
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effect of the second coordination sphere on the magnetism of [Ln(NO3)3(H2O)3]·(18-crown-6) (Ln = Dy and Er)†
Radovan Herchel *ab,
Pavel Zoufalýa and
Ivan Nemeca
*ab,
Pavel Zoufalýa and
Ivan Nemeca
aDepartment of Inorganic Chemistry, Faculty of Science, Palacký University, 17. Listopadu 12, CZ-771 46 Olomouc, Czech Republic. E-mail: radovan.herchel@upol.cz
bRegional Centre of Advanced Technologies and Materials, Faculty of Science, Palacký University, Šlechtitelů 27, CZ-783 71 Olomouc, Czech Republic
First published on 2nd January 2019
Abstract
The objective of this work was the exploration of the effect of the second coordination sphere on the magnetic properties of [Ln(NO3)3(H2O)3]·(18C6) (Ln = Dy (1) and Er (2)) compounds comprising co-crystallized 18-crown-6 ethers. Both compounds were identified as field-induced single molecule magnets (SMMs) with estimated magnetization reversal barriers Ueff = 66–71 K for 1 and Ueff = 21–24 K for 2. Theoretical calculations with the B3LYP functional revealed substantial change and redistribution of the electrostatic potential upon accounting for the second coordination sphere represented by two 18C6 molecules, which resulted in the change of the crystal-field around metal atoms. As a result, the multireference CASSCF calculations exposed significant impact of the second coordination sphere on the energy splitting of the respective 6H15/2 (DyIII) and 4I15/2 (ErIII) ground states, the magnetization reversal barrier and the magnetic anisotropy parameters. Moreover, the calculated magnetization reversal barriers, Ucalc. = 57 K for 1 and Ucalc. = 16 K for 2, are in good agreement with the experimental values accentuating the importance of the second coordination sphere on the magnetic properties of SMMs.
Introduction
The lanthanide coordination compounds are of great interest to many magnetochemists around the world since Ishikawa's seminal report about the phthalocyanine complexes [Bu4N][Ln(Pc)2] (Ln = Tb and Dy, Pc = phthalocyanine, Bu4N+ = tetrabutylammonium cation) behaving as single-molecule magnets (SMMs).1 SMMs are molecular nanomagnets exhibiting slow relaxation of the magnetization and magnetic bistability of molecular origin.2,3 SMM based materials can be potentially utilized in devices with high storage capacity, in sensors, quantum computing, spintronics, etc.4,5 The key concept of SMMs is the existence of the energy barrier acting against the reversal of the magnetization and so far many lanthanide-based SMMs have been reported.6 In the case of dysprosium(III) and erbium(III) ions, this barrier is defined by the ligand-field induced splitting of the ground state 6H15/2 and 4I15/2, respectively, into the respective Kramers sublevels of the J-manifold. Thus, the coordination number and the shape of the coordination polyhedron are crucial properties defining the energy barrier properties,7 hence SMMs performance. In last years, the enormous progress was achieved regarding the maximising the effective energy barrier (Ueff) in Dy(III) SMMs, e.g. Ueff = 1025 K was reported for [Dy(bbpen)Br] (H2bbpen = N,N′-bis(2-hydroxybenzyl)-N,N′-bis(2-methylpyridyl)ethylene-diamine),8 Ueff = 1815 K was found in [Dy(OtBu)2(py)5][BPh4], (py = pyridine)9 and the record-holding SMM is [Dy(Cpttt)2][B(C6F5)4] (Cpttt = 1,2,4-tri(tert-butyl)cyclopentadienide) with Ueff = 1837 K and magnetic hysteresis present up to 60 K (ref. 10) or [(CpiPr5)Dy(Cp*)]+ (CpiPr5 = penta-iso-propylcyclopentadienyl, Cp* = pentamethylcyclopentadienyl) which has 80 K highest magnetic blocking temperature.11 In addition, even though erbium based compounds exhibit slow relaxation of magnetization less frequently and with lower Ueff barriers than their dysprosium analogues continuous research progresses steadily to greater values of Ueff. This can be demonstrated on several examples featuring predominantly organometallic ligands: e.g. [(Cp*)Er(COT)] (Cp* = pentamethylcyclopentadienide; COT = cyclooctatetraenide dianion) with hysteresis loop from 1.8 to 5 K and two magnetic relaxation barriers 197 K and 323 K,12 [(Dsp)Er(COT)] (Dsp− = 3,4-dimethyl-2,5-bis(trimethylsilyl) phospholyl) shows slow relaxation with the zero applied magnetic field and barrier 358 K,13 lastly boron containing six membered borabenzene ligand in [(C5H5BH)Er(COT)] with Ueff = 371 K and [(C5H5BCH3)Er(COT)] which for ErIII complexes holds currently highest energy barrier of 421 K.14However, the SMMs properties are not solely defined by the effective energy barrier due to the fact that the slow relaxation magnetization can be governed by various mechanisms – one-phonon direct and two-phonon Orbach and Raman processes,15 and additionally also by the quantum tunnelling of the magnetization. However, the identification and comprehension of the relaxation processes is not an easy task and a better insight into these mechanisms is crucial for the further development of the single-molecule magnets.16,17
One of broadly used ligands in search of tuning magnetic behaviour of the lanthanide-based compounds appears to be 18-crown-6 (18C6). This hexadentate ether serves frequently in formation of half-sandwich or sandwich complexes.18 Nevertheless, only in a few works this ligand directly bonds to lanthanide atom and at the same time magnetic properties were studied. Gadolinium based complex [{Gd(18C6)(μ-OH)(CH3CN)}2][Ni(dmit)2]2 (dmit2− = 2-thioxo-1,3-dithiole-4,5-dithiolate) with the nickel encompassing complex counter anion is reported to have antiferromagnetic exchange between gadolinium atoms.19 Likewise magnetic properties of synthesized thiocyanate containing [Eu(18C6)(NCS)3] and [Tb(18C6)(NCS)3] compounds were studied. However, only Tb(III) complex was subjected to analysis of frequency dependent behaviour in AC magnetic susceptibility measurement, but no slow relaxation of magnetization was detected.20 Simple motif [Ln(NO3)3(18C6)] (Ln = CeIII, PrIII, NdIII) was explored relatively recently in 2016 probing magnetic properties of nitrato ligands with several lanthanides, where under applied external magnetic field of 1000 Oe slow relaxation of magnetization was observed for Ce(III) and Nd(III) complexes.21 The same year were published two new structures of dysprosium containing coordinated 18C6 ligand, namely [Dy(18C6)(NO3)2]BPh4 and [Dy(18C6)(NO3)2]ClO4, in which diverse coordination of nitrato ligands induced differences in the slow relaxation of the magnetization demonstrated by Ueff = 43 K and Ueff = 63 K, respectively.22 Currently, the newest research of magnetic properties in 18C6-based coordination compounds with lanthanides includes four new compounds of general formula [Ln(H2O)3(18C6)](ClO4)3 (Ln = TbIII, DyIII, ErIII, YbIII) and two of them were identified as field-induced SMMs.23
All of the previously mentioned compounds contained 18C6 bonded directly to the central lanthanide atom. However, several works also include the ether only as co-crystalized molecules. Zhao-Yang et al. explored the impact of chlorido and nitrato anions in two glycol coordinated dysprosium complexes where the purpose of crown-ether utilization was mainly to further separation of metal atoms and to promote crystallization. The prepared complexes [Dy(H2TEG)A3]·(18C6) (H2TEG = triethylene glycol; A = Cl−, NO3−) exhibit field induced two step magnetic relaxation having 28 K and 54 K energy barriers.24 Interestingly, the very simple compound Dy(NO3)3·6H2O or more precisely [Dy(NO3)3(H2O)4]·2H2O was reported recently as field-induced SMM.25 The fact that second coordination sphere and its impact on magnetic behaviour of SMMs remains partly overlooked with the exception of work by Petit et al. on Co(II) complex,26 inspired us to study the influence of the second coordination sphere on the electronic structure and magnetic properties of Kramers ions based Dy(III) and Er(III) complexes [Ln(NO3)3(H2O)3]·(18C6) for which we report the static and dynamic magnetic properties and theoretical calculations based on CASSCF multireference method.
Results and discussion
Synthesis and characterization
The infrared spectroscopy measurements confirmed the presence of the characteristic vibration bands associated to bonds in nitrato (NO3), water (OH) and crown ether (CH). The complete crystallographic information and structural description can be acquired from original literature.27 This work focuses predominantly on magnetic properties arising from covalent and noncovalent interactions in the studied compounds and thus the basic characterization of the crystal structures will be briefly described. The phase purity of the prepared compounds 1 and 2 was confirmed by powder X-ray diffraction (Fig. S1, ESI†). Both compounds of general formula [Ln(NO3)3(H2O)3]·(18C6), Ln = Dy (1), Er (2), are isostructural and the complex molecules are composed of three water and three nitrate molecules coordinated to the metal atom by the oxygen donor atoms forming thus polyhedron with coordination number nine and geometry closest to muffin (Table S1, ESI†) as evaluated by the Shape algorithm.28All three nitrato ligands coordinate metal atoms in bidentate fashion with the Ln–O bond lengths ranging from 2.38 to 2.43 Å (in 1 and 2). The metal–ligand bond lengths are slightly shorter in the case of the aqua ligands: 2.36–2.39 Å (1) and 2.34–2.39 Å (2). The crystal structures of 1 and 2 are composed of the [Ln(NO3)3(H2O)3] complex molecules assembled into 1D chains by O–H⋯O hydrogen bonding between the aqua ligands and intercalated 18C6 molecules. Two of three coordinated aqua ligands are symmetry related by the mirror plane and form a pair of rather strong O–H⋯O hydrogen bonds (d(O⋯O)) = 2.82–2.87 Å in 1 and 2 solely with one 18C6 molecule (Fig. 1). The third aqua ligand act as a bridging moiety, which interconnects two 18C6 molecules via two mirror related hydrogen bonds 2.832(11) for 1 and 2.814(11) Å for 2 (Fig. 1). Further stabilization is provided by plethora of weaker interactions including non-covalent contacts between the aliphatic part (![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) CH2 donor groups) of 18C6 and nitrate oxygen atoms forming weak C–H⋯O (nitrato) hydrogen bonds (d(C⋯O) = 3.33–3.61 Å in 1 and 2) – Fig. 1 and S2.† The above-mentioned main supramolecular chain motif is extended to 2D layer by non-covalent O⋯O interaction between the nitrates of two adjacent complex molecules. This interaction is rather close with d(O⋯O) = 2.957(16) in 1 and 2.986(16) Å in 2.
CH2 donor groups) of 18C6 and nitrate oxygen atoms forming weak C–H⋯O (nitrato) hydrogen bonds (d(C⋯O) = 3.33–3.61 Å in 1 and 2) – Fig. 1 and S2.† The above-mentioned main supramolecular chain motif is extended to 2D layer by non-covalent O⋯O interaction between the nitrates of two adjacent complex molecules. This interaction is rather close with d(O⋯O) = 2.957(16) in 1 and 2.986(16) Å in 2.
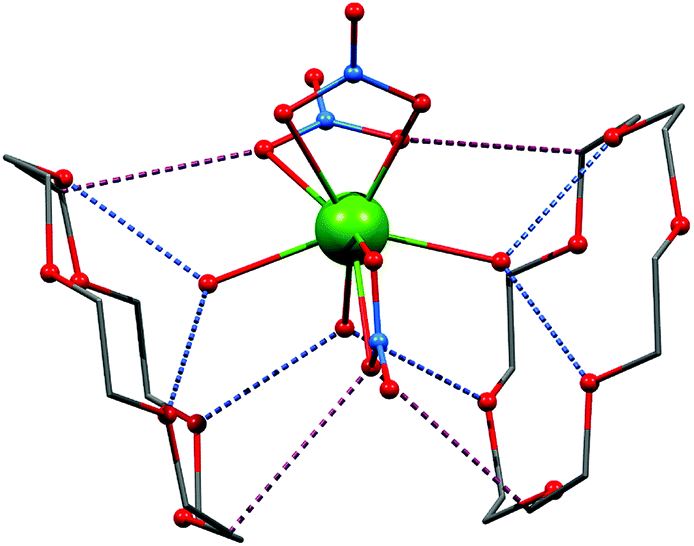 | ||
| Fig. 1 The molecular structure of 1 showing the second coordination sphere formed by the net of O–H⋯O hydrogen bonds between O–H atoms of coordinated water and oxygen of crown ether (blue dashed lines) and C–H⋯O (nitrato) contacts (violet dashed line). | ||
Magnetic properties
The temperature and field dependent static magnetic data were acquired on polycrystalline samples of 1 and 2 as depicted in Fig. 2. The value of μeff/μB at room temperature is 10.3 for 1 and 9.2 for 2, which are values close to the theoretically expected values of 10.65 (DyIII, 6H15/2) and 9.58 (ErIII, 4I15/2). The μeff/μB is gradually decreasing upon cooling the samples down to 1.9 K both for 1 and 2, which is the most likely due to depopulation of ligand field multiplets arising from ground atomic terms effected by spin–orbit coupling and ligand field of the chromophores. There are no maxima on susceptibility, which eliminates existence of substantial intermolecular contacts providing magnetic exchange interactions of the antiferromagnetic nature.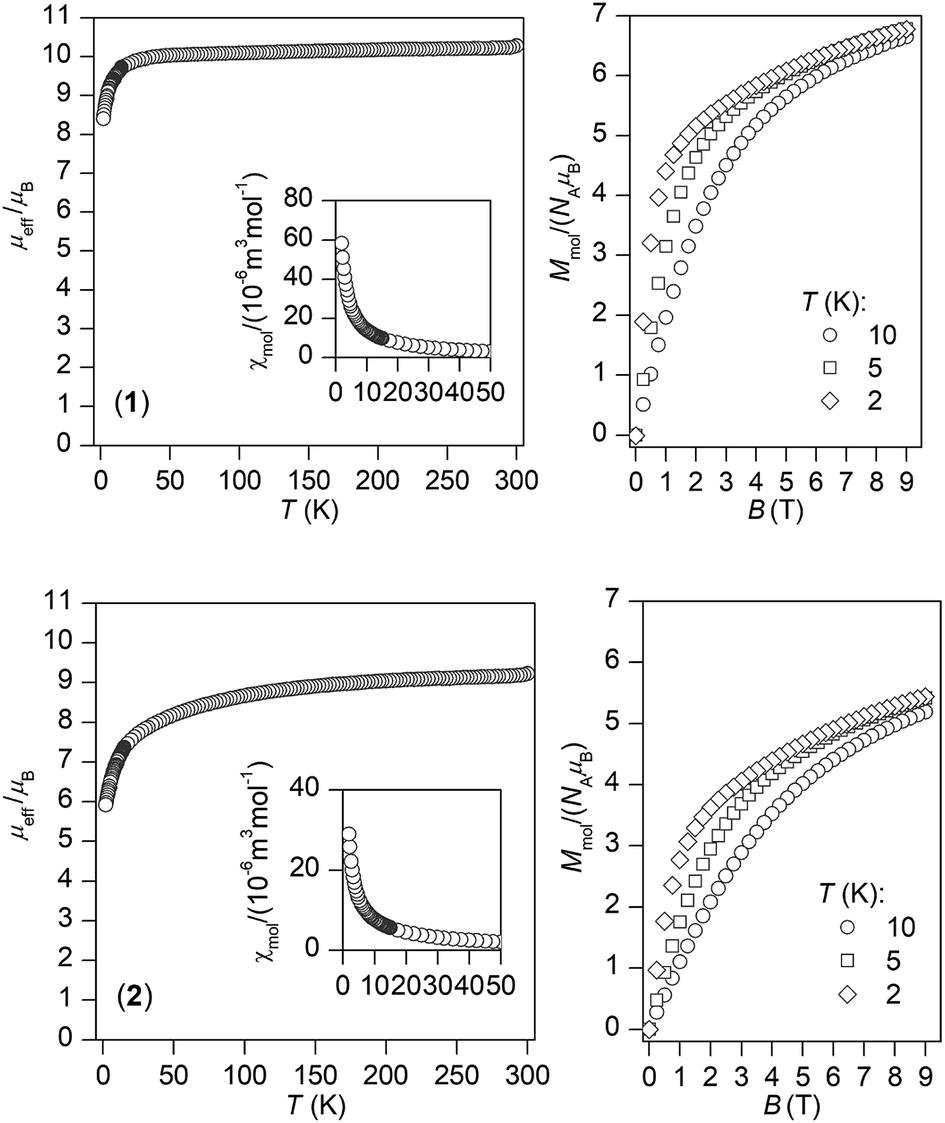 | ||
| Fig. 2 Magnetic data for complexes 1 and 2. Temperature dependence of the effective magnetic moment (left) and the isothermal molar magnetizations measured at various temperatures (right). | ||
The reciprocal susceptibilities were analysed with the Curie–Weiss law in the temperature range of 1.9–300 K for 1 and in the temperature range of 50–300 K for 2 (Fig. S3, ESI†), which resulted in C = 1.648 × 10−4 m3 mol−1 K, Θ = −2.2 K and g = 1.28 for 1 and C = 1.405 × 10−4 m3 mol−1 K, Θ = −18.8 K and g = 1.18 for 2. All the Weiss constants are of negative values and g-factors are close to theoretical Landé g-factors, i.e. 1.33, and 1.20 for DyIII and ErIII, respectively. The isothermal magnetization data, Mmol/NAμB, measured at T = 2 K saturate to 6.8 for 1 and 5.4 for 2 and these values are well below theoretically predicted values based on J and Landé g-factors, which are 10.0 for DyIII and 9.0 for ErIII. This points out to large magnetic anisotropy of these complexes.
The AC susceptibility measurements were made firstly in zero and nonzero static magnetic fields as depicted in Fig. S4 (ESI).† None of these compounds showed nonzero out-of-phase signal of AC susceptibility at zero static magnetic field, but evidently, small magnetic field is efficient to observe slow relaxation of magnetization and suppress tunnelling effect. Therefore, the temperature dependence of AC susceptibility was measured at BDC = 0.1 T for frequencies 1–1500 Hz as depicted in Fig. 3 and 4. Evidently, the relaxation of the compound 1 is rather complicated, exhibiting three relaxation pathways – there are three maxima in the frequency-dependent plot of χ′′ (Fig. 3 bottom). Usually, the existence of the multiple relaxation processes is assigned to the intermolecular interactions in solid state. However, the shortest distance between Dy atoms is quite large and equals 7.927 Å, thus is seems that such interactions should be negligible. On contrary, it was recently demonstrated that multiple relaxation processes can show up even in well isolated single-ion metal complexes due to the existence of the multilevel energy spectrum, thus they are of the intramolecular origin.29 The relaxation pattern in 2 is much simpler, exhibiting just one dominant relaxation process, but without well-defined maxima (Fig. 4). Therefore, the simplified model was utilized to analyze AC susceptibility data for both compounds, which is derived under the assumption that the adiabatic susceptibility is usually approaching zero in the single-molecule magnets, thus it holds χS → 0,
 | (1) |
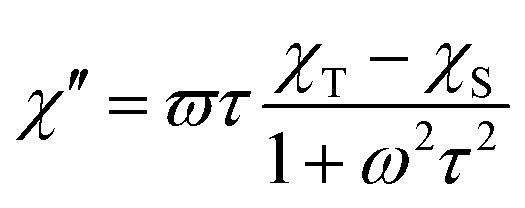 | (2) |
 | ||
| Fig. 3 Temperature and frequency dependent in-phase χ′and out-of-phase χ′′ molar susceptibilities for 1 at the applied external field BDC = 0.1 T. Lines serve as guide to the eye. | ||
 | ||
| Fig. 4 Temperature and frequency dependent in-phase χ′and out-of-phase χ′′ molar susceptibilities for 2 at the applied external field BDC = 0.1 T. Lines serve as guide to the eye. | ||
can be approximated as
| χ′′/χ′ ≅ ϖτ = 2πfτ | (3) |
Such approximation was already used to analyze magnetic data for other SMM compounds30 and parameters of Orbach relaxation processes are derived from linear regression analyses using following equation
| ln(χ′′/χ′) = ln(2πfτ0) + Ueff/kT | (4) |
The analyses of ac susceptibility data for highest frequencies resulted in the magnetic moment reversal barrier parameters Ueff = 66–71 K for 1 (Fig. S5, ESI†) and Ueff = 21–24 K for 2 (Fig. S6, ESI†).
Theoretical calculations
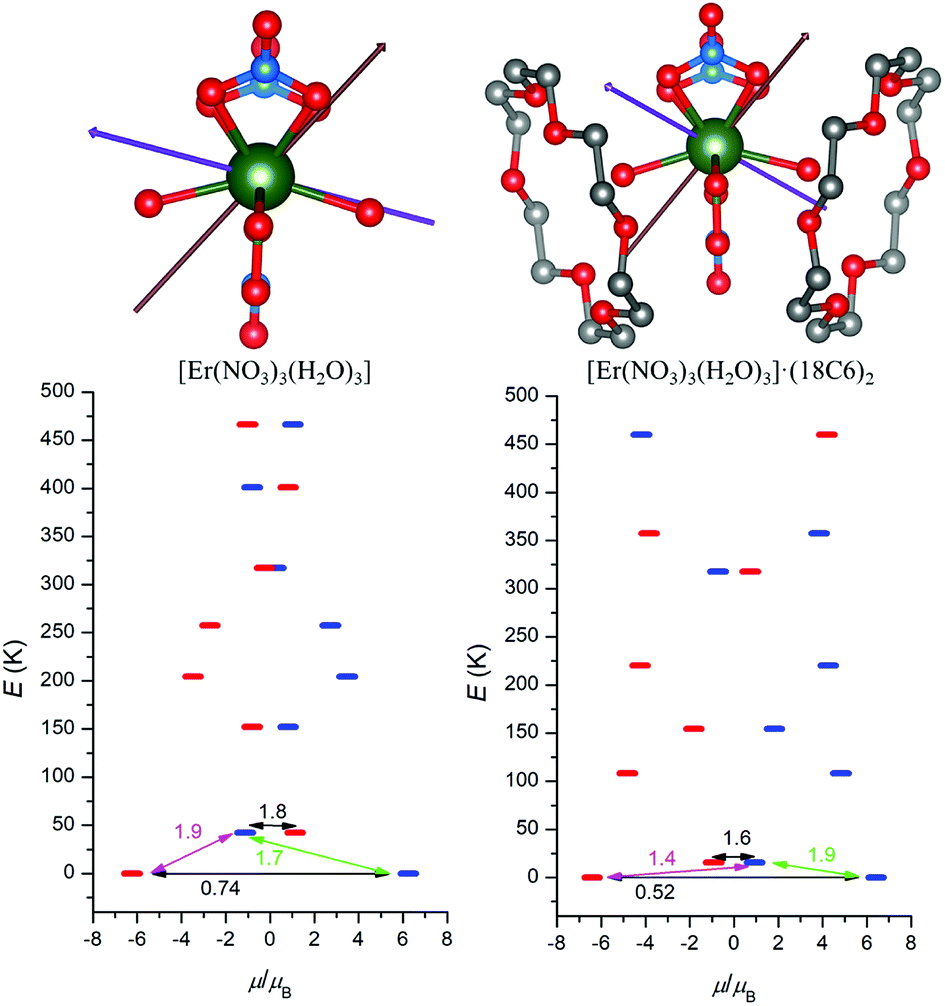
With the aim to explore the impact of the second coordination sphere on the electronic structure and magnetic properties of 1 and 2, the post-Hartree–Fock multireference calculations were done utilizing Complete Active Space Self-Consistent Field method (CASSCF). Thus, MOLCAS 8.0 computational package was employed for CASSCF calculations with respective relativistic ANO-RCC basis set and Douglas–Kroll–Hess (DKH) approximation accounting for the relativistic effects. First, the CASSCF calculations were done for the complexes [Ln(NO3)3(H2O)3] (Ln = Dy and Er), which resulted in the energies of the ligand-field multiplets showed in Fig. 5, where a scheme of the magnetization blocking barrier is depicted. | ||
| Fig. 5 Top: The molecular structures of [Dy(NO3)3(H2O)3] and [Dy(NO3)3(H2O)3]·(18C6)2 used for CASSCF calculations overlaid with the easy axes of the ground and first excited Kramers doublets coloured with violet and brown colour, respectively. Bottom: ab initio computed magnetization blocking barrier for both molecular structures. The thick blue/red bars indicate the Kramer's doublets (KDs) as a function of magnetic moment. Green lines indicate the magnetization reversal mechanism. The magenta lines show the possible pathway of the Orbach process. The black lines represent the presence of QTM/TA-QTM between the connecting pairs. | ||
The respective effective g-factors of the Kramers doublets are listed in Tables S2 and S3.† There is large axial anisotropy of the lowest Kramers doublet for both complexes (gz ≫ gx, gy) and the orientation of the respective easy axis of the magnetization is showed in Fig. 5 and 6. The energy of the first excited Kramers doublet is ε1 = 143 K for DyIII and ε1 = 42 K for ErIII complex and the easy axis of these excited states is also depicted in Fig. 5 and 6. Secondly, the similar calculations were done for the complexes [Ln(NO3)3(H2O)3]·(18C6)2 (Ln = Dy and Er), in which two non-coordinated 18-crown-6 molecules bound to [Ln(NO3)3(H2O)3] through the non-covalent interactions form the second coordination sphere. Surprisingly, the incorporating the second coordination sphere caused deep impact on the energy level splitting of 6H15/2 (DyIII) and 4I15/2 (ErIII) ground states as showed in Fig. 5 and 6. The energy of the first excited state was lowered for both compounds as ε1 = 57 K for DyIII and ε1 = 16 K for ErIII complex. Next, the ground state Kramers doublet properties were also significantly modified, the rhombic g-factors, gx and gy increased for DyIII complex (Table S4†), which resulted in the larger probabilities of the quantum tunnelling (Fig. 5).
 | ||
| Fig. 6 Top: The molecular structures of [Er(NO3)3(H2O)3] and [Er(NO3)3(H2O)3]·(18C6)2 used for CASSCF calculations overlaid with the easy axes of the ground and first excited Kramers doublets coloured with violet and brown colour, respectively. Bottom: ab initio computed magnetization blocking barrier for both molecular structures. The thick blue/red bars indicate the Kramer's doublets (KDs) as a function of magnetic moment. Green lines indicate the magnetization reversal mechanism. The magenta lines show the possible pathway of the Orbach process. The black lines represent the presence of QTM/TA-QTM between the connecting pairs. | ||
On contrary, the probability of the quantum tunnelling decreases in ErIII complex (Fig. 6) due to smaller rhombic g-factors gx and gy (Table S5†). To summarize, these quantum calculations agree with the experimental observations: (i) both compounds 1 and 2 are field-induced SMMs, which is consistent with calculated quantum tunnelling probabilities, (ii) the derived effective reversal barriers Ueff = 66–71 K for 1 and Ueff = 21–24 K for 2 are rather close to calculated ones, Ucalc. = 57 K for 1 and Ucalc. = 16 K for 2. To better understand the substantial effect of the second coordination sphere on the calculated energy levels and magnetic anisotropy, the additional DFT calculations were done. The B3LYP functional was used to calculate the electrostatic potential retaining the molecular geometry for the ligand environment of the previous calculations [Ln(NO3)3(H2O)3] and [Ln(NO3)3(H2O)3]·(18C6)2 (Ln = Dy and Er) and removing the central Ln atoms. Then, the electrostatic potential was calculated in a sphere of 1 Å radius located at the lanthanide atom Ln position. The color mapped plots are shown in Fig. 7 and S7 (ESI)† and it is evident that incorporating 18C6 molecules into the calculations results in less negative electrostatic potential, which rationalize the results of CASSCF calculations in such a way, that weakening of the crystal-field potential around central lanthanide atoms leads to the smaller energy splitting. Thus, the non-covalent contacts of the 18C6 molecules in the second coordination sphere are sufficient to induce significant changes of the electrostatic potential, hence the crystal/ligand-field resulting in the variation of the multiplets energy levels.
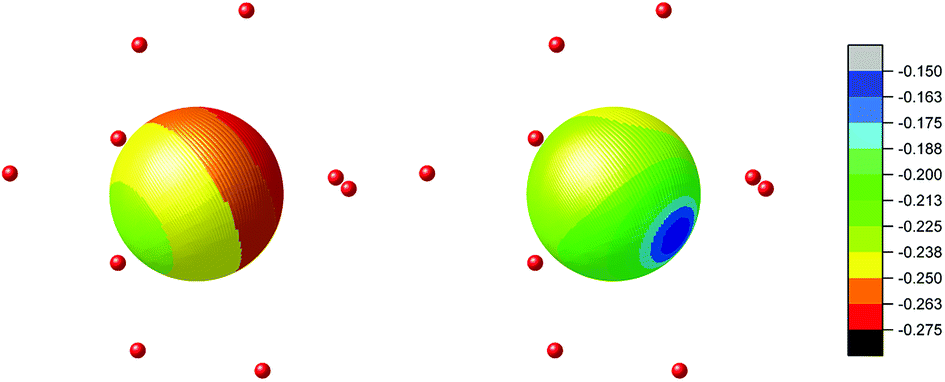 | ||
| Fig. 7 The electrostatic potentials of the ligands projected in a sphere of 1 Å radius located at the dysprosium position in molecular systems [Dy(NO3)3(H2O)3] (left) and [Dy(NO3)3(H2O)3]·(18C6)2 (right) obtained by removing the dysprosium atom. The color scale of the electrostatic potential is shown on the right. The donor oxygen atoms from nitrato and aqua ligands are shown as red balls. | ||
Experimental section
All the materials were purchased in Sigma-Aldrich and used without any purification. Basic chemical composition was determined by Flash 2000 (Thermo Scientific) analyser. IR measurements were performed on FT-IR spectrometer Thermo Nicolet Nexus 670, magnetic measurements on PPMS Dynacool (Quantum Design) and SQUID magnetometer XL-7 (Quantum Design). Structure was confirmed by X-ray diffractometer MiniFlex600 (Rigaku). The procedure for synthesis of [M(NO3)3(H2O)3]·(18C6) was performed similarly as described in literature.27Characterization
Lanthanide salt of Dy(NO3)3·6H2O (0.329 mmol) or Er(NO3)3·5H2O (0.271 mmol) was dissolved in mixture of acetonitrile and methanol 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. To this solution a stoichiometric amount of solid 18C6 was added which resulted in forming precipitate that redissolved quickly. The mixture was left stirring for approximately 15 minutes, filtrated and left to evaporate slowly. After a few days, bulky crystals were collected, washed with methanol and dried under vacuum in desiccator containing silica gel.
:1. To this solution a stoichiometric amount of solid 18C6 was added which resulted in forming precipitate that redissolved quickly. The mixture was left stirring for approximately 15 minutes, filtrated and left to evaporate slowly. After a few days, bulky crystals were collected, washed with methanol and dried under vacuum in desiccator containing silica gel.
Theoretical calculations
The respective CASSCF calculations were performed with the MOLCAS 8.0 program package31 on the [Ln(NO3)3(H2O)3] and [Ln(NO3)3(H2O)3]·(18C6)2 (Ln = Dy and Er) molecular systems using atomic coordinates derived from the experimental X-ray structures (CSD codes LEYHUS and LEYJII). The active space was defined by seven f-orbitals as CAS(9,7) for 1 and CAS(11,7) for 2. The RASSCF method was employed in CASSCF calculations32 with the following numbers of multiplets: 21 sextets, 224 quartets and 490 doublets for DyIII, 35 quartets and 112 doublets for ErIII. The spin–orbit coupling based on atomic mean field approximation (AMFI)33 was considered using RASSI-SO with the following numbers of multiplets: 21 sextets, 128 quartets and 130 doublets for DyIII, 35 quartets and 112 doublets for ErIII. The relativistic effects were treated with the Douglas–Kroll Hamiltonian.34 The following basis sets were utilized: Ln.ANO-RCC-VQZP (Ln = Dy and Er), O.ANO-RCC-VDZP, N.ANO-RCC-VDZP, C.ANO-RCC-VDZ and H.ANO-RCC-VDZ.35 Then, SINGLE_ANISO module36 was used to calculate the effective spin Hamiltonian parameters for Kramers doublets of the ground states and the respective parameters of the blocking energy barrier. The calculated easy axes were visualized with VESTA 3 program.37The DFT calculations were done with ORCA 4.0 software38 utilizing B3LYP functional39 together with relativistic basis sets DKH-def2-TZVP for N and O atoms, and DKH-def2-SVP for C and H atoms.40 Increased integration grids (Grid6 in ORCA convention) and tight SCF convergence criteria were used in all calculations.
Conclusions
The magnetic properties of the compounds [Ln(NO3)3(H2O)3]·(18C6) (Ln = Dy (1) and Er (2)) were studied both experimentally and theoretically. The AC susceptibility confirmed the onset of slow relaxation of the magnetization typical for SMMs under applied static magnetic field. The DyIII compound 1 evidenced three relaxation pathways in contrast to ErIII compound 2 for which only one relaxation pathway was found. The analysis of the experimental AC magnetic data revealed Ueff = 66–71 K for 1 and Ueff = 21–24 K for 2. The subsequent CASSCF calculations were done both for the complexes with the primary coordination sphere composed of aqua and nitrato ligands [Ln(NO3)3(H2O)3], and for complexes comprising also the secondary coordination sphere of two 18-crown-6 molecules [Ln(NO3)3(H2O)3]·(18C6)2. It was found that secondary coordination sphere significantly influenced the calculated energy splitting, hence the magnetization reversal barrier and magnetic anisotropy of the resulting Kramers doublets both for 1 and 2. Moreover, the calculated magnetization reversal barriers, Ucalc. = 57 K for 1 and Ucalc. = 16 K for 2, after incorporating the second coordination sphere are in good agreement with the experimental ones emphasizing the importance of non-covalent interactions induced by 18-crown-6 co-crystallized molecules on nitrato and aqua ligands. This was also supported by DFT calculations of the electrostatic potentials around metal atoms, which rationalized the CASSCF results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the Grant Agency of the Czech Republic (GAČR 17-08992S), the National Programme of Sustainability I (LO1305) of the Ministry of Education, Youth and Sports of the Czech Republic and from Palacký University Olomouc (PrF_2018_011).Notes and references
- N. Ishikawa, M. Sugita, T. Ishikawa, S. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef CAS PubMed.
- (a) R. Sessoli, L. Hui, A. R. Schake, S. Wang, J. B. Vincent, K. Folting, D. Gatteschi and G. Christou, J. Am. Chem. Soc., 1993, 115, 1804–1816 CrossRef CAS; (b) R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef CAS.
- D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets; Oxford University Press, Oxford, 2006 Search PubMed.
- (a) Molecular Magnets Physics and Applications, ed. J. Bartolomé, F. Luis and J. F.Fernández, Springer-Verlag, Berlin, Heidelberg, 2014 Search PubMed; (b) B. Warner, F. E. Hallak, H. Prüser, J. Sharp, M. Persson, A. J. Fisher and C. F. Hirjibehedin, Nat. Nanotechnol., 2015, 10, 259–263 CrossRef CAS PubMed; (c) A. Ardavan, O. Rival, J. J. L. Morton, S. J. Blundell, A. M. Tyryshkin, G. A. Timco and R. E. P. Winpenny, Phys. Rev. Lett., 2007, 98, 057201 CrossRef PubMed.
- M. N. Leuenberger and D. Loss, Nature, 2001, 410, 789–793 CrossRef CAS PubMed; L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179–186 CrossRef PubMed.
- (a) D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed; (b) P. Zhang, Y.-N. Guo and J. Tang, Coord. Chem. Rev., 2013, 257, 1728–1763 CrossRef CAS; (c) F. Pointillart, O. Cador, B. Le Guennic and L. Ouahab, Coord. Chem. Rev., 2017, 346, 150–175 CrossRef CAS; (d) A. Dey, P. Kalita and V. Chandrasekhar, ACS Omega, 2018, 3, 9462–9475 CrossRef CAS.
- (a) J. Long, F. Habib, P.-H. Lin, I. Korobkov, G. Enright, L. Ungur, W. Wernsdorfer, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2011, 133, 5319–5328 CrossRef CAS PubMed; (b) J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078–2085 RSC.
- J. Liu, Y.-C. Chen, J.-L. Liu, V. Vieru, L. Ungur, J.-H. Jia, L. F. Chibotaru, Y. Lan, W. Wernsdorfer, S. Gao, X.-M. Chen and M.-L. Tong, J. Am. Chem. Soc., 2016, 138, 5441–5450 CrossRef CAS PubMed.
- Y.-S. Ding, N. F. Chilton, R. E. P. Winpenny and Y.-Z. Zheng, Angew. Chem., Int. Ed., 2016, 55, 16071–16074 CrossRef CAS PubMed.
- (a) F. Guo, B. M. Day, Y. Chen, M. Tong, A. Mansikkamäki and R. A. Layfield, Angew. Chem., Int. Ed., 2017, 56, 11445–11449 CrossRef CAS PubMed; (b) C. A. P. Goodwin, F. Ortu, D. Reta, N. F. Chilton and D. P. Mills, Nature, 2017, 548, 439–442 CrossRef CAS PubMed.
- F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Science, 2018, eaav0652 Search PubMed.
- S.-D. Jiang, B.-W. Wang, H.-L. Sun, Z.-M. Wang and S. Gao, J. Am. Chem. Soc., 2011, 133, 4730–4733 CrossRef CAS PubMed.
- S.-M. Chen, J. Xiong, Y.-Q. Zhang, Q. Yuan, B.-W. Wang and S. Gao, Chem. Sci., 2018, 9, 7540–7545 RSC.
- Y.-S. Meng, C.-H. Wang, Y.-Q. Zhang, X.-B. Leng, B.-W. Wang, Y.-F. Chen and S. Gao, Inorg. Chem. Front., 2016, 3, 828–835 RSC.
- R. L. Carlin, Magnetochemistry, Springer-Verlag, Berlin, 1986 Search PubMed.
- L. Escalera-Moreno, J. J. Baldoví, A. Gaita-Ariño and E. Coronado, Chem. Sci., 2018, 9, 3265–3275 RSC.
- Y.-S. Ding, K.-X. Yu, D. Reta, F. Ortu, R. E. P. Winpenny, Y.-Z. Zheng and N. F. Chilton, Nat. Commun., 2018, 9, 3134 CrossRef PubMed.
- (a) C. Pettinari, F. Marchetti and A. Drozdov, in Comprehensive Coordination Chemistry II, 2003, vol. I, pp. 211–246 Search PubMed; (b) F. T. Edelmann, Coord. Chem. Rev., 2014, 261, 73–155 CrossRef CAS; (c) M. J. Hardie and C. L. Raston, J. Chem. Soc., Dalton Trans., 2000, 15, 2483–2492 RSC.
- S. Nishihara, T. Akutagawa, T. Hasegawa and T. Nakamura, Inorg. Chem., 2003, 42, 2480–2482 CrossRef CAS PubMed.
- S. Petrosyants, Z. Dobrokhotova, A. Ilyukhin, N. Efimov, Y. Mikhlina and V. Novotortsev, Inorg. Chim. Acta, 2015, 434, 41–50 CrossRef CAS.
- H. Wada, S. Ooka, T. Yamamura and T. Kajiwara, Inorg. Chem., 2016, 56, 147–155 CrossRef PubMed.
- Y.-S. Ding, T. Han, Y.-Q. Hu, M. Xu, S. Yang and Y.-Z. Zheng, Inorg. Chem. Front., 2016, 3, 798–807 RSC.
- L. Maxwell, M. Amoza and E. Ruiz, Inorg. Chem., 2018, 57, 13225–13234 CrossRef CAS PubMed.
- G. Peng, Y.-Y. Zhang and Z.-Y. Li, Inorg. Chem. Commun., 2017, 77, 40–43 CrossRef CAS.
- E. Mamontova, J. Long, R. Ferreira, A. Botas, D. Luneau, Y. Guari, L. Carlos and J. Larionova, Magnetochemistry, 2016, 2, 41 CrossRef.
- S. Petit, G. Pilet, D. Luneau, L. F. Chibotaru and L. Ungur, Dalton Trans., 2007, 4582–4588 RSC.
- R. D. Rogers and A. N. Rollins, J. Chem. Crystallogr., 1994, 24, 321–329 CrossRef CAS.
- (a) M. Pinsky and D. Avnir, Inorg. Chem., 1998, 37, 5575–5582 CrossRef CAS PubMed; (b) A. Ruiz-Martínez, D. Casanova and S. Alvarez, Chem. - Eur. J., 2008, 14, 1291–1303 CrossRef PubMed; (c) A. Ruiz-Martínez, D. Casanova and S. Alvarez, Dalton Trans., 2008, 19, 2583–2591 RSC.
- L. T. A. Ho and L. F. Chibotaru, Phys. Rev. B, 2016, 94, 1–5 Search PubMed.
- (a) J. Bartolomé, G. FIloti, V. Kuncser, G. Schinteie, V. Mereacre, C. E. Anson, A. K. Powell, D. Prodius and C. Turta, Phys. Rev. B: Condens. Matter, 2009, 80, 014430 CrossRef; (b) R. Ishikawa, R. Miyamoto, H. Nojiri, B. K. Breedlove and M. Yamashita, Inorg. Chem., 2013, 52, 8300–8302 CrossRef CAS PubMed; (c) I. Nemec, R. Marx, R. Herchel, P. Neugebauer, J. van Slageren and Z. Travnicek, Dalton Trans., 2015, 44, 15014–15021 RSC.
- (a) F. Aquilante, L. De Vico, N. Ferré, G. Ghigo, P. Å. Malmqvist, P. Neogrády, T. B. Pedersen, M. Pitoňák, M. Reiher, B. O. Roos, L. Serrano-Andrés, M. Urban, V. Veryazov and R. Lindh, J. Comput. Chem., 2010, 31, 224–247 CrossRef CAS PubMed; (b) J. A. Duncan, J. Am. Chem. Soc., 2009, 131, 2416 CrossRef CAS PubMed; (c) G. Karlström, R. Lindh, P. Å. Malmqvist, B. O. Roos, U. Ryde, V. Veryazov, P.-O. Widmark, M. Cossi, B. Schimmelpfennig, P. Neogrády and L. Seijo, Comput. Mater. Sci., 2003, 28, 222–239 CrossRef; (d) V. Veryazov, P.-O. Widmark, L. Serrano-Andres, R. Lindh and B. O. Roos, Int. J. Quantum Chem., 2004, 100, 626–635 CrossRef CAS.
- P. Å. Malmqvist, B. O. Roos and B. Schimmelpfennig, Chem. Phys. Lett., 2002, 357, 230–240 CrossRef CAS.
- (a) B. A. Hess, C. M. Marian, U. Wahlgren and O. Gropen, Chem. Phys. Lett., 1996, 251, 365–371 CrossRef CAS; (b) B. Schimmelpfennig, AMFI, an atomic mean-field spinorbit integral program, Stockholm University, 1996 Search PubMed.
- (a) N. Douglas and N. M. Kroll, Ann. Phys., 1974, 82, 89–155 CrossRef; (b) B. A. Hess, Phys. Rev. A, 1986, 33, 3742–3748 CrossRef CAS.
- (a) B. O. Roos, R. Lindh, P. Å. Malmqvist, V. Veryazov and P.-O. Widmark, J. Phys. Chem. A, 2008, 112, 11431–11435 CrossRef CAS PubMed; (b) B. O. Roos, R. Lindh, P. Å. Malmqvist, V. Veryazov and P.-O. Widmark, Chem. Phys. Lett., 2005, 409, 295–299 CrossRef CAS.
- (a) L. F. Chibotaru, L. Ungur and A. Soncini, Angew. Chem., Int. Ed., 2008, 47, 4126–4129 CrossRef CAS PubMed; (b) L. F. Chibotaru, L. Ungur, C. Aronica, H. Elmoll, G. Pillet and D. Luneau, J. Am. Chem. Soc., 2008, 130, 12445–12455 CrossRef CAS PubMed; (c) L. F. Chibotaru and L. Ungur, J. Chem. Phys., 2012, 137, 064112–064122 CrossRef CAS PubMed; (d) L. Ungur, M. Thewissen, J.-P. Costes, W. Wernsdorfer and L. F. Chibotaru, Inorg. Chem., 2013, 52, 6328–6337 CrossRef CAS PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–127639 CrossRef CAS.
- (a) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS; (b) F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- (a) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS; (c) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Analysis of molar susceptibilities, energy levels of the lowest ligand field multiplets, XPRD and shape calculations results. See DOI: 10.1039/c8ra09648a |
| This journal is © The Royal Society of Chemistry 2019 |