
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Base-promoted lipase-catalyzed kinetic resolution of atropisomeric 1,1′-biaryl-2,2′-diols†
Gamal A. I. Moustafaab,
Kengo Kasamaa,
Koichi Higashioa and
Shuji Akai *a
*a
aGraduate School of Pharmaceutical Sciences, Osaka University, 1-6 Yamadaoka, Suita, Osaka 565-0871, Japan. E-mail: akai@phs.osaka-u.ac.jp
bDepartment of Medicinal Chemistry, Faculty of Pharmacy, Minia University, Minia 61519, Egypt
First published on 9th January 2019
Abstract
Herein we report a dramatic acceleration of the lipase-catalyzed kinetic resolution of atropisomeric 1,1′-biaryl-2,2′-diols by the addition of sodium carbonate. This result likely originates from the increased nucleophilicity of the phenolic hydroxyl group toward the acyl-enzyme intermediate. Under these conditions, various substituted C2-symmetric and non-C2-symmetric binaphthols and biphenols were efficiently resolved with ∼50% conversion in only 13–30 h with excellent enantioselectivity.
Introduction
Axially chiral biaryls are ubiquitous structural motifs in various catalysts, natural products, and pharmaceuticals.1 In particular, optically active atropisomeric 1,1′-biaryl-2,2′-diols (e.g. 1,1′-bi-2-naphthols (BINOLs)) have been widely used as chiral ligands for asymmetric transformations and as privileged scaffolds for designing numerous important chiral catalysts/ligands.2 Consequently, the development of efficient enantioselective syntheses of 1,1′-biaryl-2,2′-diols has attracted considerable attention from various research groups.3 Although the enantioselective oxidative coupling of 2-naphthols using chiral transition metals is an atom-economical route, it is mainly applicable to homochiral binaphthols with specific substitution patterns.4 Apart from the kinetic resolution (KR) of racemic precursors via non-enzymatic routes,5 the lipase-catalyzed KR of racemic atropisomeric 1,1′-biaryl-2,2′-diols has been intensively studied.6 At the core of this, racemic BINOL and its derivatives have been resolved via either hydrolytic (in aqueous media)7 or acylative (in non-aqueous media) approaches.8,9 In the latter cases, the acylative KR of racemic BINOLs using a lipoprotein lipase from Pseudomonas sp. requires long reaction times (3–14 days) to afford the corresponding (R)-mono-esters in 32–53% yields with 90–95% ee and the recovery of (S)-BINOLs with 55–89% ee (E values:10,11 41–117).8a,b We believe that acceleration of the resolution process and the expansion of substrate scope are highly demanded to establish a more practical KR. Notably, to the best of our knowledge, none of the existing enzymatic KR protocols has been applied to C1-symmetric, i.e. non-C2-symmetric biaryl diols. Therefore, we studied the development of a modular KR that enhances the enzymatic transesterification and also covers a broader range of substituted biaryl diols. In this article, we show that the addition of sodium carbonate dramatically accelerates the KR for a broad range of C1-symmetric and C2-symmetric atropoisomeric 1,1′-biaryl-2,2′-diols, while maintaining excellent enantiocontrol.Results and discussion
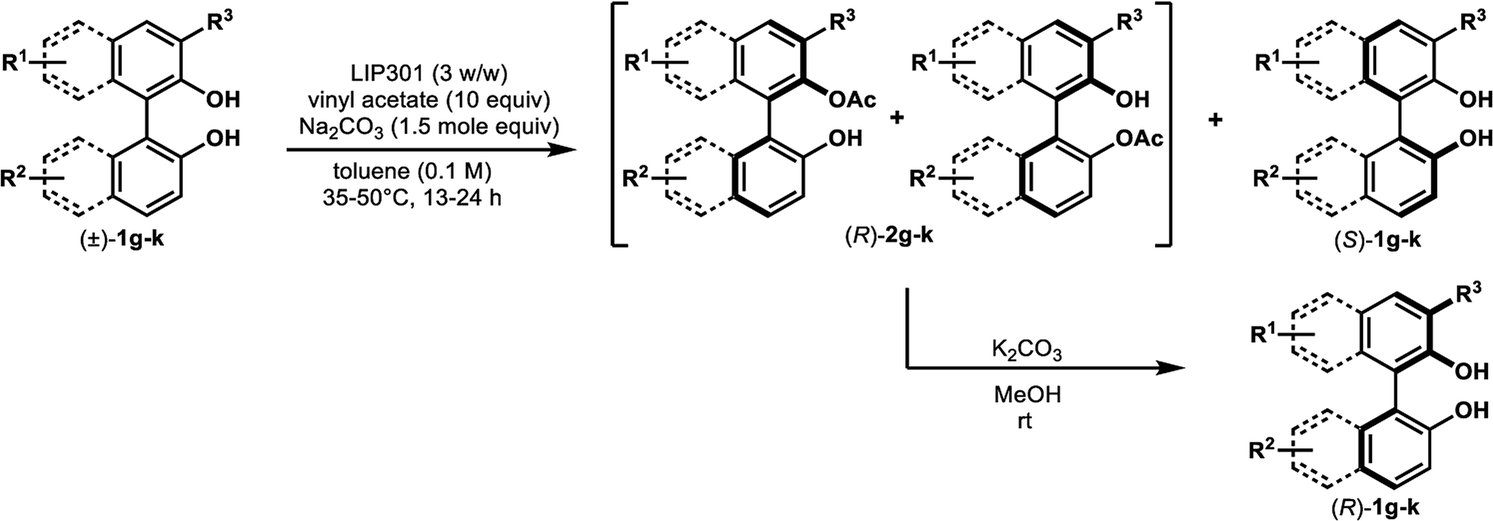
We began this study by investigating the KR parameters of BINOL (±)-1a as a model substrate using either vinyl acetate (10 equiv.) or isopropenyl acetate (10 equiv.) as an acyl donor and the commercially available immobilized Pseudomonas sp. lipoprotein lipase (Toyobo LIP301). As shown in Table 1, only 4–30% conversions were achieved after 24 h at the temperature of 35 or 50 °C with lipase loading of 1–3 w/w in spite of the high enantioselectivity (E value = 58–>200) (entries 1–6). To realize the requisite rate enhancement, we envisioned that the addition of some basic additives would promote the reaction by enhancing the nucleophilicity of the phenolic hydroxyl group towards an acyl-enzyme intermediate. To our delight, the addition of Na2CO3 (1.5 mol equiv.) at 35 °C dramatically increased the acylation rate while maintaining a substantial level of enantio-discrimination (E value = >200) giving the monoacetate (R)-2a in 50% NMR yield with 99% ee and the recovered (S)-1a in 50% NMR yield with 98% ee (entry 8). This result was reproduced on a larger scale (0.5 mmol of (±)-1a) leading to perfect conversion after 24 h with the formation of (R)-2a (51% isolated yield, 96% ee) and (S)-1a (48% isolated yield, >99% ee) (E = >200) (entry 9). Reducing the amount of Na2CO3 to 0.3 mol equiv. resulted in only 8% conversion after 24 h (entry 10). We could reduce the lipase loading further to 1 w/w and the acyl donor to 5 equiv. at 50 °C to afford both (R)-2a and (S)-1a in high optical purities with 50% conversion (entries 11–12) (cf. previous studies required 4 w/w lipase and 20 equiv. of vinyl acetate8a,b). The use of isopropenyl acetate led to a similar rate enhancement with high enantioselectivity (E value = 149) (entry 14).12 In all cases, the formation of diacetate 3a was not observed.
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Na2CO3 (mol equiv.) | LIP301b (w/w) | Acyl donor (equiv.) | Temp (°C) | Time (h) | Conv. (%)c | (R)-2a (% ee)d | (S)-1a (% ee)d | Ee |
| a Except for entry 9, the screening was done using ca. 0.03 mmol of (±)-1a.b Commercially available immobilized Pseudomonas sp. lipoprotein lipase (Toyobo LIP301).c Calculated based on the optical purities of (R)-2a and (S)-1a, see ref. 10 and 11a.d Determined by chiral HPLC.e For E value, see ref. 10 and 11a.f Reaction conducted using 0.5 mmol of (±)-1a.g Isolated yields. | |||||||||
| 1 | 0 | 1 | Vinyl acetate (10) | 35 | 24 | 4 | >99 | 4 | >200 |
| 2 | 0 | 3 | Vinyl acetate (10) | 35 | 24 | 7 | >99 | 8 | >200 |
| 3 | 0 | 3 | Vinyl acetate (10) | 50 | 24 | 30 | 95 | 40 | 58 |
| 4 | 0 | 1 | Isopropenyl acetate (10) | 35 | 24 | 7 | 98 | 7 | 106 |
| 5 | 0 | 3 | Isopropenyl acetate (10) | 35 | 24 | 8 | 98 | 9 | 108 |
| 6 | 0 | 3 | Isopropenyl acetate (10) | 50 | 24 | 16 | 98 | 18 | 118 |
| 7 | 1.5 | 1 | Vinyl acetate (10) | 35 | 24 | 36 | 99 | 56 | >200 |
| 8 | 1.5 | 3 | Vinyl acetate (10) | 35 | 24 | 50 | 99 | 98 | >200 |
| 9f | 1.5 | 3 | Vinyl acetate (10) | 35 | 24 | 51 | 96 (51% yield)g | >99 (48% yield)g | >200 |
| 10 | 0.3 | 3 | Vinyl acetate (10) | 35 | 24 | 8 | 99 | 8 | >200 |
| 11 | 1.5 | 1 | Vinyl acetate (10) | 50 | 24 | 50 | 95 | 94 | 139 |
| 12 | 1.5 | 1 | Vinyl acetate (5) | 50 | 24 | 50 | 96 | 95 | 138 |
| 13 | 1.5 | 1 | Isopropenyl acetate (10) | 35 | 24 | 33 | 98 | 49 | 161 |
| 14 | 1.5 | 3 | Isopropenyl acetate (10) | 35 | 17 | 50 | 98 | 94 | 149 |

We also examined the effects of other types of organic and inorganic bases at 35 °C for 24 h (Table 2). Both the conversions and enantioselectivities dropped off sharply when pyridine or Hünig's base were added (entries 2 and 3). Although 48% conversion was attained by adding triethylamine, the enantioselectivity was very poor (entry 4). The conversions did not exceed 13% when Li2CO3, MgCO3, and CaCO3 were examined despite the high enantioselectivity (entries 5, 7, and 9, respectively). Very low conversion and poor enantioselectivity were obtained upon using Cs2CO3 (entry 10). The use of K2CO3 resulted in 46% conversion with moderate enantiocontrol (E = 43) (entry 8). Other sodium salts like NaHCO3 and Na2HPO4 led to poor conversions after 24 h, albeit in the high E values (>200) (entries 11, 12 and 14). Although Na3PO4 afforded a 52% conversion (entry 13), the resultant enantioselectivity (E = 99) was inferior to that obtained with Na2CO3 (E = >200, entry 6).
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Conv. (%)a | (R)-2a (% ee)b | (S)-1a (% ee)b | E |
| a Calculated based on the optical purities of (R)-2a and (S)-1a, see ref. 10 and 11a.b Determined by chiral HPLC.c From Table 1, entry 2.d The formation of diacetate 3a was not observed under these conditions.e From Table 1, entry 8.f 3 mol equiv. of NaHCO3 was added. | |||||
| 1c | None | 7 | >99 | 8 | >200 |
| 2 | Pyridine | 13 | 91 | 13 | 24 |
| 3 | EtN(iPr)2 | 24 | 60 | 19 | 5 |
| 4d | Et3N | 48 | 52 | 48 | 5 |
| 5 | Li2CO3 | 4 | 99 | 4 | >200 |
| 6e | Na2CO3 | 50 | 99 | 98 | >200 |
| 7 | MgCO3 | 13 | 99 | 15 | >200 |
| 8d | K2CO3 | 46 | 90 | 75 | 43 |
| 9 | CaCO3 | 5 | 99 | 5 | >200 |
| 10d | Cs2CO3 | 10 | 18 | 2 | 1.5 |
| 11 | NaHCO3 | 12 | 99 | 13 | >200 |
| 12f | NaHCO3 | 17 | 99 | 20 | >200 |
| 13 | Na3PO4 | 52 | 90 | 99 | 99 |
| 14 | Na2HPO4 | 20 | 99 | 20 | >200 |

We found that the poor enantioselectivity caused by triethylamine, K2CO3, or Cs2CO3, in comparison to the excellent enantiocontrol in the case of Na2CO3, was due to the competitive non-enzymatic acetylation. Thus, a similar reaction of (±)-1a in the presence of these bases, while omitting the lipase, resulted in the formation of a mixture of (±)-2a and the diacetate (±)-3a (Table 3, entries 1, 3, and 4), while the formation of diacetate 3a was not detected in the presence of lipase (Table 2, entries 4, 8, and 10). On the other hand, Na2CO3 did not promote any non-enzymatic acylation (entry 2) and has been identified as the ideal base for the enzymatic transformation in terms of both reaction rate and enantioselectivity.

Under the optimal conditions obtained using Na2CO3 as the base, various substituted C2-symmetric biaryl diols (±)-1b–e were successfully resolved with ∼50% conversion in only 16–30 h with excellent enantioselectivity (E = > 100) (Table 4, entries 1, 2, 4 and 5). Particularly, 6,6′-dimethoxy-1,1′-biphenol (1d) underwent highly enantioselective KR with ∼50% conversion after 24 h using lower amounts of lipase (2 w/w) and vinyl acetate (5 equiv.), thus offering an advantage over the reported enzymatic method that required three days using 4 w/w of lipase PS-IM and 1.5 equiv. of vinyl acetate to achieve 41% conversion.8c Notably, the reaction of 1c proceeded very slowly in absence of Na2CO3; only 10% yield of (R)-2c was obtained after 24 h (entry 3), which again confirms the significance of Na2CO3 in accelerating the resolution process as seen in entry 2. All these reactions led only to the formation of monoacetates (R)-2 without any detectable amount of the corresponding diacetates 3. On the other hand, 3,3′-dibromo-1,1′-binaphthol (1f) did not react at all (entry 6); similar results were obtained using different types of immobilized lipases under different conditions probably due to the steric bulkiness imparted by the substituents at the 3,3′-positions that may retard the acyl transfer from the acyl-enzyme intermediate to the substrate.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | Temp. and time | (R)-2b–g | (S)-1b–g | E | ||
| Isolated yield (%) | % eeb | Isolated yield (%) | % eeb | ||||
| a Reaction was conducted using (±)-1b–f (0.1 mmol).b Determined by chiral HPLC.c In absence of Na2CO3.d 2 w/w LIP301 and 5 equiv. vinyl acetate were used.e i-Pr2O was used instead of toluene. | |||||||
| 1 |  |
50 °C, 29 h | 49 | 95 | 50 | 99 | >200 |
| 2 |  |
50 °C, 30 h | 54 | 97 | 43 | 99 | >200 |
| 3c | 50 °C, 30 h | 10 | 99 | 86 | 11 | >200 | |
| 4d |  |
35 °C, 24 h | 53 | 89 | 47 | >99 | 101 |
| 5d,e |  |
35 °C, 16 h | 52 | 95 | 47 | 97 | 165 |
| 6 | 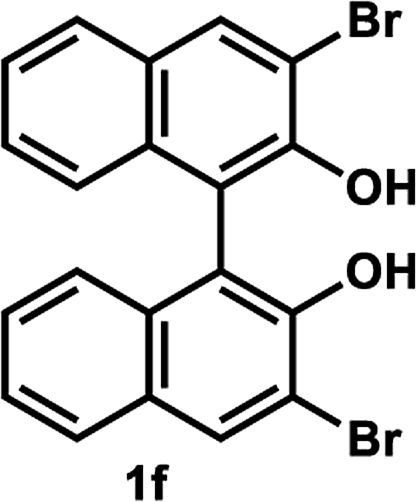 |
50 °C, 24 h | No reaction | ||||

The successful application of our method to C1-symmetric biaryl diols (±)-1g–k is noteworthy (Table 5). Although they produced a mixture of regioisomeric acetates (R)-2g–k, we found that each isomer has the same (R) absolute configuration and high optical purity. For instance, two acetates (R)-2i were obtained; each of them with 91% ee, and their mixture was subjected to methanolysis to give a single product (R)-1i with 91% ee (Table 5, entry 3). In a similar manner, simple methanolysis of the mixtures of regioisomeric acetates (R)-2g–h and (R)-2j–k afforded (R)-diols (R)-1g–h and (R)-1i–k with complete retention of optical purity (entries 1, 2, 5, and 6). While the reported asymmetric oxidative cross coupling of 2-naphthols afforded (R)-1h with a maximum optical purity of 72% ee,4e the optimized KR protocol discussed here afforded much higher optical purities for both (R)-1h (95% ee) and (S)-1h (>99%) (entry 2). Importantly, the acetylation of 1i did not proceed in absence of Na2CO3, reconfirming the necessity to add this base to promote the KR (entry 4). Notably, the reaction worked well for substrates possessing only one substituent at the 3-position (Table 5, entries 3, 5, and 6).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | Temp. and time | (R)-1g–k | (S)-1g–k | E | ||
| Isolated yield (%)b | % eeb | Isolated yield (%) | % eec | ||||
| a Reaction was conducted using (±)-1g–k (0.1 mmol).b Obtained by methanolysis of a mixture of two regioisomers (R)-2 (for details, see Experimental section), and the optical purity was determined by chiral HPLC after methanolysis.c Determined by chiral HPLC.d Using 3 mol equiv. Na2CO3.e Using 4 w/w LIP301.f In absence of Na2CO3. | |||||||
| 1d |  |
35 °C, 14 h | 48 | 95 | 51 | 93 | 126 |
| 2 | 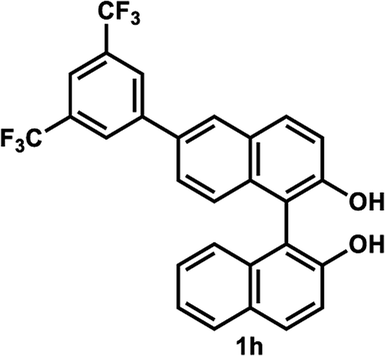 |
35 °C, 24 h | 47 | 95 | 40 | >99 | >200 |
| 3d,e |  |
35 °C, 13 h | 48 | 91 | 44 | >99 | 114 |
| 4f | 35 °C, 24 h | No reaction | |||||
| 5 | 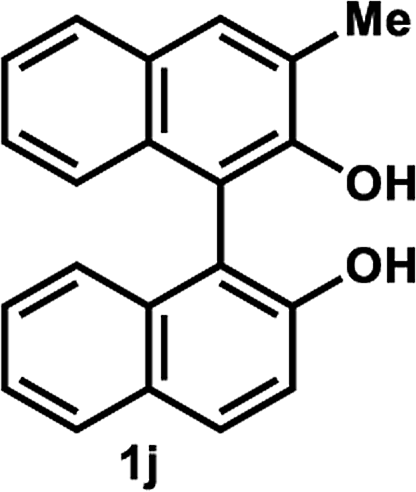 |
50 °C, 24 h | 45 | 92 | 41 | 99 | 126 |
| 6 |  |
50 °C, 14 h | 43 | 96 | 44 | 99 | >200 |
We further examined the applicability of our method to the biaryl methanol derivative 4a (Table 6). However, the addition of Na2CO3 in this case was deleterious to the enantioselectivity, affording the optically inactive product 5a (2% ee, 100% NMR yield) in which the primary hydroxyl group is acetylated (Table 6, entry 1). The 100% conversion is probably due to a concomitant non-enzymatic acetylation of the primary alcohol.13 The omission of Na2CO3 dramatically improved the enantioselectivity (E = 34) (entry 2). Lowering the reaction temperature to 25 °C afforded better E value (65) (entry 3); such high enantioselectivity was not realized in previously reported lipase-catalyzed resolution.14 The protocol was also applied successfully to diol 4b, resulting in high enantioselectivity (E = 127) with 50% conversion after 32 h (entry 4).15
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Substrate | Na2CO3 (mol equiv.) | Temp. (°C) | Time (h) | Conv. (%)b | (R)-5a–bc | (S)-4a–bc | E |
| a Reaction was conducted using (±)-4a–b (0.1 mmol).b Determined by 1H NMR analysis of the crude mixture.c Optical purity was determined by chiral HPLC. ND: not determined. | ||||||||
| 1 | 4a | 1.5 | 35 | 24 | 100 | 2% ee, quant. | ND | ND |
| 2 | 4a | 0 | 35 | 24 | 57 | 68% ee, 57% NMR yield | >99% ee, 43% NMR yield | 34 |
| 3 | 4a | 0 | 25 | 48 | 52 | 88% ee, 55% isolated yield | 97% ee, 43% isolated yield | 65 |
| 4 | 4b | 0 | 25 | 32 | 50 | 93% ee, 49% isolated yield | 98% ee, 51% isolated yield | 127 |

Based on the results that produced acceleration of the reaction by the addition of Na2CO3 in the case of phenolic substrates 1a–e and 1g–k, we considered the lipase-mediated catalytic cycle for the enantioselective esterification. In principle, the KR cycle begins with the transfer of acyl group from the acyl donor (e.g. vinyl acetate) to the hydroxyl group of serine in the reactive site of lipase to form an acyl-enzyme intermediate,16 which is subsequently attacked by the phenolic hydroxyl group of 1 in an enantioselective manner. Since Pseudomonas sp. lipoprotein lipase has been used in the KR of secondary alcohols without Na2CO3 leading to ∼50% conversion in shorter time,17 the formation of the acyl-enzyme intermediate is probably not the rate-limiting step. Instead, steric hindrance in the biaryl skeleton of 1 may decelerate the acyl transfer to 1. Moreover, the phenolic hydroxyl groups in 1 are more acidic with less nucleophilicity toward the acyl-enzyme than the alcoholic hydroxyl groups. This assumption was supported by the results obtained for alcohols 4, in which the addition of Na2CO3 was not required and the acylation occurred on their primary hydroxyl groups with perfect chemoselectivity.
It is worth mentioning that the dynamic kinetic resolution (DKR) of secondary alcohols using a combination of ruthenium complexes and lipases has often been performed in the presence of Na2CO3.18 Similarly, we also noted the importance of Na2CO3 addition in our recent chemoenzymatic DKR of certain 2,2′-dihydroxy-1,1′-biaryls using a combination of a ruthenium complex and LIP301;9 however, no clear explanation of the role of such additive has been proposed until now. Herein, we have partially elucidated the impact of this additive in enhancing the KR rate. In addition, the optical purities (76–89% ee) of the compounds 1b, 1c, 1g, and 1i, obtained by our DKR protocol,9 have been improved to 91–97% ee in this study by modification of the KR conditions.
Conclusions
In contrast to the former lipase-catalyzed KRs of atropisomeric 2,2′-dihydroxy-1,1′-biaryls that required long reaction times (3–14 days) and were of limited scope,8 we have improved the rate of enzymatic KR and expanded its applicability to a range of this class of compounds, such as 1a–e and 1g–k, with ∼50% conversions within 30 h and with excellent enantioselectivity (E = 101–>200). The rate enhancement, realized by the addition of Na2CO3, is probably due to an improvement in the nucleophilicity of the phenolic hydroxyl group of 1 towards an acyl-enzyme intermediate. Since there is only one report describing an organocatalyzed resolution of non-C2-symmetric axially chiral dihydroxy biaryl 1i with moderate enantioselectivity (S value = 38),5a our method shows broader applicability to non-C2-symmetric dihydroxy biaryls with higher enantioselectivities. The finding described here will also expand the efficiency of DKR of atropisomers particularly when combined with efficient racemization catalysts. Detailed mechanistic investigation of the effect of Na2CO3 is in progress in our laboratory.Experimental
General considerations
Melting points were determined on a Yanagimoto Melting Point Apparatus and are uncorrected. Infrared (IR) absorption spectra were recorded on a SHIMADZU FTIR-8400S spectrophotometer. 1H and 13C NMR spectra were measured on a JEOL JNM-ECA500 (1H: 500 MHz, 13C: 125 MHz) or JEOL JNM-ECS400 (1H: 400 MHz, 13C: 100 MHz) instrument with chemical shifts reported in δ (ppm) relative to the residual nondeuterated solvent signal for 1H (CHCl3: δ = 7.26 ppm) and relative to the deuterated solvent signal for 13C (CDCl3: δ = 77.0 ppm). The mass spectra (MS) were measured on a JEOL JMS-S3000 (MALDI) with TOF mass analyser. HPLC analyses were carried out using a JASCO LC-2000Plus system (HPLC pump: PU-2080, UV detector: MD-2018) equipped with a Daicel CHIRALPAK IC-3, CHIRALPAK AD-3, CHIRALCEL OD-3, CHIRALCEL OZ-3 or CHIRALPAK IE column; each with a size of 4.6 mm × 250 mm. Optical rotations were measured on a JASCO P-1020 polarimeter. Gel permeation chromatography was carried out on LaboACE LC-5060 with JAIGEL-2HR columns (Japan Analytical Industry). The lipase from Pseudomonas sp. (TOYOBO lipoprotein lipase GradeIII LPL-311) immobilized on Hyflo Super-Cel (commercial name: TOYOBO LIP301) was gifted from TOYOBO CO., LTD. Kanto silica gel 60N was used for column chromatography. In general, the reactions were carried out in anhydrous solvents. Racemic substrates 1b,19 1c,20 1d,21 1e,22 1f,23 1g,9 1h,4e 1i,24 1j,23 1k,25 4a,14 and 4b14 were prepared according to reported procedure.Kinetic resolution of (±)-1a (Table 1, entry 9)
To a stirred solution of (±)-1a (143 mg, 0.50 mmol) and vinyl acetate (0.46 mL, 5.0 mmol) in anhydrous PhCH3 (5.0 mL, 0.10 M) were added LIP301 (0.43 g, 3 w/w) and Na2CO3 (80 mg, 0.75 mmol). After being stirred for 24 h at 35 °C, the reaction mixture was filtered through a Celite pad. The Celite pad was washed with EtOAc and the combined filtrate was evaporated in vacuo. The residue was purified by flash column chromatography (hexanes/EtOAc = 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to give ester (R)-2a (84 mg, 51% yield, 96% ee) and recovered (S)-1a (68 mg, 48% yield, >99% ee).
:5; flow rate: 1.0 mL min−1; retention times: 8.0 min (R), 10.7 min (S)).:5; flow rate: 1.0 mL min−1; retention times: 14.5 min (R), 20.8 min (S)).
:1) to give ester (R)-2a (84 mg, 51% yield, 96% ee) and recovered (S)-1a (68 mg, 48% yield, >99% ee).
:5; flow rate: 1.0 mL min−1; retention times: 8.0 min (R), 10.7 min (S)).:5; flow rate: 1.0 mL min−1; retention times: 14.5 min (R), 20.8 min (S)).Kinetic resolution of C2-symmetric biaryl diols (±)-1b–f (Table 4)
:1) to give ester (R)-2c (21 mg, 54% yield, 97% ee) and recovered (S)-1c (14.8 mg, 43% yield, 99% ee).
(R)-2′-Hydroxy-7,7′-dimethoxy-(1,1′-binaphthalen)-2-yl acetate (R)-2c. White solid; mp 47–49 °C; [α]20D − 40.8 (c 0.24, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 8.00 (d, J = 9.0 Hz, 1H), 7.88 (d, J = 9.0 Hz, 1H), 7.83 (d, J = 9.0 Hz, 1H), 7.76 (d, J = 9.0 Hz, 1H), 7.28 (d, J = 1.0 Hz, 1H), 7.17–7.20 (m, 2H), 7.01 (dd, J = 9.0, 3.0 Hz, 1H), 6.59 (d, J = 2.0 Hz, 1H), 6.40 (d, J = 2.0 Hz, 1H), 5.17 (s, 1H), 3.58 (s, 3H), 3.57 (s, 3H), 1.85 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ 170.3, 158.9, 158.3, 152.1, 148.6, 134.8, 134.7, 130.4, 130.1, 129.8, 129.5, 127.6, 124.3, 121.7, 119.3, 118.8, 115.50, 115.46, 113.2, 104.0, 103.6, 55.1, 55.0, 20.4; IR (CHCl3) ν 3545, 1755 cm−1; HRMS (MALDI) m/z calcd for C24H20O5 [M]+: 388.1305, found: 388.1304. Its optical purity (97% ee) was determined by HPLC analysis at 20 °C using a CHIRALPAK IC-3 column (hexanes/2-propanol = 92.5
:7.5; flow rate: 1.0 mL min−1; retention times: 8.6 min (R), 12.4 min (S)).
(S)-7,7′-Dimethoxy-1,1′-bi-2-naphthol (S)-1c. White solid; mp 62–63 °C; [α]20D + 152.5 (c 0.31, CHCl3) (lit.28 [α]22D + 122.3 (c 1.0, CHCl3) for (S)-1c with 94% ee); 1H-NMR (400 MHz, CDCl3) δ 7.88 (d, J = 9.0 Hz, 2H), 7.79 (d, J = 9.0 Hz, 2H), 7.22 (d, J = 9.0 Hz, 2H), 7.04 (dd, J = 9.0, 3.0 Hz, 2H), 6.48 (d, J = 3.0 Hz, 2H), 5.07 (s, 2H), 3.58 (s, 6H). The spectroscopic data are in good agreement with those reported.28 Its optical purity (99% ee) was determined by HPLC analysis at 20 °C using a CHIRALPAK IC-3 column (hexanes/2-propanol = 80
:20; flow rate: 1.0 mL min−1; retention times: 7.3 min (R), 9.4 min (S)).
:1).
(R)-7,7′-Dibromo-2′-hydroxy-(1,1′-binaphthalen)-2-yl acetate (R)-2b. White solid; mp 65–67 °C; [α]23D − 75.7 (c 0.63, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 8.05 (d, J = 9.0 Hz, 1H), 7.89 (d, J = 9.0 Hz, 1H), 7.85 (d, J = 9.0 Hz, 1H), 7.73 (d, J = 9.0 Hz, 1H), 7.61 (dd, J = 9.0, 2.0 Hz, 1H), 7.33–7.44 (m, 4H), 7.13 (d, J = 2.0 Hz, 1H), 5.25 (s, 1H), 1.87 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ 170.3, 152.6, 148.9, 134.54, 134.50, 131.1, 130.73, 130.68, 130.03, 130.01, 129.8, 127.4, 127.2, 126.2, 122.4, 122.2, 121.9, 121.5, 118.9, 112.7, 20.4 (two peaks are overlapped); IR (CHCl3) ν 3529, 1754 cm−1; HRMS (MALDI) m/z calcd for C22H12O3Na79Br2 [M + Na]+: 506.9202, found: 506.9198. Its optical purity (95% ee) was determined by chiral HPLC analysis at 20 °C using a CHIRALPAK IC-3 column (hexanes/2-propanol) = 97.5
:2.5; flow rate: 1.0 mL min−1; retention times: 8.3 min for (R)-2b, 11.4 min for (S)-2b.
(S)-7,7′-Dibromo-1,1′-bi-2-naphthol (S)-1b. White solid; mp 136–138 °C; [α]23D + 263 (c 0.75, CHCl3) (lit.28 [α]21D + 129.1 (c 1.8, CHCl3) for (S)-1b with 73% ee); 1H-NMR (500 MHz, CDCl3) δ 7.95 (d, J = 9.0 Hz, 2H), 7.77 (d, J = 9.0 Hz, 2H), 7.48 (dd, J = 9.0, 2.0 Hz, 2H), 7.38 (d, J = 9.0 Hz, 2H), 7.23 (d, J = 2.0 Hz, 2H), 5.04 (s, 2H). The spectroscopic data are in good agreement with those reported.28 Its optical purity (99% ee) was determined by chiral HPLC analysis at 20 °C using a CHIRALPAK IC-3 column (hexanes/2-propanol = 97.5
:2.5; flow rate: 1.0 mL min−1; retention times: 29.2 min for (R)-1b; 31.6 min for (S)-1b).
:1).
(R)-2′-Hydroxy-6,6′-dimethoxy-(1,1′-biphenyl)-2-yl acetate (R)-2d. Colorless oil; [α]21D + 93.4 (c 0.63, CHCl3) (lit.8c [α]20D + 91.2 (c 0.70, CHCl3) for (R)-2d with 98% ee); 1H-NMR (400 MHz, CDCl3) δ 7.42 (t, J = 8.0 Hz, 1H), 7.24 (t, J = 8.0 Hz, 1H), 6.92–6.94 (m, 1H), 6.83–6.85 (m, 1H), 6.66 (d, J = 8.0 Hz, 1H), 6.55 (dd, J = 8.0, 1.0 Hz, 1H), 5.01 (s, 1H), 3.78 (s, 3H), 3.71 (s, 3H), 1.96 (s, 3H). The spectroscopic data are in good agreement with those reported.8c Its optical purity (89% ee) was determined by HPLC analysis at 20 °C using a CHIRALPAK IC-3 column (hexanes/2-propanol = 92
:8; flow rate: 1.0 mL min−1; retention times: 29.1 min (R), 32.2 min (S)).
(S)-2,2′-Dihydroxy-6,6′-dimethoxy-1,1′-biphenyl (S)-1d. White solid; mp 138–140 °C; [α]21D − 102 (c 0.64, CHCl3) (lit.21 [α]20D − 144 (c 0.77, CHCl3) for (S)-1d with 98.6% ee); 1H-NMR (400 MHz, CDCl3) δ 7.31 (t, J = 8.0 Hz, 2H), 6.72 (d, J = 8.0 Hz, 2H), 6.62 (d, J = 8.0 Hz, 2H), 5.05 (s, 2H), 3.77 (s, 6H). The spectroscopic data are in good agreement with those reported.21 Its optical purity (>99% ee) was determined by HPLC analysis at 20 °C using a CHIRALPAK IC-3 column (hexanes/2-propanol = 85
:15; flow rate: 1.0 mL min−1; retention times: 12.9 min (R), 18.9 min (S)).
:1).
(R)-2,2′-Dihydroxy-6,6′-dimethyl-1,1′-biphenyl (R)-2e. Colorless oil; [α]22D + 36.8 (c 0.63, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.37 (t, J = 8.0 Hz, 1H), 7.25 (d, J = 8.0 Hz, 2H), 7.17 (t, J = 8.0 Hz, 1H), 7.00 (d, J = 8.0 Hz, 1H), 6.83–6.86 (m, 2H), 4.82 (s, 1H), 2.04 (s, 3H), 1.94 (s, 3H), 1.91 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ 170.5, 153.0, 149.6, 140.1, 137.6, 129.5, 129.1, 128.5, 128.3, 122.7, 122.1, 120.0, 113.9, 20.3, 19.6, 19.4; IR (CHCl3) ν 3532, 1744 cm−1; HRMS (MALDI) m/z calcd for C16H16O3Na [M + Na]+: 279.0997, found: 279.0992. Its optical purity (95% ee) was determined by HPLC analysis at 20 °C using a CHIRALPAK IC-3 column (hexanes/2-propanol = 99
:1; flow rate: 1.0 mL min−1; retention times: 13.7 min (R), 16.9 min (S)).
(S)-2′-Hydroxy-6,6′-dimethyl-(1,1′-biphenyl)-2-yl acetate (S)-1e. White solid; mp 122–124 °C (lit.29 mp 159–160 °C); [α]21D − 54.3 (c 0.50, CHCl3) (lit.23 [α]20D−60.5 (c 1.0, CHCl3) for (S)-1e with >99% ee); 1H-NMR (400 MHz, CDCl3) δ 7.21–7.27 (m, 2H), 6.86–6.92 (m, 4H), 4.71 (brs, 2H), 2.01 (s, 6H). The spectroscopic data are in good agreement with those reported.29 Its optical purity (97% ee) was determined by HPLC analysis at 20 °C using a CHIRALCEL OZ-3 column (hexanes/2-propanol = 95
:5; flow rate: 1.0 mL min−1; retention times: 7.5 min (R), 10.4 min (S)).
Kinetic resolution of non C2-symmetric biaryl diols (±)-1g–k (Table 5)
:1) to give a 3:2 mixture of two regioisomeric esters (R)-2i (19.5 mg, 48% yield, 91% ee for each regioisomer) in addition to recovered (S)-1i (16.0 mg, 44% yield, >99% ee). The mixture of regioisomers (R)-2i was subjected to methanolysis as follows: to a stirred solution of (R)-2i (19.5 mg, 0.048 mmol, 91% ee) in MeOH (1.0 mL) was added K2CO3 (28 mg, 0.20 mmol). After being stirred for 15 min at room temperature, the reaction mixture was acidified with 1 N HCl and diluted with CH2Cl2. The mixture was transferred to a separating funnel and the organic layer was washed with brine. The organic layer was separated, dried over MgSO4, filtered, and concentrated in vacuo to give (R)-1i (17.4 mg, 48% yield from (±)-1i, 91% ee) as a pure pale-yellow solid.
(R)-3-Bromo-2′-hydroxy-(1,1′-binaphthalen)-2-yl acetate and (R)-3′-bromo-2′-hydroxy-(1,1′-binaphthalen)-2-yl acetate (R)-2i. Pale yellow solid; 1H-NMR (500 MHz, CDCl3) δ 8.35 (s, 0.4H), 8.22 (s, 0.6H), 8.07 (d, J = 9.0 Hz, 0.6H), 7.97 (d, J = 8.0 Hz, 0.6H), 7.92 (d, J = 9.0 Hz, 0.4H), 7.89 (d, J = 9.0 Hz, 0.4H), 7.86 (d, J = 8.0 Hz, 0.4H), 7.78 (d, J = 8.0 Hz, 0.6H), 7.51 (m, 1H), 7.43 (d, J = 9.0 Hz, 0.6H), 7.32–7.37 (m, 2.4H), 7.25–7.28 (m, 1H), 7.22 (d, J = 9.0 Hz, 0.6H) 7.20 (d, J = 9.0 Hz, 0.4H), 7.04 (d, J = 9.0 Hz, 0.6H), 7.01 (d, J = 9.0, 0.4H), 5.30–5.64 (brs, 1H), 1.97 (s, 1.2H), 1.88 (s, 1.8H). Its optical purity (91% ee) was determined by HPLC analysis at 20 °C using a CHIRALCEL IC-3 column (hexanes/2-propanol) = 95
:5; flow rate: 1.0 mL min−1; retention times for (R)-regioisomers of 2i: 6.7 min and 8.9 min; retention times for (S)-regioisomers of 2i: 8.0 min and 10.0 min.
(R)-3-Bromo-1,1′-bi-2-naphthol (R)-1i. White solid; mp 82–89 °C (lit.9 mp 130–132 °C); [α]23D + 56.5 (c 0.28, CHCl3) (lit.9 [α]20D + 38.3 (c 0.23, CHCl3) for (R)-1i with 86% ee); 1H-NMR (400 MHz, CDCl3) δ 8.28 (s, 1H), 7.98 (d, J = 9.0 Hz, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.83 (d, J = 8.0 Hz, 1H), 7.28–7.42 (m, 5H), 7.14 (d, J = 7.0 Hz, 1H), 7.10 (d, J = 9.0 Hz, 1H), 5.58 (s, 1H), 4.95 (s, 1H). The spectroscopic data are in good agreement with those reported.9 Its optical purity (91% ee) was determined by HPLC analysis at 20 °C using a CHIRALPAK IC-3 column (hexanes/2-propanol = 95
:5; flow rate: 1.0 mL min−1; retention times: 10.1 min (R), 27.0 min (S)).
(S)-3-Bromo-1,1′-bi-2-naphthol (S)-1i. White solid; mp 64–68 °C; [α]24D − 64.1 (c 0.39, CHCl3). Its spectroscopic data are in good agreement with those described above for (R)-1i. Its optical purity (>99% ee) was determined by HPLC analysis at 20 °C using a CHIRALPAK IC-3 column (hexanes/2-propanol = 95
:5; flow rate: 1.0 mL min−1; retention times: 11.4 min (R), 26.5 min (S)).
:1). Methanolysis of (R)-2g (19.2 mg, 0.048 mmol), as described above, afforded (R)-1g (17.0 mg, 48% yield from (±)-1g, 95% ee).
Ethyl (R)-2-acetoxy-2′-hydroxy-(1,1′-binaphthalene)-6-carboxylate and ethyl (R)-2′-acetoxy-2-hydroxy-(1,1′-binaphthalene)-6-carboxylate (R)-2g. Pale yellow solid; 1H-NMR (500 MHz, CDCl3) δ 8.58 (d, J = 2.0 Hz, 0.3H), 8.48 (d, J = 1.0 Hz, 0.7H), 8.04 (d, J = 9.0 Hz, 0.3H), 7.95 (d, J = 9.0 Hz, 0.7H), 7.88 (d, J = 9.0 Hz, 0.7H), 7.84 (d, J = 9.0 Hz, 0.7H), 7.76–7.79 (m, 0.6H), 7.73 (d, J = 8.0 Hz, 0.3H), 7.70 (dd, J = 9.0, 2.0 Hz, 0.7H), 7.37–7.40 (m, 0.7H), 7.33 (d, J = 9.0 Hz, 0.3H), 7.19–7.28 (m, 1.7H), 7.16 (d, J = 9.0 Hz, 0.3H), 7.07–7.13 (m, 1H), 6.93 (d, J = 9.0 Hz, 0.7H), 6.84 (d, J = 8.0 Hz, 0.3H), 5.07–5.26 (m, 1H), 4.24–4.31 (m, 2H), 1.74 (s, 0.9H), 1.72 (s, 2.1H), 1.27 (m, 3H).
Ethyl (R)-2,2′-dihydroxy-(1,1′-binaphthalene)-6-carboxylate (R)-1g. White solid; mp 83–85 °C (lit.9 mp 98–100 °C); [α]22D − 72.8 (c 0.39, CHCl3) (lit.9 [α]21D − 54.6 (c 0.7, CHCl3) for (R)-1g with 89% ee); 1H-NMR (500 MHz, CDCl3) δ 8.60 (s, 1H), 8.06 (d, J = 9.0 Hz, 1H), 7.98 (d, J = 9.0 Hz, 1H), 7.90 (d, J = 9.0 Hz, 1H), 7.84 (dd, J = 9.0, 2.0 Hz, 1H), 7.43 (d, J = 9.0 Hz, 1H), 7.36–7.39 (m, 2H), 7.31 (td, J = 7.0, 2.0 Hz, 1H), 7.16 (d, J = 9.0 Hz, 1H), 7.09 (d, J = 9.0 Hz, 1H), 5.31 (s, 2H), 5.21 (s, 1H), 4.38 (q, J = 7.0 Hz, 2H), 1.41 (t, J = 7.0 Hz, 3H). The spectroscopic data are in good agreement with those reported.9 Its optical purity (95% ee) was determined by HPLC analysis at 20 °C using a CHIRALCEL OD-3 column (hexanes/2-propanol = 85
:15; flow rate: 1.0 mL min−1; retention times: 19.5 min (R), 14.0 min (S)).
Ethyl (S)-2,2′-dihydroxy-(1,1′-binaphthalene)-6-carboxylate (S)-1g. White solid; mp 93–95 °C; [α]21D + 67.5 (c 0.82, CHCl3). Its spectroscopic data are in good agreement with those described above for (R)-1g. Its optical purity (93% ee) was determined by HPLC analysis at 20 °C using a CHIRALPAK IC-3 column (hexanes/2-propanol = 85
:15; flow rate: 1.0 mL min−1; retention times: 18.8 min (R), 12.9 min (S)).
:1). Methanolysis of (R)-2h (25.4 mg, 0.047 mmol), as described above, afforded (R)-1h (23.0 mg, 47% yield from (±)-1h, 95% ee).
(R)-6-(3,5-Bis(trifluoromethyl)phenyl)-2′-hydroxy-(1,1′-binaphthalen)-2-yl acetate and (R)-6′-(3,5-bis(trifluoromethyl)phenyl)-2′-hydroxy-(1,1′-binaphthalen)-2-yl acetate (R)-2h. Pale yellow solid. 1H-NMR (400 MHz, CDCl3) δ 7.85–8.22 (m, 7H), 7.35–7.58 (m, 5H), 7.25–7.30 (m, 1H), 7.18 (d, J = 9.0 Hz, 0.7H), 7.04 (d, J = 8.0 Hz, 0.3H), 5.32 (s, 0.7H), 5.23 (s, 0.3H), 1.91 (s, 2H), 1.90 (s, 1H).
(R)-6-(3,5-Bis(trifluoromethyl)phenyl)-1,1′-bi-2-naphthol (R)-1h. White solid; mp 94–95 °C; [α]22D − 71.2 (c 0.86, CHCl3) (lit.4e [α]22D − 39.8 (c 5.53, CHCl3) for (R)-1h with 72% ee); 1H-NMR (400 MHz, CDCl3) δ 8.06–8.12 (m, 4H), 8.01 (d, J = 8.5 Hz, 1H), 7.92 (d, J = 8.0 Hz, 1H), 7.87 (s, 1H), 7.53 (dd, J = 8.5, 1.5 Hz, 1H), 7.46 (d, J = 9.0 Hz, 1H), 7.40 (d, J = 9.0 Hz, 2H), 7.34 (t, J = 7.5 Hz, 1H), 7.28 (d, J = 8.5 Hz, 1H), 7.16 (d, J = 8.5 Hz, 1H), 5.18 (s, 1H), 5.06 (s, 1H). The spectroscopic data are in good agreement with those reported.4e Its optical purity (95% ee) was determined by HPLC analysis at 20 °C using a CHIRALPAK IC-3 column (hexanes/2-propanol = 95
:5; flow rate: 1.0 mL min−1; retention times: 9.4 min (R), 11.9 min (S)).
(S)-6-(3,5-Bis(trifluoromethyl)phenyl)-1,1′-bi-2-naphthol (S)-1h. White solid; mp 91–93 °C; [α]22D + 69.1 (c 0.97, CHCl3). Its spectroscopic data are in good agreement with those described above for (R)-1h. Its optical purity (>99% ee) was determined by HPLC analysis at 20 °C using a CHIRALPAK IC-3 column (hexanes/2-propanol = 95
:5; flow rate: 1.0 mL min−1; retention times: 9.7 min (R), 11.6 min (S)).
(R)-2-Hydroxy-3′-methyl-(1,1′-binaphthalen)-2-yl acetate and (R)-2′-hydroxy-3′-methyl-(1,1′-binaphthalen)-2-yl acetate (R)-2j. White solid; 1H-NMR (500 MHz, CDCl3) δ 8.07 (d, J = 9.0 Hz, 0.7H), 7.97 (d, J = 8.0 Hz, 0.7H), 7.85–7.92 (m, 1H), 7.78 (d, J = 8.0 Hz, 0.7H), 7.74 (s, 0.7H), 7.53–7.16 (m, 6.2H), 7.03 (d, J = 8.6 Hz, 0.3H), 6.97 (d, J = 9.0 Hz, 0.7H), 5.21 (s, 1H), 2.50 (s, 2.1H), 2.44 (s, 0.9H), 1.87 (s, 2.1H), 1.86 (s, 0.9H).
(R)-3-Methyl-1,1′-bi-2-naphthol (R)-1j. White solid; mp 94–96 °C; [α]22D + 17.4 (c 0.75, CDCl3) (lit.30 [α]20D + 22.1 (c 1.5, CDCl3) for (R)-1j with 95% ee); 1H-NMR (500 MHz, CDCl3) δ 7.98 (d, J = 9.0 Hz, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.81–7.83 (m, 2H), 7.29–7.40 (m, 4H), 7.23–7.26 (m, 1H), 7.15 (d, J = 9.0 Hz, 1H), 7.08 (d, J = 8.0 Hz, 1H), 5.12 (brs, 1H), 5.07 (brs, 1H), 2.52 (s, 3H). The spectroscopic data are in good agreement with those reported.30 Its optical purity (92% ee) was determined by HPLC analysis at 20 °C using a CHIRALPAK IC-3 column (hexanes/2-propanol = 95
:5; flow rate: 1.0 mL min−1; retention times: 6.0 min (R), 9.3 min (S)).
(S)-3-Methyl-1,1′-bi-2-naphthol (S)-1j. White solid; mp 93–99 °C; [α]22D − 19.4 (c 0.75, CDCl3). Its spectroscopic data are in good agreement with those described above for (R)-1j. Its optical purity (99% ee) was determined by HPLC analysis at 20 °C using a CHIRALPAK IC-3 column (hexanes/2-propanol = 95
:5; flow rate: 1.0 mL min−1; retention times: 6.1 min (R), 9.4 min (S)).
:1). Methanolysis of (R)-2k (19.6 mg, 0.043 mmol), as described above, afforded (R)-1k (17.9 mg, 43% yield from (±)-1k, 96% ee).
(R)-6-Bromo-1-(2,5-dichloro-6-hydroxy-3-methoxyphenyl)naphthalen-2-yl acetate and (R)-2-(6-bromo-2-hydroxynaphthalen-1-yl)-3,6-dichloro-4-methoxyphenyl acetate (R)-2k. Pale yellow solid; 1H-NMR (500 MHz, CDCl3) δ 8.08 (d, J = 2.0 Hz, 0.2H), 8.96 (d, J = 2.0 Hz, 0.8H), 7.89 (d, J = 9.0 Hz, 0.2H), 7.74 (d, J = 9.0 Hz, 0.8H), 7.49 (dd, J = 9.0, 2.0 Hz, 0.2H), 7.38–7.43 (m, 1H), 7.25–7.27 (m, 0.8H), 7.21 (s, 0.8H), 7.19 (d, J = 8.0 Hz, 0.2H), 7.10 (s, 0.2H), 7.01 (d, J = 9.0 Hz, 0.8H), 5.23–5.30 (brs, 1H), 3.99 (s, 2.4H), 3.93 (s, 0.6H), 2.11 (s, 0.6H), 1.86 (s, 2.4H).
(R)-6-Bromo-1-(2,5-dichloro-6-hydroxy-3-methoxyphenyl)naphthalen-2-ol (R)-1k. White solid; mp 72–74 °C; [α]22D + 20.2 (c 0.83, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 8.00 (d, J = 2.0 Hz, 1H), 7.80 (d, J = 9.0 Hz, 1H), 7.44 (dd, J = 9.0, 2.0 Hz, 1H), 7.30 (d, J = 9.0 Hz, 1H), 7.14 (s, 1H), 7.08 (d, J = 9.0 Hz, 1H), 5.12 (s, 1H), 5.03 (s, 1H), 3.95 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ 151.4, 149.9, 144.6, 130.9, 130.5, 130.3, 130.24, 130.20, 125.5, 123.3, 121.6, 119.0, 118.9, 117.7, 113.7, 113.0, 56.9; IR (neat) ν 3517 cm−1; HRMS (MALDI) m/z calcd for C17H1179Br35Cl2O3 [M]+: 411.9263, found: 411.9263. Its optical purity (96% ee) was determined by HPLC analysis at 20 °C using a CHIRALPAK IE column (hexanes/2-propanol = 95
:5; flow rate: 1.0 mL min−1; retention times: 22.3 min (R), 26.8 min (S)).
(S)-6-Bromo-1-(2,5-dichloro-6-hydroxy-3-methoxyphenyl)naphthalen-2-ol (S)-1k. White solid; mp 65–68 °C; [α]22D − 22.7 (c 0.81, CHCl3). Its spectroscopic data are in good agreement with those described above for (R)-1k. Its optical purity (99% ee) was determined by HPLC analysis at 20 °C using a CHIRALPAK IE column (hexanes/2-propanol = 95
:5; flow rate: 1.0 mL min−1; retention times: 22.5 min (R), 24.7 min (S)).
Kinetic resolution of biaryl methanol derivatives (±)-4a–b (Table 6)
:1) to give ester (R)-5a (18.1 mg, 55% yield, 88% ee) and recovered (S)-4a (12.0 mg, 43% yield, 97% ee).
(R)-(1-(2-Hydroxy-4,6-dimethylphenyl)naphthalen-2-yl)methyl acetate (R)-5a. Colorless oil; [α]23D − 22.9 (c 0.79, CHCl3); 1H-NMR (500 MHz, CDCl3) δ 7.95 (d, J = 8.5 Hz, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.62 (d, J = 8.5 Hz, 1H), 7.50–7.53 (m, 1H), 7.39–7.42 (m, 2H), 6.77 (s, 1H), 6.75 (s, 1H), 4.99 (d, J = 12.5 Hz, 1H), 4.96 (d, J = 12.5 Hz, 1H), 4.54 (s, 1H), 2.38 (s, 3H), 2.05 (s, 3H), 1.79 (s, 3H); 13C-NMR (125 MHz, CDCl3) δ 170.9, 153.1, 139.4, 137.9, 133.6, 133.3, 132.5, 132.1, 129.0, 128.2, 127.1, 126.6, 126.1, 125.5, 123.3, 120.3, 113.9, 64.8, 21.3, 20.8, 19.7; IR (CHCl3) ν 3548, 1737 cm−1; HRMS (MALDI) m/z calcd for C21H20O3Na [M + Na]+: 343.1310, found: 343.1305. Its optical purity (88% ee) was determined by HPLC analysis at 20 °C using a CHIRALPAK AD-3 column (hexanes/2-propanol = 95
:5; flow rate: 1.0 mL min−1; retention times: 15.5 min (R), 17.5 min (S)).
(S)-2-(2-(Hydroxymethyl)naphthalen-1-yl)-3,5-dimethylphenol (S)-4a. White solid; mp 140–141 °C; [α]23D + 32.6 (c 0.75, CHCl3) (lit.14 [α]20D + 21.9 (c 0.8, CHCl3) for (S)-4a with 79% ee); 1H-NMR (500 MHz, CDCl3) δ 7.96 (d, J = 8.5 Hz, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.72 (d, J = 8.0 Hz, 1H), 7.48–7.51 (m, 1H), 7.38–7.39 (m, 2H), 6.79 (s, 1H), 6.75 (s, 1H), 4.53 (s, 2H), 2.38 (s, 3H), 1.80 (s, 3H). The spectroscopic data are in good agreement with those reported.14 Its optical purity (97% ee) was determined by HPLC analysis at 20 °C using a CHIRALPAK AD-3 column (hexanes/2-propanol = 85
:15; flow rate: 1.0 mL min−1; retention times: 8.8 min (R), 11.2 min (S)).
:EtOAc = 8:1).
(R)-(1-(2-Hydroxy-3,6-dimethylphenyl)naphthalen-2-yl)methyl acetate (R)-5b. Colorless oil; [α]21D − 13.7 (c 0.51, CHCl3); 1H-NMR (400 MHz, CDCl3) δ 7.96 (d, J = 8.5 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.63 (d, J = 8.0 Hz, 1H), 7.50–7.54 (m, 1H), 7.35–7.42 (m, 2H), 7.14 (d, J = 7.5 Hz, 1H), 6.84 (d, J = 7.5 Hz, 1H), 4.99 (d, J = 12.5 Hz, 1H), 4.94 (d, J = 12.5 Hz, 1H), 4.58 (s, 1H), 2.28 (s, 3H), 2.04 (s, 3H), 1.78 (s, 3H); 13C-NMR (100 MHz, CDCl3) δ 170.8, 151.3, 135.3, 133.6, 133.2, 132.4, 132.3, 130.6, 129.1, 128.2, 127.1, 126.7, 126.2, 125.5, 122.7, 121.9, 121.7, 64.8, 20.8, 19.6, 16.0; IR (CHCl3) ν 3553, 1737 cm−1; HRMS (MALDI) m/z calcd for C21H20O3Na [M + Na]+: 343.1310, found: 343.1305. Its optical purity (93% ee) was determined by HPLC analysis at 20 °C using a CHIRALPAK AD-3 column (hexanes/2-propanol = 95
:5; flow rate: 1.0 mL min−1; retention times: 8.5 min (S), 10.6 min (R)).
(S)-2-(2-(Hydroxymethyl)naphthalen-1-yl)-3,6-dimethylphenol (S)-4b. Colorless oil; [α]21D + 21.7 (c 0.6, CHCl3) (lit.14 [α]20D + 27.2 (c 0.4, CHCl3) for (S)-4b with 87% ee); 1H-NMR (500 MHz, CDCl3) δ 7.98 (d, J = 8.5 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.75 (d, J = 8.5 Hz, 1H), 7.49–7.52 (m, 1H), 7.34–7.41 (m, 2H), 7.16 (d, J = 7.5 Hz, 1H), 6.86 (d, J = 7.5 Hz, 1H), 4.52 (s, 2H), 2.29 (s, 3H), 1.79 (s, 3H). The spectroscopic data are in good agreement with those reported.14 Its optical purity (98% ee) was determined by HPLC analysis at 20 °C using a CHIRALPAK AD-3 column (hexanes/2-propanol = 90
:10; flow rate: 1.0 mL min−1; retention times: 12.9 min (R), 29.8 min (S)).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by JSPS KAKENHI [grant numbers: 16H01151/18HO4411 (Middle Molecular Strategy) and 18H02556] and AMED (grant number 18am0101084j0002). We acknowledge Toyobo Co. Ltd. for supplying LIP301.Notes and references
- (a) S. T. Toenjes and J. L. Gustafson, Future Med. Chem., 2018, 10, 409–422 CrossRef CAS PubMed; (b) P. W. Glunz, Bioorg. Med. Chem. Lett., 2018, 28, 53–60 CrossRef CAS PubMed; (c) J. E. Smyth, N. M. Butler and P. A. Keller, Nat. Prod. Rep., 2015, 32, 1562–1583 RSC; (d) A. Zask, J. Murphy and G. A. Ellestad, Chirality, 2013, 25, 265–274 CrossRef CAS PubMed; (e) S. R. LaPlante, P. J. Edwards, L. D. Fader, A. Jakalian and O. Hucke, ChemMedChem, 2011, 6, 505–513 CrossRef CAS PubMed; (f) J. Clayden, W. J. Moran, P. J. Edwards and S. R. LaPlante, Angew. Chem., Int. Ed., 2009, 48, 6398–6401 CrossRef CAS PubMed.
- For reviews, see: (a) S. S. Yu and L. Pu, Tetrahedron, 2015, 71, 745–772 CrossRef CAS; (b) M. Shibasaki and S. Matsunaga, in Privileged Chiral Ligands and Catalysts, ed. Q.-L.Zhou, Wiley-VCH, Weinheim, 2011, pp. 295–332 Search PubMed; (c) J. M. Brunel, Chem. Rev., 2007, 107, PR1–PR45 CrossRef CAS; (d) Y. Chen, S. Yekta and A. K. Yudin, Chem. Rev., 2003, 103, 3155–3211 CrossRef CAS PubMed; (e) P. Kocovsky, S. Vyskocil and M. Smrcina, Chem. Rev., 2003, 103, 3213–3245 CrossRef CAS PubMed.
- For general reviews on the asymmetric synthesis of atropoisomeric biaryls, see: (a) Y. B. Wang and B. Tan, Acc. Chem. Res., 2018, 51, 534–547 CrossRef CAS PubMed; (b) P. Renzi, Org. Biomol. Chem., 2017, 15, 4506–4516 RSC; (c) P. Loxq, E. Manoury, R. Poli, E. Deydier and A. Labande, Coord. Chem. Rev., 2016, 308, 131–190 CrossRef CAS; (d) J. Wencel-Delord and F. Colobert, Synthesis, 2016, 48, 2981–2996 CrossRef CAS; (e) S. Shirakawa, S. Y. Liu and S. Kaneko, Chem. – Asian J., 2016, 11, 330–341 CrossRef CAS PubMed; (f) J. Wencel-Delord, A. Panossian, F. R. Leroux and F. Colobert, Chem. Soc. Rev., 2015, 44, 3418–3430 RSC; (g) D. Zhang and Q. R. Wang, Coord. Chem. Rev., 2015, 286, 1–16 CrossRef CAS; (h) G. Y. Ma and M. P. Sibi, Chem.–Eur. J., 2015, 21, 11644–11657 CrossRef CAS PubMed; (i) G. Bencivenni, Synlett, 2015, 1915–1922 CrossRef CAS; (j) G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563–639 CrossRef CAS PubMed; (k) G. Bringmann, A. J. P. Mortimer, P. A. Keller, M. J. Gresser, J. Garner and M. Breuning, Angew. Chem., Int. Ed., 2005, 44, 5384–5427 CrossRef CAS PubMed.
- For reviews, see: (a) M. Klussmann and D. Sureshkumar, Synthesis, 2011, 353–369 CrossRef CAS; (b) H. Wang, Chirality, 2010, 22, 827–837 CrossRef CAS PubMed; (c) M. C. Kozlowski, B. J. Morgan and E. C. Linton, Chem. Soc. Rev., 2009, 38, 3193–3207 RSC . For selected examples, see:; (d) H. Y. Kim, S. Takizawa, H. Sasai and K. Oh, Org. Lett., 2017, 19, 3867–3870 CrossRef CAS PubMed; (e) S. Narute, R. Parnes, F. D. Toste and D. Pappo, J. Am. Chem. Soc., 2016, 138, 16553–16560 CrossRef CAS PubMed; (f) H. Egami, K. Matsumoto, T. Oguma, T. Kunisu and T. Katsuki, J. Am. Chem. Soc., 2010, 132, 13633–13635 CrossRef CAS PubMed; (g) H. Egami and T. Katsuki, J. Am. Chem. Soc., 2009, 131, 6082–6083 CrossRef CAS PubMed; (h) S. Takizawa, T. Katayama, C. Kameyama, K. Onitsuka, T. Suzuki, T. Yanagida, T. Kawai and H. Sasai, Chem. Commun., 2008, 1810–1812 RSC; (i) J. B. Hewgley, S. S. Stahl and M. C. Kozlowski, J. Am. Chem. Soc., 2008, 130, 12232–12233 CrossRef CAS PubMed; (j) Q. X. Guo, Z. J. Wu, Z. B. Luo, Q. Z. Liu, J. L. Ye, S. W. Luo, L. F. Cun and L. Z. Gong, J. Am. Chem. Soc., 2007, 129, 13927–13938 CrossRef CAS PubMed; (k) X. L. Li, J. B. Hewgley, C. A. Mulrooney, J. M. Yang and M. C. Kozlowski, J. Org. Chem., 2003, 68, 5500–5511 CrossRef CAS PubMed; (l) M. Nakajima, I. Miyoshi, K. Kanayama, S. Hashimoto, M. Noji and K. Koga, J. Org. Chem., 1999, 64, 2264–2271 CrossRef CAS; (m) M. Smrcina, J. Polakova, S. Vyskocil and P. Kocovsky, J. Org. Chem., 1993, 58, 4534–4538 CrossRef CAS.
- For instances, see: (a) S. C. Lu, S. B. Poh and Y. Zhao, Angew. Chem., Int. Ed., 2014, 53, 11041–11045 CrossRef CAS PubMed; (b) G. Y. Ma, J. Deng and M. P. Sibi, Angew. Chem., Int. Ed., 2014, 53, 11818–11821 CrossRef CAS PubMed; (c) H. Aoyama, M. Tokunaga, J. Kiyosu, T. Iwasawa, Y. Obora and Y. Tsuji, J. Am. Chem. Soc., 2005, 127, 10474–10475 CrossRef CAS PubMed.
- For a review, see: B. Skrobo, J. D. Rolfes and J. Deska, Tetrahedron, 2016, 72, 1257–1275 CrossRef CAS.
- (a) P. Gupta, A. Rouf, B. A. Shah, N. Mahajan, A. Chaubey and S. C. Taneja, J. Mol. Catal. B: Enzym., 2014, 101, 35–39 CrossRef CAS; (b) T. Taniguchi, T. A. Fukuba, S. Nakatsuka, S. Hayase, M. Kawatsura, H. Uno and T. Itoh, J. Org. Chem., 2008, 73, 3875–3884 CrossRef CAS PubMed; (c) N. Aoyagi, N. Ogawa and T. Izumi, Tetrahedron Lett., 2006, 47, 4797–4801 CrossRef CAS; (d) M. Seki, T. Furutani, M. Hatsuda and R. Imashiro, Tetrahedron Lett., 2000, 41, 2149–2152 CrossRef CAS; (e) K. Tanaka, T. Furuta, K. Fuji, Y. Miwa and T. Taga, Tetrahedron: Asymmetry, 1996, 7, 2199–2202 CrossRef CAS; (f) G. L. Lin, S. J. Chen and H. L. Sun, J. Chin. Chem. Soc., 1994, 41, 459–465 CrossRef CAS; (g) R. J. Kazlauskas, Org. Synth., 1992, 70, 60 CrossRef CAS; (h) R. J. Kazlauskas, J. Am. Chem. Soc., 1989, 111, 4953–4959 CrossRef CAS; (i) S. Miyano, K. Kawahara, Y. Inoue and H. Hashimoto, Chem. Lett., 1987, 355–356 CrossRef CAS; (j) Y. Fujimoto, H. Iwadate and N. Ikekawa, J. Chem. Soc., Chem. Commun., 1985, 1333–1334 RSC . For hydrolytic enzymatic desymmetrization of biaryl esters:; (k) T. Matsumoto, T. Konegawa, T. Nakamura and K. Suzuki, Synlett, 2002, 122–124 CrossRef CAS; (l) For hydrolytic enzymatic KR of a racemic biphenol derivative, see:; (m) T. Takemura, G. Emoto, J. Satoh, Y. Kobayashi, C. Yaginuma, Y. Takahashi, T. Utsukihara and C. A. Horiuchi, J. Mol. Catal. B: Enzym., 2008, 55, 104–109 CrossRef CAS.
- (a) M. Juarez-Hernandez, D. V. Johnson, H. L. Holland, J. McNulty and A. Capretta, Tetrahedron: Asymmetry, 2003, 14, 289–291 CrossRef CAS; (b) M. Inagaki, J. Hiratake, T. Nishioka and J. Oda, Agric. Biol. Chem., 1989, 53, 1879–1884 CAS . For acylative enzymatic KR of a racemic biphenol derivative, see:; (c) C. Sanfilippo, G. Nicolosi, G. Delogu, D. Fabbri and M. A. Dettori, Tetrahedron: Asymmetry, 2003, 14, 3267–3270 CrossRef CAS.
- For our latest study on DKR of 2,2′- dihydroxy-1,1′-biaryls, see: G. A. I. Moustafa, Y. Oki and S. Akai, Angew. Chem., Int. Ed., 2018, 57, 10278–10282 CrossRef CAS PubMed.
- E value indicates the enantioselectivity in the enzymatic KRs11a and is the same as the S value that is generally used to denote the enantioselectivity in the organocatalytic KRs11b.
- (a) C. S. Chen, Y. Fujimoto, G. Girdaukas and C. J. Sih, J. Am. Chem. Soc., 1982, 104, 7294–7299 CrossRef CAS; (b) J. M. Keith, J. F. Larrow and E. N. Jacobsen, Adv. Synth. Catal., 2001, 343, 5–26 CrossRef CAS.
- We observed a similar rate enhancement in some other solvents. For example, in diisopropyl ether, 50% conversion was achieved at 35 °C after 24 h with high enantioselectivity ((R)-2a: 96% ee; (S)-1a: 97% ee; E value >200) as that shown in Table 1, entry 8..
- When (±)-4a was subjected to reaction conditions similar to those in Table 6, entry 1 while omitting the lipase, (±)-5a was obtained in 40% yield..
- B. Skrobo and J. Deska, Tetrahedron: Asymmetry, 2013, 24, 1052–1056 CrossRef CAS.
- For alternative approaches to enantioenriched derivatives of 4 via dynamic kinetic ring opening of the corresponding Bringmann’s lactones, see: (a) G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563–639 CrossRef CAS PubMed; (b) G. Bringmann, T. Gulder and T. A. M. Gulder, in Asymmetric Synthesis: The Essentials, ed. M.Christmann and S.Bräse, Wiley-VCH, Weinheim, 2nd edn, 2007, pp. 246–250 Search PubMed; (c) G. Bringmann and D. Menche, Acc. Chem. Res., 2001, 34, 615–624 CrossRef CAS PubMed.
- K. Faber, in Biotransformations in Organic Chemistry: A Textbook, Springer, Cham, 7th edn, 2018, ch. 2 Search PubMed.
- For typical examples, see: (a) K. Kato, M. Katayama, S. Fujii and H. Kimoto, Biosci., Biotechnol., Biochem., 1995, 59, 2178–2180 CrossRef CAS PubMed; (b) K. Kato, Y.-f. Gong, S. Tanaka, M. Katayama and H. Kimoto, J. Mol. Catal. B: Enzym., 2001, 11, 287–294 CrossRef CAS.
- (a) B. Martin-Matute, M. Edin, K. Bogar, F. B. Kaynak and J. E. Bäckvall, J. Am. Chem. Soc., 2005, 127, 8817–8825 CrossRef CAS PubMed; (b) B. Martin-Matute, M. Edin, K. Bogar and J. E. Bäckvall, Angew. Chem., Int. Ed., 2004, 43, 6535–6539 CrossRef CAS PubMed; (c) J. H. Choi, Y. H. Kim, S. H. Nam, S. T. Shin, M. J. Kim and J. Park, Angew. Chem., Int. Ed., 2002, 41, 2373–2376 CrossRef CAS.
- P. Lustenberger and F. Diederich, Helv. Chim. Acta, 2000, 83, 2865–2883 CrossRef CAS.
- D.-R. Hwang, C.-P. Chen and B.-J. Uang, Chem. Commun., 1999, 1207–1208 RSC.
- J.-M. Yoon, C.-Y. Lee, Y.-I. Jo and C.-H. Cheon, J. Org. Chem., 2016, 81, 8464–8469 CrossRef CAS PubMed.
- E. Alberico, S. Karandikar and S. Gladiali, ChemCatChem, 2010, 2, 1395–1398 CrossRef CAS.
- T. R. Wu, L. Shen and J. M. Chong, Org. Lett., 2004, 6, 2701–2704 CrossRef CAS PubMed.
- Y. Li and Q. Li, Org. Lett., 2012, 14, 4362–4365 CrossRef CAS PubMed.
- H. Gao, Q.-L. Xu, C. Keene, M. Yousufuddin, D. H. Ess and L. Kürti, Angew. Chem., Int. Ed., 2016, 55, 566–571 CrossRef CAS PubMed.
- D. Cai, D. L. Hughes, T. R. Verhoeven and P. J. Reider, Tetrahedron Lett., 1995, 36, 7991–7994 CrossRef CAS.
- L. V. Heumann and G. E. Keck, J. Org. Chem., 2008, 73, 4725–4727 CrossRef CAS PubMed.
- M. Sako, S. Takizawa, Y. Yoshida and H. Sasai, Tetrahedron: Asymmetry, 2015, 26, 613–616 CrossRef CAS.
- A. I. Meyers, T. D. Nelson, H. Moorlag, D. J. Rawson and A. Meier, Tetrahedron, 2004, 60, 4459–4473 CrossRef CAS.
- M. Terada and K. Dan, Chem. Commun., 2012, 48, 5781–5783 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra09070j |
| This journal is © The Royal Society of Chemistry 2019 |