
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImmobilization adjusted clock reaction in the urea–urease–H+ reaction system†
Dan Yanga,
Junhe Fana,
Fengyi Caoa,
Zuojun Denga,
John A. Pojmanb and
Lin Ji *a
*a
aDepartment of Chemistry, Capital Normal University, Beijing 100048, China. E-mail: jilin@mail.cnu.edu.cn
bDepartment of Chemistry, Louisiana State University, Baton Rouge, 70803, USA
First published on 25th January 2019
Abstract
The bell-shaped reactivity-pH curve is the fundamental reason that the temporal programmable kinetic switch in clock reactions can be obtained in bio-competitive enzymatic reactions. In this work, urease was loaded on small resin particles through ionic binding. Experimental results reveal that the immobilization not only increased the stability of the enzyme and the reproducibility of the clock reaction, but also shifted the bell-shaped activity curve to lower pHs. The latter change enables the clock reaction to occur from an initial pH of 2.3, where the free enzyme had already lost its activity. Two mechanisms explain the influence of the immobilization on the clock reaction. Immobilization modified the pH sensitive functional groups on the enzyme, shifting the activity curve to a more acidic region, and reduced diffusion alters the enzyme dynamics.
Introduction
An enzyme's bell-shaped activity dependence on pH can create autocatalytic behavior in an unbuffered solution if the product of the reaction affects the pH. One of the simplest systems to realize this is the urease-catalyzed hydrolysis of urea, which displays autocatalysis driven by the increase in pH accompanying the production of ammonia under non-buffered conditions.1–4 The transient pH jump in the clock reaction can be coupled to self-assembling systems operating in the targeted pH regime, where upon they form and decay on a pre-programmed course in closed systems as encoded by the pH profile. Heuser et al.5 coupled this transient state with dipeptides and created an autonomously self-regulating dynamic gel with programmed lifetime. A breathing gel was realized by incorporating the pH switch to tune the size and fluorescence intensity of a microgel.6 Jee et al.7 utilized the pH switch to trigger base-catalyzed polymerization fronts under mild conditions. This system creates promising prospect to install biocompatible feedback loops, kinetic switch, threshold sensing8 or programmed temporal behavior,9,10 thereby equipping classical soft matter materials with capacities for active, adaptive, and autonomous character. This urea–urease reaction has a special biological relevance because it is used by bacteria to elevate the pH and protect them from the acidic environment of the stomach.11One problem for the application of the base-catalyzed urea–urease system is the low reproducibility, which is the natural consequence of the fact that pure enzymes are unstable in water. In many situations, a practical solution is to stabilize the enzyme via immobilization. Immobilization12 is a straightforward route to re-use or continuously use the enzyme, and it can also help to enable the employment of enzymes in different solvents, at extreme temperatures, in exceptionally high substrate concentration or extremes of pH.13,14 Besides, immobilized enzymes are often loaded on small particles, which may offer special advantages in medical applications such as drug delivery devices or biosensor applications like quorum sensing.15,16 Ureases are enzymes highly desirable in immobilized form since they are used in applications with increased pH inherent in the reaction.17
In this work, commercial amino resin particles were used as a carrier to immobilize the urease used in the base-catalyzed urea hydrolysis reaction. Clock reaction behavior was found in more acidic conditions where the free enzyme system showed no evidence of a clock reaction. The stability of the enzyme and the reproducibility of the clock reaction were considerably improved. Further investigations show that the immobilization is realized through the bonding between the pH responsive group on the enzyme and the resins. Therefore, it shifted the bell-shaped activity-pH curve to lower pH. This is an important reason for the change in the clock reaction. Also, the diffusion limitation effect on small molecules induced by immobilization alters the product and substrate inhibition dynamics and further contributes to the shape change of the clock reaction evolution.
Experimental section
Urease (from Jack Bean, 2000 U mg−1, J&K Scientific), Nessler's reagent (Sinopharm Chemical Reagent Co., Ltd), weak basic amino resin (LX-1000HA, Xi'an Sunresin, China) with diameter ranging between 0.2 and 0.7 mm were used.To remove metal ions, the resins were first soaked in NaOH (2%) and HCl (5%) for 24 hours, respectively, and washed with deionized water until a neutral pH was reached. Then the urease was immobilized on the resin through ionic binding. 10 g of pre-treated resin was activated by a buffer solution (pH = 8.0) and then added into 20 mL 100 U mL−1 urease solution (dissolved in pH 8.0 phosphate buffer). The mixture was kept in the shaker for 12 h (25 ± 0.1 °C). The specific process is shown in Fig. 1a. Since the resin must be activated in a pH 8 buffer to create the negative charge, the urease should be kept in a buffered solution of the same pH before immobilization, even though this is not its optimal pH. Several other buffer pH values have been tried as shown in Fig. 1b. However, it turned out that they are less effective for the immobilization. The pH of the solution in which the enzyme is kept as 8 in all the later on experiments.
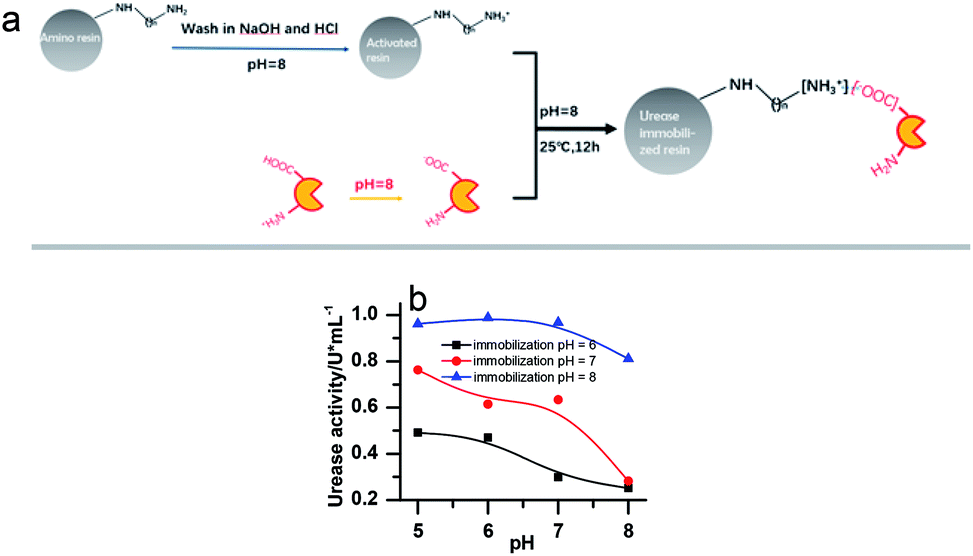 | ||
| Fig. 1 (a) Scheme of urease immobilization processes. (b) The activity of the immobilized enzyme in buffer solution of different pHs. [(NH4)2SO4] = 0.1 M; [H2SO4] = 1 M; [urea] = 0.5 M; [urease]free = [urease]IM = 4 U mL−1. | ||
The concentrations of immobilized enzyme listed in the figure captions were calculated on the assumption that all the enzyme molecules were immobilized.
To test the activity of the urease, a 1 mL urease solution was first heated in 25 °C water bath for 5 minutes. Then, the enzymatic reaction was started by adding a 1 mL solution of urea in buffer solution. Then the reactant mixture was stirred for 15 minutes at 25 °C before the reaction was terminated by 1 mL of 1 M H2SO4. The amount of NH3 liberated was determined with Nessler's reagent and a UV spectrophotometer (TU-1810) at 425 nm. The urease activity was calculated by calibrating the Nessler's reagent results with a standard (NH4)2SO4 solution. One unit activity of urease is defined as the amount of urease that liberates 1 μmol NH3 per minute at the desired pH (25 °C). The activity of the immobilized urease was also determined as described above except that the urease solution was prepared with immobilized urease.
Stock solutions of urea, urease and sulfuric acid were prepared with distilled water. For the urea–urease clock reactions, different amounts of urease solution were added into the mixture of urea and sulfuric acid to give 20 mL total volume. The reactions were performed in a well-stirred batch reactor, and the evolutions of the pH were recorded through a data acquisition system (CONSORT D230). All experiments were performed at 25 ± 0.1 °C. For the testing of immobilized urease, different masses of urease-loaded resins were used to prepare the enzyme solutions according to the putative amount of enzyme loaded. The clock time (Tclock) of the reaction is defined as the time taken to reach pH 5.
Results
The stability of the immobilized urease was compared with the stability of free enzyme by testing their activities at their optimum pH daily. The free urease samples were stored in aqueous solution, and the immobilized ones were stored wet. As shown in Fig. 2a, the activity of the free urease either stored in the refrigerator (4 °C) or at room temperature (25 °C) kept decreasing. However, the activity of the immobilized urease was almost unchanged for 9 days. The increased stability of the immobilized ureases significantly enhances the quantitative reproducibility of the clock behavior with the same enzyme solution. Bubanja et al.12 showed that the clock reaction will significantly change the Tclock and pHmax when the stock solution were stored for hours, and the system could fail to reach basic values if the solution was tens of hours old. In our work, the clock reaction curve tested four days after the solution preparation is almost the same as the one achieved by the freshly prepared enzyme solution (Fig. 2b).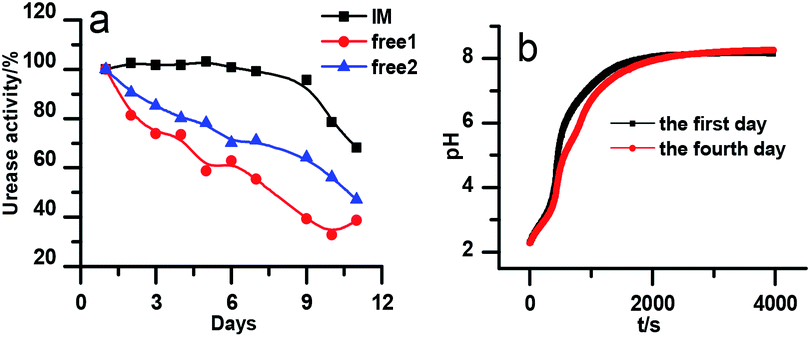 | ||
| Fig. 2 (a) Stability test of immobilized and free urease. The black line indicates the stability test of immobilized urease at 25 °C. The blue and red lines indicate the stability test of free urease at 25 °C and 4 °C, respectively. [Urease] = 15 U mL−1; [urea] = 0.5 M; [(NH4)2SO4] = 0.1 M; H2SO4 = 1 M; (b) reproducibility experiments for immobilized urease system. [Urease] = 4 U mL−1; [urea] = 0.01 M. | ||
However, with immobilized urease, the clock reaction behavior was observed only when the initial pH was very low. Under current enzyme and substrate concentration settings, the pH will not increase if the initial pH of the system is lower than 2.8 in the free enzyme system (Fig. 3b), while in the immobilized enzyme system, clock behavior was found when the pHini was as low as 2.3 (Fig. 3a). As a matter of fact, if the initial pH is >2.6 in the immobilized system, the pH evolution curve follows the normal Michaelis–Menten behavior and fails to give the clock behavior. As shown in Fig. 3c, when the pHini is within 2.3–3, the free urease was almost inactive, while most of the immobilized urease remains active. Changing the urease or substrate concentration still allows control of Tclock. Generally, in the immobilized urease system, the pHmax is lower, and the accessible range of Tclock becomes wider. It is shown in Fig. 3f that under the same conditions, the Tclock in free urease system (solid black line) can change from 0 to 4000 s, while that in the immobilized urease system (solid red line) it ranges from 0 to 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s.
000 s.
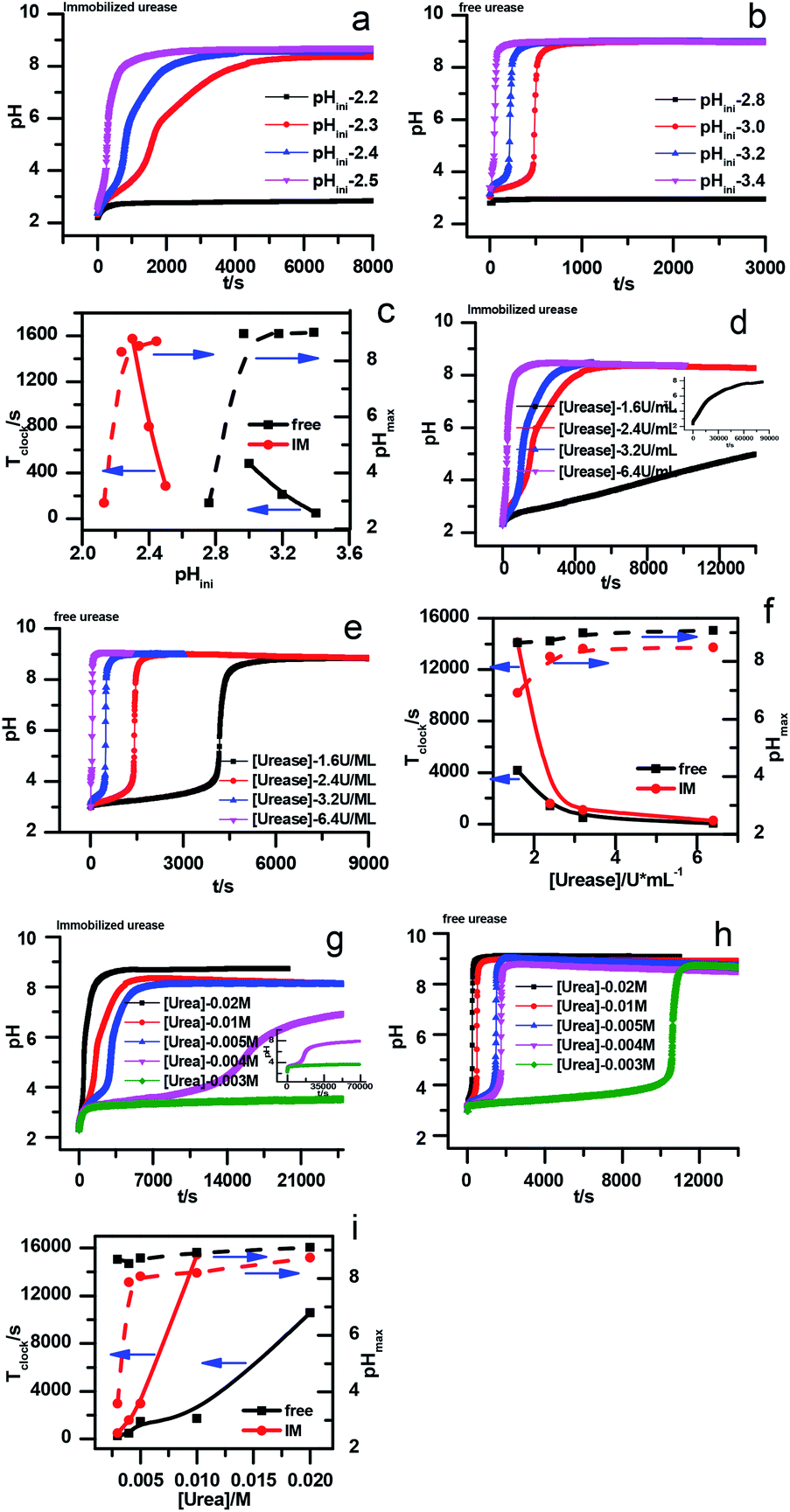 | ||
| Fig. 3 Experimental pH–time profiles of the urea–urease–H+ system. (a–c) [Urea] = 0.01 M; [urease] = 3.2 U mL−1; (d–f) [urease] = 3.2 U mL−1; (g–i) [urea] = 0.01 M. | ||
The jump in the pH evolution curve of the immobilized urease system is not as steep as in the free enzyme system; similar clock curves can also been found in Heuser's work,5 where the urease was embedded in a hydrogel. The shape change in the pH evolution curve may make one doubt whether it is still a clock reaction. In fact, it has been pointed out18 that clock reactions do not necessarily have a sudden jump. There are two types of clock reactions, the substrate-depletion clock reactions and the autocatalysis-driven ones. The former usually have a sudden jump, while the latter do not because the autocatalytic species may gradually accumulate while the accumulation rate increases.
There are two necessary conditions for a clock reaction. One is that the formation rate of at least one substance accelerates during the reaction; the other is the clock time can be reproduced. In our system, the clock time can be quantitatively reproduced (see Fig. 2b). The acceleration mechanism is based on the bell-shaped activity-pH curve, which is universal for most if not all enzymes. Enzymes usually show the highest activity at an intermediate pH, below or above which the enzyme activity will decrease. In the urea–urease–H+ system, the enzymatic reaction starts in an acidic environment when the urease activity is low, and the reaction is slow. With the hydrolysis of urea, the formation of the basic product gradually increases the pH, and at the same time increases the urease activity, which provides the accelerating mechanism. In other words, the occurrence of the reaction (generating basic substances) leads to the increase of the reaction rate (enzyme activity). The activity test of immobilized urease still showed a bell-shaped pH dependent curve (Fig. 4). Therefore, the acceleration mechanism is still present.
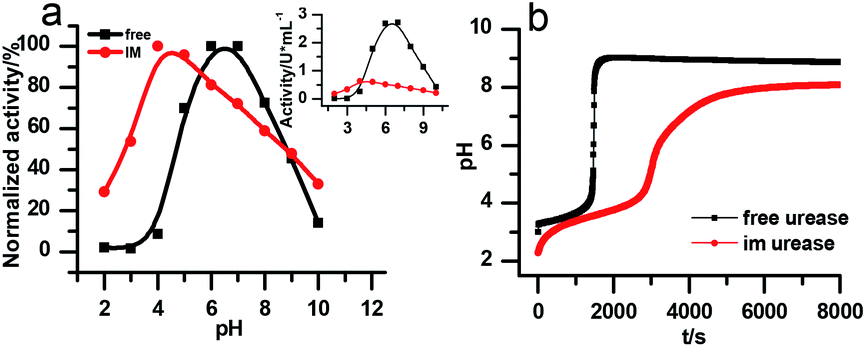 | ||
| Fig. 4 (a) The pH-dependent activity of the free and immobilized urease. The larger graph is the relative activity that is normalized to its maximum, the inset shows the change in absolute activities. [(NH4)2SO4] = 0.1 M; [H2SO4] = 1 M; [urea] = 0.5 M; [urease]free = [urease]IM = 4 U mL−1; (b) experimental results of the clock reaction of free and immobilized urease at same concentration. [Urea] = 0.005 M; [urease] = 3.2 U mL−1. | ||
Enzymes typically have two types of pH-dependent functional groups, the carboxyl group and the amino group. Both of them are protonated at low pH, forming the positively charged EnH2+ state. When pH is increased in the acidic range, the carboxyl group is deprotonated, which is controlled by its dissociation equilibrium constant KES1. At an intermediate pH, the enzyme exhibits its maximum catalytic activity. With increasing pH, the enzyme activity decreases as the amino group is deprotonated, giving the negatively charged En− state. This process is quantificationally governed by the amine dissociation equilibrium constant KES2 (Fig. 5). In the urea–urease–H+ system, pH increases with the production of ammonia, which increases the urease activity in the beginning part of time (pH range) and the system exhibits self-accelerating property. After reaching the optimum pH, the increase of pH will decrease the urease activity, giving the reaction a self-inhibiting property. The self-accelerating and self-inhibiting ranges are quantitatively controlled by the pKES1 and pKES2 of the enzyme.19
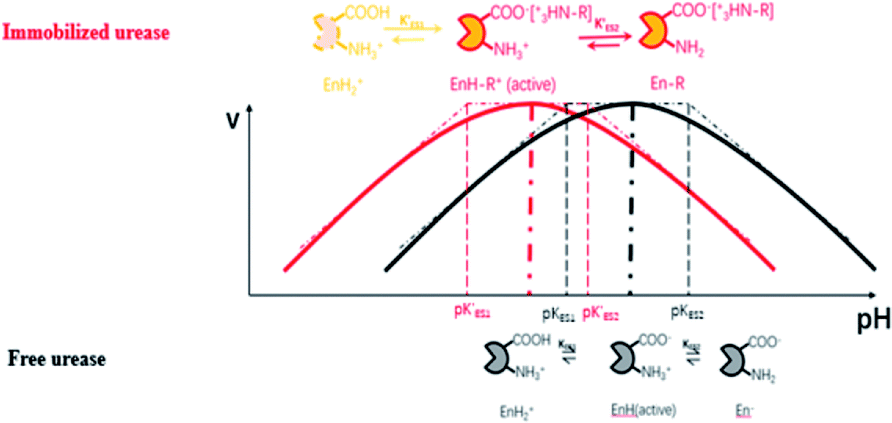 | ||
| Fig. 5 Diagram of the functional group (de)protonation basis for the bell shaped rate–pH curve for free and immobilized enzyme. | ||
In the immobilized urease system, the observed shift of the activity-pH curve to lower pH is hypothesized to be the direct consequence of ionic binding. The immobilization chemically modified the pH-dependent groups on the enzyme. In alkaline environment where the urease is immobilized on the resin, the carboxyl group and the amino group are in the –COO− and –NH2 states. The resins are positively charged after activation. The negatively charged –COO− groups on the urease are attracted by the –NH3+ groups on the resin so that the enzyme is immobilized. With this type of ionic immobilization, the combination of the deprotonated carboxyl group (–COO−), and the activated resin results in a greater degree of dissociation of the carboxyl group on the urease molecule. This increased dissociation is manifested as the apparent dissociation equilibrium constant 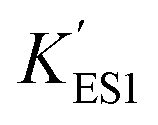 increasing after immobilization, so that the left branch of the pH dependent activation curve is shifted to lower pH. Considering the fact that enzymes usually exhibit good catalytic activity when electrically neutral, it is logically follows that the apparent
increasing after immobilization, so that the left branch of the pH dependent activation curve is shifted to lower pH. Considering the fact that enzymes usually exhibit good catalytic activity when electrically neutral, it is logically follows that the apparent 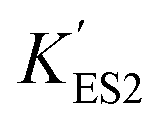 in the immobilized system is also increased. Besides, since the resin provides diffusion restriction, the high pH product may create kind of “high pH micro-environment” for the enzyme molecule. Therefore, the “effective” pH experience by the enzyme is actually higher than the solution environment. This kind of “protection” helps the enzymes to keep their activity in extremely low pH environment.
in the immobilized system is also increased. Besides, since the resin provides diffusion restriction, the high pH product may create kind of “high pH micro-environment” for the enzyme molecule. Therefore, the “effective” pH experience by the enzyme is actually higher than the solution environment. This kind of “protection” helps the enzymes to keep their activity in extremely low pH environment.
The proposed mechanism of how ionic immobilization can shift the pH dependent activation curve is shown in Fig. 5. There have been reports that the ionic binding is able to change the enzyme properties such as pH optimum or pH stability,20 and it has been used to shift the optimal condition of an enzyme towards more alkaline or acidic conditions.21 In this way, both the self-accelerating and self-inhibiting parts are shifted to lower pHs with the immobilized urease. This means the system will reach the self-suppressing state at lower pHs than with the free enzyme. Thus, it is reasonable that high pH cannot be easily achieved. In addition, the immobilized enzymes are usually less active than the free one since the diffusion is limited, and the active centers may be partially covered. These are also the reasons why the pHmax is lower, and the Tclock is longer.
Diffusion limitation of small molecules should be another way through which the immobilization influences the clock behavior (Fig. 6a). Comparison of the data in Fig. 6b verifies that to get the same Tclock requires higher substrate concentration in the immobilized urease system than with free enzyme. A possible reason that at the lower pH, the shift of pH is because the enzyme residues on immobilized resin are almost completely protonated, giving the molecule a high positive charge. This high positive potential may hinder the substrate's diffusion to the immobilized enzymes.
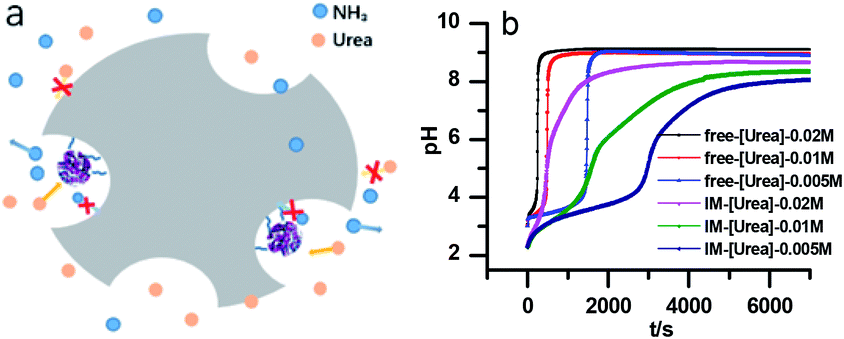 | ||
| Fig. 6 (a) Diagram of the diffusion limitation effect in the immobilized urease system. (b) The pH evolution curve in the free and immobilized urease system with different substrate concentrations. [Urease] = 3.2 U mL−1. | ||
To quantitatively test these hypotheses, dynamical simulations were performed, employing the model proposed by Hu et al.1 with modified enzymatic parameters, where the rate of enzymatic reaction is written as:
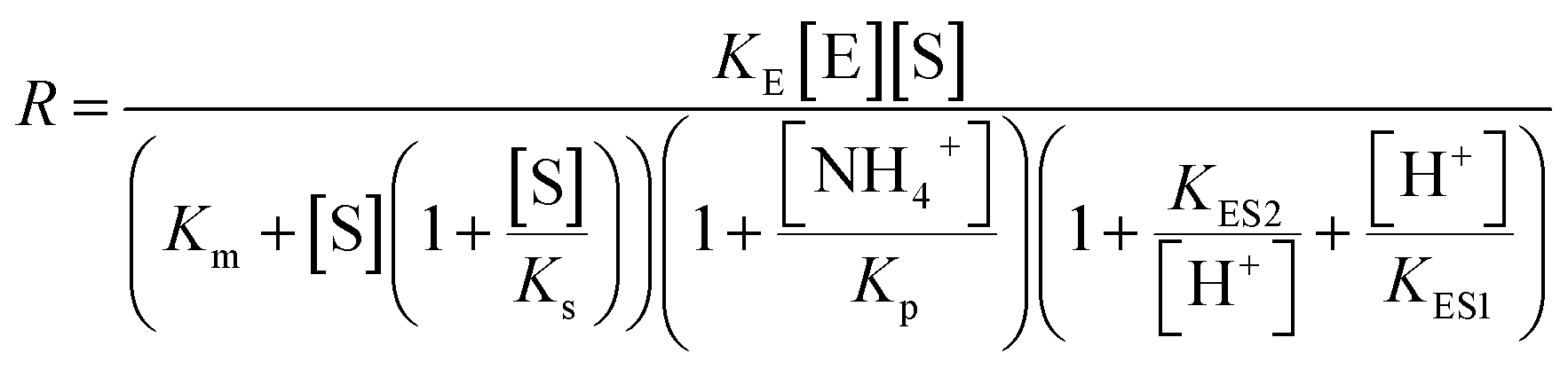
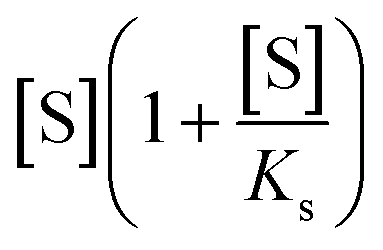 describes the substrate inhibition effect. The second term of the denominator
describes the substrate inhibition effect. The second term of the denominator 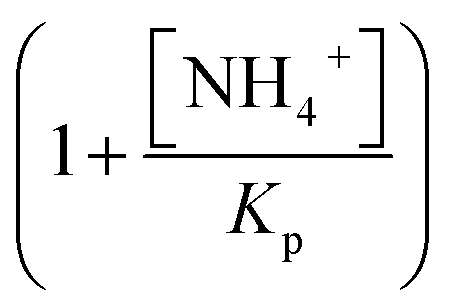 describes the product inhibition effect, and Kp is the equilibrium constant for non-competitive product inhibition. The third term
describes the product inhibition effect, and Kp is the equilibrium constant for non-competitive product inhibition. The third term 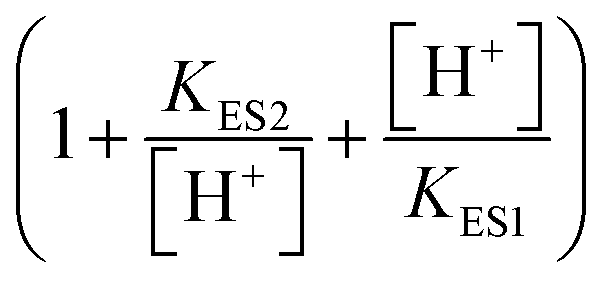 describes the pH dependence of the rate. KES1 and KES2 are the dissociation equilibrium constants of the carboxyl group and the amino group on the enzyme, respectively.19 To take the immobilization effect in to account, KES1 and KES2 are increased by an order of magnitude to reflect the ionic binding modified rate–pH curve shift. The diffusion limitation effects are reflected by the reduction of both the substrate concentration and the non-competitive product inhibition constant Kp. The adjusted parameters used in the simulations are listed in Table 1. Other dynamical equations and parameters are listed in the ESI.† The rate equations were solved by the Berkeley Madonna package with the stiff ODE solver (Rosenbrock).
describes the pH dependence of the rate. KES1 and KES2 are the dissociation equilibrium constants of the carboxyl group and the amino group on the enzyme, respectively.19 To take the immobilization effect in to account, KES1 and KES2 are increased by an order of magnitude to reflect the ionic binding modified rate–pH curve shift. The diffusion limitation effects are reflected by the reduction of both the substrate concentration and the non-competitive product inhibition constant Kp. The adjusted parameters used in the simulations are listed in Table 1. Other dynamical equations and parameters are listed in the ESI.† The rate equations were solved by the Berkeley Madonna package with the stiff ODE solver (Rosenbrock).
| [H+]/M | KES1 | KES2 | Kp/M | [S]/M | |
|---|---|---|---|---|---|
| Free urease | 3 × 10−4 | 5 × 10−6 | 2 × 10−9 | 2 × 10−3 | 5 × 10−3 |
| Im urease-1 | 10−3 | 5 × 10−5 | 2 × 10−8 | 2 × 10−3 | 5 × 10−3 |
| Im urease-2 | 10−3 | 5 × 10−5 | 2 × 10−8 | 5 × 10−4 | 2.5 × 10−3 |
Fig. 7a is the calculated rate–pH curve, with the main image showing the normalized rate data and the inset the absolute rates. It shows a clear shift to lower pH after increasing both KES1 and KES2, which is in good agreement with the experimental data in Fig. 4. Note that just changing the KES1 and KES2, the absolute value of the rate is reduced at the optimal pH. This may because that the self-inhibition occurs in an earlier stage than in the free enzyme system. In the calculated clock reaction curve (Fig. 7b), we can see that if we only take the pH–rate curve shift into consideration (red line), the pHmax, Tclock are changed, but the shape of the curve remains the same. Taking the diffusion limitation effect described by reducing Kp and substrate concentration will make the curve appear more like that in experiment (Fig. 4b).
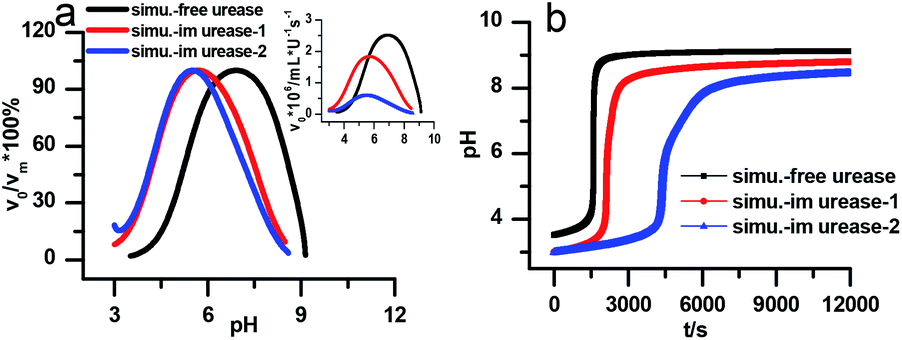 | ||
| Fig. 7 Simulated kinetic profiles in the urea–urease–H+ reaction. (a) The bell-shaped rate–pH curve is the larger graph with the relative value that are normalized to the maximum, the inset is the absolute rate data. (b) Clock reaction curves in the free and immobilized urease system. Parameters employed for each curve are listed in Table 1. Other fixed parameters value can be found in ESI.† | ||
Discussion
There are a number of autocatalytic enzyme processes that produce both clocks and propagating fronts such as hydrogenase22 or glucose oxidase.23 Maintaining these features under immobilization conditions will allow for increased use of such nonlinear reactions. The urease reaction is an ideal candidate for investigation of the effects of immobilisation on the autocatalytic reaction. To obtain stable and reproducible clock reaction results, there are actually strict requirements for both the raw materials and the immobilization conditions. Our experiments show that without pre-treating the resins with strong acid or alkali, trace amounts of metal ions will significantly hinder the immobilization. The concentration of urease is very low in our systems, which gives higher requirement than its usual usage. The source of the enzyme and specific batch significantly affect the reproducibility of results.It is possible that there is transient enzyme leaching in the initial low pH time period, since ionic immobilization strongly depends on pH value, either during the immobilization process or during the enzyme's use. Part of the carboxyl groups on the enzyme may be protonated to destroy the ionic binding. In experiments, we find that the immobilized urease is not stable when re-used. This could be the consequence of transient enzyme leaching. Some of the enzyme molecules that work in the reaction may actually be the “leached” free ones, which cannot be used in the next experiment. However, the current method successfully improves the storage stability of the enzyme and reproducibility of the clock reaction. Immobilization through covalent bonding might provide enhanced reusability of the enzyme, but the commonly used crosslinker glutaraldehyde turns out to be an inhibitor of urease,24 and it reacts with the Nessler reagent, which significantly influences the activity assay (finding other suitable crosslinkers is an ongoing effort).
There are also methods that immobilize the enzyme with physical encapsulation, gel entrapment, adsorption, affinity interaction etc. In Krajewska's25 recent review of urease immobilization, various methods had been successfully used to immobilize urease but there are various scenarios for the change of the bell shaped activity-pH curve. It can be shifted either to the right or to the left, and the overall result does not solely depend on the type of immobilization. In theory, all these immobilization methods will produce the diffusion limitation effect. However, the enzyme behavior may be related to complex factors such as the rate-determining elementary processes of the enzyme, different conformational change during the immobilization, etc.
Conclusions
In this work, the clock behavior of the urea–urease–H+ reaction with resin-immobilized urease was investigated. The urease was successfully immobilized on amino resin particles by ionic binding, which significantly improved the stability of the enzyme and the reproducibility of the clock reaction. The clock behavior was found in more acidic regions where the free enzyme was not active. The clock time could be varied by changing the enzyme or substrate concentration over a wider range of concentrations with the immobilized enzyme than with free enzyme. Further investigation revealed that the ionic immobilization shifted the bell-shaped activity-pH curve to the acidic area, which was the key factor that guaranteed the unusual behavior of the enzyme in the more acidic region. The functional group modification mechanism of the “acid shift” and its influence on the clock behavior are discussed and further verified by model simulation. The immobilization also limits the diffusion of small molecules and thus changes the enzyme kinetics. The diffusion limitation effect is a common result of various immobilization methods, and it contributes significantly to the shape change of the clock curve. Our immobilization method is simple and cost effective. The particles have controllable and uniform diameter distribution, and this kind of immobilization broadens the working area of the reaction to the acidic region, and the functional group modification and diffusion limitation factor may help us understand and control the clock behavior of such systems in more complicated and practical applications.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is supported by Natural Science Foundation of China Program (21773158, 21673175) and the Beijing Natural Science Foundation Program (2152009). JAP acknowledges support from the US National Science Foundation (CBET 1511653). We would like to thank professor Hui Luo for helpful suggestions and discussions.References
- G. Hu, J. A. Pojman, S. K. Scott, M. M. Wrobel and A. F. Taylor, Base-Catalyzed Feedback in the Urea-Urease Reaction, J. Phys. Chem. B, 2010, 114, 14059–14063 CrossRef CAS PubMed.
- M. M. Wrobel, T. Bansagi Jr, S. K. Scott, A. F. Taylor, C. O. Bounds, A. Carranzo and J. A. Pojman, pH Wave-Front Propagation in the Urea-Urease Reaction, Biophys. J., 2012, 103, 610–615 CrossRef CAS PubMed.
- T. Bansagi Jr and A. F. Taylor, Role of Differential Transport in an Oscillatory Enzyme Reaction, J. Phys. Chem. B, 2014, 118, 6092–6097 CrossRef PubMed.
- F. Muzika, T. Bansagi Jr, I. Schreiber, L. Schreiberova and A. F. Taylor, A Bistable Switch in pH in Urease-Loaded Alginate Beads, Chem. Commun., 2014, 50, 11107–11109 RSC.
- T. Heuser, E. Weyandt and A. Walther, Biocatalytic Feedback-Driven Temporal Programming of Self-Regulating Peptide Hydrogels, Angew. Chem., Int. Ed., 2015, 54, 13258–13262 CrossRef CAS PubMed.
- H. Che, B. C. Buddingh and J. C. M. van Hest, Self-Regulated and Temporal Control of a “Breathing” Microgel Mediated by Enzymatic Reaction, Angew. Chem., 2017, 56, 12581–12585 CrossRef CAS PubMed.
- E. Jee, T. Bánsági, A. F. Taylor and J. A. Pojman, Temporal Control of Gelation and Polymerization Fronts Driven by an Autocatalytic Enzyme Reaction, Angew. Chem., Int. Ed., 2016, 55, 2127–2131 CrossRef CAS PubMed.
- S. N. Semenov, A. J. Markvoort, T. F. de Greef and W. T. Huck, Threshold Sensing through a Synthetic Enzymatic Reaction-Diffusion Network, Angew. Chem., 2014, 53, 8066–8069 CrossRef CAS PubMed.
- S. G. Postma, I. N. Vialshin, C. Y. Gerritsen, M. Bao and W. T. Huck, Preprogramming Complex Hydrogel Responses Using Enzymatic Reaction Networks, Angew. Chem., 2017, 56, 1794–1798 CrossRef CAS PubMed.
- L. Heinen, T. Heuser, A. Steinschulte and A. Walther, Antagonistic Enzymes in a Biocatalytic pH Feedback System Program Autonomous DNA Hydrogel Life Cycles, Nano Lett., 2017, 17, 4989–4995 CrossRef CAS PubMed.
- K. Stingl, K. Altendorf and E. P. Bakker, Acid Survival of Helicobacter Pylori: How Does Urease Activity Trigger Cytoplasmic pH Homeostasis?, Trends Microbiol., 2002, 10, 70–74 CrossRef CAS PubMed.
- I. N. Bubanja, T. Bánsági and A. F. Taylor, Kinetics of the Urea–Urease Clock Reaction with Urease Immobilized in Hydrogel Beads, React. Kinet., Mech. Catal., 2017, 123, 177–185 CrossRef.
- U. Hanefeld, L. Gardossi and E. Magner, Understanding Enzyme Immobilisation, Chem. Soc. Rev., 2009, 38, 453–468 RSC.
- S. Datta, L. R. Christena and Y. R. S. Rajaram, Enzyme Immobilization: An Overview on Techniques and Support Materials, Biotech, 2012, 3, 1–9 Search PubMed.
- M. R. Tinsley, A. F. Taylor, Z. Huanga, F. Wanga and K. Showalter, Dynamical Quorum Sensing and Synchronization in Collections of Excitable and Oscillatory Catalytic Particles, Phys. D, 2010, 239, 785–790 CrossRef CAS.
- T. Bánsági and A. F. Taylor, Switches Induced by Quorum Sensing in a Model of Enzyme-Loaded Microparticles, J. R. Soc., Interface, 2017, 15, 20170945 CrossRef PubMed.
- B. Krajewska, Ureases. II. Properties and Their Customizing by Enzyme Immobilizations: A Review, J. Mol. Catal. B: Enzym., 2009, 59, 22–40 CrossRef CAS.
- A. K. Horváth and I. Nagypál, Classification of Clock Reactions, ChemPhysChem, 2015, 16, 588–594 CrossRef PubMed.
- R. Chang, Physical Chemistry for the Biosciences, University Science, Sausalito, CA, 2005 Search PubMed.
- J. M. Guisán, Immobilization of Enzymes and Cells (Methods in Biotechnology), 2006 Search PubMed.
- J. M. Guisan, G. Alvaro, C. M. Rosell and R. Fernandez-Lafuente, Industrial Design of Enzymic Processes Catalysed by Very Active Immobilized Derivatives: Utilization of Diffusional Limitations (Gradients of pH) as a Profitable Tool in Enzyme Engineering, Biotechnol. Appl. Biochem., 1994, 20, 357–369 CrossRef CAS PubMed.
- G. Bodo, R. M. Branca, A. Toth, D. Horvath and C. Bagyinka, Concentration-Dependent Front Velocity of the Autocatalytic Hydrogenase Reaction, Biophys. J., 2009, 96, 4976–4983 CrossRef CAS PubMed.
- D. G. Míguez, V. K. Vanag and I. R. Epstein, Fronts and Pulses in an Enzymatic Reaction Catalyzed by Glucose Oxidase, Proc. Natl. Acad. Sci. U.S.A., 2007, 104, 6992–6997 CrossRef PubMed.
- H. J. Smith and C. Simons, Enzymes and Their Inhibition, 2005 Search PubMed.
- B. Krajewska, A Combined Temperature-pH Study of Urease Kinetics. Assigning pka Values to Ionizable Groups of the Active Site Involved in the Catalytic Reaction, J. Mol. Catal. B: Enzym., 2016, 124, 70–76 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: An introductory text on the foundations of the simulation study, detailed description of the reaction mechanism and a list of simulation parameters. See DOI: 10.1039/c8ra09244c |
| This journal is © The Royal Society of Chemistry 2019 |