
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiomimetic composite scaffold from an in situ hydroxyapatite coating on cellulose nanocrystals
Chen Huangabc,
Samarthya Bhagia cf,
Naijia Haoc,
Xianzhi Mengc,
Luna Liangc,
Qiang Yong*ab and
Arthur J. Ragauskas*cde
cf,
Naijia Haoc,
Xianzhi Mengc,
Luna Liangc,
Qiang Yong*ab and
Arthur J. Ragauskas*cde
aCollege of Chemical Engineering, Nanjing Forestry University, Nanjing 210037, China. E-mail: swhx@njfu.com.cn
bCo-Innovation Center for Efficient Processing and Utilization of Forest Resources, Nanjing Forestry University, Nanjing 210037, China
cDepartment of Chemical & Biomolecular Engineering, University of Tennessee Knoxville, Knoxville, Tennessee 37996, USA. E-mail: aragausk@utk.edu
dCenter for Renewable Carbon, Institute of Agriculture, The University of Tennessee, Knoxville, TN 37996, USA
eUTK-ORNL Joint Institute for Biological Science, Biosciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
fBiosciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
First published on 15th February 2019
Abstract
A novel nanocomposite scaffold was developed by homogeneous deposition of hydroxyapatite (HAP) on a cellulose nanocrystals (CNCs) matrix suspended in a simulated body fluid (SBF). By adjusting the pH of the SBF, the HAP content in the nanocomposite could be controlled between 15 wt% and 47 wt%. Physical and chemical characteristics of the nanocomposites were analyzed by SEM, FTIR, XRD, SAED, and TEM, which confirmed the successful incorporation of HAP onto the CNCs. The nanocomposites were then freeze-casted into porous scaffolds by different solidification technologies (i.e., directional freezing (DF), plunging in liquid N2 (PL) or in a −20 °C freezer (FZ)) followed by lyophilization. Compression testing of the HAP/CNCs foams indicated that DF caused significant improvement in mechanical properties due to the specific orientation and anisotropic porous structure compared to conventional freezing methods such as PL and FZ. Moreover, the scaffold with high HAP content exhibited improved mechanical and thermal properties, which holds potential for application in bone tissue engineering.
1. Introduction
Materials mimicking the natural composite of bone in both chemical and structural aspects have been studied for decades due to its extraordinary mechanical properties and biological functionality.1 Inspired by the intrinsic relationship between hydroxyapatite (HAP) and human bone, different types of 3D scaffolds have been developed in order to remedy the drawbacks of currently applied bone substitutes.2,3 Human bone is a hierarchically organized composite made up of collagen fibrils and apatite crystals (hydroxyapatite).4 The primary role of HAP in human bone is its osteoconductivity in which osteoprogenitor cells can adhere, migrate and finally form new bone.5 HAP in bone is precipitated between the oriented collagen and thus forms mineralized collagen fibers with a hierarchical structure.6 This type of structure gives the bone great mechanical properties.7 Many polymeric materials, like polyvinyl alcohol (PVA), poly(lactic acid) (PLA) and chitosan have been combined with HAP to enhance their osteoconductivity and hemocompatibility.8–10 However, these approaches have not yet fully addressed all the requirements needed for successful bone scaffolds due to their inherent drawbacks like poor mechanical properties and lack of flexibility,11–13 and research is ongoing in several fields, including on the use of several alternative materials such as nanocellulose.14Cellulose nanocrystals (CNCs), a strong and promising natural material derived from native cellulose, have attracted tremendous interest due to their excellent physical and chemical properties and hold much potential for conversion into bio-based materials that are rigid, biocompatible, non-immunogenic and renewable.15 These rod-like crystalline cellulose structures are typically 5–20 nm in diameter and several hundred nanometers in length.16 It has been reported that the CNCs have an average Young's modulus around 130 GPa, even stronger than steel.17 In addition, CNCs are highly hydrophilic with a multitude of hydroxyl groups available, which can offer binding sites for bone cells and make it superior to other materials such as collagen and silk fibroin.18 Because of these excellent properties, CNCs have been successfully applied in many fields, such as drug delivery systems, tissue substitutes, hydrogels, conductive films, filtration membranes, and in composites as a reinforcement agent to enhance their physical properties.19,20 Although CNCs are not capable of being degraded in the human body, it is highly biocompatible which will not cause any inflammatory response after implanting.21 The use of CNCs associated with HAP as a template for developing bone substitute is a field of growing interest. Kumar et al. (2014) reported a polyvinyl alcohol/HAP/CNCs composite, showing enhanced mechanical property as with the incorporation of CNCs.22 Through a sol–gel method, Herdocia-Lluberes et al. (2015) fabricated a CNCs/HAP pellet composite which could promote the proliferation of the osteoblast cells.23 Moreover, Saska et al. (2011) conducted in vivo test with a novel bacteria cellulose/HAP membrane, and results showed that the membrane could accelerate the new bone formation.24 These results suggest a promising perspective for using this composite in bone tissue engineering.
However, almost all the studies concerning CNCs/HAP composite were prepared by just blending HAP and CNCs, resulting in a heterogeneous mixture with relatively poor mechanical properties. During the preparation of nanocellulose by sulfuric acid hydrolysis, the surface of such CNCs was activated by the introduction of sulfonic groups. It was hypothesized that ionic groups could be induced to precipitate on CNCs by electrostatic interactions if such activated CNCs was put into a solution containing various ionic groups. Thus, a novel in situ HAP coating on individual CNCs fiber by immersing into a simulated body fluid (SBF) containing calcium ions and phosphate groups was developed herein this study. The content of HAP particles was controlled by adjusting the pH of the solution. Scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray diffraction (XRD), selected area electron diffraction (SAED) and Fourier transform infrared spectroscopy (FTIR) were used to confirm the existence of such HAP coating. Moreover, the HAP coated CNCs nanocomposite was then freeze-casted into porous scaffolds by different freezing technologies, in which the directional freezing formed an orientated structure and was anticipated to improve the mechanical property.
2. Experimental section
2.1. Materials
CNCs purchased from the University of Maine were prepared by sulfuric acid hydrolysis of wood pulp, possessing a dimension of 5–20 nm in width and 150–200 nm in length. The obtained product was an aqueous slurry (12.1 wt%) and the sulfur content of the CNCs was 0.95 wt%. All the other chemicals of analytical grade were purchased from Fisher Scientific (Waltham, MA, USA) and used as received without further purification unless otherwise noted.2.2. Fabrication of CNCs/HAP composite
A simplified SBF was prepared based on a previous report by Ishikawa et al. (2015).4 NaCl, CaCl2·2H2O, Na2HPO4·12H2O, and HCl were dissolved in DI water so as to set the final Na+, Ca2+, Cl− and HPO42− concentrations to 141.0 mM, 2.5 mM, 165.0 mM and 1.0 mM, respectively. The CNCs aqueous suspension (1.044 g) was added to the prepared SBF (200.0 mL) in a flask with a calcium ion (in SBF solution) and sulfonic group (in CNCs) molar ratio of 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The suspension was then stirred until no bulk CNCs particles were apparent and then transferred to an ultrasonic water bath for 30 min to evenly disperse CNCs. The mixture was then adjusted to different pH values with tris(hydroxymethyl)aminomethane solution (50 g L−1). Next, the suspension was put in an orbital shaker at 37 °C and 150 rpm for 1 h. This process of ultrasound and subsequent mild stirring was repeated three times. The solid contents in suspensions were then collected by centrifugation at 1705g for 10 min and washed with anhydrous ethanol three times to remove the residual salts. Finally, the CNCs/HAP composites were dried for two days in a conventional oven at 45 °C.
:1. The suspension was then stirred until no bulk CNCs particles were apparent and then transferred to an ultrasonic water bath for 30 min to evenly disperse CNCs. The mixture was then adjusted to different pH values with tris(hydroxymethyl)aminomethane solution (50 g L−1). Next, the suspension was put in an orbital shaker at 37 °C and 150 rpm for 1 h. This process of ultrasound and subsequent mild stirring was repeated three times. The solid contents in suspensions were then collected by centrifugation at 1705g for 10 min and washed with anhydrous ethanol three times to remove the residual salts. Finally, the CNCs/HAP composites were dried for two days in a conventional oven at 45 °C.
2.3. Preparation of porous CNCs/HAP scaffolds
The porous scaffolds were prepared by freeze-casting 5% (w/w) CNCs or CNCs/HAP suspensions solidified by directional freezing (DF), plunging in liquid N2 (PL, −196 °C) and putting in a −20 °C freezer (FZ). This was accomplished by adding dry CNCs and CNCs/HAP sample in DI water under magnetic stirring for 2 h to obtain a homogeneous suspension with 5 wt% consistency. Then, ∼3.5 mL of the solutions were transferred into 10 mL glass vials and frozen by FZ (−20 °C for 24 h), PL (−196 °C for 10 min) or DF (−196 °C for 10 min). In the case of DF, only the bottom of the vials contacted liquid N2 and vertical heat transfer was prevented by insulation of the vials with polystyrene foam. Upon solidification, the samples were immediately put in a freeze drier (−80 °C and 12 Pa) for 48 h to obtain the porous scaffolds.2.4. Characterization of nanocomposites
The surface morphology of the CNCs/HAP composites and the longitudinal side view of the porous scaffolds were observed employing a SEM (Zeiss EVO MA15, Germany) at an accelerating voltage of 5 kV. Before SEM analysis, the samples were put in a 70 °C oven to remove the moisture and then sputter coated with gold. The dimensions of CNCs or CNCs/HAP fibers were observed by TEM. Before the TEM analysis, samples were placed in DI water to obtain a 0.02% (w/v) solution. A drop of the diluted solution was then deposited on a carbon-coated copper grid to improve the dispersion of the CNCs. The residual water was removed with a filter paper from the periphery of the copper grid and dried in the air for 1 min. The specimens were observed using a Zeiss TEM (Zeiss Libra 200 MC, Germany) with an acceleration voltage of 200 kV. The lengths and widths of the CNCs and CNCs/HAP composite were analyzed with ImageJ software, and around 100 fibers were selected for characterization.25 SAED patterns were obtained with the same TEM based on the selected areas in the TEM images. FTIR analysis was accomplished using a Nicolet 6700 spectrometer (Thermo Scientific, MA, USA). Spectral data was acquired in the transmittance mode with 16 scans at 4 cm−1 resolution over the range of 4000–500 cm−1. The XRD patterns were recorded using a D8 advanced instrument (Bruker, Germany) with Cu Kα radiation at a voltage of 40 kV and a current of 30 mA. The scanning speed was 2° min−1 within the 2θ angular range of 5–60°.Thermogravimetric analysis (TGA) was carried out using a Q5000 instrument (TA Instruments, New Castle, DE, USA). About 5–10 mg of the nanocomposites were analyzed from RT to 700 °C at a heating rate of 10 °C min−1 under high-purity nitrogen. The HAP content in the nanocomposites was also determined by TGA following a published methodology except for that technical air (79% N2 and 21% O2) was used to facilitate complete combustion of organic compounds in the samples.26 The HAP contents were then calculated based on the initial and residual weights of the samples.
The porosity of different samples was determined according to a modified displacement method in a previous report.27 In brief, the volume and weight of the cylindrical foams were measured as V0 and W0, respectively. The volume was calculated based on the diameter and height of the cylinder determined by a digital Vernier caliper. Then the specimen was immersed into anhydrous ethanol for 48 h until saturation. The weight of the saturated foam was measured as W1. Finally, the porosity of the samples was obtained as the formula of P = (W1 − W0)/(ρ × V0), where ρ is the density of the anhydrous ethanol (0.789 g mL−1). Measurements were made on three samples of each foam, and the results represent an average value. Compression strength of different nanocomposite foams was determined with an INSTRON 5567 universal test machine (Canton, MA, USA) using a load cell of 500 N. The cylindrical foams were subjected to a constant compression rate of 0.1 mm s−1 until 80% strain and the compression modulus was calculated from the initial linear region (5%) of the stress–strain curve. Samples were stored in a desiccator before analysis.
3. Results and discussion
3.1. Synthesis of CNCs/HAP nanocomposites
Simulated body fluid (SBF) is a solution containing inorganic ions similar to those in human blood plasma which includes Ca2+, Na+, PO43− and Cl− as its main ions. SBF has been widely used to evaluate the bioactivity of different materials by forming the apatite layer on its surface.18 Unlike conventional bone substitutes such as titanium and stainless steel alloys, HAP crystals formed in SBF are nanosized and tend to grow and elongate in the c axis (vertical orientation), the same as the orientation in real bones.25 The original SBF solution developed by Kokubo (1991) is composed of various inorganic ions such as Na+, K+, Mg2+, Ca2+, Cl−, HCO3−, HPO42− and SO42−.28 However, a recent study illustrated the presence of Mg2+, HCO3−, SO42− had only a slight effect on the formation and deposition of the HAP crystals.29 Thus, a simplified SBF containing Ca2+, Na+, Cl−, and HPO42− was adopted in this study and this is consistent with the literatures.4,30,31 The existence of a large amount of Na+ and Cl− is necessary to equalize the ion strength. The SBF is a metastable solution, and no HAP crystals were observed under the mild pH condition if no high-affinity substrate was introduced.29 In this study, CNCs were employed as a substrate to facilitate the deposition of HAP. To investigate the HAP coating, different pH values of 6.0 (acid condition), 7.4 (body condition) and 8.5 (basic condition) were chosen and the morphologies of CNCs after HAP coating were analyzed with SEM (as shown in Fig. 1). SEM images clearly indicated the successful synthesis of the CNCs/HAP composite. Specifically, the surface of CNCs was flat and neat (Fig. 1a), whereas after immersion in SBF at pH 6.0, a small amount of grain-like HAP particles could be seen embedded into the CNCs matrix (Fig. 1b). The deposition of HAP is primarily induced by the anionic functional groups (i.e., sulfonic and hydroxyl groups) on the surface of CNCs that can initiate the nucleation of HAP and promote the coating of HAP on CNCs matrix.4 Increasing the pH to 7.4, the particles became smaller compared to the ones accomplished at pH 6.0. At a higher pH value of 8.5, small HAP particles aggregated due to the strong electrostatic interactions with CNCs and became predominant in the SEM image (Fig. 1d). It is known that HAP is soluble in an aqueous solution under acidic conditions. On the other hand, HAP tends to aggregate at high pH values. In our study, at the pH values of 6.0 and 7.4, only a few HAP deposits on CNCs were observed. Further increasing the pH to 8.5 significantly promoted the growth of the CNCs/HAP composites. | ||
| Fig. 1 The SEM images of CNCs before and after hydroxyapatite (HAP) coating. (a) Pure CNCs; (b) at SBF of pH 6.0; (c) at SBF pH 7.4; (d) at SBF pH 8.5. | ||
3.2. Structure characterization
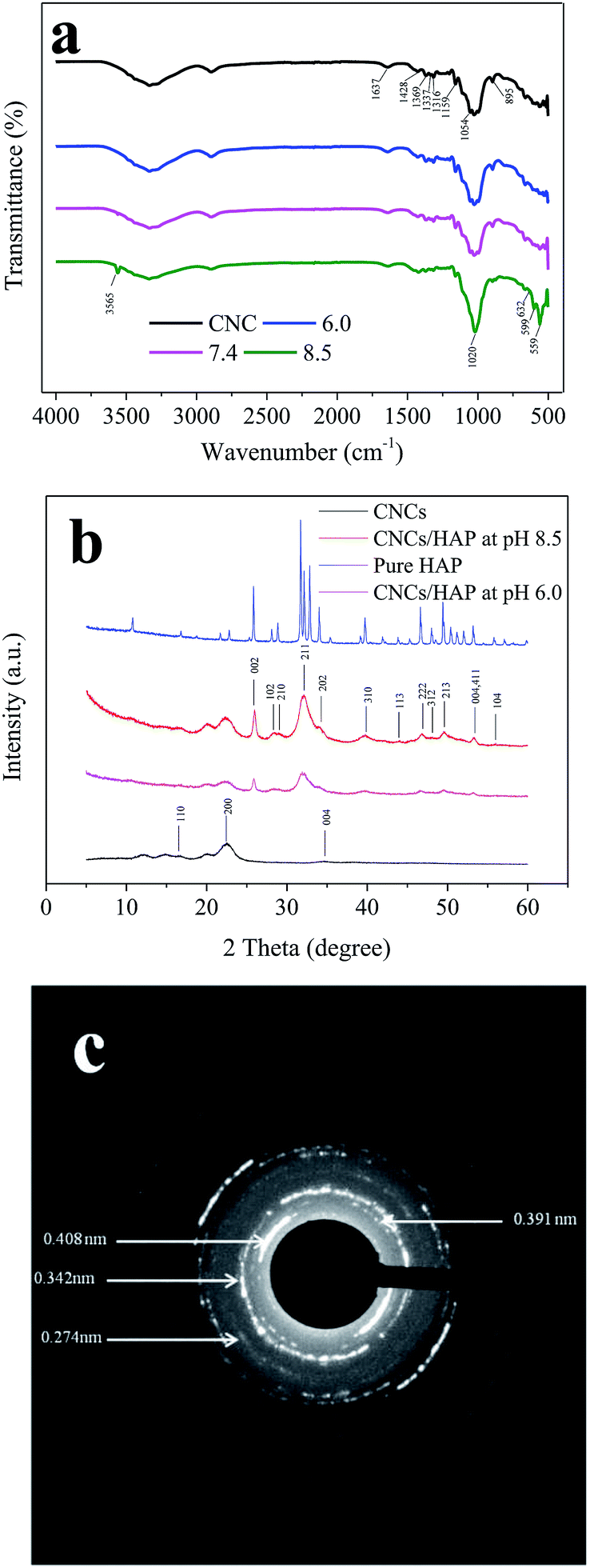 | ||
| Fig. 2 FTIR (a), XRD (b) and SAED (c) analysis of the CNCs and HAP coated CNCs. | ||
The crystalline structures of the pure CNCs, CNCs/HAP composites synthesized at pH 6.0 and 8.5, and pure HAP were analyzed by XRD (Fig. 2b). The characteristic peaks of cellulose at 16.7°, 22.5°, and 34.7° were assigned to the typical planes (110), (200) and (004) of cellulose, respectively.39 These diffraction peaks can also be found in numerous nanocellulose composites.18,22 After coating, the samples exhibited typical characteristic peaks of HAP at 2θ = 26.0° (002), 28.4° (102), 29.0° (210), 32.1° (211), 34.3° (202), 39.7° (310), 44.0° (113), 46.7° (222), 48.2° (312), 49.7° (213) and 56.1° (104), respectively. The peaks at 53.4° indicated planes (004/411) presented in the HAP.40 All of these characteristic peaks of our synthesized HAP could be found in the pure HAP (Fig. 2b). In addition, it was found that the CNCs/HAP composites obtained at different pH values almost have the same crystalline structure, indicating that the pH has very limited effect on the structure of generated composites. The crystalline structure of the coating was further analyzed with SAED (Fig. 2c). The diffraction pattern exhibited several strong and broad diffuse rings, indicating the strong crystalline structure of the HAP. From the pattern, planes of (200), (002) and (111) could be clearly seen with d200 = 0.408 nm, d002 = 0.342 nm and d111 = 0.391 nm, respectively. In addition, an interplanar spacing of 0.274 nm could be found in the diffraction pattern which could be assigned to the planes of (211), (112) or (300) in the HAP.41,42 The crystalline results of XRD and SAED supported the FTIR result and confirmed the sample was composed of CNCs and HAP.
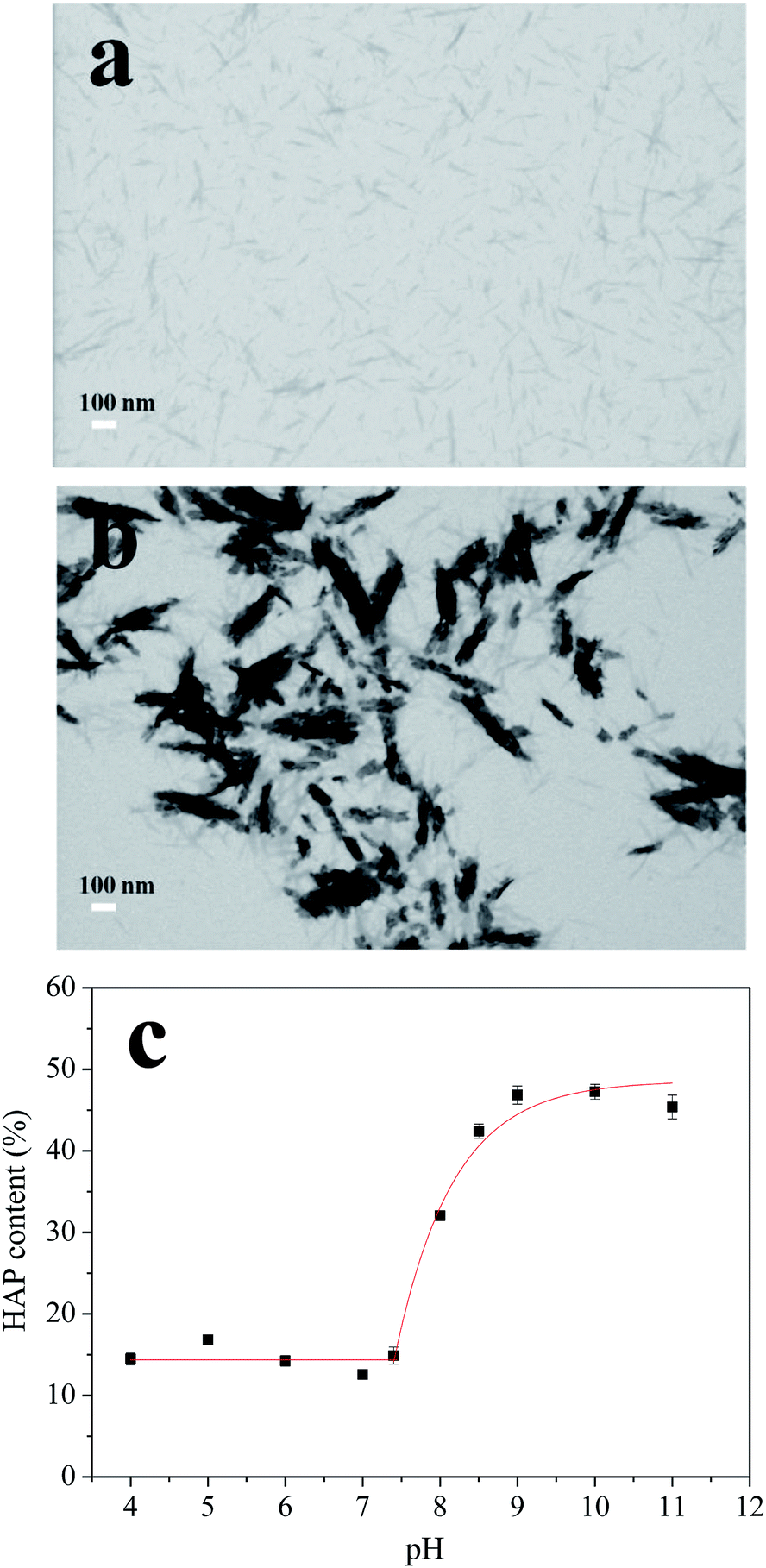 | ||
| Fig. 3 Micrographs of CNCs before (a) and after (b) HAP coating, scale bars 100 nm. The relationship between the HAP content and the pH values (c). | ||
The synthesis process was optimized by adjusting the pH of the SBF from 5.0 to 11.0. Thermogravimetric analysis (TGA) method was used to analyze the content of HAP in different nanocomposites and results were shown in Fig. 3c. When the pH value of the SBF increased from 4.0 to 7.4, the HAP contents in the nanocomposite were nearly constant (∼15%). Further adjusting the SBF system to basic conditions, the HAP content increased exponentially by increasing pH values, with the highest content of ∼47% obtained at pH 9.0. The deposition of HAP revealed a high degree of HAP supersaturation in the basic condition. Furthermore, when pH was increased beyond 9.0 to 10.0 and 11.0, HAP contents did not change that much which indicated that all of the Ca2+ and PO43− ions have deposited on CNCs at pH 9.0. These results suggested that the significant HAP coating process started at pH 7.4 and saturated at pH 9.0. Therefore, pH values of 7.4 and 8.5, which were in this deposition pH range, were chosen for further experiments.
3.3. Freeze-casting of the nanocomposite for porous scaffolds
In this study, nanocomposites prepared by HAP coating on CNCs were freeze-casted into porous foams by different freezing technologies including conventional freezing and directional freezing in liquid N2 (DF). The morphology of the freeze-casted foams is shown in Fig. 4. The samples prepared by directional freeze-casting had oriented and continuous channels (Fig. 4a–c), compared to samples frozen by PL. During directional freezing, only the bottom of the vials was in contact with liquid N2, thus ice crystals were induced to grow vertically, parallel to the imposed temperature gradient.47 Moreover, it can be found that the size of channels in the pure CNC scaffold was around 534.3 μm, while it decreased to 217.0 μm (at pH 7.4) and 227.1 μm (at pH 8.5) with the introduction of HAP. In addition, in the samples with only CNCs, pores in the channels were irregular and randomly distributed (Fig. 4a). With the addition of HAP, a more uniform and homogeneous pore structure was observed (Fig. 4b and c). This phenomenon was possibly caused by various adhesive forces (such as ionic bonds and hydrogen bonds) between CNCs and HAP that benefitted construction of a stable, uniform and oriented porous architecture.22 However, when the nanocomposites were solidified by plunging into the liquid N2, the ice crystals could nucleate at any places, and this resulted in a structure with randomly orientated pores (Fig. 4d–f).
 | ||
| Fig. 4 SEM images of the longitudinal side views of the freeze-casted porous foams. (a–c) Directionally frozen; (d–f) frozen by plunging into liquid N2. (a and d) Pure CNCs; (b and e) prepared at pH 7.4; (c and f) prepared at pH 8.5. | ||
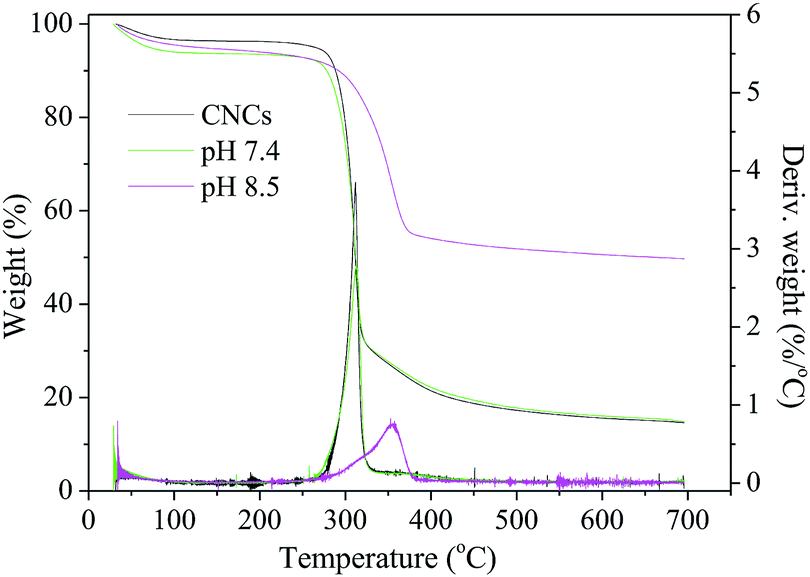 | ||
| Fig. 5 TGA-DTG curves of the CNCs/HAP scaffolds. | ||
| Samples | Solidification technology | Porosity (%) | Density (mg cm−3) | Stress at 50% strain (kPa) | Compressive modulus (kPa) |
|---|---|---|---|---|---|
| CNC | FZ (−20 °C) | 91.2 ± 3.3 | 44.7 ± 1.2 | 17.7 ± 1.6 | 29.6 ± 1.2 |
| PL (−196 °C) | 85.6 ± 2.1 | 46.4 ± 1.7 | 23.9 ± 1.1 | 42.4 ± 2.7 | |
| DF (−196 °C) | 83.2 ± 2.4 | 50.4 ± 0.9 | 37.0 ± 0.3 | 80.6 ± 1.4 | |
| CNC/HAP at pH 7.4 | FZ (−20 °C) | 90.3 ± 3.1 | 46.1 ± 2.5 | 13.6 ± 2.1 | 31.4 ± 1.2 |
| PL (−196 °C) | 86.9 ± 1.3 | 48.1 ± 1.7 | 34.0 ± 1.0 | 72.8 ± 0.7 | |
| DF (−196 °C) | 82.6 ± 0.7 | 50.8 ± 2.2 | 43.1 ± 0.9 | 119.6 ± 2.7 | |
| CNC/HAP at pH 8.5 | FZ (−20 °C) | 81.0 ± 1.3 | 46.8 ± 1.6 | 23.2 ± 1.4 | 54.1 ± 1.4 |
| PL (−196 °C) | 75.4 ± 2.2 | 47.4 ± 1.0 | 35.4 ± 1.2 | 100.8 ± 3.6 | |
| DF (−196 °C) | 73.6 ± 0.3 | 51.1 ± 1.8 | 61.7 ± 1.1 | 227.6 ± 2.7 | |
| CNC/HAP mixture | FZ (−20 °C) | 83.2 ± 0.5 | 45.9 ± 0.9 | 20.6 ± 1.2 | 35.7 ± 2.6 |
| PL (−196 °C) | 80.3 ± 1.5 | 48.2 ± 0.3 | 27.4 ± 0.7 | 56.9 ± 0.8 | |
| DF (−196 °C) | 78.2 ± 0.7 | 50.3 ± 1.2 | 40.3 ± 1.7 | 92.5 ± 2.3 |
As to density, it also varied with the solidification method (Table 1). DF caused the highest density for all the three samples. For example, for CNCs/HAP nanocomposite prepared at pH 7.4, density increased from 46.1 to 48.1 and 50.8 mg cm−3 with freezing methods of FZ, PL, and DF, respectively. This finding is consistent with the porosity results, as increased porosity results in decreased density. According to Xu et al. (2014), density variation of samples was caused by the volume shrinkage during the freeze-casting process, which indicated that DF had a higher volume shrinkage compared to other two technologies, thus leading to a higher density. Concerning the effect of HAP content on density, a slight increase was observed with increasing HAP content which could be explained by the fact that the HAP particle is heavier than CNCs.47
Scaffolds for bone substitution also need to be mechanically strong to provide enough structural stability to support cell adhesion. A recent study reported that a 20 times improvement in mechanical strength was obtained with the HAP coated CNCs, compared to the normal CNCs/HAP mixture.50 The compression results of the synthesized CNCs/HAP scaffolds from different solidification technologies are summarized in Table 1. The mechanical properties of the scaffolds were also significantly related to the freezing technologies (freezing temperature: FZ > DF > PL), including compressive stress and modulus, as shown in Table 1. For example, the compressive stress of CNCs scaffold increased from 17.7 kPa to 23.9 kPa and 37.0 kPa when FZ was replaced by PL and DF. The same trend can also be found in compressive modulus which increased from 29.6 kPa to 42.4 kPa and 80.6 kPa with different freezing methods. It could be concluded that both the freezing rate and freezing direction affected the mechanical properties. As observed, the fast freezing rate improved the mechanical properties (comparing PL to FZ). Moreover, the best compression results were obtained for the DF samples indicating that the oriented structure of the scaffold with strong anisotropy also improved the mechanical properties. During DF, ice crystals were induced to grow vertically, along with the direction of the imposed thermal gradient. The process formed an oriented and continuous channel architecture parallel to the direction of the compression force, this may explain why the DF resulted in the better mechanical performance than FZ and PL. On the other hand, with the coating of HAP, both the compression stress and compression modulus significantly increased, with highest values of 61.7 kPa and 227.6 kPa obtained with the nanocomposite at pH 8.5. This enhancement could be attributed to the improvement of stress transfer from the CNCs matrix to the strong HAP particles, and thus resulting in the superior mechanical properties. We further investigated the mechanical properties of the plain CNCs/HAP mixture (with the HAP content same to the CNCs/HAP composite prepared at pH 8.5), as shown in Table 1. The highest mechanical stress and modulus of the CNCs/HAP mixture were 40.3 kPa and 92.5 kPa (prepared by DF), respectively, which were much lower than our CNCs/HAP composite due to the heterogeneous dispersion of HAP in CNCs. This result indicated that our in situ HAP coated CNCs composite was superior to the CNCs/HAP mixture. Although the mechanical strength is still lower than a actual bone, we've made an improvement compared to some other bone scaffolds, including 8.13 times compression modulus of a chitosan–hyaluronic acid scaffold,51 6 times of a 3-D HAP composite,52 and 2.0 times of a hydrogel/fiber scaffold,53 which holds the potential for application as a substitute to the traditional metallic materials and bioceramics due to its biocompatibility. Further bone cell-related research will be done in the following experiment to test its perspective as a real bone substitute.
4. Conclusion
Our study illustrated a facile approach to fabricate a biomimetic scaffold by cultivating the CNCs in a simulated body fluid. With such methodology, the HAP was successfully coated on the CNCs, having a HAP layer size around 16.7–38.2 nm. The HAP content of the nanocomposite could be controlled by adjusting the pH of the solution, with the highest value of ∼47% prepared at pH 9.0. Moreover, after being freeze-casted into solid scaffolds, these nanocomposites all showed high porosity (higher than 70%) and light weight (density around 50 mg cm−3). Compression results indicated the introduction of HAP increased the compressive properties of the freeze-casted scaffolds. On the other hand, the scaffolds solidified by DF have a better mechanical property than by PL and FZ due to the orientated structure it caused. These results demonstrated that the use of such in situ HAP coating followed by the directional freezing technology generated a biomimetic scaffold with high porosity and improved mechanical strength that can serve as a potential substitute for native bone.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors thank the Priority Academic Program Development of Jiangsu Higher Education Institution (PAPD) and the Doctorate Fellowship Foundation of Nanjing Forestry University for supporting the work presented in this paper.References
- K. Shahzadi, I. Mohsin, L. Wu, X. Ge, Y. Jiang, H. Li and X. Mu, ACS Nano, 2017, 11, 325–334 CrossRef CAS PubMed.
- F. S. L. Bobbert and A. A. Zadpoor, J. Mater. Chem. B, 2017, 5, 6175–6192 RSC.
- S. Subramaniam, Y. H. Fang, S. Sivasubramanian, F. H. Lin and C. P. Lin, Biomaterials, 2016, 74, 99–108 CrossRef CAS PubMed.
- M. Ishikawa, Y. Oaki, Y. Tanaka, H. Kakisawa, G. Salazar-Alvarez and H. Imai, J. Mater. Chem. B, 2015, 3, 5858–5863 RSC.
- M. J. Olszta, X. Cheng, S. S. Jee, R. Kumar, Y. Y. Kim, M. J. Kaufman, E. P. Douglas and L. B. Gower, Mater. Sci. Eng., R, 2007, 58, 77–116 CrossRef.
- M. Sadat-Shojai, M. T. Khorasani, E. Dinpanah-Khoshdargi and A. Jamshidi, Acta Biomater., 2013, 98, 7591–7621 CrossRef PubMed.
- H. P. Schwarcz, D. Abueidda and I. Jasiuk, Front. Phys., 2017, 5, 39 CrossRef.
- F. Heidari, M. Razavi, M. E. Bahrololoom, M. Yazdimamaghani, M. Tahriri, H. Kotturi and L. Tayebi, Ceram. Int., 2018, 44, 275–281 CrossRef CAS.
- D. Mao, Q. Li, N. Bai, H. Dong and D. Li, Carbohydr. Polym., 2018, 180, 104–111 CrossRef CAS PubMed.
- K. H. Zuo, Y. P. Zeng and D. Jiang, Mater. Sci. Eng., C, 2010, 30, 283–287 CrossRef CAS PubMed.
- S. Chopra, S. Mahdi, J. Kaur, Z. Iqbal, S. Talegaonkar and F. J. Ahmad, J. Pharm. Pharmacol., 2010, 58, 1021–1032 CrossRef PubMed.
- K. Hashima, S. Nishitsuji and T. Inoue, Polymer, 2010, 51, 3934–3939 CrossRef CAS.
- D. Huang and A. Wang, RSC Adv., 2013, 3, 1210–1216 RSC.
- M. Holzwarth and P. X. Ma, Biomaterials, 2011, 32, 9622–9629 CrossRef PubMed.
- J. M. Dugan, J. E. Gough and S. J. Eichhorn, Biomacromolecules, 2010, 11, 2498–2504 CrossRef CAS PubMed.
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40, 3941–3994 RSC.
- A. Dufresne, Mater. Today, 2013, 16, 220–227 CrossRef CAS.
- J. Si, Z. Cui, Q. Wang, Q. Liu and C. Liu, Carbohydr. Polym., 2016, 143, 270–278 CrossRef CAS PubMed.
- T. Abitbol, A. Rivkin, Y. Cao, Y. Nevo, E. Abraham, T. Ben-Shalom, S. Lapidot and O. Shoseyov, Curr. Opin. Biotechnol., 2016, 39, 76–88 CrossRef CAS PubMed.
- B. Wicklein, A. Kocjan, G. Salazar-Alvarez, G. Carosio, G. Camino, M. Antonietti and L. Bergstrom, Nat. Nanotechnol., 2015, 10, 277–283 CrossRef CAS PubMed.
- H. Jiang, Y. Zuo, Q. Zou, H. Wang, J. Du, Y. Li and X. Yang, ACS Appl. Mater. Interfaces, 2013, 5, 12036–12044 CrossRef CAS PubMed.
- A. Kumar, Y. S. Negi, V. Choudhary and N. K. Bhardwaj, Cellulose, 2014, 21, 3409–3426 CrossRef CAS.
- C. S. Herdocia-Lluberes, S. Laboy-Lopez, S. Morales, T. J. Gonzalez-Robles, J. A. Gonzalez-Feliciano and E. Nicolau, J. Nanomater., 2015, 1–9 Search PubMed.
- S. Saska, H. S. Barud, A. M. M. Gaspar, R. Marchetto, S. J. L. Ribeiro and Y. Messaddeq, Int. J. Biomater., 2011, 2011, 175362 CAS.
- F. Lindberg, J. Heinrichs, F. Ericson, P. Thomsen and H. Engqvist, Biomaterials, 2008, 29, 3317–3323 CrossRef CAS PubMed.
- D. Gebauer, V. Oliynyk, M. Salajkova, J. Sort, Q. Zhou, L. Bergström and G. Salazar-Alvarez, Nanoscale, 2011, 3, 3563–3566 RSC.
- L. Y. Jiang, Y. Li, X. Wang, L. Zhang, J. Wen and M. Gong, Carbohydr. Polym., 2008, 74, 680–684 CrossRef CAS.
- T. Kokubo, Biomaterials, 1991, 12, 155–163 CrossRef CAS PubMed.
- T. Kobayashi, S. Ono, S. Hirakura, Y. Oaki and H. Imai, CrystEngComm, 2012, 14, 1143–1149 RSC.
- Y. He, X. Wang, L. Chen and J. Ding, J. Mater. Chem. B, 2014, 2, 2220–2227 RSC.
- J. Zhao, Z. Zhang, Z. Yu, Z. He, S. Yang and H. Jiang, Appl. Surf. Sci., 2014, 289, 89–96 CrossRef CAS.
- M. L. Troedec, D. Sedan, C. Peyratout, J. R. Bonnet, A. Smith, R. Guinebretiere, V. Gloaguen and P. Krausz, Composites, Part A, 2008, 39, 514–522 CrossRef.
- C. Huang, A. J. Ragauskas, X. Wu, Y. Huang, X. Zhou, J. He, C. Huang, C. Lai, X. Li and Q. Yong, Bioresour. Technol., 2017, 250, 365–373 CrossRef PubMed.
- J. Ryu, S. H. Ku, H. Lee and C. B. Park, Adv. Funct. Mater., 2010, 20, 2132–2139 CrossRef CAS.
- G. Wei, J. Reichert, J. Bossert and K. D. Jandt, Biomacromolecules, 2008, 9, 3258–3267 CrossRef CAS PubMed.
- X. Cai, H. Tong, X. Shen, W. Chen, J. Yan and J. Hu, Acta Biomater., 2009, 5, 2693–2703 CrossRef CAS PubMed.
- E. P. Erasmus, R. Sule, O. T. Johnson, J. Massera and I. Sigalas, Sci. Rep., 2018, 8, 3699 CrossRef CAS PubMed.
- R. Rabadan-Ros, P. Mazón, S. Serena, M. A. Sainz, L. Meseguer-Olmo and P. N. DeAza, J. Eur. Ceram. Soc., 2017, 37, 2943–2952 CrossRef CAS.
- W. Chen, H. Yu, Y. Liu, P. Chen, M. Zhang and Y. Hai, Carbohydr. Polym., 2011, 83, 1804–1811 CrossRef CAS.
- S. Koutsopoulos, J. Biomed. Mater. Res., 2002, 62, 600–612 CrossRef CAS PubMed.
- E. I. Suvorova, F. Christensson, H. E. L. Madsen and A. A. Chernov, J. Cryst. Growth, 1998, 186, 262–274 CrossRef CAS PubMed.
- E. I. Suvorova and P. A. Buffat, J. Microsc., 1999, 196, 46–58 CrossRef CAS PubMed.
- K. K. Mallick, J. Am. Ceram. Soc., 2009, 92, S85–S94 CrossRef CAS.
- E. Ebaretonbofa and J. R. G. Evans, J. Porous Mater., 2002, 9, 257–263 CrossRef CAS.
- Y. Zhou, S. Fu, Y. Pu, S. Pan, M. V. Levit and A. J. Ragauskas, RSC Adv., 2013, 3, 19272–19277 RSC.
- T. Köhnke, A. Lin, T. Elder, H. Theliander and A. J. Ragauskas, Green Chem., 2012, 14, 1864–1869 RSC.
- Z. Xu, Q. Sun, F. Huang, Y. Pu, S. Pan and A. J. Ragauskas, RSC Adv., 2014, 4, 12148–12153 RSC.
- Q. Sun, A. Mandalika, T. Elder, S. S. Nair, X. Meng, F. Huang and A. J. Ragauskas, Green Chem., 2014, 16, 3458–3462 RSC.
- L. Hu, C. A. Wang, Y. Huang, C. Sun, S. Lu and Z. Hu, J. Eur. Ceram. Soc., 2010, 30, 3389–3396 CrossRef CAS.
- C. Huang, N. Hao, S. Bhagia, M. Li, X. Meng, Y. Pu, Q. Yong and A. J. Ragauskas, Materialia, 2018, 4, 237–246 CrossRef.
- H. Tan, C. R. Chu, K. A. Payne and K. G. Marra, Biomaterials, 2009, 30, 2499–2506 CrossRef CAS PubMed.
- S. Yunoki, T. Ikoma, A. Tsuchiya, A. Tsuchiya, A. Monkawa, K. Ohta, S. Sotome, K. Shinomiya and J. Tanaka, J. Biomed. Mater. Res., Part B, 2007, 80, 166–173 CrossRef PubMed.
- S. Khorshidi and A. Karkhaneh, J. Biomed. Mater. Res., 2017, 106, 718–724 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2019 |