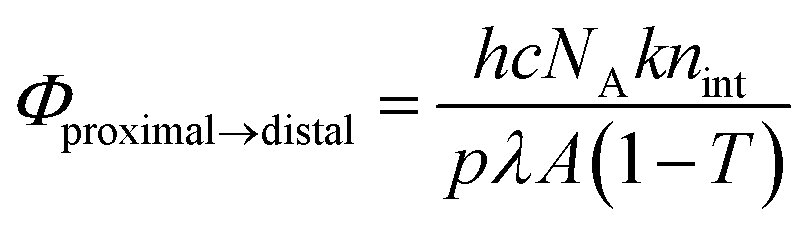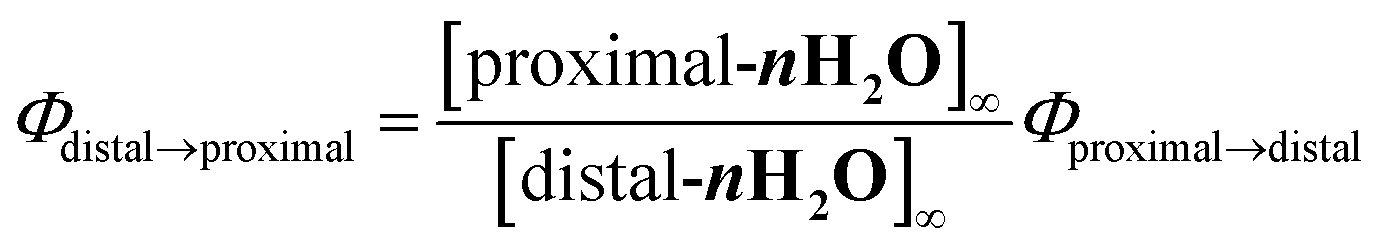DOI:
10.1039/C8RA08943D
(Paper)
RSC Adv., 2019,
9, 2002-2010
Photoisomerization and thermal isomerization of ruthenium aqua complexes with chloro-substituted asymmetric bidentate ligands†
Received
29th October 2018
, Accepted 4th January 2019
First published on 15th January 2019
Abstract
A series of ruthenium complexes with chloro-substituted bidentate ligands, proximal-[Ru(tpy)(Cl-pyqu)L]n+ [n = 1 for L = Cl, and n = 2 for L = OH2, tpy = 2,2′;6′,2′′-terpyridine, pyqu = 2-(2′-pyridyl)quinoline] were synthesized and their reversible photoisomerizations and thermal isomerizations were investigated experimentally. The crystal structures of the complexes indicated that introduction of a chloro substituent at the 4- or 4′-position of the pyqu ligand did not change the structure around the metal center from that of the non-substituted complex, proximal-[Ru(tpy)(pyqu)L]n+. In contrast, the 6′-substituted complexes had sterically hindered environments around the metal center. The ruthenium aqua complexes showed reversible photoisomerization between the proximal and distal isomers. The quantum yield for photoisomerization of the 6′-substituted ruthenium aqua complex was almost twice as large as those of the other derivatives. This is explained by weakening of the ligand field on the ruthenium center by introduction of a chloro substituent at the 6′-position. Thermal back isomerization from the distal isomer to the proximal one was observed for the 6′-substituted complex, but such reactions were not observed for the other derivatives. The steric hindrance in the 6′-substituted aqua complex enhanced both thermal isomerization and photoisomerization.
Introduction
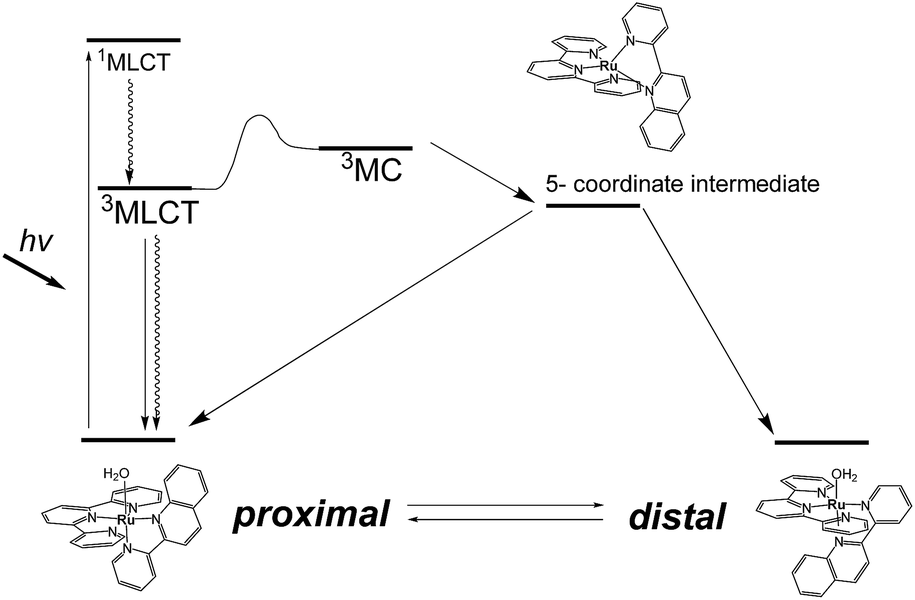
Photoresponsive ruthenium complexes with photolabile monodentate ligand(s) are of interest because of their catalytic activities, bioactivities, and photochemical and electrochemical properties.1 These complexes are excited by visible light to metal-to-ligand charge-transfer (1MLCT) states, and then relax to 3MLCT states via rapid intersystem crossing.2 The 3MLCT excited states are responsible for thermal excitation to metal-centered (3MC) excited states, which leads photodissociation of a labile ligand to give five-coordinated intermediates (Scheme 1).3 Recoordination of a ligand to the intermediate results in photosubstitution or photoisomerization reactions. Electronic and structural effects on the photolabilities of polypyridyl ruthenium complexes in photosubstitution have been extensively studied.1c,4 It has been found that the reactivity of a complex is strongly controlled by the energy differences between the 3MLCT excited states and 3MC states.5 Steric hindrance around the metal center weaken the ligand field and accelerates photosubstitution by promoting the thermal transition from 3MLCT to 3MC states.4f,6
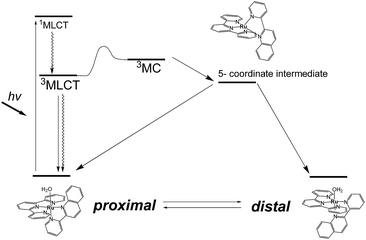 |
| | Scheme 1 Energy diagram for photoisomerization of proximal and distal [Ru(tpy)(pyqu)OH2]2+. | |
Photoisomerizations of ruthenium complexes are rarer than photosubstitutions.7 The first example was cis–trans isomerization of [Ru(2,2′-bipyridine)2(OH2)2]2+, reported by Meyer et al.8 Linkage isomerizations of dimethyl sulfoxide derivatives have been experimentally and mechanistically studied by Rack et al.,4b,9 and were recently used in materials that show light-induced macroscopic morphological changes.10
We have reported irreversible11 and reversible12 photoisomerizations of mononuclear ruthenium aqua complexes containing asymmetric bidentate ligands. These molecules are important because of their high catalytic activities in water oxidation, which is essential for the development of artificial photosynthesis.13 Our experimental and theoretical studies showed that the reversibility of the reaction was controlled by intramolecular hydrogen bonding between an aqua ligand and a pendant moiety of the bidentate ligand (Scheme 1).11b,12,14 Recently, we used these photoresponsive complexes for morphological control of large molecular assemblies. Ruthenium-complex-based amphiphiles were prepared by introducing two alkyl chains into the tridentate and bidentate ligands. Giant vesicles containing the ruthenium complexes displayed morphological changes under irradiation with red light at 635 nm, i.e., in the phototherapeutic window, and can be used in drug delivery systems.15 Investigation of structural and electronic effects on photoisomerization is therefore important for enabling control of the catalytic activities and photoresponsive properties of ruthenium complexes.
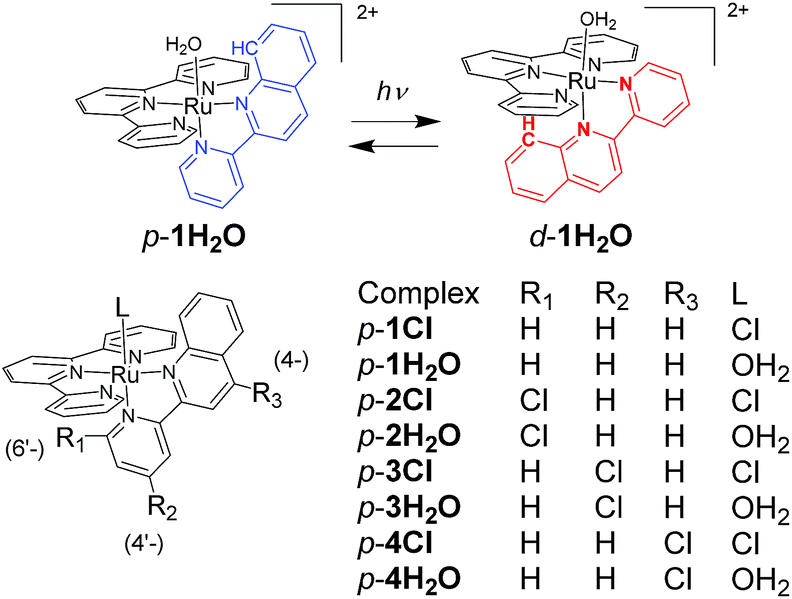
In this work, we investigated structural and electronic effects on the reversible photoisomerizations of proximal-[(tpy)(pyqu)OH2]2+ (p-1H2O) by introducing a chloro substituent on the bidentate pyqu ligand (tpy = 2,2′;6′,2′′-terpyridine, pyqu = 2-(2′-pyridyl)quinoline). As shown in Scheme 2, we prepared ruthenium aqua complexes, namely p-2H2O, p-3H2O, and p-4H2O, with a chloro substituent at the 4-, 4′-, and 6′-positions on the bidentate ligand, respectively. The electron-withdrawing chloro substituent on the pyqu ligand stabilizes the π* levels of the complexes. In contrast, based on studies of the photosubstitution of ruthenium complexes, introduction of the chloro substituent at the 6′-position (p-2H2O) destabilizes the 3MC state because of structural distortion around the ruthenium center. All the complexes synthesized in this work were fully characterized using X-ray crystallography. The results show that structural distortion strongly affected photoisomerization of the complexes. Additionally, thermal back isomerization was observed for the sterically hindered aqua complex, p-2H2O.
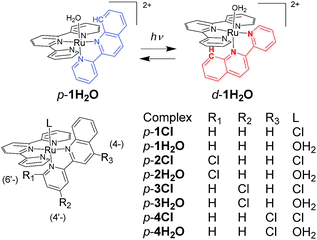 |
| | Scheme 2 Reversible photoisomerization of p-1H2O (top) and structures of p-nCl and p-nH2O (n = 1–4). | |
Results and discussion
Synthesis
Chloro-substituted bidentate ligands were obtained by oxidation of the pyqu ligand with hydrogen peroxide, followed by chlorination with POCl3. Heating a mixture of Ru(tpy)Cl3, the bidentate ligand, and a reducing reagent (triethylamine or ascorbic acid) gave a series of ruthenium chloro complexes. In the thermal reactions, the chloro complexes were obtained as mixtures of the proximal and distal isomers. The distal isomers were minor products, based on thin-layer-chromatography and 1H NMR spectra of the crude products. In the 1H NMR spectra of the crude products, the formation of distal isomers was indicated by prominent doublet peaks at 10.0–10.5 ppm with coupling constants of 5–6.5 Hz, which were assigned to the 6′-proton on the pyqu derivatives. The proximal isomer was isolated using silica-gel chromatography or the differences between the solubilities of the proximal and distal isomers in methanol (yields for p-1Cl, p-3Cl, and p-4Cl were 54%, 20%, and 45%, respectively). The proximal selectivity were better at a reaction temperature of 50 °C than at reflux temperature for p-1Cl, p-3Cl, and p-4Cl. The synthetic yields for p-1Cl, p-3Cl, and p-4Cl were poor because of unreacted bidentate ligands and Ru(tpy)Cl3. The reaction of 6′-substituted pyqu, i.e., 2-[2′-(6′-chloro)pyridyl]quinoline, and Ru(tpy)Cl3 was much slower than those of the other derivatives because of the sterically bulky bidentate ligand. A mild reaction of Ru(tpy)Cl3 with 6′-substituted pyqu at 50 °C gave no product even after 3 days. At reflux temperature, 23% of the product was obtained after 24 h; the proximal isomer was the main product. We tried to increase the synthetic yield by performing the reaction under high pressure and at high temperature (1 MPa and 150 °C), but Ru(tpy)2 was obtained as the main product. In contrast to the proximal selectivity of thermal reactions with Ru(tpy)Cl3 and pyqu derivatives observed in this work, it has been reported that the thermal reactions of Ru(tpy)Cl3 and 2-(2-pyridyl)-1,8-naphthyridine11b,11d or phenanthroline derivatives16 with a pendant moiety at the 2-position are distal selective. We previously reported that the d-1H2O would be more instable rather than the p-1H2O due steric repulsion between the pendant moiety of the pyqu ligand and a monodentate ligand as shown by the solid-state structure. The proximal selectivity of ruthenium–chloro complexes in this work could be caused by the similar reasons.
Ruthenium aqua complexes (p-nH2O) were quantitatively obtained by dissolving the corresponding ruthenium chloro complexes in water. On dissolution, the purple solid ruthenium chloro complexes immediately changed to reddish aqueous solutions of aqua complexes. 1H NMR spectra of pure proximal-ruthenium aqua complexes were obtained immediately after dissolution. The results coincide with our previous result that the aquation of p-1Cl proceeded at 9.2 × 10−2 s−1 at room temperature,12 which is much faster the aquation of ruthenium chloro complexes. The acceleration of the aquation reaction of p-nCl (n = 1–4) is caused by steric repulsion between the monodentate chloro ligand and the pendant moiety of the bidentate ligand, as shown by the crystal structure (vide infra). Acceleration of ligand exchange reactions by the steric repulsion has been discussed by Takeuchi et al.17 and Bonnet et al.18 bidentate ligand, as shown by the crystal structure (vide infra).
Crystal structures
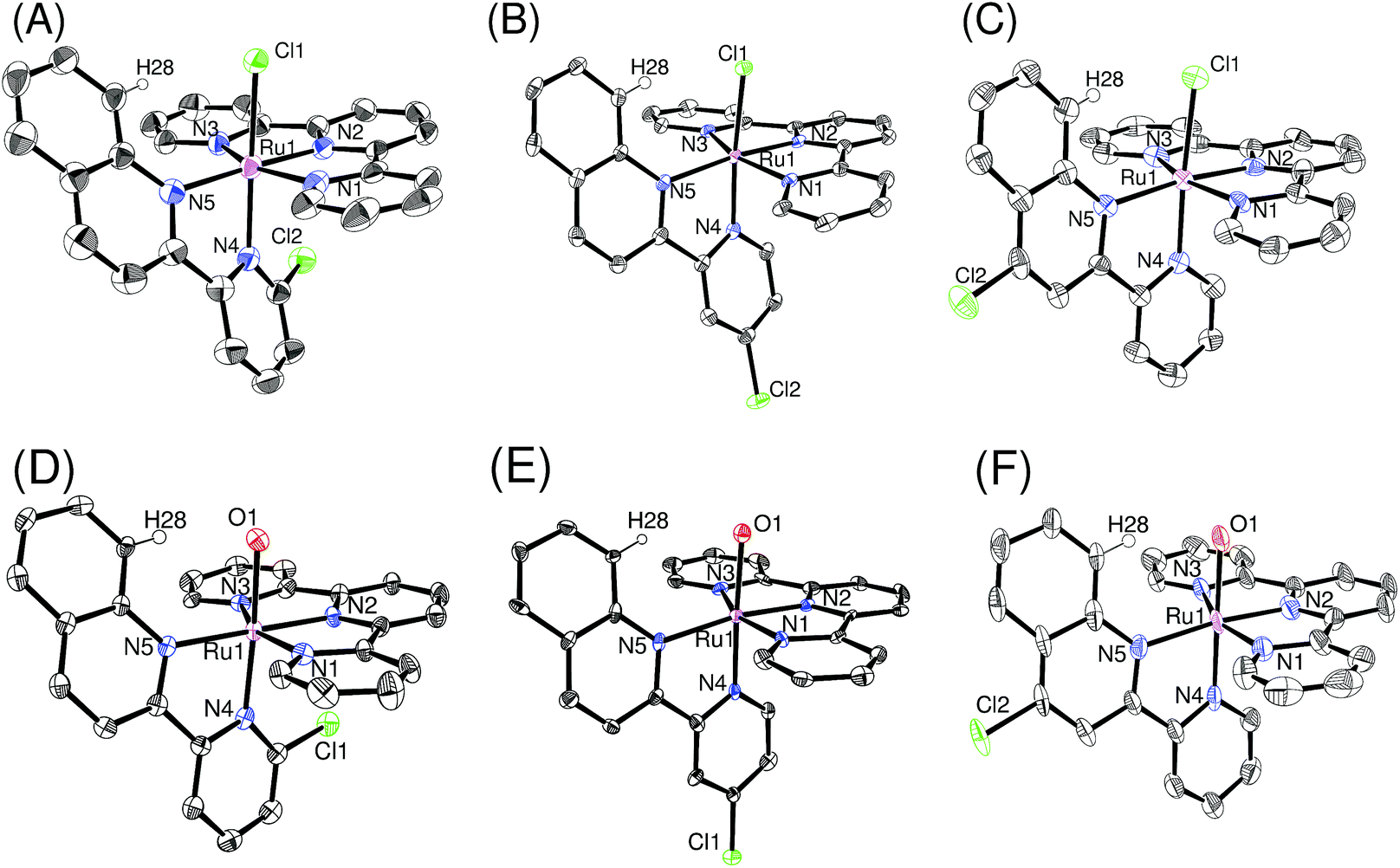
Single crystals of ruthenium chloro complexes suitable for X-ray crystallography were obtained by slow evaporation of ethanol solutions of p-nCl containing counter anions (NH4PF6 or NaBF4). Crystals of ruthenium aqua complexes were obtained by dissolving the corresponding chloro complexes in water, mixing with an ethanol solution of the counter anion and acetone, and then allowing the acetone to evaporate. Fig. 1 shows ORTEP views of the isolated complexes. The X-ray crystallographic data, and selected bond lengths and bond angles are summarized in Tables S1,† 1, and 2, respectively. For the ruthenium chloro complexes p-1Cl, 3Cl, 4Cl, the bond lengths and angles around the ruthenium center are similar to those reported by Reedijk et al.19 for [Ru(tpy)(2,2′-biquinoline)Cl]+ rather than those of derivatives of [Ru(tpy)(2,2′-bipyridine)Cl]+.20 This similarity arises because of steric repulsion between a chloro ligand and pendant moieties of pyqu derivatives; this is confirmed by the distances between H28 and Cl1 (2.42–2.57 Å). The bond length between Ru1 and N4 [2.076(4) Å] and the N2–Ru1–N4 angle [100.58(18)] in p-2Cl are larger than those in the other complexes, by ca. 0.05 Å and 5°, respectively (Table 1). This large difference mainly arises from steric repulsion between the chloro substituent at the 6′-position of the pyqu ligand and the terpyridine ligand. These large differences were also observed for the ruthenium aqua complexes. The N2–Ru1–N4 angles in p-1H2O, 3H2O, and 4H2O (93.6° to 95.6°) were similar to one another, whereas that for p-1H2O [101.93(15)°] was larger than those in the others, as shown in Table 2.
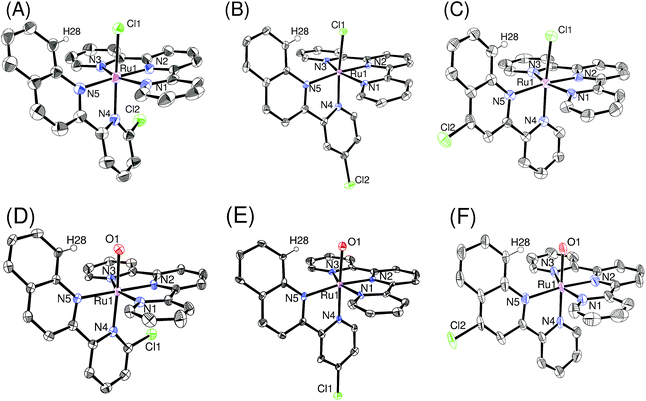 |
| | Fig. 1 ORTEP views of (A) p-[2Cl](PF6), (B) p-[3Cl](BF4), (C) p-[4Cl](PF6)·EtOH, (D) p-[2H2O](PF6)2·2H2O, (E) p-[3H2O](CF3SO3)2·H2O, and (F) p-[4H2O](NO3)2·2H2O. Counter anions and solvents are omitted for clarity. The ORTEP view of p-[2Cl](PF6) is plotted in 30% probability (measured at 296 K), and others are plotted in 50% probability (measured at 100 K). | |
Table 1 Selected bond distances (Å) and angles (°) for series of ruthenium chloro complexes
| |
p-1Cl |
p-2Cl |
p-3Cl |
p-4Cl |
| Ru1–Cl1 |
2.464(2) |
2.4058(15) |
2.4462(9) |
2.446(2) |
| Ru1–N1 |
2.087(8) |
2.090(5) |
2.092(3) |
2.057(7) |
| Ru1–N2 |
1.961(4) |
1.946(4) |
1.955(3) |
1.950(7) |
| Ru1–N3 |
2.085(8) |
2.063(4) |
2.075(3) |
2.072(7) |
| Ru1–N4 |
2.025(5) |
2.076(4) |
2.020(3) |
2.031(7) |
| Ru1–N5 |
2.138(4) |
2.099(5) |
2.115(3) |
2.148(6) |
| Cl1–Ru1–N1 |
86.16(13) |
86.18(12) |
84.93(8) |
91.00(19) |
| Cl1–Ru1–N2 |
82.39(14) |
83.36(13) |
86.14(8) |
80.33(19) |
| Cl1–Ru1–N3 |
92.01(15) |
90.47(12) |
96.03(8) |
87.31(19) |
| Cl1–Ru1–N4 |
171.94(13) |
170.54(13) |
169.14(8) |
176.08(18) |
| Cl1–Ru1–N5 |
105.20(12) |
98.03(13) |
101.12(8) |
105.82(19) |
| N1–Ru1–N2 |
79.68(18) |
79.8(2) |
79.50(11) |
80.0(3) |
| N1–Ru1–N3 |
160.20(17) |
159.7(2) |
159.02(11) |
160.3(3) |
| N1–Ru1–N4 |
85.95(18) |
86.08(17) |
84.65(11) |
87.6(3) |
| N1–Ru1–N5 |
101.48(17) |
96.50(19) |
99.99(11) |
95.5(2) |
| N2–Ru1–N3 |
80.53(18) |
79.94(19) |
79.66(11) |
80.4(3) |
| N2–Ru1–N4 |
94.51(19) |
100.58(18) |
94.96(11) |
95.8(3) |
| N2–Ru1–N5 |
172.36(18) |
175.98(18) |
172.67(11) |
172.5(3) |
| N3–Ru1–N4 |
94.83(19) |
98.66(17) |
94.80(11) |
92.7(3) |
| N3–Ru1–N5 |
98.03(17) |
103.79(18) |
100.37(11) |
103.9(3) |
| N4–Ru1–N5 |
78.09(18) |
77.48(18) |
77.71(11) |
78.0(2) |
Table 2 Selected bond distances (Å) and angles (°) for series of ruthenium aqua complexes
| |
p-1H2O |
p-2H2O |
p-3H2O |
p-4H2O |
| Ru1–O1 |
2.137(8) |
2.129(4) |
2.157(6) |
2.136(5) |
| Ru1–N1 |
2.061(8) |
2.091(4) |
2.081(8) |
2.054(6) |
| Ru1–N2 |
1.937(7) |
1.961(3) |
1.949(8) |
1.952(6) |
| Ru1–N3 |
2.060(8) |
2.070(4) |
2.068(8) |
2.066(6) |
| Ru1–N4 |
2.091(9) |
2.057(4) |
2.030(8) |
2.002(6) |
| Ru1–N5 |
2.139(9) |
2.096(3) |
2.146(8) |
2.086(5) |
| O1–Ru1–N1 |
86.9(3) |
87.51(15) |
88.2(3) |
88.5(2) |
| O1–Ru1–N2 |
84.0(3) |
84.44(14) |
84.4(3) |
85.1(2) |
| O1–Ru1–N3 |
91.0(3) |
89.52(15) |
88.1(3) |
88.0(2) |
| O1–Ru1–N4 |
175.5(3) |
170.76(14) |
177.1(3) |
177.3(2) |
| O1–Ru1–N5 |
105.1(3) |
95.33(14) |
103.9(3) |
101.1(2) |
| N1–Ru1–N2 |
79.7(3) |
79.53(15) |
79.3(3) |
79.9(2) |
| N1–Ru1–N3 |
159.5(3) |
158.81(13) |
160.1(3) |
159.7(2) |
| N1–Ru1–N4 |
88.6(3) |
87.09(16) |
89.4(3) |
89.0(2) |
| N1–Ru1–N5 |
101.3(3) |
98.12(15) |
96.5(3) |
96.8(2) |
| N2–Ru1–N3 |
79.9(3) |
79.31(15) |
80.9(3) |
79.9(2) |
| N2–Ru1–N4 |
94.4(3) |
101.93(15) |
93.6(3) |
95.6(2) |
| N2–Ru1–N5 |
170.8(3) |
177.64(17) |
170.7(3) |
172.9(2) |
| N3–Ru1–N4 |
92.9(3) |
98.20(16) |
93.7(3) |
94.7(2) |
| N3–Ru1–N5 |
98.9(3) |
103.04(14) |
103.4(3) |
103.5(2) |
| N4–Ru1–N5 |
76.6(4) |
78.03(14) |
78.0(3) |
78.1(2) |
Absorption spectra
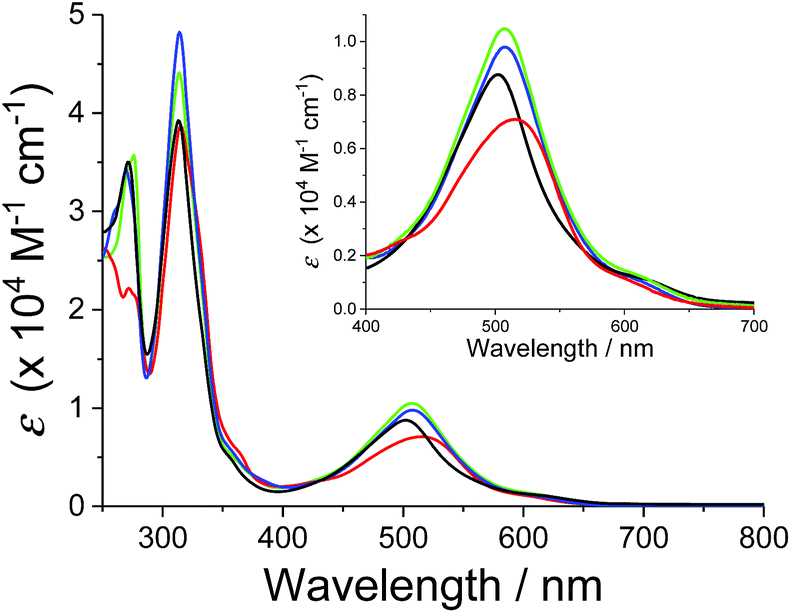
The absorption spectra of the series of ruthenium chloro and aqua complexes are shown in Fig. S8† and 2, respectively. The absorption maxima and extinction coefficients in the visible region are summarized in Table 3. All the complexes showed π–π* transitions in the UV region and MLCT transitions in the visible region. As expected from the ligand field strengths of the chloro and aqua ligands, the absorption maxima of the MLCT transitions for the ruthenium chloro complexes (p-nCl) were red-shifted from those of the corresponding ruthenium aqua complexes (p-nH2O) by 20–37 nm. The MLCT absorption bands of p-3H2O and p-4H2O (λmax = 507 nm) were red shifted by 5 nm compared with that of p-1H2O (λmax = 502 nm). The p-2H2O complex (λmax = 515 nm) showed the largest red shift among the series of aqua complexes (Fig. 2). The red shifts in the spectra can be explained by stabilization of the π* levels of the pyqu ligands by the chloro substituent, according to density functional theory (DFT) calculations (vide infra).
Table 3 Summary of absorption properties and parameters for reversible photoisomerization
| |
|
|
Photoisomerization |
| |
λmax/nm (ε/M−1 cm−1) |
Kpi |
Φp→d, Φd→p |
| p-1Cl |
528 (8700) |
— |
— |
| p-2Cl |
552 (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300) 300) |
— |
— |
| p-3Cl |
538 (10600) |
— |
— |
| p-4Cl |
537 (11900) |
— |
— |
| p-1H2O |
502 (8800) |
3.2 |
8.3 × 10−3, 2.4 × 10−2 |
| p-2H2O |
515 (8100) |
3.2 |
1.7 × 10−2, 5.7 × 10−2 |
| p-3H2O |
507 (10700) |
3.0 |
8.2 × 10−3, 2.7 × 10−2 |
| p-4H2O |
507 (12300) |
2.7 |
8.6 × 10−3, 2.0 × 10−2 |
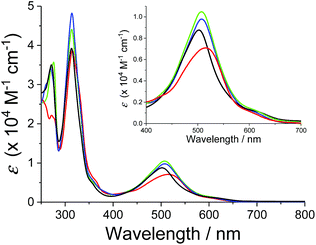 |
| | Fig. 2 Absorption spectra of p-1H2O (black), p-2H2O (red), p-3H2O (blue), and p-4 H2O (green) in water. Absorption spectrum of p-1H2O was obtained from ref. 12. | |
Photoisomerization
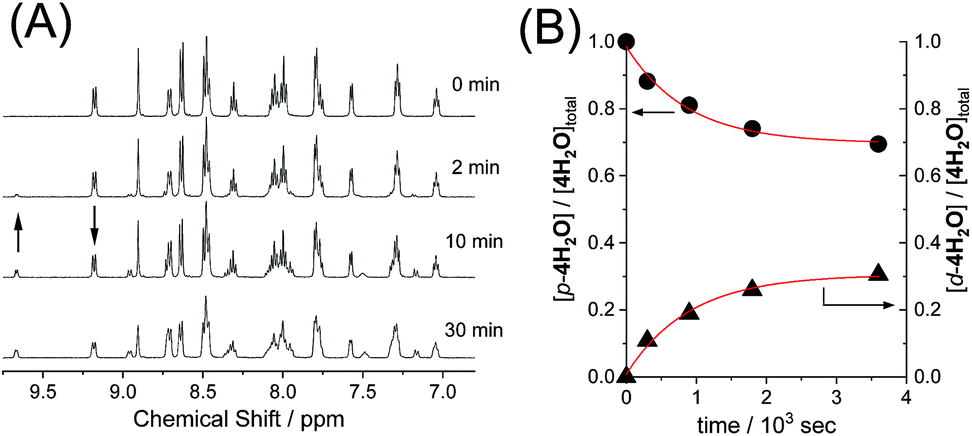
The reversible photoisomerizations of the series of ruthenium aqua complexes were investigated using 1H NMR spectroscopy. UV-vis absorption spectra of the complexes were recorded during the photochemical reactions, but the differences between the proximal and distal isomers were too small for accurate evaluation of the photoisomerization. Fig. 3 shows representative 1H NMR spectroscopic changes for p-4H2O under visible-light irradiation (λ > 380 nm, 70 mW cm−2). The doublet peak at 9.17 ppm for p-4H2O decreased with irradiation time and a new doublet peak from the distal isomer, d-4H2O, appeared at 9.66 ppm; these peaks are assigned to the protons at the 8- and 6′-positions, respectively, of the pyqu ligand. The kinetic profiles obtained by integration of the proximal and distal isomer peaks are shown in Fig. 3(B). The kinetic profiles show that reversible photoisomerization occurred to give a mixture of p-and d-4H2O in the photostationary state. Table 3 shows the kinetic parameters for the photoisomerizations. The equilibrium constants (Kpi = [p-nH2O]/[d-nH2O]) in the photostationary state are close to one another. The data in Table 3 show that the quantum yields for the photoisomerization using monochromatic light at 508 nm (full width at half maximum = 1.5 nm, 1.6 mW cm−2) were similar (0.0082–0.0086) for p-1H2O, 3H2O, and 4H2O. However, the quantum yield for p-2H2O was 0.017, which is almost twice as large as those for the other aqua complexes.
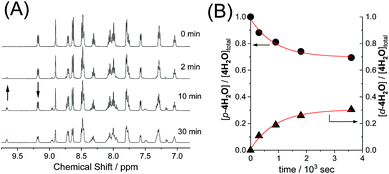 |
| | Fig. 3 (A) 1H NMR spectra of p-4H2O (4 mM) in D2O under the light irradiation (λ > 380 nm, 70 mW cm−2) at 293 K. (B) kinetic profiles of p-4H2O (●) and d-4H2O (▲) under the light irradiation, where the ratio of proximal and distal isomers was estimated from peak areas at 9.17 ppm and 9.66 ppm, respectively. | |
DFT calculations
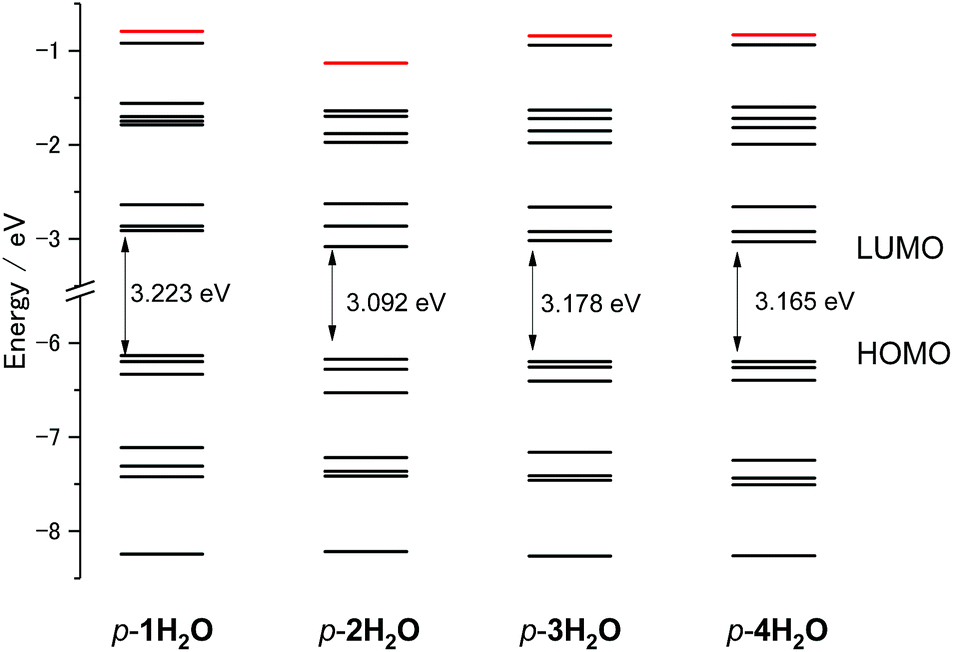
DFT calculations for the series of ruthenium aqua complexes were performed with the B3LYP functional and LanL2DZ basis set using the polarizable continuum model in water. The energy-optimized structures and molecular orbitals of p-nH2O are shown in Fig. S10–S13.† The highest occupied molecular orbitals (HOMOs) of the complexes are mainly localized on the metal center and their levels lie in the range −6.134 to −6.197 eV, as shown in Fig. S10–S13.† The lowest unoccupied molecular orbitals (LUMOs) of p-1H2O are localized on both the tpy and the pyqu ligands, whereas those of p-2H2O, p-3H2O, and p-4H2O are localized on the chloro-substituted pyqu ligands because of stabilization of their π* orbitals by the electron-withdrawing chloro substituents. The LUMO levels of the chloro-substituted complexes are more stabilized than that of p-1H2O, by 0.107 to 0.171 eV. The red shift in the absorption spectra of the chloro-substituted complexes is ascribed to stabilization of the LUMO levels, according to the time-dependent DFT calculations, as shown in Fig. S14.† In the calculated spectra, the absorption maximum in the MLCT transition for p-2H2O (482 nm) was more red-shifted than those for p-1H2O (463 nm), p-3H2O (475 nm), and p-4H2O (477 nm); this shows good agreement with the experimental results in absorption spectroscopy. Fig. 4 shows the energy level diagram of the molecular orbitals (MOs) of p-nH2O. The MO levels of p-3H2O and p-4H2O are almost identical. The energies of the metal-centered unoccupied orbitals (dσ*, shown in red in Fig. 4) for p-1H2O, p-3H2O, and p-4H2O are −0.794, −0.841, and −0.833 eV, respectively, indicating that the MC levels among the three complexes are almost identical. The energy level of the dσ* orbital (LUMO+7) for p-2H2O (−1.132 eV) is more stabilized than those of the other isomers by 0.195–0.338 eV. This indicates that the steric repulsion between the tridentate ligand and the chloro substituent of the pyqu ligand in p-2H2O weakens the ligand field. It should be noted that stabilization of dσ* by steric repulsion occurs in the distal isomers. The energy level of the dσ* orbital for p-2H2O (−0.99 eV) is lower than those for the other derivatives by 0.07–0.13 eV (see Fig. S15†). Several groups have reported rate enhancement by steric effects in photosubstitution reactions of ruthenium(II) complexes.4f,6 Our calculation results support the suggestion that steric hindrance accelerates the photoisomerization of ruthenium aqua complexes, including the photodissociation of an aqua ligand.
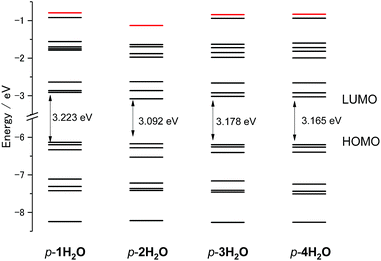 |
| | Fig. 4 Energy level diagram of molecular orbitals of p-nH2O (n = 1–4). The energy level of Ru–N antibonding dσ* orbitals (LUMO+8 for p-1H2O, 3H2O, and 4H2O, and LUMO+7 for p-2H2O) are marked in red. | |
Thermal isomerization
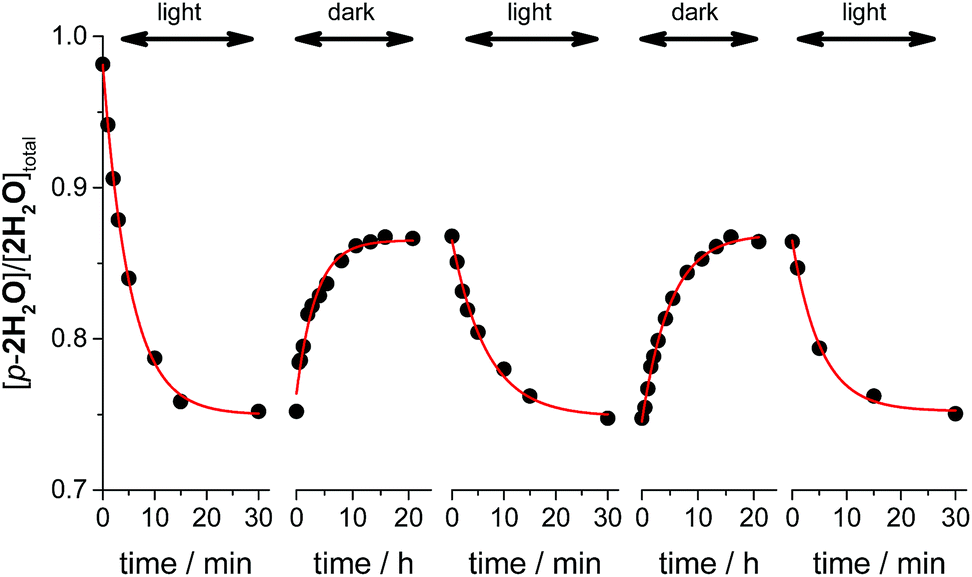
Thermal isomerizations of the series of ruthenium aqua complexes were examined by variable temperature (VT) NMR spectroscopy. The proximal isomer was irradiated in room temperature to reach photostationary state prior to the measurement. The mixtures of proximal and distal isomers were heated at 323 K overnight in the dark. Under the conditions used, proximal/distal ratio for 1H2O, 3H2O, and 4H2O were unchanged. In contrast, during heating of the mixture of p-2H2O and d-2H2O, the proportion of the proximal isomer increased from 0.76 to 0.86. We repeated the cycle of light irradiation at room temperature and incubation in the dark at 323 K. The changes in the proportion of p-2H2O are shown in Fig. 5. The proportion of p-2H2O increased during incubation in the dark and decreased under light irradiation. The proportion of the proximal isomer did not change when the sample was left at room temperature in the dark for several months. We consider that thermal isomerization of 2H2O occurs because of steric hindrance around the metal center. As shown in the crystal structures of the ruthenium aqua complex, the enhanced steric repulsion between tridentate and bidentate ligands in the p-2H2O by 6′-substituted chloro ligand leads to the distortion from octahedral geometry. The enhancement of thermal isomerization by distortion around the metal center has been reported for a ruthenium alloxazine complex by Kojima et al.7a and for a ruthenium complex with a 1,10-phenanthroline ligand with bulky moieties.21 We thus assume that decrease of ligand field in p-2H2O assists the bond dissociation of Ru–oxygen bond upon heating to give five-coordinated intermediates, followed by formation of the isomerized complexes.
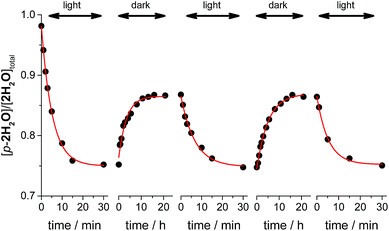 |
| | Fig. 5 Kinetic profiles of proximal-2H2O (total concentration, [2H2O]total = 2.7 mM) in the repeated cycles of photoisomerization and thermal isomerization. The ratios of the proximal isomer were calculated from the peak integration of the proximal isomer at 8.72 ppm (1H) and the distal isomer at 8.15 ppm (4H). Conditions: light source λ > 380 nm, T = 293 K under the light irradiation, T = 323 K in the dark reaction, solvent D2O. | |
Conclusion
In the present study, we demonstrated photoisomerization and thermal isomerization of a series of ruthenium aqua complexes with asymmetric bidentate ligands. The introduction of an electron-withdrawing chloro moiety at the 4- and 4′-positions of the pyqu ligand affected the absorption properties of the complexes (p-3H2O and 4H2O). Their proximal–distal photoisomerization reactions were similar and no thermal isomerization was observed. In contrast, the sterically hindered complex with a 6′-substituted pyqu ligand gave a high photoisomerization quantum yield and showed thermal back isomerization. These results will contribute to the design of photoresponsive molecules and molecular catalysts utilized under the light irradiation.
Experimental section
Materials
Ru(tpy)Cl3,22 2-(2′-pyridyl)quinoline N-oxide,15 2-[2′-(6′-chloro)-pyridyl]quinoline,15 2-[2′-(4′-chloro)pyridyl]quinoline,15 p-1Cl,12 and p-1H2O12 were prepared as previously described. Silica-gel column chromatography was performed using silica gel 60 (Kanto Chemical Co., Inc., 100–210 μm). The metal complexes were purified using Sephadex LH-20 (GE Healthcare). All other reagents were purchased, and used without further purification.
Measurements and DFT calculations
1H NMR spectra were recorded using a Bruker 500 MHz spectrometer. 1H NMR spectra were referenced using tetramethylsilane in organic solvents or sodium 3-(trimethylsilyl)propionate-2,2,3,3-d4 in D2O as an internal standard. X-ray diffraction data were recorded using a Bruker SMART APEXII CCD area detector diffractometer; the detector was positioned at a distance of 6.0 cm from the crystal and monochromated Mo Kα radiation (λ = 0.71073 Å) from a rotating anode with a mirror focusing apparatus operated at 1.2 kW (50 kV, 24 mA) was used. A sample crystal was mounted on a glass fiber for X-ray diffraction measurements. The structures were solved by direct methods and expanded using Fourier techniques. The non-hydrogen atoms were refined anisotropically. Hydrogen atoms were included but not refined. The final cycle of full-matrix least-squares refinement was based on the observed reflections [I > 2.00σ(I)] and variable parameters, and converged with unweighted and weighted agreement factors R and Rw. UV-vis absorption spectra were recorded using a Hitachi U-3310 UV-vis spectrometer and a quartz cell with a light pass length of 1 cm. DFT calculations were performed using the Gaussian 09 package of programs.23 Molecular structures were fully optimized using the B3LYP method, which uses the hybrid Becke's three-parameter exchange functional24 with the correlation energy functional of Lee, Yang, and Parr.25 Calculations were performed using the standard double-ζ-type LanL2DZ basis set implemented in Gaussian 09.
The internal quantum yields (Φproximal→distal) for photoisomerization were obtained from integrations of the resonances for p-nH2O and distal-nH2O in the 1H NMR spectra under light irradiation. Monochromic light (508.4 nm, full width at half maximum = 1.5 nm, 1.6 mW cm−2) passed through a band pass filter (Spectro-Film Inc.) was used. The internal quantum yields were calculated according to the following equation:
where
k,
nint,
h,
p,
λ,
c,
NA,
T, and
A are the rate constant for photoisomerization (p-
nH2O to d-
nH2O), initial amount (
nint = 2 μmol) of p-
nH2O, Plank's constant, photon flux (
p = 1.6 mW cm
−2), wavelength (
λ = 5.084 × 10
−7 m), the speed of light, Avogadro's number, transmittance, and irradiated area (
A = 1.2 cm
2), respectively. The internal quantum yield for the back reaction (
Φdistal→proximal) was calculated according to the following equation:
15where [proximal-
nH2O]
∞ and [distal-
nH2O]
∞ are the concentrations of p-
nH2O and d-
nH2O at the photochemical steady state.
Syntheses
2-(2′-Pyridyl)-4-chloroquinoline. 2-(2′-Pyridyl)quinoline N-oxide (272 mg, 1.22 mmol) was dissolved in POCl3 (3 mL) and the solution was refluxed under nitrogen for 3 h. The reaction mixture was slowly added to an aqueous solution of Na2CO3. After cooling to room temperature, the suspension was extracted with CHCl3 (30 mL × 2). The organic layer was dried with MgSO4 and evaporated to dryness. The crude product (123 mg) was purified with silica gel chromatography using a mixed solvent (CHCl3:AcOEt:hexane = 50:15:135, v/v/v). The first band was collected and evaporated to dryness. Yield 94 mg (32%). Anal. calcd for 2-(2′-pyridyl)-4-chloroquinoline, C14H9ClN2: C, 69.86; H, 3.77; N, 11.64. Found: C, 69.88; H, 3.62, N, 11.28. 1H NMR (500.13 MHz, CDCl3) δ ppm 8.75 (d, J = 4.7 Hz, 1H, H6′), 8.71 (s, 1H, H3), 8.66 (d, J = 8.0 Hz, 1H, H3′), 8.28 (d, J = 8.3 Hz, 1H), 8.20 (d, J = 8.4 Hz, 1H), 7.89 (dt, J = 7.8, 1.7 Hz, 1H), 7.80 (t, J = 7.1 Hz, 1H), 7.66 (t, J = 7.6 Hz, 1H), 7.39 (dd, J = 7.4, 4.8 Hz, 1H). 13C NMR (125.77 MHz, CDCl3) δ ppm 156.1, 155.3, 149.2, 148.7, 143.4, 137.1, 130.4, 130.2, 127.7, 126.4, 124.5, 124.2, 121.9, 119.2.
Proximal-[Ru(tpy)(6′-Clpyqu)Cl]Cl (p-[2Cl]Cl). To a 50 mL round bottom flask, Ru(tpy)Cl3 (45.4 mg, 0.10 mmol), 2-(2′-(6′-chloro)-pyridyl)quinoline (21.3 mg, 0.089 mmol), triethylamine (0.1 mL), EtOH (12 mL), water (4 mL), and LiCl (100 mg) were added and the mixture was refluxed for 24 h. The purple solution was filtered through Celite in order to remove unreacted Ru(tpy)Cl3. The filtrate was evaporated to ca. 3 mL and left in a refrigerator overnight. The purple precipitate was filtered and washed with ether. The precipitate was dissolved in methanol and then filtered, and the filtrate was evaporated to dryness. The crude product was purified with Sephadex LH-20 (1.5 cm × 20 cm) using methanol as an eluent. The purple band was collected and purity of the product was checked by TLC (eluent – saturated methanol solution of NaCl). Yield 15.1 mg (23%). Anal. calcd for p-[2Cl]Cl·5H2O, C29H20Cl3N5Ru·5H2O: C, 47.33; H, 4.11; N, 9.52. Found: C, 47.20; H, 3.97, N, 9.30. UV-vis: λ, nm (ε, 103 mol−1 L cm−1) in EtOH, 241 (31.1), 260 (32.4), 280 (sh), 321 (44.5), 552 (10.3). 1H NMR (500.13 MHz, CDCl3:CD3OD = 9:1) δ ppm 9.43–9.37 (m, 1H, H8,pyqu), 8.82 (d, J = 8.9 Hz, 1H, H3,pyqu), 8.74–8.67 (m, 2H, H3′,pyqu, H4,pyqu), 8.29 (d, J = 8.1 Hz, 2H, H3′,tpy, H5′,tpy), 8.22 (d, J = 8.0 Hz, 2H, H3,tpy, H3′′,tpy), 8.14–8.07 (m, 1H, H5,pyqu), 8.02 (t, J = 8.1, 8.1 Hz, 1H, H4′,tpy), 7.84 (td, J = 7.9, 1.5 Hz, 2H, H4,tpy, H4′′,tpy), 7.80 (t, J = 8.0 Hz, 1H, H4′,pyqu), 7.77–7.73 (m, 2H, H6,pyqu, H7,pyqu), 7.71 (d, J = 5.5 Hz, 2H, H6,tpy, H6′′,tpy), 7.24 (ddd, J = 7.4, 5.8, 1.3 Hz, 2H, H5,tpy, H5′′,tpy), 7.04 (dd, J = 7.9, 1.0 Hz, 1H, H5′,pyqu). 13C NMR (125.77 MHz, CDCl3:CD3OD = 9:1) δ ppm 161.5, 159.9, 159.6, 158.9, 157.6, 152.9, 151.1, 139.1, 138.7, 137.5, 135.3, 131.4, 131.11, 131.09, 129.8, 129.5, 128.8, 127.5, 127.3, 124.44, 124.42, 124.39, 124.35, 123.5, 122.1, 120.1.
Proximal-[Ru(tpy)(4′-Clpyqu)Cl]Cl, (p-[3Cl]Cl). To a 50 mL round bottom flask, Ru(tpy)Cl3 (90.2 mg, 0.20 mmol), 2-(2′-(4′-chloro)-pyridyl)quinoline (47.2 mg, 0.20 mmol), ascorbic acid (137 mg), EtOH (18 mL), water (6 mL), and LiCl (200 mg) were added and the mixture was heated at 50 °C for 6 h. The purple solution was filtered through Celite in order to remove unreacted Ru(tpy)Cl3. The filtrate was evaporated to ca. 3 mL and left in a refrigerator overnight. The purple microcrystalline was collected by filtration and washed with an aqueous solution of HCl (3 mol L−1, 1 mL × 2) and ether (3 mL × 2). The crude product was dissolved in minimum amount of methanol, and then purified on Sephadex LH-20 using methanol as an eluent. The reddish purple band was collected, evaporated to ca. 3 mL, and then left in refrigerator overnight. The purple microcrystals were collected by filtration. Yield 29.0 mg (20%). Anal. calcd for p-[3Cl]Cl·3.5H2O, C29H20Cl3N5Ru·3.5H2O: C, 49.13; H, 3.84; N, 9.88. Found: C, 49.40; H, 3.52, N, 9.53. UV-vis: λ, nm (ε, 103 mol−1 L cm−1) in EtOH, 235 (37.7), 260 (sh), 271 (38.4), 319 (49.7), 538 (10.6). 1H NMR (500.13 MHz, CDCl3:CD3OD = 9:1) δ ppm 10.30 (d, J = 8.8 Hz, 1H), 8.74 (d, J = 8.7 Hz, 1H), 8.60 (d, J = 8.7 Hz, 1H), 8.54 (d, J = 8.1 Hz, 2H), 8.47 (d, J = 2.1 Hz, 1H), 8.41 (d, J = 8.0 Hz, 2H), 8.24–8.15 (m, 2H), 7.96 (ddd, J = 8.6, 6.9, 1.4 Hz, 1H), 7.89 (t, J = 7.7 Hz, 3H), 7.61 (d, J = 5 Hz, 2H), 7.41 (d, J = 6.3 Hz, 1H), 7.26–7.19 (m, 2H), 7.16 (dd, J = 6.3, 2.2 Hz, 1H). 13C NMR (125.77 MHz, CDCl3:CD3OD = 9:1) δ ppm 160.8, 158.6, 158.3, 155.6, 152.7, 152.3, 151.0, 143.5, 138.5, 137.4, 135.7, 132.1, 131.2, 129.8, 129.7, 128.4, 127.3, 126.6, 125.2, 124.0, 123.0, 118.7.
Proximal-[Ru(tpy)(4-Clpyqu)Cl]Cl (p-[4Cl]Cl). To a 50 mL round bottom flask, Ru(tpy)Cl3 (42.8 mg, 0.097 mmol), 2-(2′-pyridyl)-4-chloroquinoline (23.3 mg, 0.097 mmol), ascorbic acid (100 mg), EtOH (9 mL), water (3 mL), and LiCl (100 mg) were added and the mixture was heated at 50 °C for 4 h. The purple solution was filtered through Celite in order to remove unreacted Ru(tpy)Cl3. The filtrate was evaporated to ca. 3 mL and left in a refrigerator overnight. The purple microcrystalline was collected by filtration and washed with an aqueous solution of HCl (3 mol L−1, 1 mL × 2) and ether (3 mL × 2). Yield 32.8 mg (45%). Anal. calcd for p-[4Cl]Cl·0.5EtOH·5H2O, C29H20Cl3N5Ru·CH3O0.5·5H2O: C, 47.47; H, 4.38; N, 9.23. Found: C, 47.58; H, 4.21, N, 8.99. UV-vis: λ, nm (ε, 103 mol−1 L cm−1) in EtOH, 234 (36.3), 273 (39.0), 279 (39.3), 318 (49.2), 377 (sh), 537 (11.9). 1H NMR (500.13 MHz, CDCl3:CD3OD = 9:1) δ ppm 10.30 (d, J = 8.7 Hz, 1H), 8.65 (s, 1H), 8.56–8.35 (m, 4H), 8.30 (d, J = 7.7 Hz, 2H), 8.11 (t, J = 7.3, 7.3 Hz, 1H), 8.02–7.86 (m, 2H), 7.81 (t, J = 7.4, 7.4 Hz, 1H), 7.75 (t, J = 5.5, 5.5 Hz, 1H), 7.59 (d, J = 5.0 Hz, 1H), 7.37 (s, 1H), 7.21–7.06 (m, 3H). 13C NMR (125.77 MHz, CDCl3:CD3OD = 9:1) δ ppm 158.9, 158.7, 158.5, 156.4, 152.9, 152.1, 151.5, 144.9, 137.5, 135.9, 135.7, 132.9, 131.8, 130.6, 127.5, 127.5, 126.9, 125.4, 124.9, 124.0, 123.0, 119.1.
Proximal-[Ru(tpy)(6′-Clpyqu)OH2](PF6)2 (p-[2H2O](PF6)2). p-[2Cl]Cl (7.41 mg, 10.1 μmol) was dissolved in water (6 mL) and left at room temperature for 1 h. An aqueous solution of NH4PF6 was added to the solution and the precipitate was filtered and dried in vacuum. Yield 5.95 mg (63%). Anal. calcd for p-[2H2O](PF6)2·3H2O, C29H22ClF12N5OP2Ru·3H2O: C, 37.17; H, 3.01; N, 7.47. Found: C, 37.23; H, 3.04, N, 7.18. UV-vis of p-[2H2O]Cl2: λ, nm (ε, 103 mol−1 L cm−1) in water, 232 (sh), 251 (29.9), 272 (25.3), 315 (44.1), 515 (8.1) 1H NMR of p-[2H2O]Cl2 (500.13 MHz, D2O:CD3OD = 4:1) δ ppm 8.90 (d, J = 8.8 Hz, 1H), 8.77 (d, J = 8.8 Hz, 1H), 8.69–8.55 (m, 4H), 8.51 (d, J = 7.87 Hz, 2H), 8.36 (d, J = 8.1 Hz, 1H), 8.28 (t, J = 8.1 Hz, 1H), 8.05 (t, J = 8.1 Hz, 2H), 7.99–7.82 (m, 4H), 7.78 (t, J = 8.1 Hz, 1H), 7.39 (t, J = 6.6 Hz, 2H), 7.22 (d, J = 8.1 Hz, 1H). 13C NMR p-[2H2O]Cl2 (125.77 MHz, D2O:CD3OD = 4:1) δ ppm 163.9, 162.3, 162.2, 162.0, 160.2, 155.8, 152.8, 141.2, 141.1, 140.5, 139.5, 134.2, 131.9, 131.72, 131.67, 130.0, 129.7, 129.5, 126.2, 126.0, 125.3, 122.2.
Proximal-[Ru(tpy)(4′-Clpyqu)OH2](PF6)2 (p-[3H2O](PF6)2). p-[3Cl]Cl (8.63 mg, 12.5 μmol) was dissolved in water (6 mL) and left at room temperature for 1 h. An aqueous solution of NH4PF6 was added to the solution and the precipitate was filtered and dried in vacuum. Yield 7.84 mg (76%). Anal. calcd for p-[Ru(tpy)(4-Clpyqu)(μ-O2H3)Ru(tpy)(4-Clpyqu)](PF6)3 2H2O, C29H22ClF12N5OP2Ru·H2O: C, 42.03; H, 2.86; N, 8.46. Found: C, 42.02; H, 2.89, N, 8.31. UV-vis of p-[3H2O]Cl2: λ, nm (ε, 103 mol−1 L cm−1) in water, 230 (37.3), 258 (sh), 270 (37.5), 314 (52.9), 508 (10.7) 1H NMR of p-[3H2O]Cl2 (500.13 MHz, D2O) δ ppm 9.40–9.25 (m, 1H), 8.83 (d, J = 8.7 Hz, 1H), 8.63 (d, J = 8.7 Hz, 1H), 8.60–8.53 (m, 3H), 8.44 (d, J = 8.1 Hz, 2H), 8.34 (dd, J = 6.4, 3.3 Hz, 1H), 8.22 (t, J = 8.1 Hz, 1H), 8.02–7.88 (m, 4H), 7.70 (d, J = 5.5 Hz, 2H), 7.43 (d, J = 6.4 Hz, 1H), 7.29–7.17 (m, 2H), 7.02 (dd, J = 6.3, 1.8 Hz, 1H).
Proximal-[Ru(tpy)(4-Clpyqu)OH2](PF6)2 (p-[4H2O](PF6)2). p-[4Cl]Cl (6.76 mg, 8.9 μmol) was dissolved in water (6 mL) and left at room temperature for 1 h. An aqueous solution of NH4PF6 was added to the solution and the precipitate was filtered and dried in vacuum. Yield 6.2 mg (76%). Anal. calcd for p-[4H2O](PF6)2·2H2O, C29H22ClN5ORuP2F12·2H2O: C, 37.90; H, 2.85; N, 7.62. Found: C, 37.70; H, 2.88, N, 7.42. UV-vis of p-[4H2O]Cl2: λ, nm (ε, 103 mol−1 L cm−1) in water, 232 (sh), 243 (31.7), 260 (32.4), 276 (42.0), 314 (51.8), 507 (12.3) 1H NMR of p-[4H2O]Cl2 (500.13 MHz, D2O) δ ppm 9.17 (d, J = 8.3 Hz, 1H), 8.90 (s, 1H), 8.70 (d, J = 8.3 Hz, 1H), 8.62 (d, J = 8.2 Hz, 2H), 8.52–8.41 (m, 3H), 8.31 (t, J = 8.2, 8.2 Hz, 1H), 8.11–7.90 (m, 4H), 7.83–7.70 (m, 3H), 7.56 (d, J = 5.8 Hz, 1H), 7.29 (t, J = 6.6 Hz, 2H), 7.03 (t, J = 6.6 Hz, 1H). 13C NMR of p-[4H2O]Cl2 (125.77 MHz, D2O) δ ppm 161.7, 161.3, 161.2, 159.4, 156.0, 155.1, 153.5, 146.9, 141.0, 139.3, 138.3, 135.2, 132.6, 130.2, 129.93, 129.91, 128.5, 127.9, 127.7, 126.6, 125.8, 122.2.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the JST PRESTO program, JSPS KAKENHI Grant Numbers 24107003, 24350028, 17K18433, 16K05762, and JP17M03044.
References
-
(a) S. Bonnet, B. Limburg, J. D. Meeldijk, R. J. M. K. Gebbink and J. A. Killian, J. Am. Chem. Soc., 2011, 133, 252–261 CrossRef CAS PubMed;
(b) B. S. Howerton, D. K. Heidary and E. C. Glazer, J. Am. Chem. Soc., 2012, 134, 8324–8327 CrossRef CAS PubMed;
(c) B. A. Albani, B. Peña, N. A. Leed, N. A. B. G. de Paula, C. Pavani, M. S. Baptista, K. R. Dunbar and C. Turro, J. Am. Chem. Soc., 2014, 136, 17095–17101 CrossRef CAS PubMed.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. Von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- Y. Liu, D. B. Turner, T. N. Singh, A. M. Angeles-Boza, A. Chouai, K. R. Dunbar and C. Turro, J. Am. Chem. Soc., 2009, 131, 26–27 CrossRef CAS PubMed.
-
(a) D. V. Pinnick and B. Durham, Inorg. Chem., 1984, 23, 1440–1445 CrossRef CAS;
(b) J. J. Rack, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 2001, 123, 2432–2433 CrossRef CAS PubMed;
(c) B. Durham, J. L. Walsh, C. L. Carter and T. J. Meyer, Inorg. Chem., 1980, 19, 860–865 CrossRef CAS;
(d) C. R. Hecker, P. E. Fanwick and D. R. McMillin, Inorg. Chem., 1991, 30, 659–666 CrossRef CAS;
(e) B. A. Albani, C. B. Durr and C. Turro, J. Phys. Chem. A, 2013, 117, 13885–13892 CrossRef CAS PubMed;
(f) S. Bonnet, J.-P. Collin, J.-P. Sauvage and E. Schofield, Inorg. Chem., 2004, 43, 8346–8354 CrossRef CAS PubMed;
(g) C. E. Welby, G. K. Armitage, H. Bartley, A. Wilkinson, A. Sinopoli, B. S. Uppal, C. R. Rice and P. I. P. Elliott, Chem.–Eur. J., 2014, 20, 8467–8476 CrossRef CAS PubMed;
(h) P. A. Scattergood, U. Khushnood, A. Tariq, D. J. Cooke, C. R. Rice and P. I. P. Elliott, Inorg. Chem., 2016, 55, 7787–7796 CrossRef CAS PubMed;
(i) F. Weisser, S. Plebst, S. Hohloch, M. van der Meer, S. Manck, F. Führer, V. Radtke, D. Leichnitz and B. Sarkar, Inorg. Chem., 2015, 54, 4621–4635 CrossRef CAS PubMed;
(j) D. Unjaroen, J. B. Kasper and W. R. Browne, Dalton Trans., 2014, 43, 16974–16976 RSC;
(k) D. Unjaroen, J. Chen, E. Otten and W. R. Browne, Inorg. Chem., 2017, 56, 900–907 CrossRef CAS PubMed.
- Q. Sun, S. Mosquera-Vazquez, Y. Suffren, J. Hankache, N. Amstutz, L. M. Lawson Daku, E. Vauthey and A. Hauser, Coord. Chem. Rev., 2015, 282–283, 87–99 CrossRef CAS.
-
(a) J. D. Knoll, B. A. Albani, C. B. Durr and C. Turro, J. Phys. Chem. A, 2014, 118, 10603–10610 CrossRef CAS PubMed;
(b) D. A. Lutterman, A. A. Rachford, J. J. Rack and C. Turro, J. Phys. Chem. Lett., 2010, 1, 3371–3375 CrossRef CAS.
-
(a) S. Miyazaki, T. Kojima and S. Fukuzumi, J. Am. Chem. Soc., 2008, 130, 1556–1557 CrossRef CAS PubMed;
(b) S. K. Padhi, R. Fukuda, M. Ehara and K. Tanaka, Inorg. Chem., 2012, 51, 5386–5392 CrossRef CAS PubMed;
(c) S. Bonnet, J.-P. Collin and J.-P. Sauvage, Inorg. Chem., 2006, 45, 4024–4034 CrossRef CAS PubMed.
- B. Durham, S. R. Wilson, D. J. Hodgson and T. J. Meyer, J. Am. Chem. Soc., 1980, 102, 600–607 CrossRef CAS.
-
(a) A. A. Rachford and J. J. Rack, J. Am. Chem. Soc., 2006, 128, 14318–14324 CrossRef CAS PubMed;
(b) B. A. McClure and J. J. Rack, Angew. Chem., Int. Ed., 2009, 48, 8556–8558 CrossRef CAS PubMed;
(c) J. J. Rack, A. A. Rachford and A. M. Shelker, Inorg. Chem., 2003, 42, 7357–7359 CrossRef CAS PubMed;
(d) B. A. McClure and J. J. Rack, Inorg. Chem., 2011, 50, 7586–7590 CrossRef CAS PubMed;
(e) A. W. King, L. Wang and J. J. Rack, Acc. Chem. Res., 2015, 48, 1115–1122 CrossRef CAS PubMed.
-
(a) Y. Jin, S. I. M. Paris and J. J. Rack, Adv. Mater., 2011, 23, 4312–4317 CrossRef CAS PubMed;
(b) Y. Jin, D. Harrington, A. A. Rachford and J. J. Rack, RSC Adv., 2014, 4, 62920–62925 RSC.
-
(a) H. Yamazaki, T. Hakamata, M. Komi and M. Yagi, J. Am. Chem. Soc., 2011, 133, 8846–8849 CrossRef CAS PubMed;
(b) M. Hirahara, M. Z. Ertem, M. Komi, H. Yamazaki, C. J. Cramer and M. Yagi, Inorg. Chem., 2013, 52, 6354–6364 CrossRef CAS PubMed;
(c) M. Hirahara, S. Nagai, K. Takahashi, K. Saito, T. Yui and M. Yagi, Inorg. Chem., 2015, 54, 7627–7635 CrossRef CAS PubMed;
(d) K. Takahashi, X. Zhang, M. Hirahara, T. Sato, K. Saito, T. Yui and M. Yagi, J. Photochem. Photobiol., A, 2015, 313, 117–125 CrossRef CAS.
- M. Hirahara, H. Tomoya, A. B. League, M. Z. Ertem, K. Takahashi, S. Nagai, K. Inaba, H. Yamazaki, K. Saito, T. Yui, C. J. Cramer and M. Yagi, Eur. J. Inorg. Chem., 2015, 2015, 3892–3903 CrossRef CAS.
-
(a) S. W. Gersten, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4029–4030 CrossRef CAS;
(b) T. Wada, K. Tsuge and K. Tanaka, Angew. Chem., Int. Ed., 2000, 39, 1479–1482 CrossRef CAS;
(c) C. Sens, I. Romero, M. Rodriguez, A. Llobet, T. Parella and J. Benet-Buchholz, J. Am. Chem. Soc., 2004, 126, 7798–7799 CrossRef CAS PubMed;
(d) R. Zong and R. P. Thummel, J. Am. Chem. Soc., 2005, 127, 12802–12803 CrossRef CAS PubMed;
(e) Y. Xu, A. Fischer, L. Duan, L. Tong, E. Gabrielsson, B. Åkermark and L. Sun, Angew. Chem., Int. Ed., 2010, 49, 8934–8937 CrossRef CAS PubMed;
(f) J. J. Concepcion, J. W. Jurss, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2008, 130, 16462–16463 CrossRef CAS;
(g) M. Yagi, S. Tajima, M. Komi and H. Yamazaki, Dalton Trans., 2011, 40, 3802–3804 RSC;
(h) D. J. Wasylenko, C. Ganesamoorthy, B. D. Koivisto, M. A. Henderson and C. P. Berlinguette, Inorg. Chem., 2010, 49, 2202–2209 CrossRef CAS PubMed;
(i) S. Masaoka and K. Sakai, Chem. Lett., 2009, 38, 182–183 CrossRef CAS;
(j) J. L. Boyer, D. E. Polyansky, D. J. Szalda, R. Zong, R. P. Thummel and E. Fujita, Angew. Chem., Int. Ed., 2011, 50, 12600–12604 CrossRef CAS.
- S. Tanaka, K. Takahashi, M. Hirahara, M. Yagi and K. Onda, J. Photochem. Photobiol., A, 2015, 313, 87–98 CrossRef CAS.
- M. Hirahara, A. Tsukamoto, H. Goto, S. Tada, M. Yagi and Y. Umemura, Chem.–Eur. J., 2016, 22, 2590–2594 CrossRef CAS.
- Y. Jahng, R. P. Thummel and S. G. Bott, Inorg. Chem., 1997, 36, 3133–3138 CrossRef CAS.
- M. H. V. Huynh, J. M. Lasker, M. Wetzler, B. Mort, L. F. Szczepura, L. M. Witham, J. M. Cintron, A. C. Marschilok, L. J. Ackerman, R. K. Castellano, D. L. Jameson, M. R. Churchill, A. J. Jircitano and K. J. Takeuchi, J. Am. Chem. Soc., 2001, 123, 8780–8784 CrossRef CAS.
-
(a) A. Bahreman, B. Limburg, M. A. Siegler, R. Koning, A. J. Koster and S. Bonnet, Chem.–Eur. J., 2012, 18, 10271–10280 CrossRef CAS PubMed;
(b) A. Bahreman, B. Limburg, M. A. Siegler, E. Bouwman and S. Bonnet, Inorg. Chem., 2013, 52, 9456–9469 CrossRef CAS PubMed.
- A. L. Spek, A. Gerli and J. Reedijk, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1994, 50, 394–397 CrossRef.
-
(a) N. Chanda, D. Paul, S. Kar, S. M. Mobin, A. Datta, V. G. Puranik, K. K. Rao and G. K. Lahiri, Inorg. Chem., 2005, 44, 3499–3511 CrossRef CAS PubMed;
(b) Z. Deng, H.-W. Tseng, R. Zong, D. Wang and R. Thummel, Inorg. Chem., 2008, 47, 1835–1848 CrossRef CAS PubMed;
(c) G. Sathyaraj, M. Kiruthika, T. Weyhermuller and B. U. Nair, Dalton Trans., 2012, 41, 8460–8471 RSC.
- S. Bonnet, J.-P. Collin and J.-P. Sauvage, Inorg. Chem., 2007, 46, 10520–10533 CrossRef CAS PubMed.
- B. P. Sullivan, J. M. Calvert and T. J. Meyer, Inorg. Chem., 1980, 19, 1404–1407 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford, CT, USA, 2009.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Molecular orbitals, absorption and 1H NMR spectra, and crystallographic data. CCDC 1546577–1546582. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra08943d |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Hiroki Gotoa,
Rei Yamamotoa,
Masayuki Yagi
*a,
Hiroki Gotoa,
Rei Yamamotoa,
Masayuki Yagi