
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure evolution of chromium-doped boron clusters: toward the formation of endohedral boron cages†
Xuecheng Shao‡
a,
Xin Qu‡ *abcd,
Siyu Liua,
Lihua Yang*cd,
Jinghai Yangcd,
Xiaohui Liue,
Xin Zhong*cd,
Shuai Sunf,
G. Vaitheeswarang and
Jian Lv*ab
*abcd,
Siyu Liua,
Lihua Yang*cd,
Jinghai Yangcd,
Xiaohui Liue,
Xin Zhong*cd,
Shuai Sunf,
G. Vaitheeswarang and
Jian Lv*ab
aState Key Laboratory for Superhard Materials, College of Physics, Jilin University, Changchun, 130012, China. E-mail: quxin@calypso.cn
bCollege of Materials Science and Engineering, Jilin University, Changchun 130012, China
cKey Laboratory of Functional Materials Physics and Chemistry of the Ministry of Education, Jilin Normal University, Changchun 130103, China. E-mail: ylh@calypso.cn
dNational Demonstration Center for Experimental Physics Education, Jilin Normal University, Siping 136000, China
eNetwork Information Center, Supercomputing Center, University of Science and Technology of China, Hefei 230026, China
fEngineering Training Center, Institute of Mechanical Science and Engineering, Jilin University, Changchun, 130012, China
gAdvanced Center of Research in High Energy Materials (ACRHEM), University of Hyderabad, Hyderabad, 500046, India
First published on 23rd January 2019
Abstract
The electron-deficient nature of boron endows isolated boron clusters with a variety of interesting structural and bonding properties that can be further enriched through metal doping. In the current work, we report the structural and electronic properties of a series of chromium-doped boron clusters. The global minimum structures for CrBn clusters with an even number of n ranging from 8 to 22 are proposed through extensive first-principles swarm-intelligence structure searches. Half-sandwich structures are found to be preferred for CrB8, CrB10, CrB12 and CrB14 clusters and to transform to a drum-like structure at CrB16 cluster. Endohedral cage structures with the Cr atom located at the center are energetically most favorable for CrB20 and CrB22 clusters. Notably, the endohedral CrB20 cage has a high symmetry of D2d and a large HOMO–LUMO gap of 4.38 eV, whose stability is attributed to geometric fit and formation of an 18-electron closed-shell configuration. The current results advance our understanding of the structure and bonding of metal-doped boron clusters.
1. Introduction
Boron is an element of fascinating structural and chemical complexity, leading to topics of considerable interest in chemistry. It has three valence electrons that are deficient compared with the four valence orbitals, and it prefers to share rather than donate the valence electrons. These characteristics make it difficult for B to achieve filled octets through classical 2c–2e bonds, giving rise to a rich variety of structures along with electron-deficient multicentered bonds, both in its elemental form and chemical compounds.1 Sixteen polymorphs have been discovered for bulk B with B12 icosahedron being a prevalent motif.2 1D nanotubes3–5 and 2D sheets6–8 have been fabricated in which triangular planar B lattices with hexagonal holes are found to be energetically favorable.9For 0D B clusters, the situation is even more interesting. Joint photoelectron spectroscopy and theoretical studies carried out over the past decades show that anionic Bn− clusters up to n = 38 are planar or quasi-planar,10,11 in which delocalized multicentered bonds are responsible for the stabilities.12 Theoretical calculations suggested a planar-to-tubular structure transition taking place at B20 for neutrals.13 Subsequently, combined collision cross section measurements and theoretical calculations confirmed the existence of tubular structures for cationic Bn+ clusters with n = 16–25.14 Most strikingly, the long-sought B fullerene analogue (borospherenes) was first observed at B40 (ref. 15) after extensive theoretical investigations,16–19 and a series of axially chiral borospherenes were subsequently identified at B39−,20 B41+ and B422+.21 For larger B clusters, recent theoretical studies have suggested quasi-planar,22,23 tubular,24 cage-like25 and bilayered23 structures as ground states at certain sizes, and core–shell structures are generally expected to be energetically most favorable for n > ∼68.26–28
The structural diversity of B clusters can be further enriched through metal doping. Metal-centered monocyclic B rings can be formed by transition metal doping of small B8–10 anionic clusters29 in which NbB10− (ref. 30) and TaB10− (ref. 30) hold the highest coordination number of 10 in planar molecular species. Interesting half-sandwich structures have been found in Co/Rh doped B12 anionic clusters,31 and drum-like structures have been observed in CoB16−,32 MnB16− (ref. 33) and TaB20− (ref. 34) clusters, in which the TaB20− cluster possesses the highest coordination number (20) heretofore known in chemistry. Motivated by the discovery of borospherenes in our previous work, we have designed symmetric endohedral B cages, such as MnB20+,35 MoB24 (ref. 36) and WB24,36 whose stabilities are attributed to the formation of 18-electron closed-shell configurations, as well as geometric fits between the sizes of the transition metal atoms and the cavities of the B cages. It was further suggested that, by reasonable choice of the transition metal atom and number of B atoms, other-sized boron cages are also likely to be stabilized. Recently, a theoretical study found that even the small B12 cluster can show interesting transitions from quasi-planar to tubular and cage-like structures through interactions with lithium atoms, in which charge transfer plays a critical role.37
Metal doping has been proven to be an effective avenue to achieve intriguing structure motifs in B clusters. Given the large number of possible combinations between metal atoms and the number of B atoms yet to be investigated, it not unreasonable to expect more fascinating phenomena in this group of chemical species. As such, in this paper, we report systematic investigations on the structure and bonding of a series of chromium (Cr) doped B clusters by means of the swarm-intelligent CALYPSO structure searching method and first-principles density functional calculations. Ground-state structures are proposed for CrBn clusters with an even number of n ranging from 8 to 22, revealing an intriguing transition from half-sandwich to drum-like and then endohedral cage-like structures. In particular, a symmetric D2h endohedral cage is revealed as the ground-state structure for the CrB20 cluster. The rest of the manuscript is organized as follow. The second section describes the computational details. Section 3 presents the results and discussion, and the conclusions from the present results are given in Section 4.
2. Calculation details
The unbiased structure searches of CrBn clusters with an even number of n ranging from 8 to 22 are based on the global minimization of the potential energy surfaces, merging ab initio total energy calculations via the CALYPSO (Crystal structure AnaLYsis by Particle Swarm Optimization) package.38–41 Several major techniques are included in the algorithm to achieve high efficiency, e.g., point group symmetry constraints in structural generation, bond characterization matrix technique for fingerprinting structures, and a local version of the particle swarm optimization algorithm enabling simultaneous search in different energy funnels.39 Its validity has been manifested by successful identification of the ground-state structures for a large number of systems. More than 2000 trial structures were generated for each cluster.During the structure searches, the underlying energy calculations and structure relaxations are carried out in the framework of density functional theory (DFT) with the PBE functional42 implemented in the ABACUS (Atomic-orbital Based Ab initio Computation at USTC) package.43 ABACUS was developed to perform large-scale DFT simulations using linear combinations of atomic orbitals.44,45 The recently developed systematically improvable optimized numerical atomic orbitals44,45 were found to be an excellent choice to describe various materials, such as molecules, crystalline solids, surfaces, and defects.43 The atomic orbitals basis set of B includes two s, two p and one polarized d orbitals (2s2p1d), whereas the basis set of Cr includes four s, two p, two d and one polarized f orbitals (4s2p2d1f). The radii of the numerical atomic orbitals are set to 7 bohr for B and 8 bohr for Cr in the first-round local structure optimization, while 8 bohr for B and 9 bohr for Cr are used in the second-round local structure optimization. We adopt the SG15 Optimized Norm-Conserving Vanderbilt pseudopotentials,46 and the energy cutoff for charge density is 240 Ry.
The low-lying isomers obtained from the structure searches were reoptimized at different spin states at PBE0/Cr/Stuttgart/B/6-311+G* level of theory using the Gaussian 09 Package.47 The calculation of harmonic vibrational frequencies ensures that the cluster geometries are true local minima on the potential energy surface (no imaginary frequencies obtained). At this step, singlet, triplet, quintet and septet states were considered for all of these even-number-electrons clusters. In a previous benchmark calculation, the PBE0 functional was confirmed to be suitable for describing the energy difference of isomers of medium-sized boron clusters (e.g., B20) compared to the high-level CCSD(T) results.28 The natural bond orbital (NBO) and adaptive natural density partitioning (AdNDP) analytical methods were carried out with the Multiwfn package48 in order to achieve a better understanding of the bonding mechanism.
3. Results and discussion
The global minimum structures for CrBn (n = 8, 10, 12, 14, 16, 18, 20 and 22) clusters obtained from the current structure searches are depicted in Fig. 1. To facilitate understanding of the structures via visualization, top and side views are given along with their point group symmetries, spin multiplicity values (M) and HOMO–LUMO energy gaps (Eg). Other low-lying isomers are shown in the ESI, Fig. S1–S8.† Generally, the effect of Cr doping on the structures of B clusters gradually is enhanced as the number of B atoms increases. For small-sized CrBn (n = 8, 10 and 12) clusters, the B structures are similar to those in bare B clusters, while larger CrBn clusters exhibit B structures different from the corresponding bare B clusters. This leads to interesting transitions from half-sandwich to drum-like and then to endohedral cage-like structures as the number of B atoms increases. High spin states (triplet and quintet states) are preferred for small-sized CrBn clusters with n < 14, and the magnetism is completely quenched for CrBn clusters with n ≥ 16.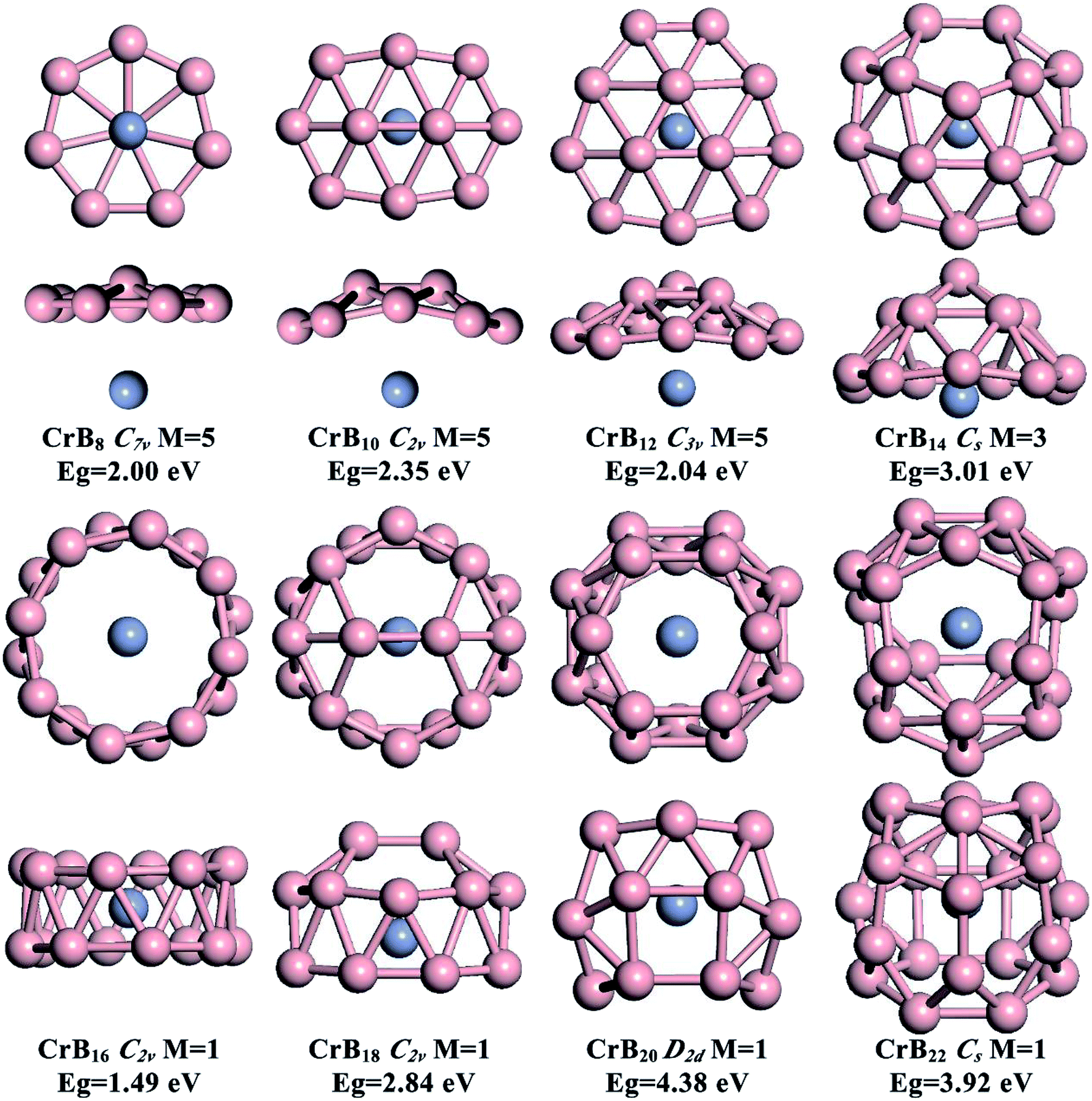 | ||
| Fig. 1 The lowest-energy ground-state isomers of CrBn (n = 8, 10, 12, 14, 16, 18, 20 and 22) clusters derived from the global minimum structure search. For each system, two sides of the views are given. For each structure, the point group symmetry, spin multiplicity (M) and LUMO–HOMO gap (Eg) are indicated. | ||
3.1 CrB8, CrB10, CrB12 and CrB14 clusters with half-sandwich structures
As depicted in Fig. 1, small-sized CrBn clusters with n = 8, 10, 12 and 14 exhibit half-sandwich structures, whereas quasi-planar or bowl-like Bn moieties are coordinated to the Cr atom. High spin states are found to be the ground states for these global-minimum structures (quintets for CrB8, CrB10, CrB12, and triplets for CrB14). Other low-lying isomers are given in Fig. S1–S8 in the ESI.† Spin density distributions shown in ESI, Fig. S9† indicates that the magnetism mainly originates from the unpaired 3d electrons of the Cr atom.The B structures in CrB8, CrB10 and CrB12 are very similar to those in bare B clusters, which are quasi-planar with one, two and three interior B atoms surrounded by seven-, eight- and nine-membered B rings, respectively. Due to the existence of the Cr atom, the interior B atoms in the B moieties display slight out-of-plane distortions. Note that, although CrB10 is isovalent to NbB10− and TaB10−, it does not adopt the metal-centered monocyclic B rings in NbB10− and TaB10− as the ground state. This may be due to the smaller size of the Cr atom (1.39 Å) compared with those of Nb (1.64 Å) and Ta (1.70 Å), which is not optimal for fitting the cavity of the ten-membered B ring. The half-sandwich structure of CrB12 is the same as those in experimental CoB12− and RhB12−, further indicating that the double aromatic B12 moiety is a promising inorganic ligand.
Inserting two B atoms into the B12 moiety in the CrB12 cluster leads to the formation of CrB14. The B14 moiety in CrB14 has a bowl-like structure with five interior B atoms surrounded by a nine-membered B ring, which is different from the structures of neutral or charged bare B14 clusters.10,49 In contrast to CrB8, CrB10 and CrB12, the Cr atom is half encircled by the bowl-like B14 moiety in CrB14, leading to enhanced Cr–B interactions and partially quenched magnetism. The significant curving of the B moiety in CrB14 indicates the gradually enhanced effect of Cr doping on the structure evolution and onset of structure transition in CrBn clusters.
3.2 The CrB16 cluster with a drum-like structure
The well-known drum-like structure with the Cr atom located at the center of a B16 double-ring tube occurs with the CrB16 cluster, having a point group of C2v (Fig. 1). Within this structure, the magnetism is completely quenched due to the strong coordination interactions between the Cr atom and the B16 tube. This type of structure was initially observed in CoB16− (ref. 32) and MnB16− (ref. 33) by joint photoelectron spectroscopy measurement and ab initio calculations. Later, studies found that larger B18 and B20 tubes can be stabilized by doping larger transition metal atoms (Rh and Ta), leading to drum-like RhB18− (ref. 50) and TaB20−,34 respectively. Interestingly, recent theoretical studies have found that the smaller B14 tube can be formed by the doping of an Fe atom,51 though the size of Fe (1.32 Å) is larger than that of Co (1.26 Å). Thus, it seems that whether drum-like structures can be formed in metal-doped B clusters is closely related to the size of the doping atom and the cavity of the B tube, as well as the charge and spin states of the metal-doped B clusters.The chemical bonding of the current drum-like CrB16 cluster was analyzed using the Adaptive Natural Density Partitioning (AdNDP)52 method, which is an extension of the Natural Bond Orbital method.53 AdNDP analyses can display both localized and delocalized bonding in molecules simultaneously, providing relatively simple bonding pictures for complicated molecular structures.33 The AdNDP analyses revealed a similar bonding character for CrB16 as those of CoB16− and MnB16−, where the 54 valence electrons in CrB16 can be divided into four bonding types as shown in Fig. 2. The occupation numbers (ON) of all the identified bonds range from 1.71 to 2.00 |e|. The first type represents localized bonds (Fig. 2a), which can be described in two manners: (1) as sixteen 3c–2e σ bonds on the sixteen B3 triangles on the drum surface with ON = 1.94 |e| or (2) as sixteen 2c–2e σ bonds within the two B8 rings with ON = 1.73 |e|. In fact, the sixteen 3c–2e σ bonds can also be represented by sixteen 2c–2e σ bonds on the two B8 rings on the drum surface are shown in Fig. S10 in ESI.†
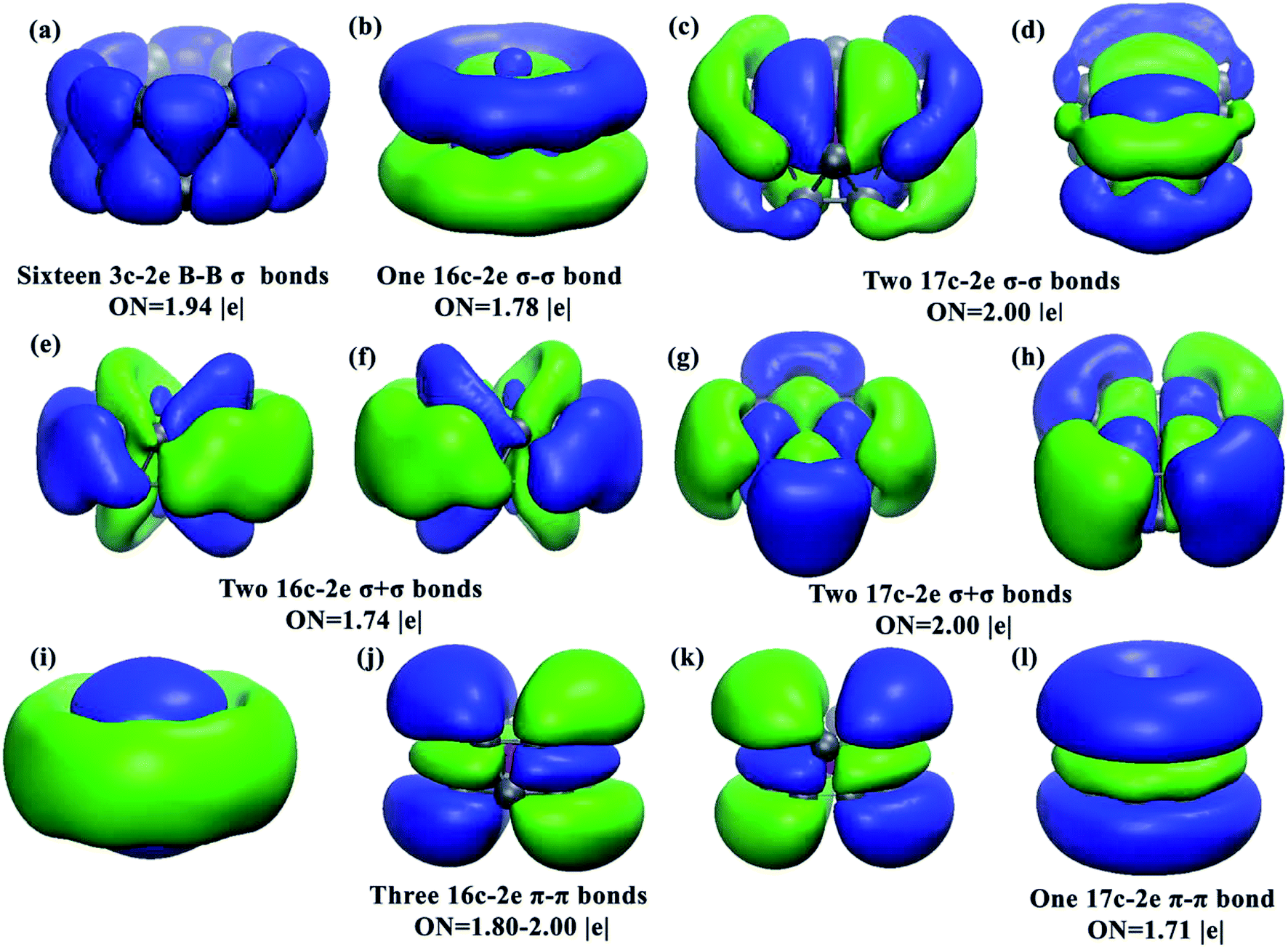 | ||
| Fig. 2 Images of the chemical bonding of CrB16 obtained from AdNDP analyses. ON stands for occupation number. | ||
The remaining three bonding types describe totally delocalized bonds (Fig. 2b–d, e–i and j–m) and account for bonding between the two B8 rings and between the Cr atom and the B16 tube. Following the previous work on CoB16− and MnB16−, the “+” sign is used to denote that the delocalized bonds between the two B8 rings overlap positively, while the “−” sign means a negative overlap. The second bonding type (Fig. 2b–d) consists of one 16c–2e σ − σ bond with ON = 1.78 |e| and two 17c–2e σ − σ bonds with ON = 2.00 |e|. The one 16c–2e σ − σ bond represents a bonding interaction within each B8 ring and an antibonding interaction between the two B8 rings, while the two 17c–2e σ − σ bonds represent mainly covalent bonding between Mn (3dxz and 3dyz) and the B16 tube. The third bonding type (Fig. 2e–h) contains two 16c–2e σ + σ bonds with ON = 1.74 |e| and two 17c–2e σ + σ bonds with ONs = 2.00 |e|. The two 16c–2e bonds represent delocalized σ bonding in the B16 frame, and the two 17c–2e σ + σ bonds represent covalent bonding between Cr (3dxy and 3dx2−y2) and the B16 tube. The fourth bonding type consists of three 16c–2e π − π bonds and one 17c–2e π − π bond. These four bonds account for π bonding interactions between the two B8 rings.
3.3 Transition from drum-like to endohedral cage-like structures in CrB18, CrB20 and CrB22 clusters
As depicted in Fig. 1, capping two B atoms on one side of the drum-like CrB16 structure leads to the global minimum structure of the CrB18 cluster, which can be seen as an intermediate structure between drum-like and endohedral cage-like structures. Notably, a symmetric endohedral cage with the Cr atom located at the center emerges as the global minimum structure for the CrB20 cluster. The structure is composed of twenty B triangles, four B quadrangles and two B hexagons in D2d symmetry. It has the largest HOMO–LUMO gap of 4.38 eV among CrBn clusters considered in the current work. The high symmetry and large HOMO–LUMO gap imply highly degenerate electronic states and the potentially high chemical stability of the CrB20 cluster. Further addition of two B atoms into the CrB20 cluster also leads to an endohedral cage-like structure in the CrB22 cluster, suggesting that endohedral cage-like motifs may be prevalent for larger CrBn clusters with n > 20. However, the endohedral CrB22 cage is rather irregular, and its HOMO–LUMO gap (3.92 eV) is smaller than that of CrB20.Early theoretical studies have demonstrated metal doping as a viable route for stabilizing cage-like B structures. A series of transition-metal-centered endohedral B cages have been proposed, such as FeB18, FeB20, MnB20+, MoB24, and WB24. However, the transition metal should possess both geometric and electronic states that can fit in high symmetric endohedral B cage-like clusters that eventually lead to the high stability structures that can be formed. The current endohedral D2d CrB20 cage should be one such paradigm. The geometric factor responsible for the stability is straightforward. Our previous calculations have demonstrated that the Cr atom is too small to fit a large B24 cage, indicating smaller B cages are suitable for accommodating one Cr atom.
To understand electronic factors responsible for the stability of the endohedral CrB20 cage, the chemical bonding of the bare D2d B20 cage and endohedral D2d CrB20 cage were analyzed based on canonical molecular orbitals (CMOs). Fig. 3 shows the comparison of eigenvalue spectra for the D2d cage without (a) and with (b) Cr encapsulation. One can clearly note that the Cr encapsulation significantly increases the HOMO–LUMO gap from 0.87 eV for the bare D2d B20 cage to 4.38 eV for the D2d CrB20. For bare D2d B20 (Fig. 3a), there are 6 occupied π-orbitals (HOMO, HOMO−1, HOMO−4, two HOMO−7, and HOMO−11) and 3 unoccupied π-orbitals (LUMO, LUMO+1 and LUMO+2). The moderate HOMO–LUMO gap in bare D2d B20 is attributed to the mid-lying binding energies of these out-of-surface delocalized π-orbitals, and they will interact with the electronic orbitals of Cr atoms in the endohedral CrB20 cages. Cr has an electronic configuration of [Ar]4s13d5 with 6 valence electrons, adding the 12 π-electrons (from 6 occupied π-orbitals) of bare D2d B20 gives a total of 18 electrons. This special electron counting number of 18 is favorable for forming a stable 18-electron closed-shell configuration, similar to that of previous MnB20+, MoB24 and WB24 clusters. Indeed, nine CMOs involved in the “spd–π interaction” have been identified for the D2d CrB20 endohedral cage as depicted in Fig. 3b, i.e., HOMO−13 (s-like), HOMO−11 (dx2−y2-like), HOMO−8 (double degenerate, px-like and py-like), HOMO−7 (dxy-like), HOMO−6 (pz-like), HOMO−5 (double degenerate, dxz-like and dyz–π like) and HOMO−1 (3dz2-like). Thus, the CrB20 cluster represents another example of having a symmetric endohedral cage configuration stabilized by the 18-electron configuration.
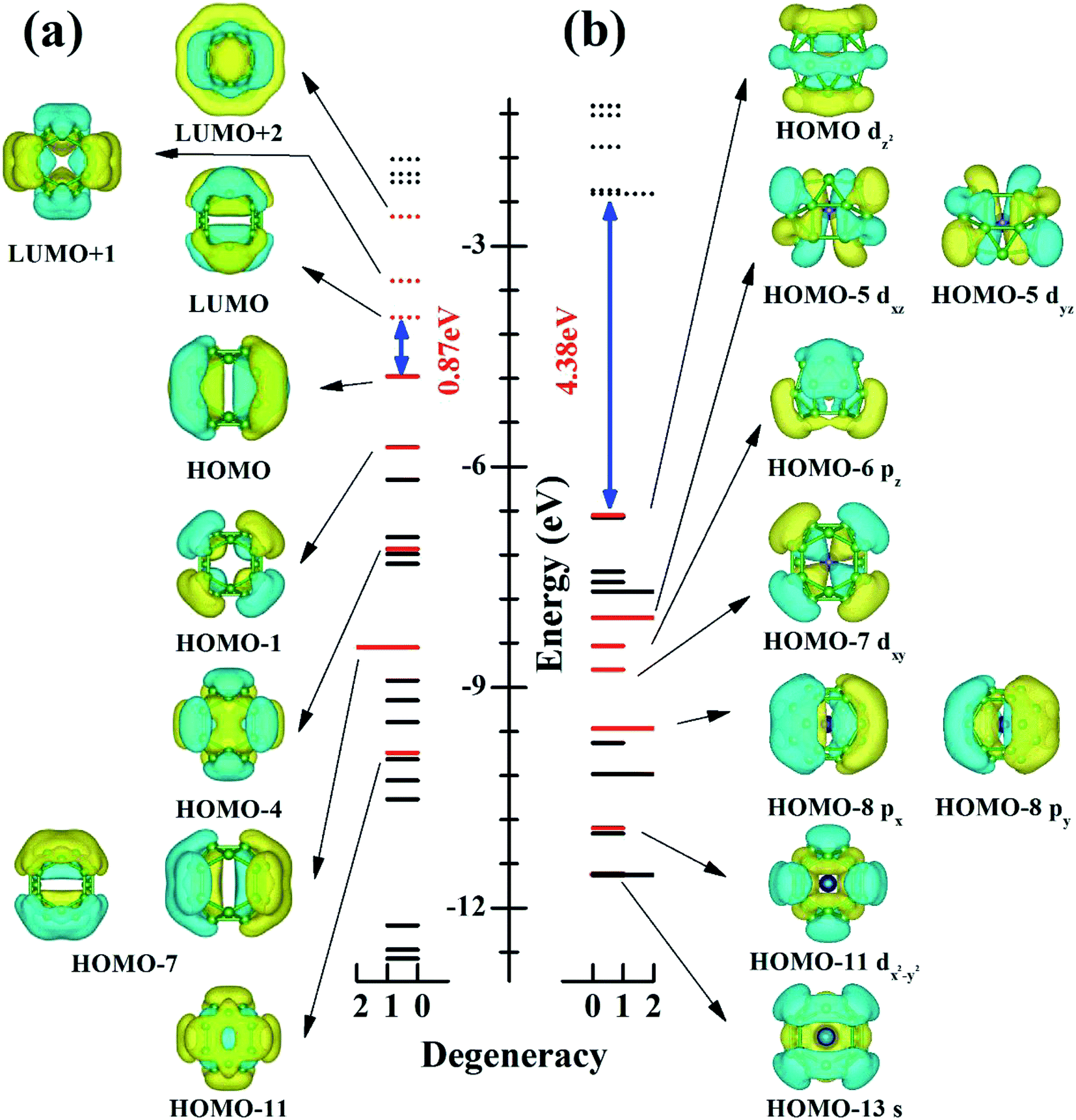 | ||
| Fig. 3 Eigenvalue spectrum vs. electronic state degeneracy of (a) bare D2d–B20 cage and (b) D2d–CrB20. For each case, the HOMO–LUMO gap is indicated (in blue). The π-orbitals (red lines) (a) and the orbitals involving the 18-electron closed-shell configuration (b) are shown. | ||
4. Conclusions
In summary, we systematically investigated the structural and electronic properties of CrBn clusters with n = 8, 10, 12, 14, 16, 18, 20 and 22 through extensive swarm-intelligent structure searches and first-principles calculations. It is found that Cr doping significantly modifies the structural evolution of B clusters. Intriguing transitions from half-sandwich to drum-like and then to endohedral cage-like structures are revealed as the number of B atoms increases. CrB8, CrB10 and CrB12 clusters exhibit half-sandwich structure with quasi-planar B moieties similar to the bare B cluster, indicating that small-sized B clusters are promising inorganic ligands. A drum-like structure is formed with CrB16 clusters, while endohedral cage structures emerge with the larger CrB20 and CrB22 clusters. The endohedral CrB20 cage has a high symmetry of D2d and the largest HOMO–LUMO gap among CrBn in the current work, indicating its high chemical stability, which is attributed to the geometric fit between the size of the Cr atom and the void of the B cage as well as the formation of the 18-electron configuration.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors acknowledge the funding support from National Natural Science Foundation of China (Grants No. 11534003, 11604117, 11404128, 11704151 and 61775081), the Science Challenge Project, No. TZ2016001, National Key Research and Development Program of China (Grant No. 2016YFB0201200), and Program for JLU Science and Technology Innovative Research Team. Part of the calculations were performed in the high performance computing center of Jilin University and Tianhe2-JK in the Beijing Computational Science Research Center.References
- X. Sun, X. Liu, J. Yin, J. Yu, Y. Li, Y. Hang, X. Zhou, M. Yu, J. Li, G. Tai and W. Guo, Adv. Funct. Mater., 2017, 27, 1603300 CrossRef.
- B. Albert and H. Hillebrecht, Angew. Chem., Int. Ed., 2009, 48, 8640–8668 CrossRef CAS PubMed.
- F. Liu, C. Shen, Z. Su, X. Ding, S. Deng, J. Chen, N. Xu and H. Gao, J. Mater. Chem., 2010, 20, 2197 RSC.
- V. Bezugly, J. Kunstmann, B. Grundkötter-Stock, T. Frauenheim, T. Niehaus and G. Cuniberti, ACS Nano, 2011, 5, 4997–5005 CrossRef CAS PubMed.
- J. Kunstmann, V. Bezugly, H. Rabbel, M. H. Rümmeli and G. Cuniberti, Adv. Funct. Mater., 2014, 24, 4127–4134 CrossRef CAS.
- G. Tai, T. Hu, Y. Zhou, X. Wang, J. Kong, T. Zeng, Y. You and Q. Wang, Angew. Chem., Int. Ed., 2015, 54, 15473–15477 CrossRef CAS PubMed.
- B. Feng, J. Zhang, Q. Zhong, W. Li, S. Li, H. Li, P. Cheng, S. Meng, L. Chen and K. Wu, Nat. Chem., 2016, 8, 563–568 CrossRef CAS PubMed.
- A. J. Mannix, X. F. Zhou, B. Kiraly, J. D. Wood, D. Alducin, B. D. Myers, X. Liu, B. L. Fisher, U. Santiago, J. R. Guest, M. J. Yacaman, A. Ponce, A. R. Oganov, M. C. Hersam and N. P. Guisinger, Science, 2015, 350, 1513–1516 CrossRef CAS PubMed.
- H. Tang and S. Ismail-Beigi, Phys. Rev. Lett., 2007, 99, 115501 CrossRef PubMed.
- A. N. Alexandrova, A. I. Boldyrev, H. J. Zhai and L. S. Wang, Coord. Chem. Rev., 2006, 250, 2811–2866 CrossRef CAS.
- Z. Zhang, E. S. Penev and B. I. Yakobson, Chem. Soc. Rev., 2017, 46, 6746–6763 RSC.
- A. P. Sergeeva, I. A. Popov, Z. A. Piazza, W. L. Li, C. Romanescu, L. S. Wang and A. I. Boldyrev, Acc. Chem. Res., 2014, 47, 1349–1358 CrossRef CAS PubMed.
- B. Kiran, S. Bulusu, H.-J. Zhai, S. Yoo, X. C. Zeng and L.-S. Wang, Proc. Natl. Acad. Sci., 2005, 102, 961–964 CrossRef CAS PubMed.
- E. Oger, N. R. M. Crawford, R. Kelting, P. Weis, M. M. Kappes and R. Ahlrichs, Angew. Chem., Int. Ed., 2007, 46, 8503–8506 CrossRef CAS PubMed.
- H. J. Zhai, Y. F. Zhao, W. L. Li, Q. Chen, H. Bai, H. S. Hu, Z. A. Piazza, W. J. Tian, H. G. Lu, Y. B. Wu, Y. W. Mu, G. F. Wei, Z. P. Liu, J. Li, S. D. Li and L. S. Wang, Nat. Chem., 2014, 6, 727–731 CrossRef CAS PubMed.
- J. Lv, Y. Wang, L. Zhu and Y. Ma, Nanoscale, 2014, 6, 11692–11696 RSC.
- T. B. Tai, L. Van Duong, H. T. Pham, D. T. T. Mai and M. T. Nguyen, Chem. Commun., 2014, 50, 1558–1560 RSC.
- N. Gonzalez Szwacki, A. Sadrzadeh and B. I. Yakobson, Phys. Rev. Lett., 2007, 98, 166804 CrossRef PubMed.
- J. T. Muya, G. Gopakumar, M. T. Nguyen and A. Ceulemans, Phys. Chem. Chem. Phys., 2011, 13, 7524 RSC.
- Q. Chen, W.-L. Li, Y.-F. Zhao, S.-Y. Zhang, H.-S. Hu, H. Bai, H.-R. Li, W.-J. Tian, H.-G. Lu, H.-J. Zhai, S.-D. Li, J. Li and L.-S. Wang, ACS Nano, 2015, 9, 754–760 CrossRef CAS PubMed.
- Q. Chen, S. Y. Zhang, H. Bai, W. J. Tian, T. Gao, H. R. Li, C. Q. Miao, Y. W. Mu, H. G. Lu, H. J. Zhai and S. D. Li, Angew. Chem., Int. Ed., 2015, 54, 8160–8164 CrossRef CAS PubMed.
- A. B. Rahane and V. Kumar, Nanoscale, 2015, 7, 4055–4062 RSC.
- L. Sai, X. Wu, N. Gao, J. Zhao and R. B. King, Nanoscale, 2017, 9, 13905–13909 RSC.
- F. Y. Tian and Y. X. Wang, J. Chem. Phys., 2008, 129, 024903 CrossRef PubMed.
- T. B. Tai and M. T. Nguyen, Chem. Commun., 2016, 52, 1653–1656 RSC.
- D. L. V. K. Prasad and E. D. Jemmis, Phys. Rev. Lett., 2008, 100, 100–103 CrossRef PubMed.
- H. Li, N. Shao, B. Shang, L.-F. Yuan, J. Yang and X. C. Zeng, Chem. Commun., 2010, 46, 3878 RSC.
- F. Li, P. Jin, D. E. Jiang, L. Wang, S. B. Zhang, J. Zhao and Z. Chen, J. Chem. Phys., 2012, 136, 074302 CrossRef PubMed.
- C. Romanescu, T. R. Galeev, W. L. Li, A. I. Boldyrev and L. S. Wang, Acc. Chem. Res., 2013, 46, 350–358 CrossRef CAS PubMed.
- T. R. Galeev, C. Romanescu, W. L. Li, L. S. Wang and A. I. Boldyrev, Angew. Chem., Int. Ed., 2012, 51, 2101–2105 CrossRef CAS PubMed.
- I. A. Popov, W.-L. Li, Z. A. Piazza, A. I. Boldyrev and L.-S. Wang, J. Phys. Chem. A, 2014, 118, 8098–8105 CrossRef CAS PubMed.
- I. A. Popov, T. Jian, G. V. Lopez, A. I. Boldyrev and L. S. Wang, Nat. Commun., 2015, 6, 8654 CrossRef CAS PubMed.
- T. Jian, W. L. Li, I. A. Popov, G. V. Lopez, X. Chen, A. I. Boldyrev, J. Li and L. S. Wang, J. Chem. Phys., 2016, 144, 154310 CrossRef PubMed.
- W.-L. Li, T. Jian, X. Chen, H.-R. Li, T.-T. Chen, X.-M. Luo, S.-D. Li, J. Li and L.-S. Wang, Chem. Commun., 2017, 53, 1587–1590 RSC.
- L. Zhao, X. Qu, Y. Wang, J. Lv, L. Zhang, Z. Hu, G. Gu and Y. Ma, J. Phys. Condens. Matter, 2017, 29, 265401 CrossRef PubMed.
- J. Lv, Y. Wang, L. Zhang, H. Lin, J. Zhao and Y. Ma, Nanoscale, 2015, 7, 10482–10489 RSC.
- X. Dong, S. Jalife, A. Vásquez-Espinal, E. Ravell, S. Pan, J. L. Cabellos, W. Liang, Z. Cui and G. Merino, Angew. Chem., Int. Ed., 2018, 57, 4627–4631 CrossRef CAS PubMed.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Phys. Rev. B, 2010, 82, 094116 CrossRef.
- J. Lv, Y. Wang, L. Zhu and Y. Ma, J. Chem. Phys., 2012, 137, 084104 CrossRef PubMed.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Comput. Phys. Commun., 2012, 183, 2063–2070 CrossRef CAS.
- H. Wang, Y. Wang, J. Lv, Q. Li, L. Zhang and Y. Ma, Comput. Mater. Sci., 2016, 112, 406–415 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. Li, X. Liu, M. Chen, P. Lin, X. Ren, L. Lin, C. Yang and L. He, Comput. Mater. Sci., 2016, 112, 503–517 CrossRef CAS.
- M. Chen, G. C. Guo and L. He, J. Phys. Condens. Matter, 2010, 22, 445501 CrossRef PubMed.
- M. Chen, G. C. Guo and L. He, J. Phys. Condens. Matter, 2011, 23, 325501 CrossRef PubMed.
- D. R. Hamann, Phys. Rev. B, 2013, 88, 085117 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- C. Longjiu, J. Chem. Phys., 2012, 136, 104301 CrossRef PubMed.
- T. Jian, W.-L. Li, X. Chen, T.-T. Chen, G. V. Lopez, J. Li and L.-S. Wang, Chem. Sci., 2016, 7, 7020–7027 RSC.
- N. M. Tam, H. T. Pham, L. Van Duong, M. P. Pham-Ho and M. T. Nguyen, Phys. Chem. Chem. Phys., 2015, 17, 3000–3003 RSC.
- D. Y. Zubarev and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2008, 10, 5207 RSC.
- E. D. Glendening, C. R. Landis and F. Weinhold, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 1–42 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra09143a |
| ‡ Equally share the first authorship. |
| This journal is © The Royal Society of Chemistry 2019 |