa activity (anti-fXa, D). ***p < 0.001, unpaired Student's t-test. Results are shown as mean ± SD. | ||
In the same animal model, we investigated the effect of HS alone and followed by HBC on platelet aggregation. HS induced statistically significant activation of collagen-induced platelet aggregation expressed as MaxA and AUC. HBC administered after HS did not affect the platelet aggregation compared to PBS-injected rats, but significantly reduced the slope of platelet aggregation curve compared to HS-injected rats (Fig. 6). HBC did not potentiate the increase of WBC induced by HS (2.6 ± 0.7 × 103/mm−3, 3.7 ± 0.7 × 103/mm−3, and 4.1 ± 0.7 × 103/mm−3 in the vehicle, HS, and HS + HBC, respectively; p < 0.05 vehicle vs. HS).
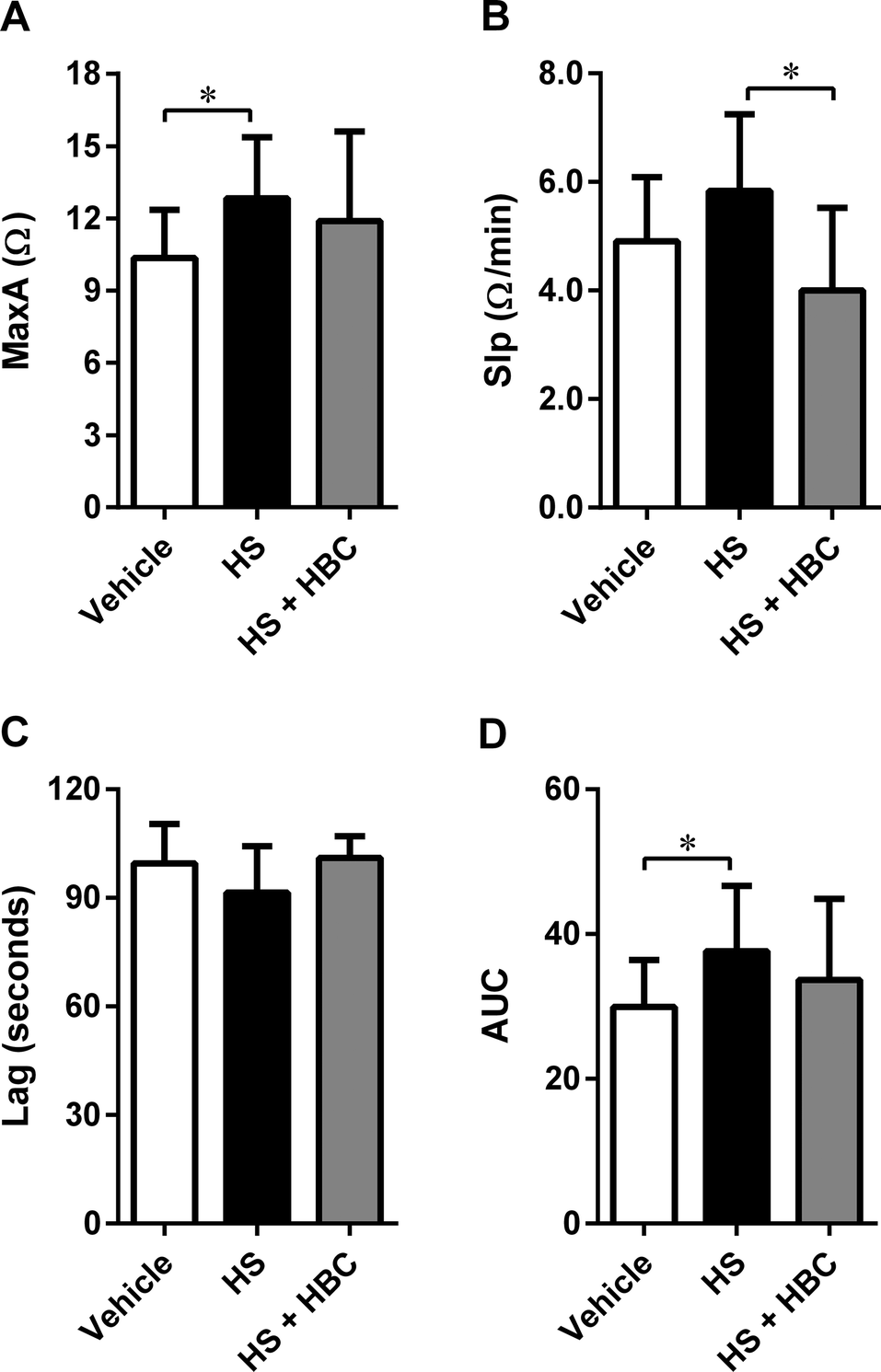 | ||
| Fig. 6 Neutralization of heparan sulfate (HS) (6 mg kg−1, 1 mL kg−1) by heparin-binding copolymer (HBC) (4.5 mg kg−1, 1 mL kg−1) in Wistar rats. Neutralization was assessed by measuring platelet aggregation expressed as the maximal extension (MaxA, A), the slope of platelet aggregation (Slp, B), lag time (Lag, C), and area under the curve (AUC, D) in rats. *p < 0.05, unpaired Student's t-test. Results are shown as mean ± SD. | ||
The size of HS–HBC complexes and heat of HS binding by HBC
Using DLS technique, we found that the diameter of the objects formed in the PBS solution of HS and in the mixtures of PBS solutions of HS and HBC at HBC/HS mass ratios from 0.3 to 1.5 was below 500 nm (Fig. 7, Table 1).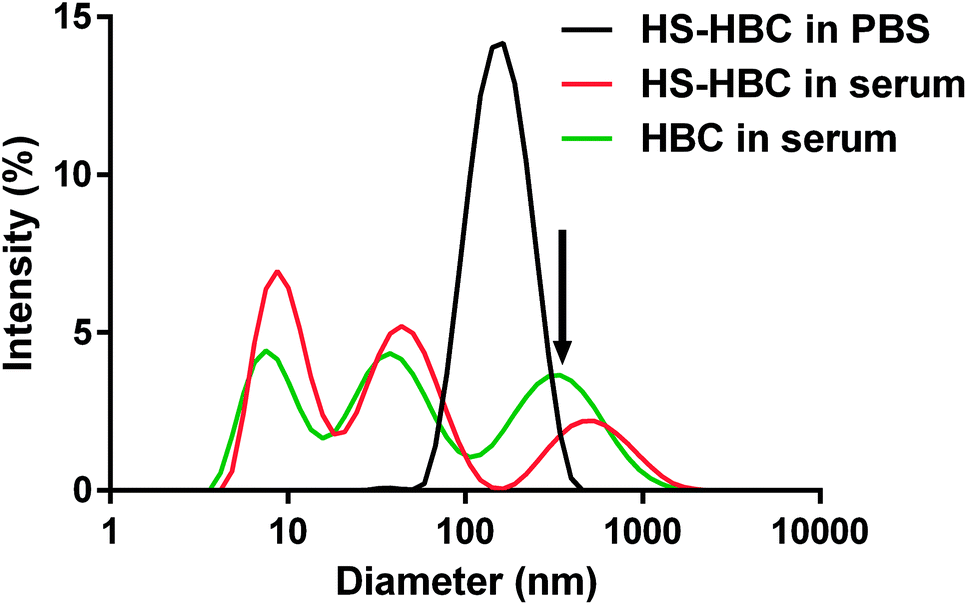 | ||
| Fig. 7 The size distribution of heparin-binding copolymer-heparan sulfate (HBC–HS) complexes measured by dynamic light scattering (DLS) analysis. The concentrations of HS and HBC were 0.4 mg mL−1 and 12 mg mL−1, respectively. | ||
| HBC/HS mass ratio | 0 (pure HS) | 0.3 | 0.6 | 0.75 | 0.9 | 1.2 | 1.5 |
|---|---|---|---|---|---|---|---|
| Zeta potential [mV] | −10.3 ± 0.7 | −8.6 ± 1.4 | −6.5 ± 0.1 | −4.7 ± 0.6 | −2.7 ± 0.8 | 0.9 ± 0.1 | 2.3 ± 0.2 |
| Z-average size [nm] | <10.0 | 89.0 | 169.7 | 154.0 | 143.8 | 371.3 | 500.7 |
Both HS and HBC are strong polyelectrolytes therefore one may expect that they form complexes with various proteins present in serum.52 Indeed, we observed that HBC formed complexes of very different sizes with serum proteins as indicated by the three maxima present in the DLS spectrum of HBC in serum (Fig. 7, green line). Because of the multitude of serum proteins, it was not possible to ascribe the maxima to the particular HBC-protein complexes. Moreover, the polycations and polyanions also form complexes called polyion complexes (PICs). In our case the respective polycation and polyanion are HBC and HS, which in PBS (i.e. in the absence of proteins) formed complexes smaller than 200 nm (Fig. 7, black line). The PICs may also interact with proteins to form even larger complexes.53 This might explain why the size of PICs in serum was more disperse and extended to greater sizes than PICs in PBS (Fig. 7, black line vs. green line) and why the PICs formed by HS and HBC in serum were greater than those in PBS (Fig. 7, red line vs. green line).
The measurements of the zeta potential of HS in the systems with increasing concentrations of HBC (Table 1) indicated that the surface charge of the HS–HBC complexes became more and more positive with increasing concentration of HBC. The zeta potential increased monotonically from −8.6 mV to +2.3 mV for HBC/HS mass ratio in the range of 0.3 to 1.5. These experiments allowed also to find out that the zeta potential of the complexes at the HBC/HS mass ratio corresponding to complete neutralization of anticoagulant activity of HS (about 0.6) was equal to −6.5 mV, while their size was about 170 nm. The negative zeta potential of the HS–HBC complexes at this particular mass ratio indicates that these complexes interact mostly with cationic serum proteins.
Most importantly, the measurement of the size of HBC–HS complexes indicated that the diameter of the complexes formed in the blood vessels would not exceed 300 nm (Fig. 7). This is a safe size concerning in vivo use and will not cause congestion in blood vessels.
In the next step, we applied ITC technique to measure the heat of the interaction between HS and HBC. We tried to determine binding affinity, stoichiometry, and enthalpy of in PBS solution and elucidate the mechanisms underlying molecular interactions. The obtained data show that the enthalpy ΔH (kcal g−1) of interaction was (9.8 ± 0.2) × 10−2 (Fig. 8) confirming the formation of the complex. The ITC measurement indicated that the process of HS complexation by HBC continued until the HBC/HS mass ratio reached about 1, what is indicated by the sudden drop of the evolved heat occurring at this ratio down to values close to zero. Since the measurement gave the net thermal effect of the interaction of HS with HBC (the solvent was the same for both polymers so the heat of mixing of both solutions was zero) it could be concluded that further addition of HBC did not result in the formation of complexes. Comparison of these results with those obtained from the measurement of the zeta potential indicated also that at HBC/HS mass ratio of 1 the zeta potential of the complexes reached zero. Thus, the positive net zeta potential of aggregates observed at HBC/HS mass ratio above 1 resulted from the excess of positively charged unbound HBC macromolecules present in the solution together with the HS–HBC complexes whose zeta potential was close to zero.
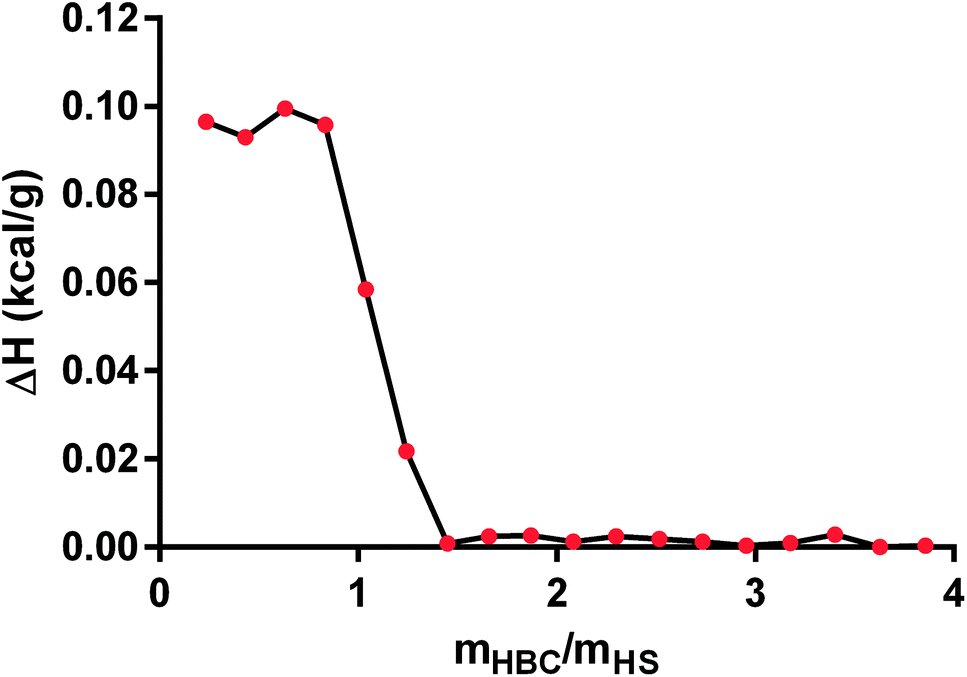 | ||
| Fig. 8 The estimated heat of binding for the titration of heparan sulfate (HS) with heparin-binding copolymer (HBC) measured by isothermal titration calorimetry (ITC) method. | ||
Discussion
In the present study, we aimed to check if HBC can bind and neutralize HS. We showed that HBC binds and forms complexes with HS, and as a result, it reverses all the tested parameters changed by HS. Importantly, the reversal of the anticoagulant activity of HS by HBC was also observed in the living organism.The binding of HS and HBC was characterized by a colorimetric Azure A method, ITC and DLS techniques in the saline or serum. We found that HBC binds this HS by forming complexes below 200 nm with less than 1:1 stoichiometry. The size of complexes is important for the safety issues: particles higher than 1000 nm may block the smallest human pulmonary arterioles. The neutralization was confirmed in vitro, and then in vivo by measuring aPTT, PT, anti-fXa activity, anti-fIIa activity, and platelet aggregation. HBC reversed the enhancement of all tested parameters caused by HS.
We used HS with N-acetylglucosamine (ANac) content of 25% (which was almost twice higher than that of heparin), as found from HS-QC NMR spectra (data not shown) while the content of iduronic acid 2-O-sulphate groups was 45% which can be compared with 74% for heparin and 21% for HS reported by Guerrini54 suggesting that the HS we have studied was quite a highly sulfated variant of this GAG.
We chose these coagulation parameters because it is well-documented that they provide useful information related to HS and HS-mimetic monitoring.1,55,56 The anticoagulant activity of HS, although different to UFH, is also mediated via its ability to activate the inhibition of factor Xa and factor IIa by antithrombin (AT).1,55 Others showed that the significant prolongation of aPTT by HS was achieved at concentrations or doses much higher than those of UFH. Similar effects, in favor of UFH, were obtained on the inactivation of factor IIa and factor Xa by AT.55,57 On the other hand, a better safety index was observed for HS compared to UFH.58,59 Similarly to others,1,55,59 we found the maximum anticoagulant effect of HS in plasma at 0.05 mg mL−1 as measured by aPTT, anti-fXa, and anti-fIIa activity tests. The slight differences in the anticoagulant response to HS between our findings and other studies may result from non-identical procedures and structural heterogeneity of HS obtained from various sources and species.
The binding and the inhibition of HS by protamine was previously demonstrated,41 but HS was inactive in the Xa assay, unless AT was exogenously added. The neutralization was even weaker in the experimental conditions more closely resembling real blood circulation (in the presence of bovine albumin), and no experiments with protamine were performed in vivo. In the beginning, we checked the binding of HS by HBC in a colorimetric method using Azure A, a cationic dye. The experiment revealed that HBC completely bound HS with an optimal ratio of 0.9:1. Next, we used well-documented coagulation parameters (aPTT, PT, anti-factor IIa activity, and anti-factor Xa activity) for HS monitoring. The in vitro results of the coagulation tests indicated that the full neutralization of HS required an HBC to HS ratio of about 0.75:1. In our recently published studies,43,44 we observed that the degree of inhibition of anticoagulant by antidote in the in vitro studies correlated positively with this degree in the in vivo studies. Therefore, we used the same ratio (0.75:1) in an animal model. Rats were intravenously injected with HBC (4.5 mg kg−1) after injection with HS (6 mg kg−1) (ratio 0.75:1). We showed that HBC completely neutralized prolonged aPTT and PT, and increased activity of anti-fXa and anti-fIIa in the living organism. Our results demonstrate that HBC could bind and inhibit HS in humans with higher efficacy than protamine, which ensures that it could be a good drug candidate. Small molecules antagonizing efficiently heparins, such as surfen or ciraparantag, could also neutralize HS, but no such experiments were performed. HBC could have a therapeutic value in bleeding related to HS, which accompany various disorders such as cancer or MPS. Probably, other functions of HS could also be blocked by HBC, as we showed in the platelet aggregation assay. The literature data describing the effect of HS on the platelet aggregation is not evident. GAGs isolated from the bovine aorta (containing only 1.7% of HS) and HS alone inhibited thrombin-induced platelet aggregation,60,61 but collagen-induced platelet aggregation was not affected. Danaparoid, a mixture of sulphated glycosaminoglycorunans derived from hog intestinal mucosa, also did not inhibit the release of serotonin from collagen-activated rabbit platelets.62 In our study, HS slightly potentiated collagen-induced platelet aggregation. We conducted an experiment in rats treated with HS, not in the exogenous mixture of platelets and HS. There is also evidence showing that heparin may increase collagen-induced platelet aggregation.63,64 In our experiment, HBC reversed the platelet activating effect of HS. Inhibition of HS activity by HBC is probably a result of binding and formation of inactive HBC–HS complexes as we showed in the colorimetric method with Azure A and ITC technique. We may expect that the virus infection through complexed HS could be interrupted.
We are aware that, besides efficacy, the new drug candidate should possess a beneficial safety profile, ensuring its successful entry into initial clinical phases of development. The example of protamine shows clearly that cationic macromolecules show cytotoxicity, which limits their usefulness as human medicines.42 However, HBC was designed to be better tolerated than protamine. HBC, consisting of nonionic PEG block and cationic block PMAPTAC assembles with anionic HS forming PICs when mixed in aqueous solution. PICs are composed of a PEG shell and segregated cation–anion PIC core.46 The characteristics of HBC, the neutralization of UFH, enoxaparin and fondaparinux, and the toxicity data were described by us previously.43 We showed that HBC, when infused into rats in the therapeutic doses, did not change: blood pressure, heart rate, tissue perfusion of rat's paw, blood oxygen saturation, respiratory rate, peak exhaled CO2, the serum concentration of aspartate aminotransferase, alanine transaminase, alkaline phosphatase, creatine phosphokinase, creatinine and the number of white blood cells, red blood cells, hemoglobin, hematocrit, mean corpuscular volume, mean corpuscular hemoglobin, mean corpuscular hemoglobin concentration or blood platelets. Similarly, in the present manuscript, administration of HBC did not potentiate, but rather inhibited WBC increase induced by HS. Nevertheless, we are still evaluating the nonclinical safety of HBC, and future experiments in the small and large animal models of specific diseases are needed before entering clinical tests.
Conclusions
In the current study, we presented a new pharmacological tool with a clear target of action (the neutralization of HS). The principle of HS neutralization indicates promising therapeutic potential of HBC in various diseases involving overproduction of HS.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by National Science Centre, Poland grant number 2016/21/B/ST5/00837. The efficacy studies were also supported by Medical University of Bialystok project number N/ST/ZB/18/003/2211.References
- N. W. Shworak, T. Kobayashi, A. D. Agostini and N. C. Smits, Prog. Mol. Biol. Transl. Sci., 2010, 93, 153–178 CrossRef CAS PubMed.
- U. Lindahl and L. Kjellén, J. Intern. Med., 2013, 273, 555–571 CrossRef CAS PubMed.
- A. Oldberg, L. Kjellen and M. Hook, J. Biol. Chem., 1979, 254, 8505–8510 CAS.
- U. Lindahl and J. Li, Int. Rev. Cell Mol. Biol., 2009, 276, 105–159 Search PubMed.
- M. M. Fuster, L. Wang, J. Castagnola, L. Sikora, K. Reddi, P. H. A. Lee, K. A. Radek, M. Schuksz, J. R. Bishop, R. L. Gallo, P. Sriramarao and J. D. Esko, J. Cell Biol., 2007, 177, 539–549 CrossRef CAS PubMed.
- D. A. Simon Davis and C. R. Parish, Front. Immunol., 2013, 4, 470 Search PubMed.
- M. Forsberg, K. Holmborn, S. Kundu, A. Dagälv, L. Kjellén and K. Forsberg-Nilsson, J. Biol. Chem., 2012, 287, 10853–10862 CrossRef CAS PubMed.
- A. Parra, N. Veraldi, M. Locatelli, M. Fini, L. Martini, G. Torri, L. Sangiorgi and A. Bisio, Glycobiology, 2012, 22, 248–257 CrossRef CAS PubMed.
- P. Tátrai, K. Egedi, Á. Somorácz, T. H. Van Kuppevelt, G. Ten Dam, M. Lyon, J. A. Deakin, A. Kiss, Z. Schaff and I. Kovalszky, J. Histochem. Cytochem., 2010, 58, 429–441 CrossRef PubMed.
- S. R. Pallerla, R. Lawrence, L. Lewejohann, Y. Pan, T. Fischer, U. Schlomann, X. Zhang, J. D. Esko and K. Grobe, J. Biol. Chem., 2008, 283, 16885–16894 CrossRef CAS PubMed.
- G. S. Kumar, A. K. Shetty and P. V. Salimath, J. Ethnopharmacol., 2008, 115, 276–283 CrossRef CAS PubMed.
- M. A. Soares, F. C. Teixeira, M. Fontes, A. L. Arêas, M. G. Leal, M. S. G. Pavão and M. P. Stelling, BioMed Res. Int., 2015, 2015, 253801 Search PubMed.
- E. H. Knelson, J. C. Nee and G. C. Blobe, Trends Biochem. Sci., 2014, 39, 277–288 CrossRef CAS PubMed.
- F. Cecchi, D. Pajalunga, C. A. Fowler, A. Üren, D. Rabe, B. Peruzzi, N. MacDonald, D. Blackman, S. Stahl, R. A. Byrd and D. Bottaro, Cancer Cell, 2012, 22, 250–262 CrossRef CAS PubMed.
- K. Fjeldstad and S. O. Kolset, Curr. Drug Targets, 2005, 6, 665–682 CrossRef CAS PubMed.
- C. Ts'ao, T. S. Galluzzo, S. J. Hart, A. S. Ng, P. G. Sorenson and V. Subbarao, Thromb. Haemostasis, 1985, 54, 768–772 CrossRef.
- M. Nishio, T. Yamanaka, K. Matsumoto, H. Kimura, K. Sakai, A. Sakai, T. Sone, A. Horiike, F. Koizumi, K. Kasahara, T. Ohira, N. Ikeda, N. Saijo, T. Arao and K. Nishio, J. Thorac. Oncol., 2011, 6, 1889–1894 CrossRef PubMed.
- R. N. Palmer, M. E. Rick, P. D. Rick, J. A. Zeller and H. R. Gralnick, N. Engl. J. Med., 1984, 310, 1696–1699 CrossRef CAS PubMed.
- S. A. Khan, R. W. Mason, R. Giugliani, K. Orii, T. Fukao, Y. Suzuki, S. Yamaguchi, H. Kobayashi, T. Orii and S. Tomatsu, Mol. Genet. Metab., 2018, 125, 44–52 CrossRef CAS PubMed.
- J. Tolar, P. J. Orchard, N. S. Key and B. R. Blazar, J. Thromb. Haemostasis, 2008, 6, 893–895 CrossRef CAS PubMed.
- A. Jinno and P. W. Park, Methods Mol. Biol., 2015, 1229, 567–585 CrossRef CAS PubMed.
- P. Hilgard and R. Stockert, Hepatology, 2000, 32, 1069–1077 CrossRef CAS PubMed.
- Y. Chen, T. Maguire, R. E. Hileman, J. R. Fromm, J. D. Esko, R. J. Linhardt and R. M. Marks, Nat. Med., 1997, 3, 866–871 CrossRef CAS PubMed.
- T. H.-C. Tang, S. Alonso, L. F.-P. Ng, T.-L. Thein, V. J.-X. Pang, Y.-S. Leo, D. C.-B. Lye and T.-W. Yeo, Sci. Rep., 2017, 7, 46191 CrossRef CAS PubMed.
- V. H. Pomin, F. F. Bezerra and P. A. G. Soares, Curr. Pharm. Des., 2017, 23, 3405–3414 CrossRef CAS PubMed.
- J. Jiang, X. Wu, H. Tang and G. Luo, PLoS One, 2013, 8, e67982 CrossRef CAS PubMed.
- M. Yoon, A. Zago, D. Shukla and P. G. Spear, J. Virol., 2003, 77, 9221–9231 CrossRef CAS PubMed.
- Y.-J. Chien, W.-J. Chen, W.-L. Hsu and S.-S. Chiou, Virology, 2008, 379, 143–151 CrossRef CAS PubMed.
- W. B. Klimstra, K. D. Ryman and R. E. Johnston, J. Virol., 1998, 72, 7357–7366 CAS.
- X. Bai, H. Bao, P. Li, W. Wei, M. Zhang, P. Sun, Y. Cao, Z. Lu, Y. Fu, B. Xie, Y. Chen, D. Li, J. Luo and Z. Liu, Virol. J., 2014, 11, 132 CrossRef PubMed.
- P. Merilahti, E. Karelehto and P. Susi, PLoS One, 2016, 11, e0147168 CrossRef PubMed.
- M. Nasimuzzaman and D. A. Persons, Mol. Ther., 2012, 20, 1158–1166 CrossRef CAS PubMed.
- M. Tamhankar, D. M. Gerhardt, R. S. Bennett, N. Murphy, P. B. Jahrling and J. L. Patterson, Virol. J., 2018, 15, 135 CrossRef PubMed.
- L. Martin, S. Schmitz, R. De Santis, S. Doemming, H. Haase, J. Hoeger, L. Heinbockel, K. Brandenburg, G. Marx and T. Schuerholz, PLoS One, 2015, 10, e0127584 CrossRef PubMed.
- K. F. Hofmann-Kiefer, G. I. Kemming, D. Chappell, M. Flondor, H. Kisch-Wedel, A. Hauser, S. Pallivathukal, P. Conzen and M. Rehm, Eur. J. Med.Res., 2009, 14, 526–531 CAS.
- M. Schuksz, M. M. Fuster, J. R. Brown, B. E. Crawford, D. P. Ditto, R. Lawrence, C. A. Glass, L. Wang, Y. Tor and J. D. Esko, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13075–13080 CrossRef CAS PubMed.
- M. L. Huang, A. L. Michalak, C. J. Fisher, M. Christy, R. A. A. Smith and K. Godula, Stem Cells, 2018, 36, 45–54 CrossRef CAS PubMed.
- S. M. A. Naini, C. Yanicostas, R. Hassan-Abdi, S. Blondeel, M. Bennis, R. J. Weiss, Y. Tor, J. D. Esko and N. Soussi-Yanicostas, Transl. Neurodegener., 2018, 7, 6 CrossRef PubMed.
- V. Ferro, Expert Opin. Ther. Targets, 2013, 17, 965–975 CrossRef CAS PubMed.
- R. Kisilevsky, W. A. Szarek, J. Ancsin, R. Vohra, Z. Li and S. Marone, J. Mol. Neurosci., 2004, 24, 167–172 CrossRef CAS PubMed.
- A. R. Hubbard and C. A. Jennings, Thromb. Haemostasis, 1985, 53, 86–89 CrossRef CAS.
- E. Sokolowska, B. Kalaska, J. Miklosz and A. Mogielnicki, Expert Opin. Drug Metab. Toxicol., 2016, 12, 897–909 CrossRef CAS PubMed.
- B. Kalaska, K. Kaminski, J. Miklosz, S.-I. Yusa, E. Sokolowska, A. Blazejczyk, J. Wietrzyk, I. Kasacka, K. Szczubialka, D. Pawlak, M. Nowakowska and A. Mogielnicki, Transl. Res., 2016, 177, 98–112 CrossRef CAS PubMed.
- B. Kalaska, K. Kamiński, J. Miklosz, K. Nakai, S. I. Yusa, D. Pawlak, M. Nowakowska, A. Mogielnicki and K. Szczubiałka, Biomacromolecules, 2018, 19, 3104–3118 CrossRef CAS PubMed.
- Y. Mitsukami, M. S. Donovan, A. B. Lowe and C. L. McCormick, Macromolecules, 2001, 34, 2248–2256 CrossRef CAS.
- S.-I. Yusa, Y. Yokoyama and Y. Morishima, Macromolecules, 2009, 42, 376–383 CrossRef CAS.
- B. Kalaska, K. Kaminski, E. Sokolowska, D. Czaplicki, M. Kujdowicz, K. Stalinska, J. Bereta, K. Szczubialka, D. Pawlak, M. Nowakowska and A. Mogielnicki, PLoS One, 2015, 10, e0119486 CrossRef PubMed.
- B. Kalaska, K. Kaminski, E. Sokolowska, D. Czaplicki, M. Kujdowicz, K. Stalinska, K. Szczubialka, J. Bereta, D. Pawlak, M. Nowakowska and A. Mogielnicki, in NSTI: Advanced Materials - TechConnect Briefs 2015, 2015, vol. 3 Search PubMed.
- K. Kamiński, M. Płonka, J. Ciejka, K. Szczubiałka, M. Nowakowska, B. Lorkowska, R. Korbut and R. Lach, J. Med. Chem., 2011, 54, 6586–6596 CrossRef PubMed.
- K. Kamiński, B. Kałaska, P. Koczurkiewicz, M. Michalik, K. Szczubiałka, A. Mogielnicki, W. Buczko and M. Nowakowska, MedChemComm, 2014, 5, 489–495 RSC.
- B. Kalaska, E. Sokolowska, K. Kaminski, K. Szczubialka, K. Kramkowski, A. Mogielnicki, M. Nowakowska and W. Buczko, Eur. J. Pharmacol., 2012, 686, 81–89 CrossRef CAS PubMed.
- X. Xu, S. Angioletti-Uberti, Y. Lu, J. Dzubiella and M. Ballauff, Langmuir, 2018 DOI:10.1021/acs.langmuir.8b01802.
- S. Young, M. Wong, Y. Tabata and A. G. Mikos, J. Controlled Release, 2005, 109, 256–274 CrossRef CAS PubMed.
- M. Guerrini, A. Naggi, S. Guglieri, R. Santarsiero and G. Torri, Anal. Biochem., 2005, 337, 35–47 CrossRef CAS PubMed.
- A. N. Teien, U. Abildgaard and M. Höök, Thromb. Res., 1976, 8, 859–867 CrossRef CAS PubMed.
- H. Zhou, S. Roy, E. Cochran, R. Zouaoui, C. L. Chu, J. Duffner, G. Zhao, S. Smith, Z. Galcheva-Gargova, J. Karlgren, N. Dussault, R. Y. Q. Kwan, E. Moy, M. Barnes, A. Long, C. Honan, Y. W. Qi, Z. Shriver, T. Ganguly, B. Schultes, G. Venkataraman and T. K. Kishimoto, PLoS One, 2011, 6, e21106 CrossRef CAS PubMed.
- F. A. Ofosu, M. R. Buchanan, N. Anvari, L. M. Smith and M. A. Blajchman, Ann. N. Y. Acad. Sci., 1989, 556, 123–131 CrossRef CAS PubMed.
- P. A. Ockelford, C. J. Carter and J. Hirsh, Pathology, 1985, 17, 78–81 CrossRef CAS PubMed.
- D. Hoppensteadt, J. M. Walenga and J. Fareed, Thromb. Res., 1990, 60, 191–200 CrossRef CAS PubMed.
- Y. Nakashima, T. Matsushima, T. Sadayasu, K. Takahara, J. Segawa and A. Kuroiwa, Artery, 1992, 19, 256–270 CAS.
- G. Luzzatto and R. Paolini, Angiology, 1989, 40, 170–174 CrossRef CAS PubMed.
- D. G. Meuleman, P. M. J. Hobbelen, G. van Dedem and H. C. T. Moelker, Thromb. Res., 1982, 27, 353–363 CrossRef CAS.
- S. F. Mohammad, W. H. Anderson, J. B. Smith, H. Y. Chuang and R. G. Mason, Am. J. Pathol., 1981, 104, 132–141 CAS.
- J. Hirsh, Am. J. Surg., 1991, 161, 512–518 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |