
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExpanded graphene oxide fibers with high strength and increased elongation†
Ashok D. Ugale a,
LinLin Chib,
Min-Kyu Kima,
Sudong Chaeb,
Jae-Young Choi*b and
Ji-Beom Yoo*ab
a,
LinLin Chib,
Min-Kyu Kima,
Sudong Chaeb,
Jae-Young Choi*b and
Ji-Beom Yoo*ab
aSungkyunkwan Advanced Institute of Nanotechnology (SAINT), Sungkyunkwan University, Suwon, 440-746, Republic of Korea. E-mail: jbyoo@skku.edu
bAdvanced Materials Science and Engineering, Sungkyunkwan University, Suwon, 440-746, Republic of Korea. E-mail: jy.choi@skku.edu
First published on 31st January 2019
Abstract
We report the role of chemically expanded graphite in the fabrication of high-performance graphene oxide fibers by wet spinning. X-ray diffraction peak showed that the interplanar distance of the expanded graphene oxide (EGO) fiber was more than that of graphene oxide (GO) fiber due to the expanded graphite. X-ray photon spectroscopy analysis revealed that EGO was more oxidized than GO. The hydrogen bonding network and secondary intermolecular interaction made the EGO aqueous solution more stable and crystalline, and it was able to be stretched in the coagulation bath. Morphological analysis showed the excellent alignment and compactness of EGO sheets in the fibers. The increased interplanar distance between the EGO sheets favored the edge-to-edge interaction more than the basal plane interaction within the fiber, thus resulting in high mechanical strength (492 MPa) and increased elongation (6.1%).
A Introduction
In recent years, many studies have focused on graphene oxide liquid crystals (GOLC) due to their large-scale applications. Graphene oxide (GO) is a chemically-modified graphene in the oxygenated form, having colloidal liquid crystalline properties.1,2 Colloidal liquid crystal fluid is the structure of the anisotropically shaped colloids retaining liquid-like fluidity and crystal-like ordering.3 The long-range structural alignment and fluidic nature of GOLC make it a key building block for diverse graphene-assembled structures, as it enables the adoption of the excellent properties of graphene to microscopic carbon materials.4,5 These macroscopic materials including fibers, hydrogels, aerogels, have been found to be excellent in extensive applications.6–8 Thus, GOLC material has become a major scientific and technological issue in the field of material science and nanotechnology.1,9 In the wet spinning of GOLC, the liquid crystalline behavior of GO dispersion can lead to the orientation of GO sheets under shear force, which can form ordered assembly in microscopic fibers.10–12 The first fabrication method of graphene fibers via wet spinning was reported by Gao et al. in 2011 and the tensile strength reached 102 MPa.13 Since then, many efforts have been devoted to improving the mechanical properties of fibers, with some of these including using phenolic carbon,5 introducing chemical bonds to cross-link GO sheets,14 using giant GO sheets,12 and intercalating small-sized graphene sheets into fibers consisting of large-sized graphene sheets.15 Recently, T. H. Han et al. reported a novel method to improve the strength and stiffness of GOLC fibers by using trivalent metal ions as a coagulant.16 Summary of different methods to improve the mechanical properties of GO fibers is given in Table S1 in ESI.† However, while the strength of the fibers was achieved, the elongation at break was too poor. If the elongation at break is too short, the application of the fibers will be limited. In recent years, many scientists have been working on developing strong fibers with increased elongation so that they can be used for wide applications. Therefore, it is important and also a big challenge to simultaneously increase the strength and elongation of GO fibers.The orientation of GO sheets, the interaction between the sheets, and defects or packing density within the fibers are the key factors for determining the properties of the fibers.17 Chao et al. explained the fracture mechanism of the fibers based on the tensile shear model. According to this model, under tensile force in the fiber axial direction, the graphene oxide sheets endure a pulling force to slide from the stacked sheet blocks.12 Thus, controlling the sliding among the GO sheets can prevent the fracture of the fibers, thereby simultaneously improving the strength and elongation. Expanded graphene oxide (EGO) sheets can control the sliding among themselves under axial force by enhancing the edge-to-edge interaction rather than the basal plane interaction. Here we present the fabrication of fibers with simultaneous high strength (492 MPa) and increased elongation (6.1%) by using EGO. The expanded graphite (chemical treated) having large interplanar distance was used as a precursor to synthesized EGO by modified Hummer's method. The strong edge-to-edge interaction and weak basal plane interaction between EGO sheets due to large interplanar distance could lead to enhanced load transfer and fiber strength. The defects within the fiber could be reduced by stretching the fiber in the coagulation bath. The underlying advantage is that EGO modulates the chemical interactions between the EGO sheets, which consequently improves the mechanical strength and elongation simultaneously.
B Results and discussion
In order to evaluate the effect of chemical expansion, chemically treated graphite and normal graphite were used to synthesize EGO and GO, respectively, by modified Hummer's method. Further, EGO and GO aqueous solution were made and used to fabricate the EGO and GO fibers, respectively, by the wet spinning process. The X-ray diffraction (XRD) plot shows that the diffraction peaks (2θ) for graphite (G) and expanded graphite (EG) are 26.460 and 26.230, respectively. In contrast, in the cases of GO and EGO fibers, they are 9.60 and 9.20 correspondingly (Fig. 1a). XRD analysis reveals that the interplanar distance between the layers of EG is 3.40 Å, while in the case of graphite it is 3.35 Å. This shows that after the chemical expansion of graphite, the interplanar distance between graphene layers has increased by 0.05 Å. A photograph of the expansion of the graphite after chemical treatment is shown in Fig. 1b and c, and the detailed process are given in the Experimental section. The calculated interplanar distances of the EGO and GO fibers are 9.55 Å and 9.2 Å respectively.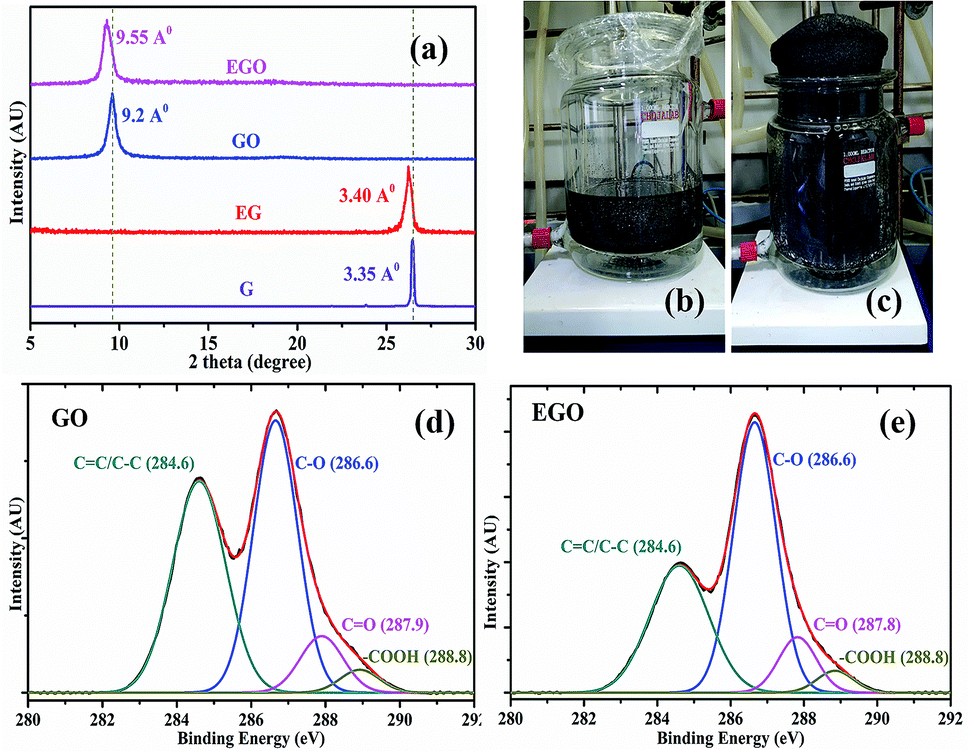 | ||
| Fig. 1 (a) XRD plot of EGO and GO fibers, expanded graphite (EG) and graphite (G) powders showing interplanar distance. Chemical treatment of graphite (b) before expansion (c) after expansion. XPS spectra of deconvoluted C 1s peaks in (d) GO and (e) EGO. | ||
Dimiev et al. explained the mechanism of graphene oxide formation in three distinct steps: the first step is the conversion of graphite into a stage-1 graphite intercalation (GIC) compound followed by the conversion of the stage-1 GIC into pristine graphite oxide (PGO). Finally, PGO reacts with water to yield graphene oxide.18 As the interplanar distance of the expanded graphite grows, it becomes easier for acid and oxidizing agents to go in-between the layers of graphite for oxidation. In Fig. 1d and e, qualitative analysis of the results of X-ray photoelectron spectroscopy (XPS) of GO and EGO sheets confirmed the effect of chemical treatment of graphite on oxidation degree. The summary of the deconvoluted C 1s peak having different functional groups and its atomic fractions are presented in Table 1. XPS analysis reveals that the EGO (65.6%) is more oxidized than GO (57.6%) due to its larger d-spacing. The degree of functionalization is the important factor for forming a liquid crystal. The colloids in aqueous solution retain their stability to electric charges (functional groups on graphene sheets) so that each particle will repel others before they come into actual contact, effectively forming a liquid crystal.19,20 The strong hydrogen bonding networks between graphene sheets control the stabilization of the graphene oxide aqueous solution for a long period.21 The high number of hydroxyl groups in EGO enhanced the secondary intermolecular interaction between EGO sheets, which allowed for the excellent dispersion stability compared to GO. Highly crystalline thermodynamically stable aqueous expanded graphene oxide liquid crystal (EGOLC) solution provided organized prealigned sheets for the spinning of a strong and flexible fiber. The larger d-spacing and increased oxidation degree of EGO makes for easy and stable dispersion in water as compared to GO. The EGO and GO aqueous solutions, after four days, prepared by the respective dispersion of EGO and GO powder in water by sonication are shown in Fig. S1 of the ESI.†
| GO | EGO | ||||||
|---|---|---|---|---|---|---|---|
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/C–C (sp2/sp3) C/C–C (sp2/sp3) |
C–O | CO |
O–CO |
C–C | C–O | CO |
O–CO |
| 42.2% | 45.2% | 9.1% | 3.3% | 34.2% | 52.5% | 9.5% | 3.6% |
The morphologies of EGO and GO flakes are examined by scanning electron microscopy (SEM), as shown in Fig. 2a and b. The lateral size of the EGO (20–45 μm) sheets is bigger than GO (10–25 μm). A number of FESEM micrographs were recorded and provided in ESI.† The two-dimensional anisotropic morphology of the graphene oxide sheets explains the formation of GOLC by analogy with the plate-like colloidal model.22 Large graphene sheets excluded volume for small sheets giving rise to entropic rearrangement to form long ordered assembly to form a liquid crystal of aqueous solution.10 The asymmetric colloids of sheets in a liquid crystal could easily form a regular alignment due to a shear force applied during spinning of fibers.3 In the UV-visible spectra, the high absorption intensity of EGO as compared to GO reveals that EGO sheets are stable and well dispersed in the aqueous solution, as indicated in Fig. 2c. Due to the strong hydrogen bonding networks and superior secondary intramolecular interaction between EGO sheets, fibers made from EGO were able to stretch in the coagulation bath as well as while drying (Videos, ESI†). While GO fibers were not able to stretch like EGO due to less intermolecular interactions (oxidation) and bad crystalline solution. The stretching of fibers in the bath and while drying made fibers more compact, uniform (Fig. 2d), aligned and reduced the defects and voids within the fibers resulting in good mechanical properties. The wet spinning of EGO and GO fibers with and without stretching is shown schematically in Fig S4 (ESI†).
 | ||
| Fig. 2 SEM images of (a) EGO and (b) GO sheets. (c) UV-visible of GO and EGO. (d) Optical image of EGO and GO fibers. | ||
The section and lateral morphology of fibers were characterized by SEM as shown in Fig. 3. Stretching has improved the alignment and compactness of EGO sheets in the EGO fibers as compared to those of the GO sheets in the GO fibers, and this is advantageous for improving the mechanical properties. The effect of stretching is on EGO and GO fibers is schematically represented in Fig. 4c. Further, stretching of fibers in the coagulation bath has made the EGO fiber more uniform with good surface morphology.
 | ||
| Fig. 3 SEM images of EGO and GO fibers. Cross-sections of (a and b) EGO, (c and d) GO fibers, surface morphologies of (e) EGO and (f) GO fibers. | ||
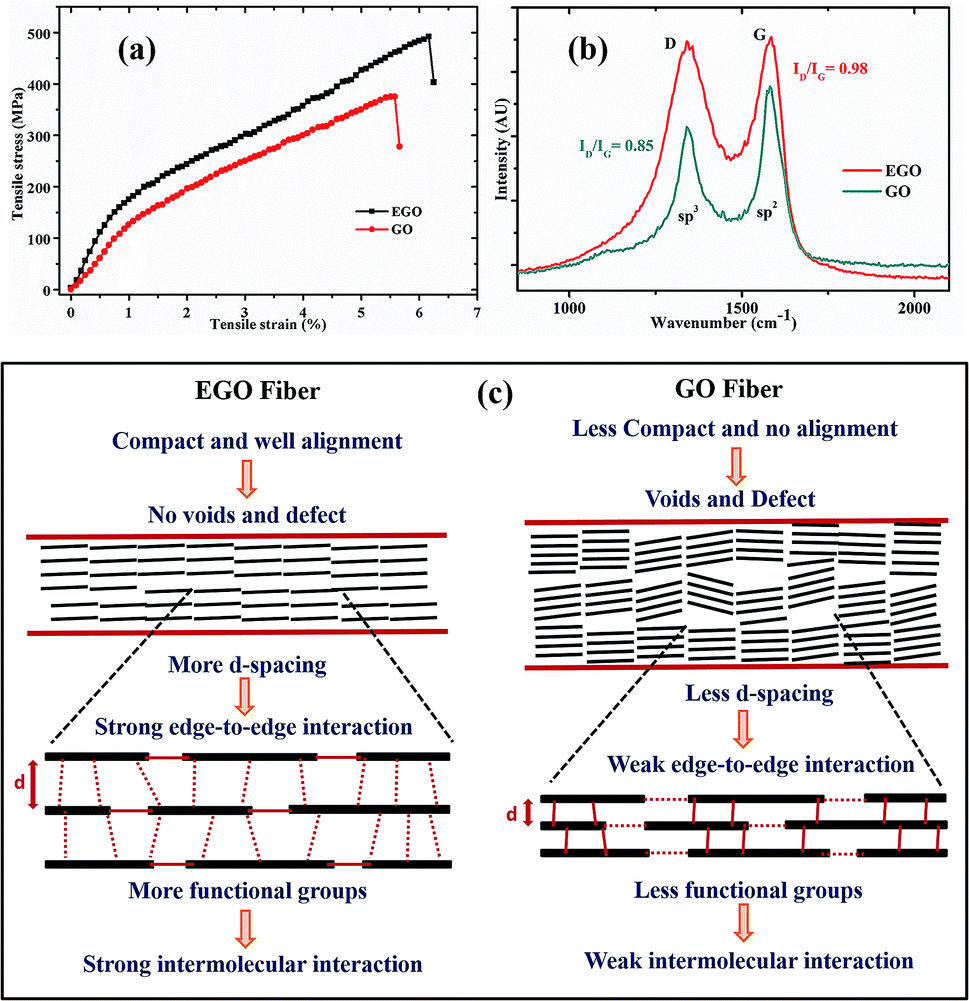 | ||
| Fig. 4 (a) Mechanical properties of spun EGO and GO fibers. (b) Raman spectra of EGO and GO fibers. (c) Schematic illustration showing alignment, defects and molecular level interaction within GO and EGO fibers. | ||
The sheet alignment, compactness, and interaction between the sheets eventually determine the mechanical properties of the fibers. The mechanical strengths of the EGO and GO fibers are shown in Fig. 4a and their mechanical properties are summarised in Table 2. Under tensile stress fibers have shown notable mechanical strength (492 MPa) and increased elastic elongation (6.1%). The area under the stress–strain curve is used to express the toughness of the material. The strength, Young's modulus, toughness, and elongation at break of the EGO fiber were increased after using chemically expanded graphite as a precursor to synthesize the expanded graphene oxide. The intermolecular interaction and hydrogen bonding are strong when the distance is small. The more interplanar distance between the EGO sheets and comparatively less distance between the edges of the sheets favored the edge-to-edge interaction rather than the basal plane interaction within the sheets of EGO fibers.23 Therefore, the edge-to-edge binding force became more dominant to achieve high elongation break and mechanical strength by controlling the sliding of EGO sheets from stacked blocks under axial force. The schematic illustration of the edge-to-edge and basal molecular interaction in between graphene sheets, which are responsible for the mechanical properties, are shown in Fig. 4c. Moreover, the stretching of EGO fibers also contributed to the improvement of the mechanical properties of the fibers by reducing the defect and voids.
| Fiber | d-spacing [Å] | Tensile strength [MPa] | Elongation break [%] | Young's Modulus [GPa] | Toughness [MJ m−3] |
|---|---|---|---|---|---|
| EGO | 9.55 | 492 | 6.1 | 14.5 | 18.77 |
| GO | 9.2 | 375.5 | 5.5 | 12.4 | 12.64 |
Raman spectroscopy is a tool that is widely used to characterize carbon-based materials. Fig. 4b shows the recorded Raman spectra of EGO and GO fibers. They consisted of two peaks, D and G. Broadly, they were assigned to the sp3 and sp2 phases of graphene, respectively. The D peak originates from the disorder of graphene, and the G peak originates from the hexagonal carbon ring.24 The integrated intensity ratios ID/IG of EGO and GO are 0.98 and 0.85, respectively. This reveals that EGO has a more disordered structure than GO, due to the increased oxidation and chemical expansion of precursor graphite, which is in accordance with the XRD and XPS data.
C Experimental
a Materials
To begin, 5 g of graphite (Asbury 3061) is mixed in the reactor containing sulfuric acid (160 ml) and hydrogen peroxide (40 ml) in the ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. The mixture is then stirred for 30 min and kept at water circulation for a day at a temperature of 50 °C. The next day, chemically expanded graphite is washed with acetone to get expanded graphite (EG). Then graphite and EG were used as a precursor to synthesize EGO and GO, respectively, by modified Hummer's method.
:2. The mixture is then stirred for 30 min and kept at water circulation for a day at a temperature of 50 °C. The next day, chemically expanded graphite is washed with acetone to get expanded graphite (EG). Then graphite and EG were used as a precursor to synthesize EGO and GO, respectively, by modified Hummer's method.
b Wet spinning of fibers
The obtained EGO and GO were used to make the liquid crystalline solution (10 mg ml−1) by adding EGO and GO, respectively, in DI water, followed by 1 h sonication and shaking for 5 h. The prepared EGO and GO solutions were centrifuged at 3000 rpm so as to remove the visible particles. The wet spinning of EGO and GO fibers was achieved by injecting the aqueous solution in the coagulation bath of ethanol/water (1:3 v/v) solutions of 5 wt% of CaCl2. The stretching of the fiber was achieved by adjusting the rpm of the collecting roller. A video of the spinning process and drawing is available in the ESI.† GO fibers were washed with DI water and dried at room temperature.
c Characterizations
The prepared G and EG powder, GO and EGO fibers were characterized by XRD (D8 Discover Bruker) using Cu Kα radiation (λ = 1.54 Å). XPS was performed using a VG Microtech ESCA2000 System operated at 14 kV and all binding energies referenced to the C 1s neutral carbon peak at 284.6 eV. SEM images were taken on a Jeol JSM-6701F field emission system. UV-visible characterization was measured using Agilent 89090A spectrometer. Optical microscopic images were taken by Olympus BX51M Microscope. The tensile test was carried on Instron 3343 at the load rating of 5 mm min−1 with the fiber length of 5 mm. The reported results are the average value of 10 samples. Raman spectra were recorded with a Witec confocal Raman microscope using 532 nm laser wavelength.D Conclusions
In summary, the role of expanded graphite is to fabricate high-performance fibers by wet spinning. In order to study the effect of expansion, chemically treated graphite (with increased interplanar distance) and normal graphite were used to synthesize the EGO and GO, respectively. The hydrogen bonding network and secondary intermolecular interaction made the EGO aqueous solution more stable and crystalline. The stable liquid crystalline EGO solution was able to continuously spin the fiber with stretching in the coagulation bath. The high alignment and compactness of the EGO sheets in the fiber along with the uniform diameter of the fibers improved the mechanical properties. The increased interplanar distance between the EGO sheets favored the edge-to-edge interaction rather than the basal plane interaction, which led to high mechanical strength (492 MPa) and increased elongation break (6.1%). Chemical treatment of the graphite was particularly advantageous for developing the intermolecular level interaction, which led to improved strength and stiffness of the fibers. These EGO fibers with simultaneous strength and elongation can be reduced to make graphene fibers with a high toughness which can be used for great technological applications in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to Professor Seunghyun Baik and Seonghyun Bae for giving access to measure the mechanical properties of the fibers. This work was supported by Korea Electric Power Corporation (KEPCO, No. R17XH01). Authors are thankful to Gil-Seon Kang for taking SEM images and Ki-Bong Nam for recording Raman spectrum.Notes and references
- S. Padmajan Sasikala, J. Lim, I. H. Kim, H. J. Jung, T. Yun, T. H. Han and S. O. Kim, Chem. Soc. Rev., 2018, 47, 6013–6045 RSC.
- K. E. Lee and S. O. Kim, Part. Part. Syst. Charact., 2017, 34, 1–2 Search PubMed.
- Z. Xu and C. Gao, ACS Nano, 2011, 5, 2908–2915 CrossRef CAS PubMed.
- C. Xiang, C. C. Young, X. Wang, Z. Yan, C. C. Hwang, G. Cerioti, J. Lin, J. Kono, M. Pasquali and J. M. Tour, Adv. Mater., 2013, 25, 4592–4597 CrossRef CAS PubMed.
- M. Li, X. Zhang, X. Wang, Y. Ru and J. Qiao, Nano Lett., 2016, 16, 6511–6515 CrossRef CAS PubMed.
- Y. Hu, H. Cheng, F. Zhao, N. Chen, L. Jiang, Z. Feng and L. Qu, Nanoscale, 2014, 6, 6448–6451 RSC.
- S. Y. Bae, I. Y. Jeon, J. Yang, N. Park, H. S. Shin, S. Park, R. S. Ruoff, L. Dai and J. B. Baek, ACS Nano, 2011, 5, 4974–4980 CrossRef CAS PubMed.
- D. Kang, Y.-E. Shin, H. J. Jo, H. Ko and H. S. Shin, Part. Part. Syst. Charact., 2017, 34, 1600382 CrossRef.
- J. Y. Kim and S. O. Kim, Nat. Mater., 2014, 13, 325–326 CrossRef CAS PubMed.
- R. Jalili, S. H. Aboutalebi, D. Esrafilzadeh, R. L. Shepherd, J. Chen, S. Aminorroaya-Yamini, K. Konstantinov, A. I. Minett, J. M. Razal and G. G. Wallace, Adv. Funct. Mater., 2013, 23, 5345–5354 CrossRef CAS.
- J. E. Kim, T. H. Han, S. H. Lee, J. Y. Kim, C. W. Ahn, J. M. Yun and S. O. Kim, Angew. Chem., Int. Ed., 2011, 50, 3043–3047 CrossRef CAS PubMed.
- Z. Xu, H. Sun, X. Zhao and C. Gao, Adv. Mater., 2013, 25, 188–193 CrossRef CAS PubMed.
- Z. Xu and C. Gao, Nat. Commun., 2011, 2, 571–579 CrossRef PubMed.
- X. Hu, Z. Xu, Z. Liu and C. Gao, Sci. Rep., 2013, 3, 1–9 Search PubMed.
- G. Xin, T. Yao, H. Sun, S. M. Scott, D. Shao, G. Wang and J. Lian, Sci. 80, 2015, 349, 1083–1087 CrossRef CAS PubMed.
- W. Eom, H. Park, S. H. Noh, K. H. Koh, K. Lee, W. J. Lee and T. H. Han, Part. Part. Syst. Charact., 2017, 34, 1–6 CrossRef.
- L. Chen, Y. He, H. Chai, F. Chen and Q. Fu, Nanoscale, 2013, 5, 5809–5815 RSC.
- A. M. Dimiev and J. M. Tour, ACS Nano, 2014, 8, 3060–3068 CrossRef CAS PubMed.
- L. Onsager, Ann. N. Y. Acad. Sci., 1949, 51, 627–659 CrossRef CAS.
- T. H. Han, J. Kim, J. S. Park, C. B. Park, H. Ihee and S. O. Kim, Adv. Mater., 2007, 19, 3924–3927 CrossRef CAS.
- J. Y. Oh, J. Park, Y. C. Jeong, J. H. Kim, S. J. Yang and C. R. Park, Part. Part. Syst. Charact., 2017, 1–8 Search PubMed.
- M. A. Bates, J. Chem. Phys., 1999, 110, 6553–6559 CrossRef CAS.
- H. Park and T. H. Han, Bull. Korean Chem. Soc., 2013, 34, 3269–3273 CrossRef CAS.
- A. D. Ugale, R. V. Jagtap, D. Pawar, S. Datar, S. N. Kale and P. S. Alegaonkar, RSC Adv., 2016, 6, 97266–97275 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra09464k |
| This journal is © The Royal Society of Chemistry 2019 |