
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolybdenum imidazole citrate and bipyridine homocitrate in different oxidation states – balance between coordinated α-hydroxy and α-alkoxy groups†
Si-Yuan Wang and
Zhao-Hui Zhou *
*
State Key Laboratory for Physical Chemistry of Solid Surfaces, College of Chemistry and Chemical Engineering, Xiamen University, Xiamen, 361005, China. E-mail: zhzhou@xmu.edu.cn; Fax: +86-592-2183047; Tel: +86-592-2184531
First published on 2nd January 2019
Abstract
Oxo and thiomolybdenum(IV/VI) imidazole hydrocitrates K2{MoIV3O4(im)3[MoVIO3(Hcit)]2}·3im·4H2O (1), (Him)2{MoIV3SO3(im)3[MoVIO3(Hcit)]2}·im·6H2O (2), molybdenum(V) bipyridine homocitrate trans-[(MoVO)2O(H2homocit)2(bpy)2]·4H2O (3) and molybdenum(VI) citrate (Et4N)[MoVIO2Cl(H2cit)]·H2O (4) (H4cit = citric acid, H4homocit = homocitric acid, im = imidazole and bpy = 2,2′-bipyridine) with different oxidation states were prepared. 1 and 2 are the coupling products of [MoVIO3(Hcit)]3− anions and incomplete cubane units [MoIV3O4]4+ ([MoIV3SO3]4+) with monodentate imidazoles, respectively, where tridentate citrates coordinate with α-hydroxy, α-carboxy and β-carboxy groups, forming pentanuclear skeleton structures. The molybdenum atoms in 1 and 2 show unusual +4 and +6 valences based on charge balances, theoretical bond valence calculations and Mo XPS spectrum. The coordinated citrates in 1 and 2 are protonated with α-hydroxy groups, while 3 and 4 with higher oxidation states of +5 and +6 are deprotonated with α-alkoxy group even under strong acidic condition, respectively. This shows the relationship between the oxidation state and protonation of the α-alkoxy group in citrate or homocitrate, which is related to the protonation state of homocitrate in FeMo-cofactor of nitrogenase. The homocitrate in 3 chelates to molybdenum(V) with bidentate α-alkoxy and monodentate α-carboxy groups. Molybdenum(VI) citrate 4 is only protonated with coordinated and uncoordinated β-carboxy groups. The solution behaviours of 1 and 2 are discussed based on 1H and 13C NMR spectroscopies and cyclic voltammograms, showing no decomposition of the species.
Introduction
Molybdenum is widely present in water, soil, various animals and plants, and constitutes a variety of metalloenzymes with redox functions.1,2 Homocitrate as a chelating ligand bound to MoIV/III of FeMo-cofactor plays a critical role in nitrogen fixation.3–11 Homocitric acid is also a key intermediate in the biosynthetic pathway to the essential amino acid lysine in fungi and euglenids.12–14 Molybdenum homocitrate is also considered to be involved in the final biosynthesis of FeMo-cofactor by the coupling reaction with iron clusters.15,16 R-Homocitrate, whose long and flexible CH2CO2 arm can form a hydrogen bond with the NH group of the imidazole ligand, may play an important role in the process of reducing nitrogen.17,18 The source and path of protons in homocitrate have been an important topic in nitrogenase.19–25As a homolog of homocitrate, citrate is also a complexing agent used in the preparations of Co(Ni)–Mo catalyst precursors of hydrosulfurization or baths for the electrodeposition of molybdenum–iron metal alloys, which assure a stable pH value during the deposition process in the electrolyte.26–31 Molybdenum citrate has also been discovered in nifV nitrogenase and the sensor kinase CitA.32,33 The incomplete cuboidal clusters Mo3S4, Mo3S3O, Mo3SO3 and Mo3O4 can form complexes with various types of ligands as a classic low-valence Mo skeleton structure,34–43 which exists in Mo storage protein.44 The latter also contains octanuclear molybdenum complexes [Mo8O26(im)2]4− with imidazole from histidine residue as polyoxometalates.44–46
It is noted that most of the mixed-valence molybdenum complexes are in VI/V and V/IV,47–54 while molybdenum(VI/IV) complex is less common.55 Here two pentanuclear imidazole hydrocitrato molybdenum(VI/IV) complexes K2{Mo3IVO4(im)3[MoVIO3(Hcit)]2}·3im·4H2O (1) and (Him)2{MoIV3SO3(im)3[MoVIO3(Hcit)]2}·im·6H2O (2) have been obtained with protonated α-hydroxy groups, while dimeric bipyridine oxomolybdenum(V) homocitrate trans-[(MoVO)2O(H2homocit)2(bpy)2]·4H2O (3) and mononuclear oxomolybdenum(VI) hydrocitrate (Et4N)[MoVIO2Cl(H2cit)]·H2O (4) were captured with coordinate α-alkoxy groups in acidic solution.
Results and discussion
Until now, a number of citrate or imidazole molybdenum complexes have been reported, while there is less interest on the mixed-ligand complexes of citrate and imidazole.45,47,56–59 Most of the mixed-valence molybdenum complexes focus on Mo(VI)/Mo(V) and Mo(V)/Mo(IV),47–54 fewer examples of Mo(VI)/Mo(IV) have been reported.55 Here pentanuclear molybdenum(IV/VI) imidazole hydrocitrates K2{MoIV3O4(im)3[MoVIO3(Hcit)]2}·3im·4H2O (1) and (Him)2{MoIV3SO3(im)3[MoVIO3(Hcit)]2}·im·6H2O (2) were obtained from an aqueous solution. The valences of the molybdenum atoms in 1 and 2 are +4 or +6 based on the charge balances, theoretical bond valence calculations and Mo XPS spectrum, which will be discussed later. They are different from the common mixed-valence complexes in the adjacent valence state, which are coordinated with citrate and imidazole.47,60Increasing the temperature can enhance the coordination abilities of citric acid and imidazole. High-valent molybdenum atoms are reduced in a closed Teflon-lined stainless steel container with the aid of a reducing agent. Low-valent molybdenum atoms and weak alkaline conditions can avoid the protonation of imidazole, making it easier for coordination with molybdenum citrate simultaneously. Possible routes for molybdenum citrates 1, 2 and 4 are outlined in Scheme 1 in three steps. First, the molybdates are reduced to tetravalent molybdenum cations [MoIV3O4(H2O)9]4+ or [MoIV3SO3(H2O)9]4+ respectively.61 The imidazoles as monodentate ligands coordinate to the molybdenum(IV) atoms in [MoIV3O4(H2O)9]4+ or [MoIV3SO3(H2O)9]4+ ions. Free β-carboxy and α-alkoxy groups in [MoVIO3(cit)]4− further coordinate with the molybdenum atom(IV) atoms in [MoIV3O4(H2O)9]4+ ion, forming a pentanuclear structure {MoIV3O4(im)3[MoVIO3(Hcit)]2}2− with α-hydroxy groups. When Na2S2O4 was used instead of hydrazine dihydrochloride as a reducing agent, the μ3-O of [MoIV3O4]4+ in 1 is replaced by μ3-S to form [MoIV3SO3]4+. Moreover, one of the terminal oxygen in [MoVIO3(H2cit)]2− could be replaced by chloride under strong acidic conditions. The [MoVIO2Cl(H2cit)]− ion was further captured by the large cation (Et4N)+ as 4 without protonation in α-alkoxy group.
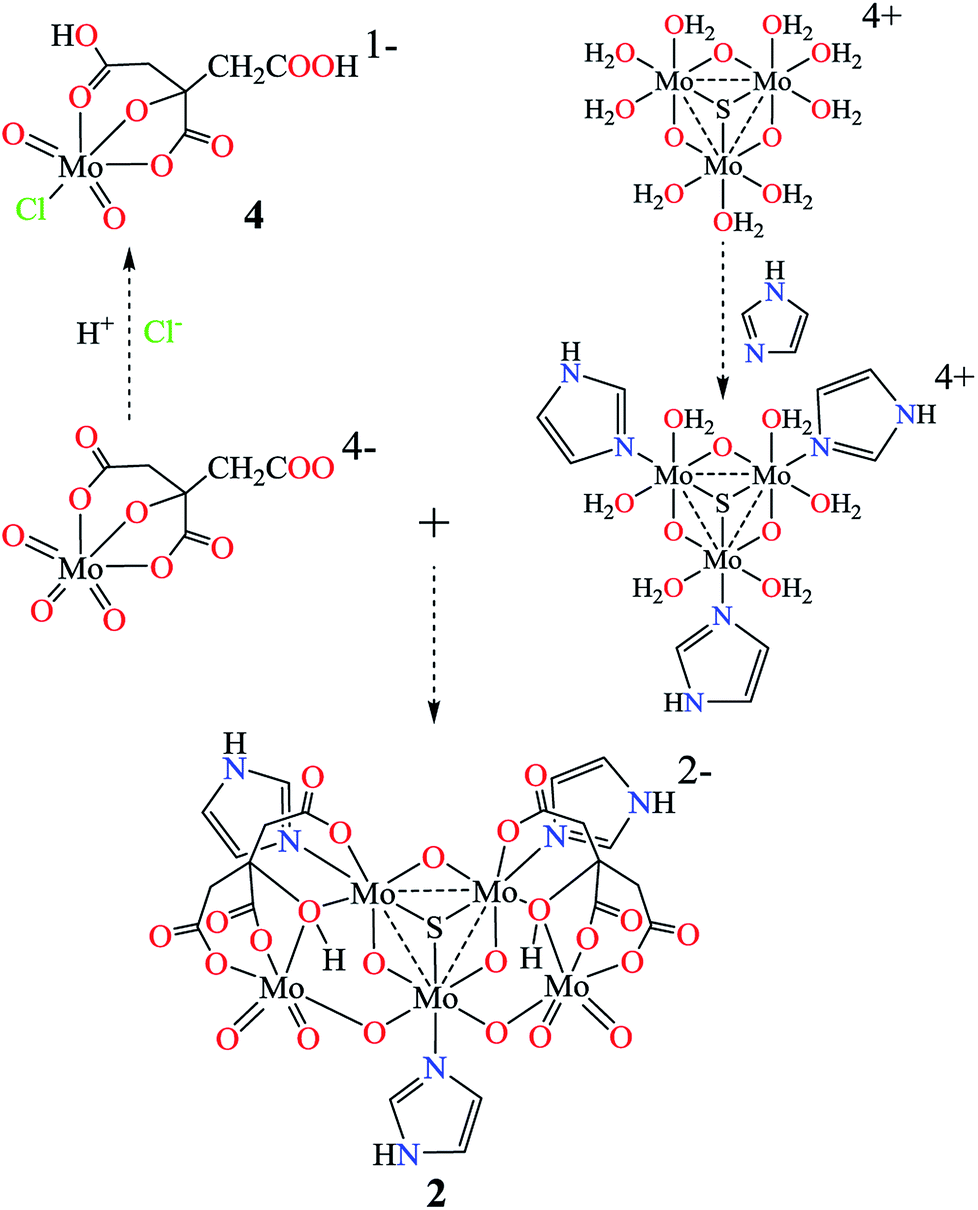 | ||
| Scheme 1 Possible routes for molybdenum(IV/VI) imidazole hydrocitrates 1 and 2 and (μ3-X = S or O) molybdenum(VI) citrate 4. | ||
Previously, a series of molybdenum(VI) homocitrates have been synthesized.62–65 The strong chelating agent, 2,2′-bipyridine, assisted the formation of reduced molybdenum(V) homocitrate trans-[(MoO)2O(H2homocit)2(bpy)2]·4H2O (3) in the presence of hydrazine. The insoluble complex 3 crystallized immediately with a decrease in temperature. Attempts to prepare molybdenum(IV) homocitrate and molybdenum(IV/VI) imidazole homocitrate were not yet successful.
As shown by the X-ray crystal structures (Fig. 1 and S1–S4†), K2{MoIV3O4(im)3[MoVIO3(Hcit)]2}·3im·4H2O (1) and (Him)2{MoIV3SO3(im)3[MoVIO3(Hcit)]2}·im·6H2O (2) are pentanuclear imidazole hydrocitrato molybdenum(IV/VI) complexes. An incomplete cubane cluster anion [MoIV3O4]4+ or [MoIV3SO3]4+ in 1 or 2 is joined by imidazole to form the circular species, respectively. Each molybdenum atom(IV) is surrounded octahedrally by two μ2-O, one μ3-O/S and one N atom of the imidazole ligand. The three imidazole ligands occupy a cis position to the μ2-O. This is similar to the reported Mo3O4(his)3 cluster in the Mo storage protein with an incomplete cubane anion, where each molybdenum atom is octahedrally coordinated to five O atoms and to the imidazole Nε2 atom of hisα140.44 Moreover, molybdenum(VI) atoms in 1 or 2 are hexa-coordinated in approximate octahedral geometries respectively. The citrate ligand uses its α-hydroxy, α-carboxy and one β-carboxy groups to act as a tridentate ligand. The other β-carboxy and α-hydroxy groups of the citrate further coordinate with the other molybdenum atom(IV). Each molybdenum(VI) citrate connects to [MoIV3O4]4+ or [MoIV3SO3]4+ unit with an O- or S-bridge to constitute novel pentanuclear skeleton structures.15
 | ||
| Fig. 1 ORTEP plots of the anion structures in K2{MoIV3O4(im)3[MoVIO3(Hcit)]2}·3im·4H2O (1, a) and (Him)2{MoIV3SO3(im)3[MoVIO3(Hcit)]2}·im·6H2O (2, b) at the 20% probability levels. | ||
Hydrogen bonds of the imidazoles played important roles in the crystal packing of 1 and 2 (Fig. S5–S8, Tables S1 and S2†). The free imidazole as well as the imidazole ligand can act as providers for the two types of hydrogen bonds as N–H⋯O and N–H⋯N. N–H⋯O interactions, which are found in imidazole with bridging or terminal oxygen atoms, and oxygen atoms from citrate or water molecules. Hydrogen bonds in the form of N–H⋯N are only observed in 1.
Trans-[(MoVO)2O(H2homocit)2(bpy)2]·4H2O (3) is dimeric bipyridine homocitrato oxomolybdenum(V) complex as shown in Fig. 2 and S9.† Mo atoms in 3 are hexacoordinate in distorted geometries and surrounded octahedrally by one μ2-O, one terminal O, two O atoms of homocitrate and two N atoms of 2,2′-bipyridine. The homocitrate anions chelate to the Mo via α-alkoxy and monodentate α-carboxy groups, while the β- and γ-carboxylic acidic groups remain free. The homocitrate coordination in 3 is similar to those in FeMo-cofactors of nitrogenases.5–7 Water molecules act as proton sources around the α-alkoxy and carboxy groups of homocitrate. It should be highlighted that one water molecule acts as a hydrogen donor for the α-alkoxy group among the crystallized water molecules, which shows strong hydrogen bonding between the water molecule and the α-alkoxy group.
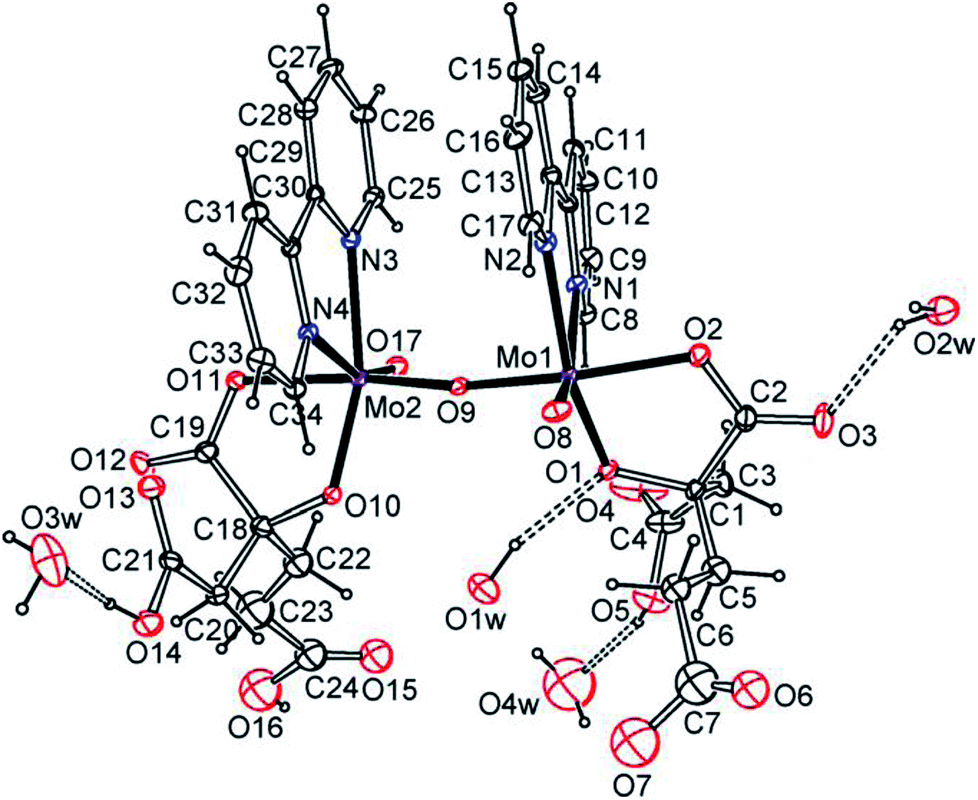 | ||
| Fig. 2 ORTEP plot of the molecular structure of trans-[(MoVO)2O(H2homocit)2(bpy)2]·4H2O (3) at the 20% probability levels. [O1⋯O1w 2.74(2), O3⋯O2w 2.74(2), O14⋯O3w 2.65(2), O5⋯O4w 2.60(4) Å]. | ||
As shown by X-ray structural analyses (Fig. 3 and S10†), (Et4N)[MoVIO2Cl(H2cit)]·H2O (4) is a monomeric oxomolybdenum(VI) complex. 4 is a product that is captured by a large cation (Et4N)+, while binuclear or tetranuclear molybdenum citrato complexes were usually isolated in acidic conditions in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio.57,66,67 The molybdenum atom is hexa-coordinated in an approximate octahedral geometry. The citrate ligand uses its α-alkoxy, α-carboxy and one β-carboxy acidic groups as a tridentate ligand, while the other β-carboxy acidic group is free. Unlike the coordinated α-hydroxy groups of citrates in 1 and 2, no protonation was observed for the α-alkoxy group even under strongly acidic conditions (pH = 0.5). The tridentate coordination mode of citrate in 4 is similar to those of K4[MoVIO3(cit)]·2H2O (6) and (NH4)4[MoVIO3(cit)]·2H2O (7),58,66 while both β-carboxy groups of citrate are protonated.
:1 ratio.57,66,67 The molybdenum atom is hexa-coordinated in an approximate octahedral geometry. The citrate ligand uses its α-alkoxy, α-carboxy and one β-carboxy acidic groups as a tridentate ligand, while the other β-carboxy acidic group is free. Unlike the coordinated α-hydroxy groups of citrates in 1 and 2, no protonation was observed for the α-alkoxy group even under strongly acidic conditions (pH = 0.5). The tridentate coordination mode of citrate in 4 is similar to those of K4[MoVIO3(cit)]·2H2O (6) and (NH4)4[MoVIO3(cit)]·2H2O (7),58,66 while both β-carboxy groups of citrate are protonated.
 | ||
| Fig. 3 ORTEP plot of the anion structure in (Et4N)[MoVIO2Cl(H2cit)]·H2O (4) at the 20% probability levels. | ||
Water cluster and hydrogen bonds also play important roles in the stabilization of the host structures. A crystallized water molecule and a carboxy oxygen atom alternately connect to each other to form a water chain, and two water chains connect to form a band by hydrogen bonds in 4 as shown in Fig. S11.† The average O⋯O distance of water layer is 2.825 Å shown in Table S3.† This is longer than the corresponding value in ice Ih (2.759 Å)68 and shorter than in liquid water (2.854 Å),69,70 but within the values in the ice II phase (2.77–2.84 Å).71
Previously, the mimic complexes [MoIV3SO3(glyc)2(im)5]·im·H2O (13) and Na2[MoIV3SO3(R,S-lact)3(im)3]·10H2O (14) in Table 1 have been used for comparison with FeMo-cofactors in nitrogenases, which provide indirect evidence for the protonation of homocitrate in the FeMo-cofactor MoIII/IVFe7S9C(S-cys)(N-His)(homocit).72 Here the longer MoIV–O distances in 1 and 2 [2.192(4)av Å in 1 and 2.201(4)av Å in 2] further support this proposal that the oxygen atoms in the MoIII/IV–O (α-alkoxy/hydroxy) of FeMo-co (16 and 17) should be protonated,20–25,73 which are obviously longer than the MoVI–O (α-alkoxy) distances [1.975(7)av Å] in 3–14 and similar to the MoIV–O (α-hydroxy) distances [2.204(4)av Å] in 15. The oxygen atom coordinated with molybdenum(IV/VI) was protonated, while the α-alkoxy group coordinated with molybdenum(VI) in 3 and 4 was deprotonated even under extreme acidic condition. This indicates that the O (α-alkoxy) coordinated with Mo(III/IV) atom had a higher charge density and was more easily protonated. The MoIV/VI–O (α-carboxy) and MoVI–O (β-carboxy) distances in 1 and 2 were shorter than those of 4 and 6–14. The characteristic MoIV–O–MoVI bridges were in apparent asymmetric configurations [1.831(4)av and 2.039(4)av Å]. The Mo–N, Mo–μ2-O, Mo–μ3-O, Mo–Mo distances in 1, 2, 3, 5, 13–15 and 18–34 have also been listed for comparison in Table S4.†42,43,47,59,72,74–81
| Complexes (Mon+) | Mo–Oα-alkoxy/hydroxy | Mo–Oα-carboxy | Mo–Oβ-carboxy |
|---|---|---|---|
| 1(IV/VI) | 2.151(4)av(IV) | 2.101(5)av | 2.071(5)av(IV) |
| 2.192(4)av(VI) | 2.259(5)av(VI) | ||
| 2(IV/VI) | 2.189(4)av(IV) | 2.104(4)av | 2.079(4)av(IV) |
| 2.201(4)av(VI) | 2.269(4)av(VI) | ||
| 5(V)81 | 1.982(3)av | 2.076(3)av | |
| 3(V) | 1.952(1)av | 2.068(2)av | |
| 4(VI) | 1.931(4) | 2.178(3) | 2.433(4) |
| 6(VI)66 | 2.052(2) | 2.237(7) | 2.411(3) |
| 7(VI)58 | 2.054(1) | 2.213(1) | 2.307(2) |
| 8(VI)56 | 1.957(7)av | 2.219(7)av | |
| 9(VI)58 | 1.919(2) | 2.178(3) | 2.538(2) |
| 10(VI)65 | 1.984(1)av | 2.209(1)av | |
| 11(VI)64 | 1.942(4)av | 2.188(4)av | 2.322(4)av |
| 12(VI)64 | 1.939(3)av | 2.197(3)av | 2.304(3)av |
| 13(IV)72 | 1.986(2)av | 2.127(2)av | |
| 14(IV)72 | 1.999(7)av | 2.133(6)av | |
| 15(IV)80 | 2.092(3)/2.204(4)av | 2.083(4)/2.118(4)av | |
| 16a82 | 2.351 | 2.293 | |
| 16b8 | 2.199 | 2.181 | |
| 16c83 | 2.250 | 2.171 | |
| 16d9 | 2.178 | 2.212 | |
| 1732 | 2.252 | 2.292 |
Solution 13C and 1H NMR spectra
Solution 13C and 1H NMR spectra are shown in Fig. 4, S12–S21 and Table S5,† and provide valuable information on the coordination environments of 1, 2 and 4. Only one set of species was observed for 1 and 2 respectively, showing no decomposition over three months. The coordinated α-carboxy and β-carboxy carbons in 1 and 2 are at around 185.5–185.2 ppm and 181.7–180.5 ppm respectively. Large downfield shifts (Δα-carboxy = 5.9–6.2 ppm; Δβ-carboxy = 4.5–5.7 ppm) are observed compared to those of the free citrate (α-carboxy 179.3 ppm; β-carboxy 176.0 ppm). The coordinated α-hydroxy (83.5 ppm in 1 and 82.9 ppm in 2) and α-alkoxy carbons (87.0 ppm in 4) also show large downfield shifts compared to those of the free citrate (75.9 ppm), respectively. The 13C NMR spectral signals for protonated α-hydroxy groups in 1 and 2 show an obvious shift to the highfield compared with the α-alkoxy in 4 and other molybdenum citrates, showing the influence of protonation. The α-carboxy and methylene carbons in 4(VI) show downfield shifts compared to those in 1(IV/VI) and 2(IV/VI).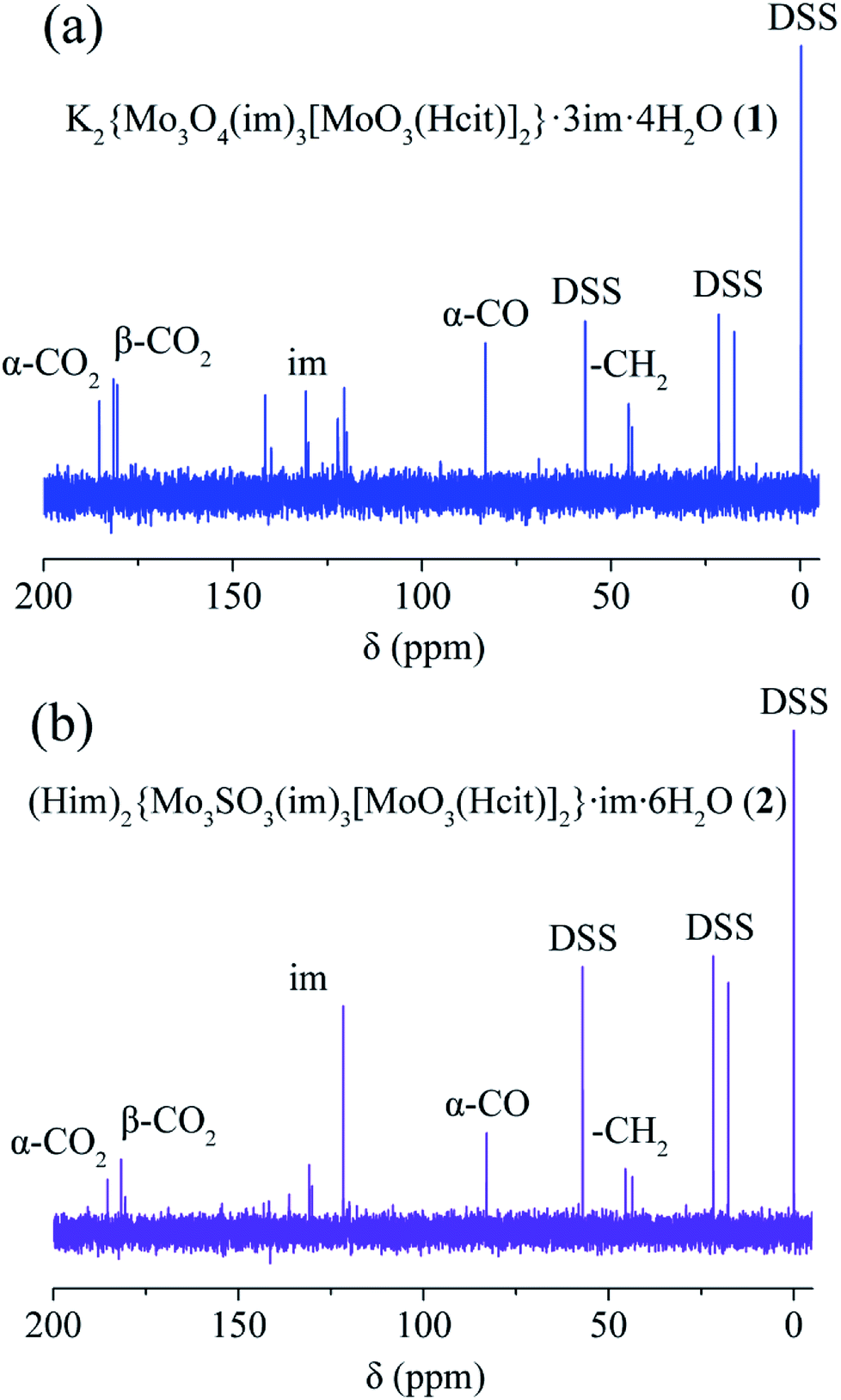 | ||
| Fig. 4 13C NMR spectra of K2{MoIV3O4(im)3[MoVIO3(Hcit)]2}·3im·4H2O (1, a), (Him)2{MoIV3SO3(im)3[MoVIO3(Hcit)]2}·im·6H2O (2, b). | ||
Mo XPS spectra and bond valence calculations
As shown in Table 2, the theoretical bond valence calculations for 1–3 show 4.274av and 5.984av for the molybdenum atoms in 2, respectively, which are close to charge balances +4 and +6. The Mo 3d5/2–Mo 3d3/2 doublet was registered at binding energies (BE) 227.3 eV, 230.3 eV and 232.5 eV in the XPS spectrum of 2 as shown in Fig. 5 and S22.† The XPS spectrum of bulk MoO3 in the Mo 3d region showed a single spin–orbit doublet with a Mo 3d5/2 binding energy of 233.0 eV.84,85 The widening of the Mo 3d level is associated with the presence of more than one molybdenum states.86–89 The Mo 3d5/2 peak at BE = 232.5 eV is characteristic of the MoVI presence, while Mo 3d5/2 and Mo 3d3/2 peaks at BE = 230.3 eV and 227.3 eV are attributed to the MoIV species.| Complexes | Atom | N | ∑Sij | Δ |
|---|---|---|---|---|
| 1 | Mo(1) | 4+ | 4.341 | 0.341 |
| Mo(2) | 4+ | 4.263 | 0.263 | |
| Mo(3) | 4+ | 4.283 | 0.283 | |
| Mo(4) | 6+ | 5.993 | 0.007 | |
| Mo(5) | 6+ | 5.970 | 0.030 | |
| 2 | Mo(1) | 4+ | 4.439 | 0.439 |
| Mo(2) | 4+ | 4.207 | 0.207 | |
| Mo(3) | 4+ | 4.177 | 0.177 | |
| Mo(4) | 6+ | 5.944 | 0.056 | |
| Mo(5) | 6+ | 6.023 | 0.023 | |
| 3 | Mo(1) | 5+ | 5.208 | 0.208 |
| Mo(2) | 5+ | 5.256 | 0.256 | |
| 4 | Mo(1) | 6+ | 6.012 | 0.012 |
 | ||
| Fig. 5 XPS spectrum (Mo 3d level) of (Him)2{MoIV3SO3(im)3[MoVIO3(Hcit)]2}·im·6H2O (2). | ||
EPR spectrum and magnetic susceptibility
Theoretical bond valence calculations give the valences 5.232av for molybdenum atoms in 3, which is close to a charge balance of +5. The two molybdenum atoms in 3 are independently paramagnetic, based on the X-band of solid state EPR spectrum at 90 K as shown in Fig. 6.90,91 The EPR spectrum of 3 appeared to be anisotropic, and the g value was 1.9089. The bands which exhibit S = 1/2 signals are consistent with a 4d1 metal center Mo5+, while neither Mo4+ or Mo6+ in 2 had an EPR signal. 2 and 3 with valences of Mo(IV/VI) or Mo(V) atoms exhibit diamagnetism as shown in Fig. S23 and S24† respectively.92–94 | ||
| Fig. 6 EPR spectrum of trans-[(MoVO)2O(H2homocit)2(bpy)2]·4H2O (3). | ||
IR and UV-visible spectra
1–4 were also characterized by IR and UV-Vis spectroscopies respectively. Based on the IR spectra shown in Fig. S25–S27,† vibrations of the symmetric and asymmetric carboxy groups, Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and Mo–O–Mo bonds appeared at 1683–1612 cm−1, 1427–1364 cm−1, 949–850 cm−1 and 722–704 cm−1, respectively. The absorptions of carboxy acidic groups for 3 and 4 appeared at 1724 cm−1 and 1732 cm−1. Due to the chelation of the five-membered rings with molybdenum atoms, the symmetric and asymmetric absorption peaks of the carboxy groups were red shifted to low wave numbers. In the UV-Vis spectra, the strong absorption peaks were due to ligand-based π–π* transitions (206 nm in 1 and 207 nm in 2) and intra ligand transitions (230 nm in 1 and 231 nm in 2) as shown in Fig. S28.† The strong absorption peaks of bipyridine appeared at 280 nm in 3 due to the N-chelating ligand-based π–π* transition as shown in Fig. S29.†
O and Mo–O–Mo bonds appeared at 1683–1612 cm−1, 1427–1364 cm−1, 949–850 cm−1 and 722–704 cm−1, respectively. The absorptions of carboxy acidic groups for 3 and 4 appeared at 1724 cm−1 and 1732 cm−1. Due to the chelation of the five-membered rings with molybdenum atoms, the symmetric and asymmetric absorption peaks of the carboxy groups were red shifted to low wave numbers. In the UV-Vis spectra, the strong absorption peaks were due to ligand-based π–π* transitions (206 nm in 1 and 207 nm in 2) and intra ligand transitions (230 nm in 1 and 231 nm in 2) as shown in Fig. S28.† The strong absorption peaks of bipyridine appeared at 280 nm in 3 due to the N-chelating ligand-based π–π* transition as shown in Fig. S29.†
Cyclic voltammograms and mass spectra
Cyclic voltammograms of 1, 2 and 4 scanning from −1.600 V to 1.600 V were performed in 1 mol L−1 Na2SO4 aqueous solution as shown in Fig. S30–S32.† The CV scan measurements were made in the positive direction, and then back to the cathode reduction region. As for the cyclic voltammograms of 1 and 2, the reductive waves of MoVI appeared at −0.567 V and −0.564 V, respectively. The oxidized peak of MoIV in 1 was at +0.776 V, while there was no oxidized peak in 2. 2 with a central sulfur atom is more easily reduced than 1 with its central oxygen atom. The fact indicates that MoIV atoms in [MoIV3SO3]4+ were more stable than those in [MoIV3O4]4+ and not easily oxidized. All redox processes were single and irreversible, which are different from those previously reported for [MoIV3O4(ox)3]2−.47,95 Mass spectra of major anions in 1, 2 and 3 are shown in Fig. 7 and S33.† As the ion source bombarded the complexes, the imidazoles and chlorides that are weakly coordinated with the metal dissociated, and there were no peaks associated with the complete anions in the spectra, except for {MoIV3O4[MoVIO3(Hcit)]2}2− ({MoIV3SO3[MoVIO3(Hcit)]2}2−) and [MoVIO2(Hcit)]1−. | ||
| Fig. 7 Mass spectra of {Mo3O4[MoVIO3(Hcit)]2}2− and {Mo3SO3[MoVIO3(Hcit)]2}2− in K2{MoIV3O4(im)3[MoVIO3(Hcit)]2}·3im·4H2O (1, a) and (Him)2{MoIV3SO3(im)3[MoVIO3(Hcit)]2}·im·6H2O (2, b) respectively. | ||
Conclusions
A series of molybdenum(IV/V/VI) citrates and homocitrate K2{MoIV3O4(im)3[MoVIO3(Hcit)]2}·3im·4H2O (1), (Him)2{MoIV3SO3(im)3[MoVIO3(Hcit)]2}·im·6H2O (2), trans-[(MoVO)2O(H2homocit)2(bpy)2]·4H2O (3) and (Et4N)[MoVIO2Cl(H2cit)]·H2O (4) were prepared from aqueous solutions. Pentanuclear imidazole hydrocitrato molybdenum(IV/VI) complexes 1 and 2 were obtained with incomplete cubane units [MoIV3SO3]4+ and [MoIV3O4]4+ units. The charge balances, theoretical bond valence calculations and XPS spectrum indicate molybdenum atoms with valences of +4 or +6 respectively, which are different from the mixed-valence complexes in the adjacent valence state. The incomplete cubane cluster units, [MoIV3SO3]4+ and [MoIV3O4]4+, with monodentate imidazoles were coordinated by two [MoVIO3(Hcit)]3− anions on both sides to form pentanuclear skeleton structures. It is worth noting that the α-alkoxy groups of coordinated citrates in 1 and 2 were protonated, indicating O (α-hydroxy) atoms in molybdenum(IV) complexes with higher charge densities are more easily protonated. The structures in 1 and 2 are important for the further study of imidazole molybdenum complexes with α-hydroxycarboxylates. The homocitrate anions in 3 chelate to Mo via the α-alkoxy and monodentate α-carboxy groups, while the remaining β- and γ-carboxylic acid groups remained free. Moreover, the monomeric oxomolybdenum citrate complex (Et4N)[MoVIO2Cl(H2cit)]·H2O (4) is captured by large cations [Et4N]+ with α-alkoxy coordination in strong acidic conditions. The solution behaviors of 1 and 2 were discussed based on 1H and 13C NMR spectroscopies giving only one set of species, and showing no decomposition of 1 and 2. The coordinated α-alkoxy/hydroxyl and carboxy carbons in 1, 2 and 4 show large downfield shifts compared with those of the free citrate. The cyclic voltammograms of 1 and 2 indicate that MoIV atoms in [MoIV3SO3]4+ were more stable than that in [MoIV3O4]4+. 2 and 3 exhibited diamagnetically.Experimental
Preparations of K2{MoIV3O4(im)3[MoVIO3(Hcit)]2}·3im·4H2O (1) and (Him)2{MoIV3SO3(im)3[MoVIO3(Hcit)]2}·im·6H2O (2)
(NH4)6Mo7O24·4H2O (0.35 g, 0.29 mmol), citric acid (0.42 g, 2.0 mmol), imidazole (0.136 g, 2.0 mmol) were dissolved in 10 mL water, then excess hydrazine dihydrochloride (0.42 g, 4.0 mmol) was added in an anaerobic environment. The pH value was adjusted to 9.5 by the addition of KOH solution (5.0 M). The reactants were heated at 120 °C for 24 h in a 10 mL Teflon-lined stainless steel container under aqueous reaction conditions and cooled to room temperature. Product 1 was isolated as yellow needle crystals after one month in 6.7% yields (0.042 g) based on molybdenum. Found (calcd for C30H42N12O28Mo5K2): C, 22.7 (22.9); H, 3.0 (2.7); N, 10.4 (10.7). IR (KBr, cm−1): ν(C–H) 3124w, 2954w, 2852w; νas(CO2) 1640vs; νs(CO2) 1397s; νs(MoO) 916s; νas(Mo–O–Mo) 704s. UV-Vis (H2O, nm): 206, 230. Solution 1H NMR (400 MHz, D2O, 25 °C, DSS): δ = 8.64 (s, 2H; CH), 8.48 (s, 1H; CH), 8.31 (s, 2H; CH), 7.73 (s, 3H; CH), 7.31 (t, J = 3 Hz, 3H; CH), 7.26 (d, J = 2 Hz, 7H; CH), 2.91 (t, J = 8 Hz, 8H; CH2); solution 13C NMR (100 MHz, D2O, 25 °C, DSS): δ = 185.2 ppm, 181.5 ppm, 180.5 ppm, 141.4 ppm, 130.7 ppm, 120.6 ppm, 83.3 ppm, 45.4 ppm, 44.5 ppm.
Similarly, the synthesis of 2 was performed, except Na2S2O4 (0.35 g, 2.0 mmol) was used instead of hydrazine dihydrochloride and the pH value was adjusted to 6.0 with the addition of NaOH solution (5.0 M). Product 2 was isolated as green plates after one month in 10.5% yields (0.065 g) based on molybdenum. Found (calcd for C30H48N12O29SMo5): C, 22.9 (23.2); H, 3.3 (3.1); N, 10.7 (10.8). IR (KBr, cm−1): ν(C–H) 3128w, 2957w, 2853w; νas(CO2) 1626vs; νs(CO2) 1399s; νs(MoO) 921s; νas(Mo–O–Mo) 712s. UV-Vis (H2O, nm): 207, 231. Solution 1H NMR (400 MHz, D2O, 25 °C, DSS): δ = 8.69 (d, J = 10 Hz, 4H; CH), 8.61 (s, 1H; CH), 7.86 (s, 1H; CH), 7.77 (s, 2H; CH), 7.45 (s, 7H; CH), 7.35 (s, 2H; CH), 7.30 (s, 1H; CH), 2.81 (t, J = 8 Hz, 8H; CH2); solution 13C NMR (100 MHz, D2O, 25 °C, DSS): δ = 185.4 ppm, 181.7 ppm, 130.8 ppm, 121.7 ppm, 82.9 ppm, 45.5 ppm, 43.6 ppm.
Preparation of trans-[(MoVO)2O(H2homocit)2(bpy)2]·4H2O (3)
The homocitric acid γ-lactone (0.060 g, 0.30 mmol)96 synthesized by five steps was dissolved in a minimal amount of water. The pH was adjusted to 11 by the addition of potassium hydroxide to generate the acyclic homocitrate. The mixture was stirred for two days to complete the hydrolysis at room temperature. (NH4)6Mo7O24·4H2O (0.026 g, 0.021 mmol) and 1.0 mL 2,2′-bipyridine ethanol solution (0.15 mol mL−1) was added and the solution was stirred for one hour, then excess hydrazine dihydrochloride (0.060 g, 0.60 mmol) was added. The pH value was adjusted to 2.5 with the addition of hydrochloric acid. A dark purple solution was obtained when 1.0 mL 2,2′-bipyridine ethanol solution (0.30 mmol mL−1) was added. Product 3 was isolated as purple plates in 20.7% yields (0.016 g) based on molybdenum after one day. Found (calcd for C34H40N4O21Mo2): C, 39.3 (39.6); H, 4.0 (3.9); N, 5.3 (5.4). IR (KBr, cm−1): ν(β-CO2H) 1724vs; νas(CO2) 1639vs; νs(CO2) 1427s, 1402s; νs(MoO) 947m, 871w, 850m; νas(Mo–O–Mo) 722m. UV (DMSO, nm): 280, 315, 422, 593.
Preparation of (Et4N)[MoVIO2Cl(H2cit)]·H2O (4)
(NH4)4[MoVIO3(cit)]·2H2O58 (0.44 g, 1.0 mmol) and tetraethylammonium chloride (0.17 g, 1.0 mmol) were mixed in 10 mL water. The pH value was adjusted to 0.5 with dilute hydrochloric acid. The resulting solution was allowed to stand at room temperature and slow evaporation afforded colorless crystals of 4 with 52.5% yield (0.026 g) based on molybdenum. Found (calcd for C14H28NO10ClMo): C, 33.4 (33.5); H, 5.4 (5.6); N, 2.5 (2.8). IR (KBr, cm−1): ν(β-CO2H) 1732vs; νas(CO2) 1683vs, 1612vs; νs(CO2) 1364vs; νs(MoO) 949vs, 909vs, 850s. Solution 1H NMR (400 MHz, D2O, 25 °C, DSS): δ = 3.25 (qAB, J = 7 Hz, 8H; CH2–N), 3.03–2.86 (d–d, J = 16 Hz, 4H; CH2), 1.28–1.24 (m, J = 2 Hz, 12H; CH3); solution 13C NMR (100 MHz, D2O, 25 °C, DSS): δ = 185.8 ppm, 175.8 ppm, 87.0 ppm, 54.5 ppm, 45.6 ppm, 9.2 ppm.
X-ray structure determinations
Crystals 1–4 were measured on an Oxford Gemini CCD diffractometer with graphite monochromatic Mo-Kα radiation (λ = 0.71073 Å) or Cu-Kα radiation (λ = 1.54184 Å) at 173 K. The structures were primarily solved by Olex2 and ShelXT in WinGX program97 and refined by full-matrix least-squares procedures with anisotropic thermal parameters for all of the nonhydrogen atoms with SHELX-2014/7.98–100 Crystallographic data and structural refinements for 1–4 are listed in Table S6.† Selected bond distances and angles are listed in Tables S7–S10.†Physical measurements
Infrared spectra were recorded as Nujol mulls between KBr plates on a Nicolet 380 FT-IR spectrometer. Elemental analyses were performed with a Vario EL III elemental analyzer. UV-visible spectra were recorded on a Shimadzu UV-Vis 2550 spectrophotometer. Solution 1H NMR and 13C NMR spectra were recorded on a Bruker AV 500 NMR spectrometer with D2O using DSS (sodium 2,2-dimethyl-2-silapentane-5-sulfonate) as an internal reference. The magnetic susceptibility of microcrystalline sample restrained in parafilm was measured on a Quantum Design MPMS.XL-7 magnetometer with an applied field of 5 kOe. Cyclic voltammetry measurements were carried out on a CHI 720E electrochemical station using a conventional three-electrode single compartment cell at room temperature under a nitrogen atmosphere. The working electrode was glassy carbon, a platinum plate was used as the counter electrode and Hg/Hg2Cl2 as the reference electrode. Mass spectra were recorded on an Agilent 6224 mass spectrometer. The XPS spectra were recorded on a Qtac-100 LEISS-XPS spectrometer with a hemispherical detector operating at constant pass energy (PE = 40 eV) and pressure 1 × 10−8 Pa. An X-ray source of power 300 W (I = 20 mA, U = 15 kV) and Al Kα radiation (Eq = hν = 1486.6 eV) were used. The sample temperature was 25 °C. All the binding energies (BE) were referenced to the C 1s line at 285 eV. Electron paramagnetic resonance (EPR) was recorded on Bruker EMX-10/12 electron paramagnetic resonance spectrometer. The magnetic susceptibility of microcrystalline samples restrained in parafilm was measured on a Quantum Design MPMS.XL-7 magnetometer with an applied field of 5 kOe.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the supports from the National Science Foundation (21773196) and the National Science Foundation of Fujian (2016I0101) for their generous financial supports. Thanks to engineers Shen-Shui Yu, Shui-Chao Lin, Hai-Yan Shi, Yan-Ping Zheng, Dr An-Ni Feng and Dr Ying-Zi Han for their helps in spectral measurements.Notes and references
- K. Heinze, Coord. Chem. Rev., 2015, 300, 121–141 CrossRef CAS.
- R. Hille, J. Hall and P. Basu, Chem. Rev., 2014, 114, 3963–4038 CrossRef CAS PubMed.
- T. R. Hoover, A. D. Robertson, R. L. Cerny, R. N. Hayes, J. Imperial, V. K. Shah and P. W. Ludden, Nature, 1987, 329, 855–857 CrossRef CAS PubMed.
- T. R. Hoover, J. Imperial, P. W. Ludden and V. K. Shah, Biochemistry, 1989, 28, 2768–2771 CrossRef CAS PubMed.
- J. Kim and D. C. Rees, Nature, 1992, 360, 553–560 CrossRef CAS PubMed.
- J. Kim and D. C. Rees, Science, 1992, 257, 1677–1682 CrossRef CAS PubMed.
- M. M. Georgiadis, H. Komiya, P. Chakrabarti, D. Woo, J. J. Kornuc and D. C. Rees, Science, 1992, 257, 1653–1659 CrossRef CAS PubMed.
- O. Einsle, F. A. Tezcan, S. L. A. Andrade, B. Schmid, M. Yoshida, J. B. Howard and D. C. Rees, Science, 2002, 297, 1696–1700 CrossRef CAS PubMed.
- T. Spatzal, M. Aksoyoglu, L. Zhang, S. L. A. Andrade, E. Schleicher, S. Weber, D. C. Rees and O. Einsle, Science, 2011, 334, 940 CrossRef CAS PubMed.
- K. M. Lancaster, M. Roemelt, P. Ettenhuber, Y. Hu, M. W. Ribbe, F. Neese, U. Bergmann and S. DeBeer, Science, 2011, 334, 974–977 CrossRef CAS PubMed.
- B. Schmid, M. W. Ribbe, O. Einsle, M. Yoshida, L. M. Thomas, D. R. Dean, D. C. Rees and B. K. Burgess, Science, 2002, 296, 352–356 CrossRef CAS PubMed.
- F. Fazius, E. Shelest, P. Gebhardt and M. Brock, Mol. Microbiol., 2012, 86, 1508–1530 CrossRef CAS PubMed.
- T. M. Zabriskie and M. D. Jackson, Nat. Prod. Rep., 2000, 17, 85–97 RSC.
- H. Quezada, A. Marin Hernandez, D. Aguilar, G. Lopez, J. C. Gallardo Perez, R. Jasso Chavez, A. Gonzalez, E. Saavedra and R. Moreno Sanchez, Mol. Microbiol., 2011, 82, 578–590 CrossRef CAS PubMed.
- Y. Hu, M. C. Corbett, A. W. Fay, J. A. Webber, K. O. Hodgson, B. Hedman and M. W. Ribbe, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17125–17130 CrossRef CAS PubMed.
- Y. Hu and M. W. Ribbe, Microbiol. Mol. Biol. Rev., 2011, 75, 664–677 CrossRef CAS PubMed.
- K. L. C. Groenberg, C. A. Gormal, M. C. Durrant, B. E. Smith and R. A. Henderson, J. Am. Chem. Soc., 1998, 120, 10613–10621 CrossRef CAS.
- M. A. Walters, S. K. Chapman and W. H. Orme Johnson, Polyhedron, 1986, 5, 561–565 CrossRef CAS.
- I. Dance, Dalton Trans., 2008, 5977–5991 RSC.
- C. Y. Chen, M. L. Chen, H. B. Chen, H. Wang, S. P. Cramer and Z. H. Zhou, J. Inorg. Biochem., 2014, 141, 114–120 CrossRef CAS PubMed.
- Q. Chen, H. Chen, Z. Cao, Z. Zhou, H. Wan, Y. Li and J. Li, Sci. Sin.: Chim., 2014, 44, 1849–1864 CAS.
- B. M. Hoffman, D. Lukoyanov, Z. Y. Yang, D. R. Dean and L. C. Seefeldt, Chem. Rev., 2014, 114, 4041–4062 CrossRef CAS PubMed.
- B. Benediktsson and R. Bjornsson, Inorg. Chem., 2017, 56, 13417–13429 CrossRef CAS PubMed.
- L. Cao, O. Caldararu and U. Ryde, J. Phys. Chem. B, 2017, 121, 8242–8262 CrossRef CAS PubMed.
- C. N. Morrison, T. Spatzal and D. C. Rees, J. Am. Chem. Soc., 2017, 139, 10856–10862 CrossRef CAS PubMed.
- J. Bergwerff, M. Jansen, B. Leliveld, T. Visser, K. Dejong and B. Weckhuysen, J. Catal., 2006, 243, 292–302 CrossRef CAS.
- O. V. Klimov, A. V. Pashigreva, M. A. Fedotov, D. I. Kochubey, Y. A. Chesalov, G. A. Bukhtiyarova and A. S. Noskov, J. Mol. Catal. A: Chem., 2010, 322, 80–89 CrossRef CAS.
- M. A. Lélias, P. J. Kooyman, L. Mariey, L. Oliviero, A. Travert, J. van Gestel, J. A. R. van Veen and F. Maugé, J. Catal., 2009, 267, 14–23 CrossRef.
- Y. Zeng, Z. Li, M. Ma and S. Zhou, Electrochem. Commun., 2000, 2, 36–38 CrossRef CAS.
- L. S. Sanches, S. H. Domingues, C. E. B. Marino and L. H. Mascaro, Electrochem. Commun., 2004, 6, 543–548 CrossRef CAS.
- E. Gómez, E. Pellicer, M. Duch, J. Esteve and E. Vallés, Electrochim. Acta, 2006, 51, 3214–3222 CrossRef.
- S. M. Mayer, C. A. Gormal, B. E. Smith and D. M. Lawson, J. Biol. Chem., 2002, 277, 35263–35266 CrossRef CAS PubMed.
- S. Reinelt, E. Hofmann, T. Gerharz, M. Bott and D. R. Madden, J. Biol. Chem., 2003, 278, 39189–39196 CrossRef CAS PubMed.
- A. Bino, F. A. Cotton and Z. Dori, J. Am. Chem. Soc., 1978, 100, 5252–5253 CrossRef CAS.
- T. Shibahara, H. Hattori and H. Kuroya, J. Am. Chem. Soc., 1984, 106, 2710–2711 CrossRef CAS.
- S. Duval, S. Floquet, C. Simonnet Jegat, J. Marrot, R. N. Biboum, B. Keita, L. Nadjo, M. Haouas, F. Taulelle and E. Cadot, J. Am. Chem. Soc., 2010, 132, 2069–2077 CrossRef CAS PubMed.
- T. F. Beltran, R. Llusar, M. Sokolov, M. G. Basallote, M. J. Fernandez Trujillo and J. A. Pino Chamorro, Inorg. Chem., 2013, 52, 8713–8722 CrossRef CAS PubMed.
- M. G. Basallote, M. J. Fernandez Trujillo, J. A. Pino Chamorro, T. F. Beltran, C. Corao, R. Llusar, M. Sokolov and C. Vicent, Inorg. Chem., 2012, 51, 6794–6802 CrossRef CAS PubMed.
- N. Avarvari, K. Kiracki, R. Llusar, V. Polo, I. Sorribes and C. Vicent, Inorg. Chem., 2010, 49, 1894–1904 CrossRef CAS PubMed.
- A. G. Algarra, M. G. Basallote, M. J. Fernandez Trujillo, M. Feliz, E. Guillamon, R. Llusar, I. Sorribes and C. Vicent, Inorg. Chem., 2010, 49, 5935–5942 CrossRef CAS PubMed.
- C. Alfonso, M. Feliz, V. S. Safont and R. Llusar, Dalton Trans., 2016, 45, 7829–7835 RSC.
- P. A. Abramov, A. Yu Laricheva, E. V. Peresypkina, I. V. Mirzaeva, N. K. Moroz and M. N. Sokolov, Inorg. Chim. Acta, 2012, 383, 7–12 CrossRef CAS.
- B. Modec and P. Bukovec, Inorg. Chim. Acta, 2015, 424, 226–234 CrossRef CAS.
- B. Kowalewski, J. Poppe, U. Demmer, E. Warkentin, T. Dierks, U. Ermler and K. Schneider, J. Am. Chem. Soc., 2012, 134, 9768–9774 CrossRef CAS PubMed.
- B. Chen, Z. Lin, B. Wang, X. Feng, L. Fan, S. Yang, X. Huang and C. Hu, CrystEngComm, 2013, 15, 7410–7413 RSC.
- R. G. Carden, J. J. Ohane, R. D. Pike and P. M. Graham, Organometallics, 2013, 32, 2505–2508 CrossRef CAS.
- Q. L. Chen, H. B. Chen, Z. X. Cao and Z. H. Zhou, Dalton Trans., 2013, 42, 1627–1636 RSC.
- D. A. Kuznetsov, T. A. Bazhenova, I. V. Fedyanin, V. M. Martynenko, A. F. Shestakov, G. N. Petrova and N. S. Komarova, Dalton Trans., 2016, 45, 16309–16316 RSC.
- A. Alberola, M. Fourmigué, C. J. Gómez García, R. Llusar and S. Triguero, New J. Chem., 2008, 32, 1103–1109 RSC.
- Q. L. Chen, W. Huang, M. L. Chen, J. Lin, Z. X. Cao and Z. H. Zhou, RSC Adv., 2014, 4, 26499–26507 RSC.
- G. Y. Zhu, M. Meng, Y. N. Tan, X. Xiao and C. Y. Liu, Inorg. Chem., 2016, 55, 6315–6322 CrossRef CAS PubMed.
- M. K. Wojnar, J. W. Ziller and A. F. Heyduk, Eur. J. Inorg. Chem., 2017, 2017, 5571–5575 CrossRef CAS.
- J. S. Maass, M. Zeller, D. Holmes, C. A. Bayse and R. L. Luck, J. Cluster Sci., 2011, 22, 193–210 CrossRef CAS.
- H. Akashi, N. Machida, T. Kamada, H. Nihashi, D. Aya, K. Yamaguchi, H. Takagi and T. Shibahara, Bull. Chem. Soc. Jpn., 2017, 90, 728–738 CrossRef CAS.
- Q. He, E. Wang, C. Hu, L. Xu, Y. Xing, Y. Lin and H. Jia, J. Mol. Struct., 1999, 484, 139–143 CrossRef CAS.
- Z. H. Zhou, H. L. Wan and K. R. Tsai, Dalton Trans., 1999, 4289–4290 RSC.
- Z. H. Zhou, Y. F. Deng, Z. X. Cao, R. H. Zhang and Y. L. Chow, Inorg. Chem., 2005, 44, 6912–6914 CrossRef CAS PubMed.
- R. H. Zhang, X. W. Zhou, Y. C. Guo, M. L. Chen, Z. X. Cao, Y. L. Chow and Z. H. Zhou, Inorg. Chim. Acta, 2013, 406, 27–36 CrossRef CAS.
- Y. W. Lin, Y. P. Tong, C. Yang and Y. R. Lin, Inorg. Chem. Commun., 2009, 12, 252–254 CrossRef CAS.
- L. Wang, P. Yin, J. Zhang, J. Hao, C. Lv, F. Xiao and Y. Wei, Chem.–Eur. J., 2011, 17, 4796–4801 CrossRef CAS PubMed.
- D. Shriver, M. Weller, T. Overton, J. Rourke and F. Armstrong, Inorganic Chemistry, W. H. Freeman and Company, Great Britain, 2014 Search PubMed.
- D. Li, J. Xu, Z. Li, Y. Xing, R. Wang, S. Liu, H. Sun and T. Wang, Synth. React. Inorg. Met.-Org. Chem., 2000, 30, 319–333 CrossRef CAS.
- D. M. Li, Y. H. Xing, Z. C. Li, J. Q. Xu, W. B. Song, T. G. Wang, G. D. Yang, N. H. Hu, H. Q. Jia and H. M. Zhang, J. Inorg. Biochem., 2005, 99, 1602–1610 CrossRef CAS PubMed.
- Z. H. Zhou, S. Y. Hou, Z. X. Cao, K. R. Tsai and Y. L. Chow, Inorg. Chem., 2006, 45, 8447–8451 CrossRef CAS PubMed.
- Z. H. Zhou, H. Wang, P. Yu, M. M. Olmstead and S. P. Cramer, J. Inorg. Biochem., 2013, 118, 100–106 CrossRef CAS PubMed.
- Z. H. Zhou, H. L. Wan and K. R. Tsai, Inorg. Chem., 2000, 39, 59–64 CrossRef CAS PubMed.
- X. Zhang, M. Hejazi, S. J. Thiagarajan, W. R. Woerner, D. Banerjee, T. J. Emge, W. Xu, S. J. Teat, Q. Gong, A. Safari, R. Yang, J. B. Parise and J. Li, J. Am. Chem. Soc., 2013, 135, 17401–17407 CrossRef CAS PubMed.
- D. Eisenberg and W. Kauzmann, The Structure and Properties of Water, Oxford Univ. Press, 1969 Search PubMed.
- R. J. Speedy, J. D. Madura and W. L. Jorgensen, J. Phys. Chem., 1987, 91, 909–913 CrossRef CAS.
- A. C. Belch and S. A. Rice, J. Chem. Phys., 1987, 86, 5675–5682 CrossRef CAS.
- An introduction to hydrogen bonding, ed. G. A. Jeffrey, Oxford Univ. Press, 1997 Search PubMed.
- S. Y. Wang, W. T. Jin, H. B. Chen and Z. H. Zhou, Dalton Trans., 2018, 47, 7412–7421 RSC.
- P. E. M. Siegbahn, Inorg. Chem., 2018, 57, 1090–1095 CrossRef CAS PubMed.
- S. F. Gheller, T. W. Hambley, R. T. C. Brownlee, M. J. O'Connor, M. R. Snow and A. G. Wedd, J. Am. Chem. Soc., 1983, 105, 1527–1532 CrossRef CAS.
- Z. H. Zhou, Q. Xu, J. Lin and S. W. Ng, Inorg. Chem. Commun., 2007, 10, 1461–1464 CrossRef CAS.
- B. Modec, D. Dolenc and M. Kasunic, Inorg. Chem., 2008, 47, 3625–3633 CrossRef CAS PubMed.
- A. Bino, F. A. Cotton and Z. Dori, J. Am. Chem. Soc., 1979, 101, 3842–3847 CrossRef CAS.
- J. Zhao and L. Xu, Eur. J. Inorg. Chem., 2011, 2011, 4096–4102 CrossRef CAS.
- H. Kawasaki, G. Sakane and T. Shibahara, Inorg. Chem. Commun., 2005, 8, 777–781 CrossRef CAS.
- M. N. Sokolov, S. A. Adonin, A. V. Virovets, P. A. Abramov, C. Vicent, R. Llusar and V. P. Fedin, Inorg. Chim. Acta, 2013, 395, 11–18 CrossRef CAS.
- Z. H. Zhou, C. Y. Chen, Z. X. Cao, K. R. Tsai and Y. L. Chow, Dalton Trans., 2008, 2475–2479 RSC.
- S. M. Mayer, D. M. Lawson, C. A. Gormal, S. M. Roe and B. E. Smith, J. Mol. Biol., 1999, 292, 871–891 CrossRef CAS PubMed.
- R. Sarma, B. M. Barney, S. Keable, D. R. Dean, L. C. Seefeldt and J. W. Peters, J. Inorg. Biochem., 2010, 104, 385–389 CrossRef CAS PubMed.
- D. McKay, J. S. J. Hargreaves and R. F. Howe, Catal. Lett., 2006, 112, 109–113 CrossRef CAS.
- J. S. J. Hargreaves, R. F. Howe, D. McKay, E. Morrison, J. L. Rico and M. Stockenhuber, Top. Catal., 2009, 52, 1559–1565 CrossRef CAS.
- R. I. Declerck Grimee, P. Canesson, R. M. Friedman and J. J. Fripiat, J. Phys. Chem., 1978, 82, 885–888 CrossRef CAS.
- S. Kasztelan, J. Grimblot, J. P. Bonnelle, E. Payen, H. Toulhoat and Y. Jacquin, Appl. Catal., 1983, 7, 91–112 CrossRef CAS.
- D. Nikolova, R. Edreva Kardjieva, G. Gouliev, T. Grozeva and P. Tzvetkov, Appl. Catal., A, 2006, 297, 135–144 CrossRef CAS.
- L. Portela, P. Grange and B. Delmon, J. Catal., 1995, 156, 243–254 CrossRef CAS.
- J. Mitra and S. Sarkar, Inorg. Chem., 2013, 52, 3032–3042 CrossRef CAS PubMed.
- A. S. Gowda, J. L. Petersen and C. Milsmann, Inorg. Chem., 2018, 57, 1919–1934 CrossRef CAS PubMed.
- J. A. McCleverty and M. D. Ward, Acc. Chem. Res., 1998, 31, 842–851 CrossRef CAS.
- M. K. Schmitt, O. Janka, R. Poettgen, C. Benndorf, M. de Oliveira, Jr., H. Eckert, F. Pielnhofer, A. S. Tragl, R. Weihrich, B. Joachim, D. Johrendt and H. Huppertz, Angew. Chem., Int. Ed., 2017, 56, 6449–6453 CrossRef CAS PubMed.
- N. A. Pushkarevsky, N. A. Semenov, A. A. Dmitriev, N. V. Kuratieva, A. S. Bogomyakov, I. G. Irtegova, N. V. Vasilieva, B. E. Bode, N. P. Gritsan, L. S. Konstantinova, J. D. Woollins, O. A. Rakitin, S. N. Konchenko, V. I. Ovcharenko and A. V. Zibarev, Inorg. Chem., 2015, 54, 7007–7013 CrossRef CAS PubMed.
- M. T. Paffett and F. C. Anson, Inorg. Chem., 1983, 22, 1347–1355 CrossRef CAS.
- H. B. Chen, L. Y. Chen, P. Q. Huang, H. K. Zhang, Z. H. Zhou and K. R. Tsai, Tetrahedron, 2007, 63, 2148–2152 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- G. M. Sheldrick, SHELXS-97, SHELXL-97, and SHELXTL/PC, Programs for solution and refinement of crystal structures, University of Göttingen, Göttingen, 1997 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: IR, UV-Vis, NMR, XPS, CV and mass spectra. CCDC 1571186 and 1837477–1837479. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra09134j |
| This journal is © The Royal Society of Chemistry 2019 |