
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmino-functionalized magnetic chitosan beads to enhance immobilization of potassium copper hexacyanoferrate for selective Cs+ removal and facile recovery†
Hyelin Roha,
Yonghwan Kima,
Yun Kon Kima,
David Harbottleb and
Jae W. Lee *a
*a
aDepartment of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 305-701, Republic of Korea. E-mail: jaewlee@kaist.ac.kr; Tel: +82-42-350-3940
bSchool of Chemical and Process Engineering, University of Leeds, Leeds LS2 9JT, UK
First published on 9th January 2019
Abstract
Potassium copper hexacyanoferrate (KCuHCF)-incorporated magnetic chitosan beads (HMC) were synthesized for both selective Cs+ removal in aqueous solutions and facile recovery of the spent adsorbent. To disperse and immobilize large amounts of the KCuHCF, methyl acrylate and diethylenetriamine were sequentially grafted onto the one-step synthesized magnetic chitosan beads. The additional introduction of amino functionality led to the enriched Cu2+ ions on the bead surface to incorporate KCuHCF into the grafting matrix. Consequently, the HMC exhibited a high Cs+ capacity calculated to be 136.47 mg g−1 from the Langmuir model, and the equilibrium was established within 4 h. Moreover, the HMC exhibited excellent stability in a wide pH range from 4 to 11 and an outstanding Cs+ selectivity (>97%) in seawater (1.11 mg L−1 Cs+). From a practical point of view, the HMC was stable during five successive adsorption cycles and easily recovered by magnets, enabling continuous operation to decontaminate a large volume of wastewater.
1. Introduction
As the demands for more efficient and economical energy increase, nuclear power has become a promising candidate instead of thermal power derived from fossil fuels due to low CO2 emissions.1 Although nuclear power has become an important energy source providing approximately 11% of the worldwide electricity under stringent government regulations, it is considered a threat that needs substantial environmental regulations, especially after accidents such as the Fukushima Daiichi nuclear power plant accident.2 This disaster caused the release and dispersion of a significant amount of radioactive nuclides into different environments such as the soil and water.3,4 Among the radionuclides, 137Cs was the most problematic fission product because of its long half-life around 30 years, high-energy gamma-ray emissions, and high solubility in water.5 Moreover, its chemical similarity with potassium ions causes Cs+ ions to spread into the ecosystem; hence, the radioactive Cs+ ions should be decontaminated safely and effectively from the environment.6A variety of purification methods such as co-precipitation, solvent extraction, adsorption, and chromatography have been investigated to separate Cs+ ions from the contaminated water. However, traditional methods such as thermal treatment or solvent extraction are limited due to the operational cost and the inability of a large-area remediation of water in the nuclear industry.7 Therefore, adsorption is one of the most efficient methods for large-scale applications of wastewater treatment, and especially for Cs+ adsorption, a number of organic and inorganic materials such as carbon,3,8 zeolite,9 clays,10,11 and layered metal thiophosphate12 have been used. Nevertheless, the presence of large amounts of competing cations such as Na+, K+, Ca2+, and Mg2+ still makes it a challenge to separate diluted Cs+ ions from wastewater using these common inorganic adsorbents.
Meanwhile, transition metal hexacyanoferrates (MHCFs), which can simply be synthesized by a precipitation method, have been widely reported for efficient Cs+ removal with their high selectivity even in competitive aqueous environments.7,13 The high affinity of the MHCFs for Cs+ ion originates from their lattice spacing which is similar to the size of hydrated Cs+ ion (3.25 Å).14,15 However, the MHCFs are a very fine powder that makes it difficult to recover the adsorbent particles from water by either centrifugation or filtration; thus, immobilizing the MHCFs in supporting materials is required for practical applications. For this approach, various polymers such as carboxymethyl cellulose,16,17 polyvinyl alcohol,18–21 alginate,22 and chitosan,23,24 have been studied as supporting materials. These polymeric materials have abundant functional groups on their surface which are highly effective for both incorporating the MHCF nanoparticles and further processing for commercial use.
Among these polymer matrices, chitosan-based adsorbents have strong advantages of cost-effectiveness, resource abundance, and many functional groups for desalination by ion adsorption.25–27 Particularly, numerous amino functional groups in chitosan contribute to the coordination of various metal ions.28,29 This unique property enables copper ions to be well dispersed, and thus, potassium copper hexacyanoferrate (KCuHCF) can be immobilized by precipitation on the surface of chitosan-based adsorbents for effective Cs+ removal. However, pure chitosan has a poor mechanical strength and low acidic stability, making it difficult to use in a real adsorption process.30–33 Previous studies have mainly focused on the crosslinking of chitosan to enhance the stability of the material,34 but this approach has a disadvantage of a reduced amino functionality due to the crosslinking reaction, eventually limiting the introduction of active sites and the adsorption performance of the chitosan.
In this work, KCuHCF-incorporated magnetic chitosan beads functionalized with amine were fabricated to improve the Cs+ removal performance by the introduction of additionally available ion binding sites. Instead of crosslinking, the magnetic Fe3O4 nanoparticle-containing chitosan beads were synthesized by a one-step co-precipitation method to improve the chemical stability.35,36 This strategy has an advantage in that the used adsorbent can be efficiently recovered by simple magnetic separation after the water purification process.37,38 Additionally, simple grafting of methyl acrylate and diethylenetriamine was done by the Michael reaction and amidation,39–42 in sequence, to increase the amino functionality in the chitosan matrix. The high content of amino groups in the chitosan beads contributed to the enhancement of the Cu2+ coordination, and this is advantageous for the subsequent immobilization of large amounts of KCuHCF, which is synthesized by simple co-precipitation. As a result, well-dispersed KCuHCF and Fe3O4 nanoparticles were successfully co-immobilized in the final chitosan bead structure to achieve both a high performance and facile recovery, respectively.43 The Cs+ removal efficiency of the adsorbent was evaluated in terms of the adsorption capacity, selectivity, kinetics, pH stability, and reusability. Moreover, various physicochemical characterizations were conducted to determine the effect of both the magnetic property and amino-functionalization on the excellent Cs+ separation performance of the adsorbent.
2. Experimental
Chemicals, fabrication of the magnetic nanoparticle-containing chitosan beads (MC), characterization, and Cs+ adsorption test depending on the adsorbent dose, pH, and reusability test are provided in the ESI.†2.1 Synthesis of the amino-functionalized MC (AMC)
The amino-functionalized MC (AMC) was synthesized by modifying the procedures reported in a previous study (Fig. 1).44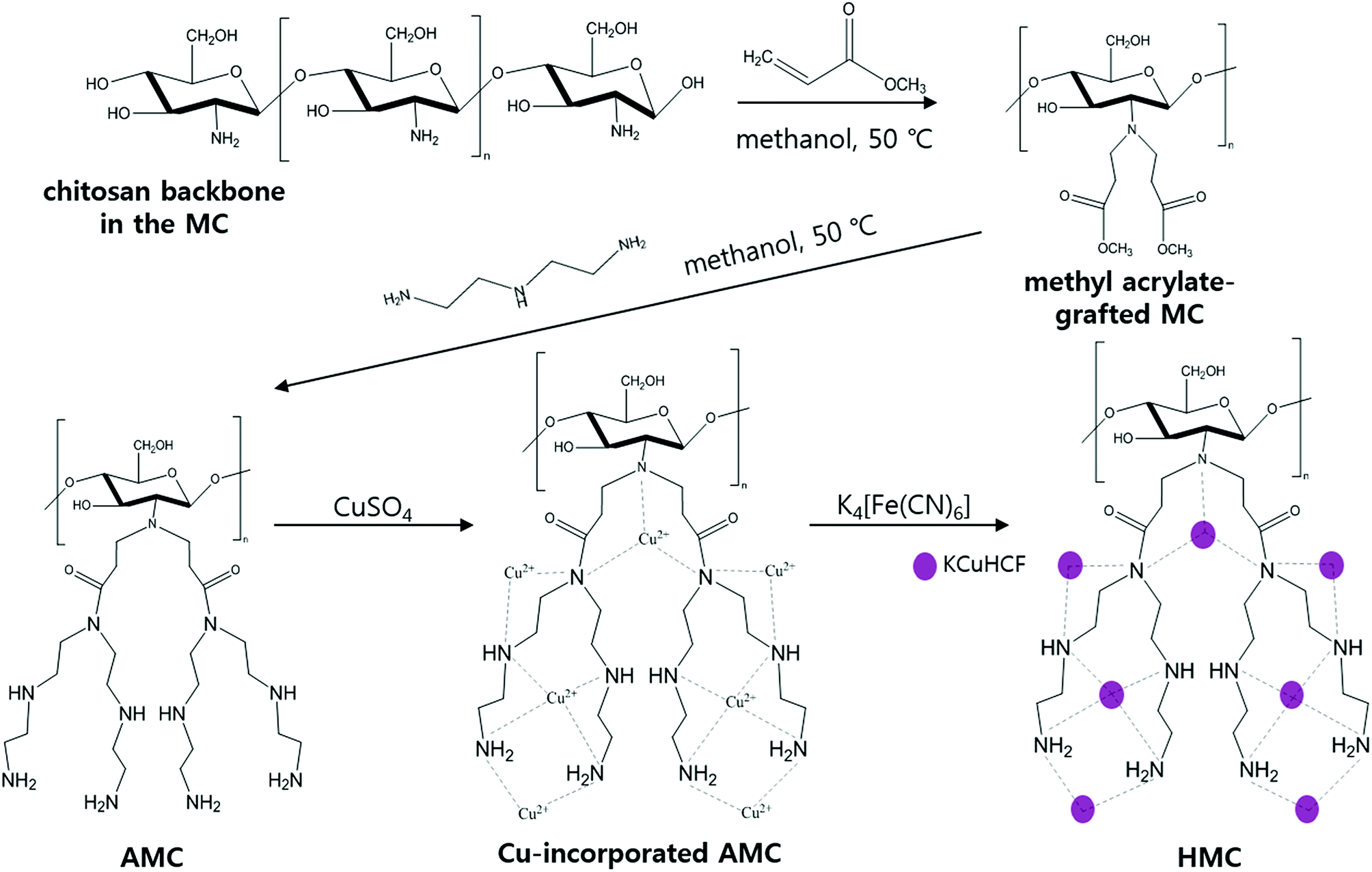 | ||
| Fig. 1 Schematic representation for the HMC synthesis. | ||
To introduce more amino groups on the surface of the adsorbent, methyl acrylate was used to connect between the MC and DETA. Methyl acrylate was grafted to the amino groups on the surface of the MC by the Michael addition reaction.41 Briefly, 1 g of the MC and 120 mL of methanol were mixed into a 2-neck-round flask. Then, the mixture was continuously stirred and heated to 50 °C under reflux with purging nitrogen gas, and 6 mL of methyl acrylate was added to the flask using a syringe. After 24 h, the methyl acrylate-grafted MC beads were washed with methanol several times and dried at 35 °C. Then, DETA was grafted to the terminal ester groups on the surface of the methyl acrylate-grafted MC by the amidation reaction.39 All the methyl acrylate-grafted MC and 150 mL of methanol were added into a 2-neck-round flask, and the mixture was agitated at 50 °C under reflux in a nitrogen atmosphere. After stabilizing this reaction condition for 1 h, 20 mL of DETA was added to the flask, and the reaction was carried out for 36 h. Finally, the prepared AMC was washed with methanol and subsequently freeze-dried.
2.2 Synthesis of the KCuHCF-immobilized AMC (HMC)
The as-synthesized AMC was immersed in 0.16 mol L−1 of copper(II) sulfate solution under stirring for 10 h to immobilize the Cu2+ ions to the amino groups on the AMC surface. The Cu-incorporated AMC was separated by a magnet and washed with DI water several times. Then, the Cu-incorporated AMC was put into 0.16 mol L−1 of a prepared potassium hexacyanoferrate solution under stirring to conduct KCuHCF formation via co-precipitation. After 10 h of contact, the KCuHCF-immobilized AMC (HMC) was collected by a magnet and washed with DI water several times and finally freeze-dried.2.3 Cs+ adsorption experiment
All the cesium (inactive cesium, 133Cs) adsorption studies were performed using a batch-shaking method. Thus, 20 mg of the HMC was put into the Cs+ solution which had a specific concentration. For the Cs+ ion-exchange experiments, the mixture was shaken for 24 h at room temperature. The initial and specific concentrations of the Cs+ ions in the aqueous phase were precisely measured using an inductively coupled plasma mass spectrometer (ICP-MS, Agilent ICP-MS 7700S).To determine the adsorption isotherm, the HMC was shaken in a Cs+ solution by varying the initial Cs+ concentration (C0) from 5 to 500 mg L−1 (m/V = 1 g L−1, m (g) is the mass of the dried adsorbent, and V (L) is the volume of the Cs+ solution.). After 24 h, the residual Cs+ concentration (Ce) in the filtrate was quantified with an ICP-MS.
The adsorption kinetics of cesium from DI water was investigated by immersing 200 mg of the HMC in 8.99 mg L−1 of the Cs+ solution (m/V = 1 g L−1) at room temperature. The shaking sample was stopped at a specific time from 1 min up to 24 h, and 1 mL of the solution was extracted, and the Cs+ concentration was determined.
The Cs+ adsorption amount (Qe, mg g−1), the removal efficiency (RE, %), and the distribution coefficient (Kd, mL g−1) were calculated using the following equations:
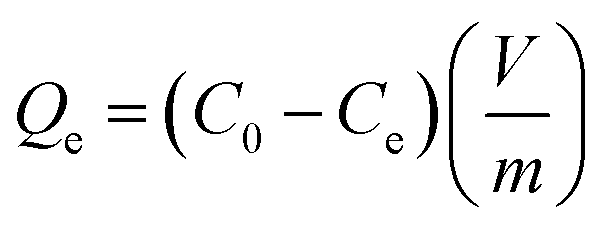 | (1) |
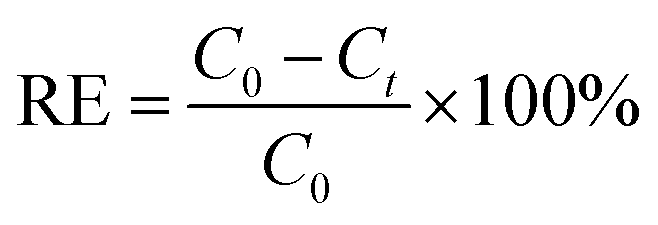 | (2) |
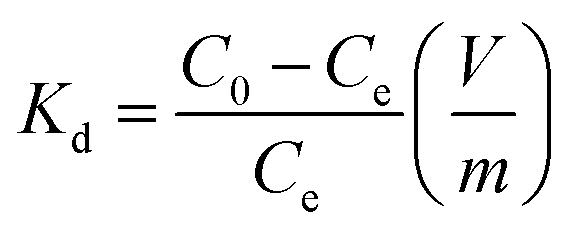 | (3) |
3. Results and discussion
3.1 Characterization: textural properties
The morphologies of the beads are shown in the SEM images (Fig. 2). Fig. 2(a) and (b) show the network structure of the chitosan polymer and the rough surface of the MC. After grafting the methyl acrylate and DETA onto the surface of the beads, the KCuHCF nanoparticles were successfully immobilized onto the surface of the AMC evident by the SEM images of the HMC (Fig. 2(c) and (d)). EDS analysis was conducted to investigate the composition of the MC and HMC (Fig. S1†). The EDS spectra of the MC showed peaks for C, O, N, and Fe, which are the four main components of the chitosan and Fe3O4. On the other hand, the EDS spectra of the HMC showed peaks for C, O, N, Fe, Cu, and K, indicating that the KCuHCF was incorporated into the beads. | ||
| Fig. 2 SEM images of (a) and (b) the MC, and (c) and (d) the HMC. | ||
The thermal stability of the MC and HMC was investigated by thermogravimetric analysis (TGA) under a nitrogen atmosphere (Fig. 3(a)). There were three stages of weight loss in the TGA curve for the MC, and the first stage with about 2.5% weight loss to 200 °C was due to the elimination of adsorbed water. The other two major weight losses occurred at 250–400 °C and 600–750 °C, which indicate the degradation of the backbone chains of the chitosan and further slow decomposition of the residual chitosan or the intermediates from the pre-degraded chitosan.45,46 After the decomposition of the organic phase in the MC over 800 °C, it is certain that the residual components are due to the incorporated Fe3O4. As shown in Fig. 3(a), the TGA profile of the HMC was similar to the curve for the MC, but there was a shift of the second step around 200 °C in the degradation and another weight loss at 300–550 °C. The shift is due to the presence of the KCuHCF,47 and the weight loss corresponds to the decomposition of the cyano groups in the KCuHCF.16 Furthermore, the mass fraction of the organic components in the HMC was approximately estimated to be 60.75%, and the rest is related to the inorganic elements from the Fe3O4 and KCuHCF in the beads.
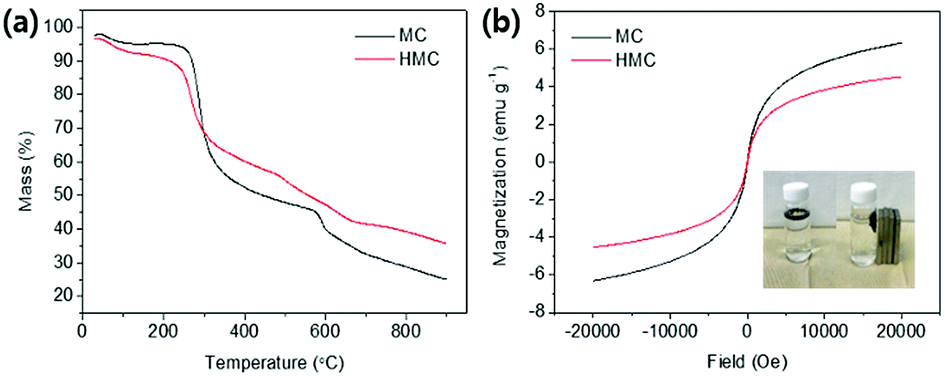 | ||
| Fig. 3 (a) TGA profiles of the MC and HMC. (b) Magnetization curves of the MC and HMC depends on field (Oe), and images of the HMC before and after separation by magnets (inset). | ||
The content of the Fe3O4 and KCuHCF in the MC and HMC was analyzed by measuring the weight percentages of Fe, K and Cu using ICP-OES. Table 1 shows the calculated amounts of the Fe3O4 and KCuHCF in the beads. Furthermore, based on the charge neutrality and stoichiometric ratio of the elements, the estimated chemical composition of the KCuHCF in the HMC structure was K0.57Cu1.71[Fe(CN)6].
| Cu | Fe | K | Fe3O4 | KCuHCF | |
|---|---|---|---|---|---|
| MC | — | 14.222 | — | 19.646 | — |
| HMC | 6.3045 | 11.801 | 1.298 | 11.827 | 19.881 |
The magnetic properties of the MC and HMC were investigated using the magnetization curves acquired from the vibrating-sample magnetometer (VSM) analysis. As shown in Fig. 3(b), the magnetization curves for both the MC and HMC beads represented superparamagnetic characteristics with zero coercivity and zero remanence.46 The magnetization values of the MC and HMC were 6.32 and 4.54 emu g−1 at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Oe, respectively. The lower mass normalized magnetization value of the HMC rather than that of the MC is associated with the incorporation of KCuHCF. As shown in the inset of Fig. 3(b), however, the HMC was attracted to the wall of the vial immediately by placement of the magnets, indicating that the HMC could be easily separated from the aqueous solution after the Cs+ adsorption.
000 Oe, respectively. The lower mass normalized magnetization value of the HMC rather than that of the MC is associated with the incorporation of KCuHCF. As shown in the inset of Fig. 3(b), however, the HMC was attracted to the wall of the vial immediately by placement of the magnets, indicating that the HMC could be easily separated from the aqueous solution after the Cs+ adsorption.
3.2 Characterization: chemical properties
The co-immobilization of crystalline Fe3O4 and KCuHCF was confirmed by X-ray diffraction (XRD) (Fig. 4(a)). The characteristic peaks at 30.5°, 35.5°, 43.4°, 57.2°, and 62.7° indicate the presence of Fe3O4 nanoparticles in the MC.48 These peaks show that the co-precipitation of chitosan and Fe3O4 for the fabrication of the magnetic beads did not change the phase of the Fe3O4 nanoparticles. Compared to the MC, the HMC has eight distinguishable peaks at 2θ = 17.7°, 25°, 36°, 40°, 44°, 52°, 55°, and 58°, and these peaks are well matched with the KCuHCF pattern (PDF no. 01-075-0023). Although some major peaks of the Fe3O4 nanoparticles are overlapped with those of the KCuHCF, the characteristic peak at 30° supports the presence of Fe3O4, along with the magnetic properties revealed by the VSM result. Thus, the XRD patterns show the successful incorporation of the Fe3O4 nanoparticles and KCuHCF into the chitosan bead structure.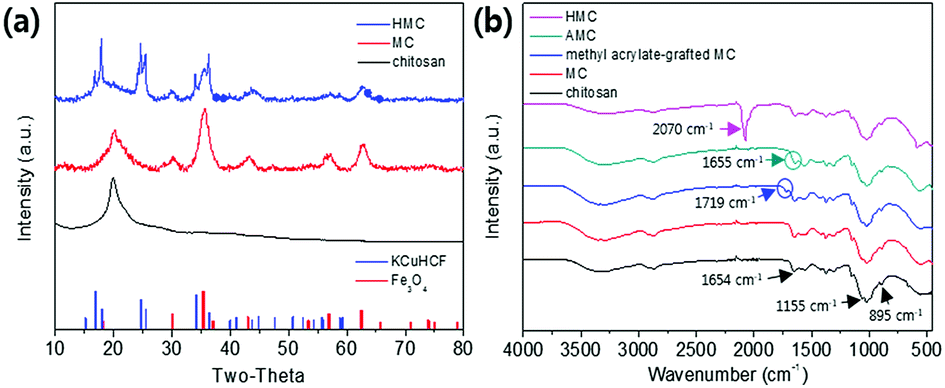 | ||
| Fig. 4 (a) XRD patterns of the MC and HMC and (b) FT-IR spectra of beads in each synthesis step. | ||
Moreover, the average crystal size of the Fe3O4 and KCuHCF in the beads was determined by the Scherrer equation:
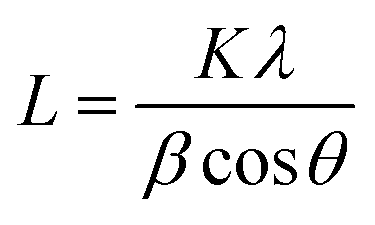 | (4) |
The FT-IR spectra of the chitosan powder, MC, methyl acrylate-grafted MC, AMC, and HMC were obtained to verify the successful reactions in each step (Fig. 4(b)). For the chitosan powder, the main peaks at 895 cm−1 and 1155 cm−1 are attributed to the saccharide, and the peak at 1654 cm−1 is assigned to the –NH absorption peak of the amine group (–NH2) in the chitosan polymer backbone. The spectrum obtained from the MC revealed a characteristic band at 560 cm−1, corresponding to the Fe–O stretching vibrational mode of Fe3O4.49 The MC showed a new absorption peak at 1719 cm−1 which was related to the formation of the ester group (–O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O–) resulting from the grafting of methyl acrylate by the Michael reaction. In addition, after grafting DETA to the beads, the newly generated peak at 1655 cm−1 is related to the formation of the amide bond (–CONH–), and simultaneously, the disappearance of peak at 1719 cm−1 showed the successful reaction between the ester group of the methyl acrylate-grafted MC and the amino group of the DETA.44 The presence of a strong peak at 2070 cm−1 for the HMC is due to C
O–) resulting from the grafting of methyl acrylate by the Michael reaction. In addition, after grafting DETA to the beads, the newly generated peak at 1655 cm−1 is related to the formation of the amide bond (–CONH–), and simultaneously, the disappearance of peak at 1719 cm−1 showed the successful reaction between the ester group of the methyl acrylate-grafted MC and the amino group of the DETA.44 The presence of a strong peak at 2070 cm−1 for the HMC is due to C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching vibration from the immobilized KCuHCF. Importantly, the HMC also included the peaks for the chitosan and Fe3O4, revealing the successful introduction of the amino functionality and KCuHCF in the magnetic chitosan structure.
N stretching vibration from the immobilized KCuHCF. Importantly, the HMC also included the peaks for the chitosan and Fe3O4, revealing the successful introduction of the amino functionality and KCuHCF in the magnetic chitosan structure.
XPS analyses were conducted to identify the active sites for the formation of KCuHCF because the HMC has immense functional groups including N and O atoms such as amine, hydroxyl, and amide, which Cu2+ ions can be coordinated to. The XPS survey results show that the HMC contained C, O, N, Cu, Fe, and K (refer to Fig. S2(a)†). The N 1s peaks at 399.34 and 400.11 eV (Fig. 5(a)) are assigned to free amine groups (NH2) and NH2–O in the MC, respectively.50 Compared to the peaks of Fig. 5(a), the N 1s spectrum of the HMC in Fig. 5(b) has peaks at 397.7, 399.62 and 401.89 eV which correspond to the CN groups in the KCuHCF, –N–, and protonated amine groups because of the chelation with Cu2+ ions after incorporation of the KCuHCF, respectively.51,52 However, the O 1s spectra (Fig. S2(b) and (c)†) for both the MC and HMC did not have any changes after the KCuHCF immobilization. In other words, Cu2+ ions, which are the precursors of the KCuHCF, chelated with the N atoms of the bead surface; thus, the introduction of amino groups using methyl acrylate and DETA onto the beads has a crucial role in the immobilization of KCuHCF.
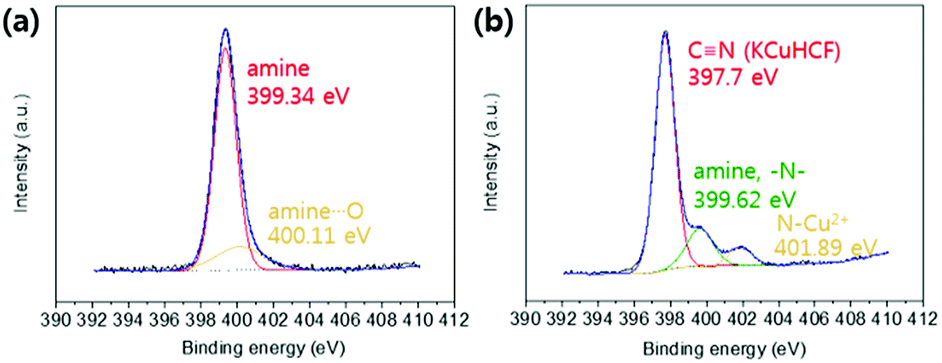 | ||
| Fig. 5 XPS N 1s spectra of the (a) MC and (b) HMC. | ||
3.3 Cs+ adsorption studies
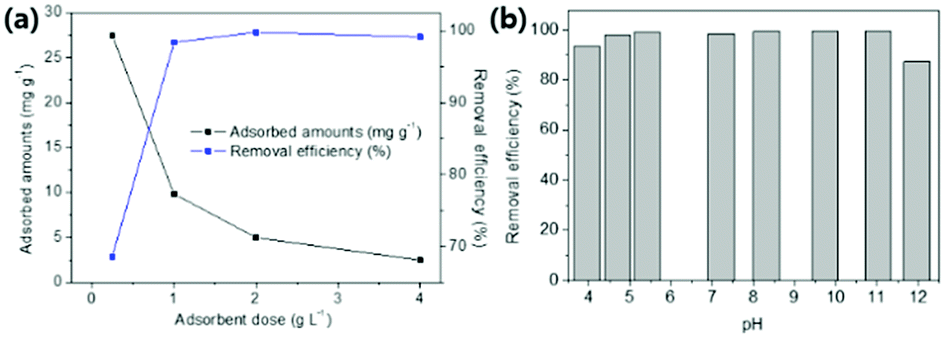 | ||
| Fig. 6 (a) Effect of adsorbent dose on removal efficiency (%) and adsorbed amounts (mg g−1). (b) Effect of pH of solution on Cs+ adsorption in DI water. | ||
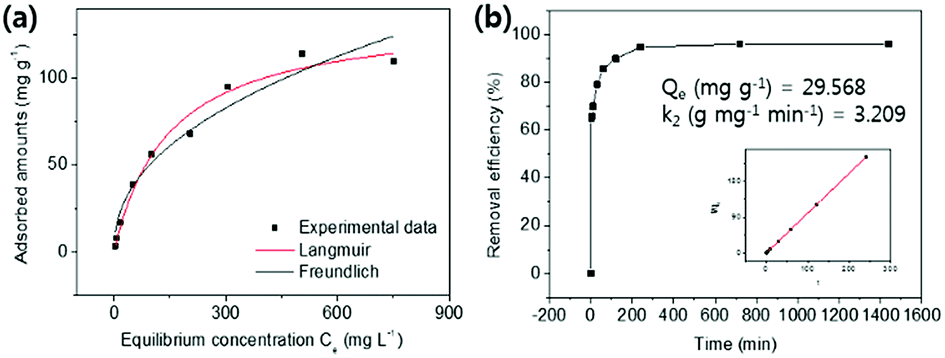 | ||
| Fig. 7 (a) The Cs+ adsorption isotherm curve and (b) adsorption kinetics and plot of t/Qt vs. t for the HMC. | ||
Langmuir isotherm
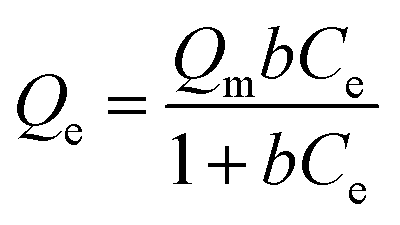 | (5) |
Freundlich isotherm
| Qe = KFCe1/n | (6) |
As presented in Table 2, the isotherm parameters were calculated by the non-linear curve fitting of the above equations. The adsorption process of the HMC agreed better with the Langmuir model with a higher correlation coefficient of 0.98, and the Freundlich heterogeneity parameter is around 2.27 (n > 1). These results suggest the uniform distribution of the KCuHCF nanoparticles in the HMC and the chemical binding of Cs+ by the active sites. Based on the Langmuir isotherm model, the HMC provided a superior maximum adsorption capacity of 136.47 mg g−1 compared to those of the other Cs+ adsorbents containing magnetic nanoparticles (Table 3). It can be attributed to the increase of amino functionality, which resulted in the incorporation of a comparatively great quantity of KCuHCF particles, leading to the superior Cs+ adsorption performance.
| Langmuir isotherm parameters | Freundlich isotherm parameters | |||||
|---|---|---|---|---|---|---|
| Qm (mg g−1) | b (L mg−1) | R2 | n | KF (L g−1) | R2 | |
| HMC | 136.47 | 0.007 | 0.98 | 2.27 | 6.69 | 0.95 |
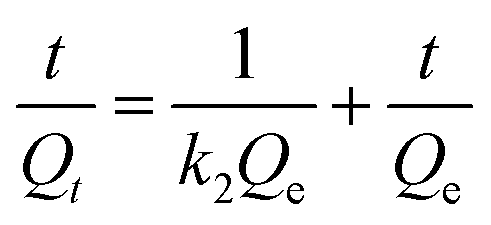 | (7) |
| C0 (mg L−1) | Removal efficiency (%) | Kd (mL g−1) | |
|---|---|---|---|
| DI water | 8.01 | 99.1 | 1.13 × 105 |
| 0.56 | 99.5 | 1.85 × 105 | |
| Seawater | 10.56 | 94.6 | 1.75 × 104 |
| 1.11 | 97.0 | 3.27 × 104 |
The Kd values of Cs+ on the HMC in seawater were as good as those in the DI water, and the other excess cations (e.g., Na+, K+, Ca2+, and Mg2+) did not highly affect the binding of Cs+. In terms of the removal efficiency, more than 94% of the Cs+ was removed even in the seawater condition, indicating that the HMC is a highly efficient candidate to remediate nuclear-contaminated water in which accidentally released Cs+ ions are diluted. These promising results show that the well-dispersed KCuHCF nanoparticles on the amino groups in the HMC could contribute to the remarkable Cs+ selectivity even in highly competitive conditions.
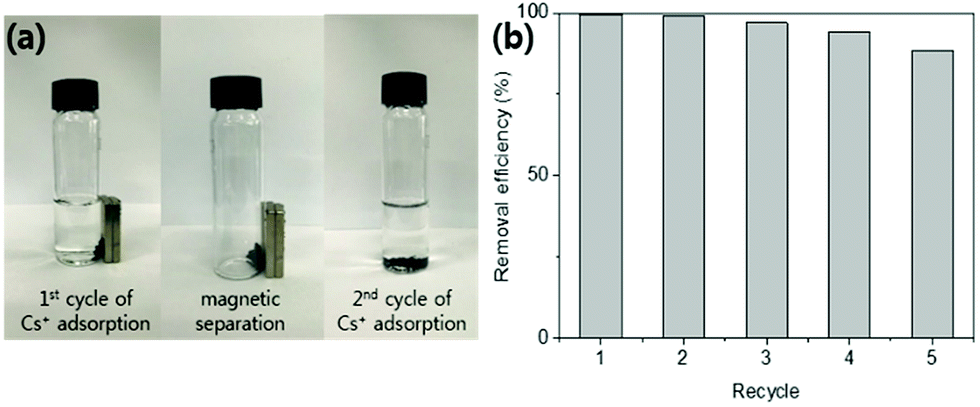 | ||
| Fig. 8 (a) Images of cyclic Cs+ adsorption and magnetic separation of the HMC. (b) Removal efficiency (%) of the HMC in seawater (1–5 cycles). | ||
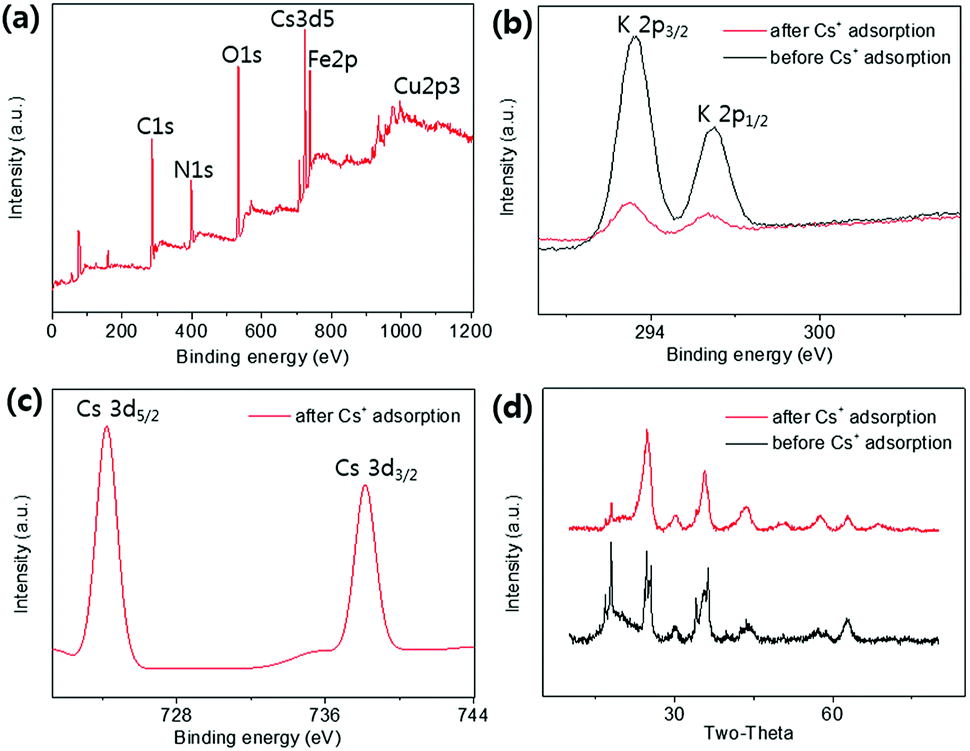 | ||
| Fig. 9 (a) Full range XPS spectrum of the HMC after Cs+ adsorption, and (b) the K 2p and (c) Cs 3d XPS spectra. (d) XRD patterns before and after Cs+ adsorption of the HMC. | ||
4. Conclusions
KCuHCF nanoparticles were incorporated into magnetic chitosan beads by coordination between amino groups and copper ions to remove Cs+ from contaminated aqueous solutions. The grafting of amino groups to magnetic chitosan beads and the subsequent immobilization of a large quantity of KCuHCF nanoparticles on the HMC were confirmed in the FT-IR and SEM-EDS spectra, respectively. The Cs+ adsorption performance of the HMC was evaluated regarding the pH stability, capacity, kinetics, selectivity, and reusability. In a broad range of pHs from 4 to 10, the HMC showed a stable removal efficiency above 94%, and the Cs+ ions were quickly removed from the aqueous solution (8.99 mg L−1, m/V = 1 g L−1) within 4 h. The maximum adsorption capacity was 136.47 mg g−1, which is one of the highest Cs+ capacities among the previous reported magnetic Cs+ adsorbents. In addition, the HMC could remove Cs+ selectively (>97%) from seawater (1.11 mg L−1), and this result is related to the well-dispersed KCuHCF nanoparticles in the HMC. Furthermore, the repetitive use of the HMC was possible with highly selective Cs+ removal in seawater, and the Cs+ adsorbed HMC could be quickly recovered by magnets. This is important in terms of industrial applications to purify a large volume of contaminated water without secondary contamination. The comparison results of XPS and XRD between the pristine and Cs+ exchanged HMC showed that this effective Cs+ removal performance was due to the ion exchange mechanism between the K+ in the immobilized KCuHCF and the Cs+ from the aqueous phase. Therefore, the HMC is one of the most favorable Cs+ adsorbents with respect to not only economic cost and eco-friendly preparation but also its rapid and selective recovery of Cs+ from an aqueous solution.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the UK-Korea Joint Research Program through NRF grants (NRF-2015M2A7A1000219) and the Advanced Biomass R&D Center as the Global Frontier Project (ABC-2010-0029728) funded by the Ministry of Science, ICT and Future Planning, South Korea. D. Harbottle acknowledges the support of Engineering and Physical Sciences Research Council grant number EP/M026426/1.References
- Y.-X. Wang, J.-R. Li, J.-C. E. Yang, B. Yuan and M.-L. Fu, RSC Adv., 2015, 5, 91431–91435 RSC.
- International Energy Agency, Key World Energy Statistics, http://www.iea.org/publications/freepublications/publication/KeyWorld2017.pdf Search PubMed.
- S. Baik, H. Zhang, Y. K. Kim, D. Harbottle and J. W. Lee, RSC Adv., 2017, 7, 54546–54553 RSC.
- H. Zhang, S. Tangparitkul, B. Hendry, J. Harper, Y. Kon Kim, T. N. Hunter, J. W. Lee and D. Harbottle, Chem. Eng. J., 2019, 355, 797–804 CrossRef CAS.
- P. J. Faustino, Y. Yang, J. J. Progar, C. R. Brownell, N. Sadrieh, J. C. May, E. Leutzinger, D. A. Place, E. P. Duffy, F. Houn, S. A. Loewke, V. J. Mecozzi, C. D. Ellison, M. A. Khan, A. S. Hussain and R. C. Lyon, J. Pharm. Biomed. Anal., 2008, 47, 114–125 CrossRef CAS PubMed.
- E. Calabrese, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 19447–19448 CrossRef CAS PubMed.
- A. Nilchi, R. Saberi, M. Moradi, H. Azizpour and R. Zarghami, Chem. Eng. J., 2011, 172, 572–580 CrossRef CAS.
- Y. K. Kim, J. H. Park and J. W. Lee, Carbon, 2018, 126, 215–224 CrossRef CAS.
- H. Yang, M. Luo, L. Luo, H. Wang, D. Hu, J. Lin, X. Wang, Y. Wang, S. Wang, X. Bu, P. Feng and T. Wu, Chem. Mater., 2016, 28, 8774–8780 CrossRef CAS.
- D. Alby, C. Charnay, M. Heran, B. Prelot and J. Zajac, J. Hazard. Mater., 2018, 344, 511–530 CrossRef CAS PubMed.
- Y. Kim, Y. K. Kim, J. H. Kim, M.-S. Yim, D. Harbottle and J. W. Lee, Appl. Surf. Sci., 2018, 450, 404–412 CrossRef CAS.
- E. Rathore, P. Pal and K. Biswas, Chemistry, 2017, 23, 11085–11092 CrossRef CAS PubMed.
- Lalhmunsiama, C. Lalhriatpuia, D. Tiwari and S.-M. Lee, Appl. Surf. Sci., 2014, 321, 275–282 CrossRef CAS.
- T. Vincent, C. Vincent, Y. Barré, Y. Guari, G. Le Saout and E. Guibal, J. Mater. Chem. A, 2014, 2, 10007–10021 RSC.
- T. Vincent, C. Vincent and E. Guibal, Molecules, 2015, 20, 20582–20613 CrossRef CAS PubMed.
- Y. Kim, Y. K. Kim, S. Kim, D. Harbottle and J. W. Lee, Chem. Eng. J., 2017, 313, 1042–1050 CrossRef CAS.
- Y. Zong, Y. Zhang, X. Lin, D. Ye, D. Qiao and S. Zeng, RSC Adv., 2017, 7, 31352–31364 RSC.
- Y. K. Kim, T. Kim, Y. Kim, D. Harbottle and J. W. Lee, J. Hazard. Mater., 2017, 340, 130–139 CrossRef CAS PubMed.
- Y. K. Kim, Y. Kim, S. Kim, D. Harbottle and J. W. Lee, J. Environ. Chem. Eng., 2017, 5, 975–986 CrossRef CAS.
- Y. K. Kim, K. Bae, Y. Kim, D. Harbottle and J. W. Lee, J. Ind. Eng. Chem., 2018, 68, 48–56 CrossRef.
- J. Y. Yoon, H. Zhang, Y. K. Kim, D. Harbottle and J. W. Lee, J. Environ. Chem. Eng., 2019, 7, 102824 CrossRef.
- K.-M. Lee, T. Kawamoto, K. Minami, A. Takahashi, D. Parajuli, G. Kido, K. Yoshino and H. Tanaka, RSC Adv., 2016, 6, 16234–16238 RSC.
- B. Folch, J. Larionova, Y. Guari, K. Molvinger, C. Luna, C. Sangregorio, C. Innocenti, A. Caneschi and C. Guerin, Phys. Chem. Chem. Phys., 2010, 12, 12760–12770 RSC.
- D. Dechojarassri, S. Asaina, S. Omote, K. Nishida, T. Furuike and H. Tamura, Int. J. Biol. Macromol., 2017, 104, 1509–1516 CrossRef CAS PubMed.
- R. Wang, S. Gao, Z. Yang, Y. Li, W. Chen, B. Wu and W. Wu, Adv. Mater., 2018, 30 Search PubMed.
- E. Szymanska and K. Winnicka, Mar. Drugs, 2015, 13, 1819–1846 CrossRef CAS PubMed.
- S. Yang, D. Shao, X. Wang, G. Hou, M. Nagatsu, X. Tan, X. Ren and J. Yu, Mar. Drugs, 2015, 13, 3116–3131 CrossRef CAS PubMed.
- W. Jiang, W. Wang, B. Pan, Q. Zhang, W. Zhang and L. Lv, ACS Appl. Mater. Interfaces, 2014, 6, 3421–3426 CrossRef CAS PubMed.
- S. Yang, N. Okada and M. Nagatsu, J. Hazard. Mater., 2016, 301, 8–16 CrossRef CAS PubMed.
- P. S. Barber, S. P. Kelley, C. S. Griggs, S. Wallace and R. D. Rogers, Green Chem., 2014, 16, 1828–1836 RSC.
- S. Chatterjee, M. W. Lee and S. H. Woo, Bioresour. Technol., 2010, 101, 1800–1806 CrossRef CAS PubMed.
- J. Duan, X. Liang, Y. Cao, S. Wang and L. Zhang, Macromolecules, 2015, 48, 2706–2714 CrossRef CAS.
- L. Lv, N. Chen, C. Feng, Y. Gao and M. Li, J. Taiwan Inst. Chem. Eng., 2017, 78, 485–492 CrossRef CAS.
- M. K. Sureshkumar, D. Das, M. B. Mallia and P. C. Gupta, J. Hazard. Mater., 2010, 184, 65–72 CrossRef CAS PubMed.
- W. Zhang, S. Jia, Q. Wu, S. Wu, J. Ran, Y. Liu and J. Hou, Mater. Sci. Eng., C, 2012, 32, 381–384 CrossRef CAS.
- A. Mohseni-Bandpi, B. Kakavandi, R. R. Kalantary, A. Azari and A. Keramati, RSC Adv., 2015, 5, 73279–73289 RSC.
- S. Naeimi and H. Faghihian, Sep. Purif. Technol., 2017, 175, 255–265 CrossRef CAS.
- S. Zavareh, Z. Behrouzi and A. Avanes, Int. J. Biol. Macromol., 2017, 101, 40–50 CrossRef CAS PubMed.
- M. Rajiv Gandhi and S. Meenakshi, Carbohydr. Polym., 2013, 91, 631–637 CrossRef CAS PubMed.
- H. Li, S. Bi, L. Liu, W. Dong and X. Wang, Desalination, 2011, 278, 397–404 CrossRef CAS.
- B. D. Mather, K. Viswanathan, K. M. Miller and T. E. Long, Prog. Polym. Sci., 2006, 31, 487–531 CrossRef CAS.
- E. S. Read, K. L. Thompson and S. P. Armes, Polym. Chem., 2010, 1, 221–230 RSC.
- C. Zhang, H. Zhang, R. Li and Y. Xing, RSC Adv., 2017, 7, 48189–48198 RSC.
- H. Zhang, Q. Dang, C. Liu, D. Cha, Z. Yu, W. Zhu and B. Fan, ACS Appl. Mater. Interfaces, 2017, 9, 11144–11155 CrossRef CAS PubMed.
- Z. Zhou, F. Jiang, T.-C. Lee and T. Yue, J. Alloys Compd., 2013, 581, 843–848 CrossRef CAS.
- C. Cao, L. Xiao, C. Chen, X. Shi, Q. Cao and L. Gao, Powder Technol., 2014, 260, 90–97 CrossRef CAS.
- C. Dwivedi, S. K. Pathak, M. Kumar, S. C. Tripathi and P. N. Bajaj, Environ. Sci.: Water Res. Technol., 2015, 1, 153–160 RSC.
- A. Anuradha, S. Kumari, S. Layek and D. D. Pathak, New J. Chem., 2017, 41, 5595–5604 RSC.
- H.-M. Yang, C. W. Park, P. K. Bae, T. Ahn, B.-K. Seo, B. H. Chung and J.-D. Kim, J. Mater. Chem. B, 2013, 1, 3035–3043 RSC.
- C. Dong, W. Chen, C. Liu, Y. Liu and H. Liu, Colloids Surf., A, 2014, 446, 179–189 CrossRef CAS.
- M. E. A. Ali, F. M. Hassan and X. Feng, J. Mater. Chem. A, 2016, 4, 6620–6629 RSC.
- H. Yang, H. Li, J. Zhai, L. Sun, Y. Zhao and H. Yu, Chem. Eng. J., 2014, 246, 10–19 CrossRef CAS.
- H. Yang, L. Sun, J. Zhai, H. Li, Y. Zhao and H. Yu, J. Mater. Chem. A, 2014, 2, 326–332 RSC.
- K. S. Hwang, C. W. Park, K.-W. Lee, S.-J. Park and H.-M. Yang, Colloids Surf., A, 2017, 516, 375–382 CrossRef CAS.
- H. Zhang, X. Zhao, J. Wei and F. Li, Nucl. Eng. Des., 2014, 275, 322–328 CrossRef CAS.
- Y.-W. Chen and J.-L. Wang, Nucl. Sci. Tech., 2016, 27, 43 CrossRef.
- C. Thammawong, P. Opaprakasit, P. Tangboriboonrat and P. Sreearunothai, J. Nanopart. Res., 2013, 15, 1689 CrossRef.
- Y. Zhu, W. Wang, H. Zhang, X. Ye, Z. Wu and A. Wang, Chem. Eng. J., 2017, 327, 982–991 CrossRef CAS.
- J. L. Mertz, Z. H. Fard, C. D. Malliakas, M. J. Manos and M. G. Kanatzidis, Chem. Mater., 2013, 25, 2116–2127 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed description for experimental and characterization (EDS and XPS). See DOI: 10.1039/c8ra09386e |
| This journal is © The Royal Society of Chemistry 2019 |