
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombretastatin A4-camptothecin micelles as combination therapy for effective anticancer activity
Mohyeddin Assali *a,
Naim Kittanab,
Sahar Alhaj Qasema,
Raghad Adasa,
Doaa Saleha,
Asala Arara and
Osayd Zohudb
*a,
Naim Kittanab,
Sahar Alhaj Qasema,
Raghad Adasa,
Doaa Saleha,
Asala Arara and
Osayd Zohudb
aDepartment of Pharmacy, Faculty of Medicine and Health Sciences, An Najah National University, P. O. Box 7, Nablus, Palestine. E-mail: m.d.assali@najah.edu
bDepartment of Biomedical Sciences, Faculty of Medicine & Health Sciences, An Najah National University, P. O. Box 7, Nablus, Palestine
First published on 9th January 2019
Abstract
Cancer is a major worldwide health problem, for which chemotherapy is a common treatment option. However drug toxicity and the development of resistance to chemotherapy are two main challenges associated with the traditional anticancer drugs. Combined pharmacological therapy based on different mechanisms might be an effective strategy in cancer treatment, and could exhibit a synergistic therapeutic efficacy. Herein, we aim to combine combretastatin A4 (CA4) and camptothecin (Cpt) chemically into a codrug through two hydrophilic linkers utilizing click chemistry to improve their water solubility and anticancer activity. The synthesized amphiphilic structure could self-assemble into a micelle structure as confirmed by atomic force microscopy (AFM) and dynamic light scattering (DLS), which showed a high stability and improved water solubility at pH 7.4, with a low critical micelle concentration (CMC) value of 0.9 mM. Moreover, in vitro hydrolysis was observed upon incubation of the hybrid compound with an esterase enzyme, which suggested a complete disassembly into the starting active drugs. Finally, cytotoxicity studies on HeLa cancer cells showed that the codrug demonstrated an enhanced (five fold) cytotoxicity as compared with the free drugs. In addition the combination index (CI) was <1, which suggests a synergistic activity for the codrug. Moreover, the tested concentrations of the codrug were not significantly cytotoxic to a noncancerous fibroblast cell line. The imaging of HeLa cells treated with FITC-loaded micelles showed a rapid internalization. In conclusion, the codrug of CA4 and Cpt might be a potential novel anticancer drug as it demonstrated a synergistic cytotoxic activity that might spare noncancerous cells.
1. Introduction
Cancer is considered one of the complicated diseases that presents as a major leading cause of death worldwide.1 Cancer chemotherapy is among one of the major treatment options for cancer, besides surgery and radiotherapy when applicable. Chemotherapy can be used as a supplemental treatment to attack micro-metastases following surgery and radiation. It can also be given prior to the surgical procedure in order to shrink the tumor.2 Anticancer drugs are normally administered systemically, so that they distribute throughout the body tissues and may affect not only the cancerous cells but also many normal cells leading to various side effects on the heart, kidneys, bladder, digestive system, bone marrow, lungs and many others.3–5 Drug toxicity and the development of drug resistance are among the main challenges associated with the use of traditional anticancer drugs. Toxicity can cause life-threatening consequences that may require the cessation of the treatment. On the other hand, the development of drug resistance by tumors might be a reason for the treatment failure in many cases. Therefore, there is a great demand to develop a new strategy to fight this lethal disease.6,7 The combination of multiple drugs with various mechanisms could be an effective strategy that might exhibit a synergistic activity.8–13 However, the proper delivery of the combined therapy has some limitations and a proper chemical combination is highly desired due to the various physiological barriers and enzymatic processes in the body.Combretastatin A4 (CA4) and camptothecin (Cpt) are among many clinically used anticancer drugs. CA4 is a natural compound that is extracted from an African willow tree called Combretum caffrum. It acts as a vascular disrupting agent (VDA). In addition it can inhibit the polymerization of tubulin resulting in cell death.14–16 However, it has two major disadvantages; the low water solubility and the short half life.10,17,18 On the other hand, Cpt is extracted from a Chinese plant called Camptotheca acuminata.19 It inhibits topoisomerase I enzyme, which is important in the DNA replication, resulting in cell death.20 Similar to CA4, Cpt also suffers from low water solubility, but in addition it has low stability and preponderance of a less active carboxylate form at physiologic pH.21,22
Solid tumors unendingly grow in size. In many times this increase in tumor size is not only associated with an increase in tumor tissue hypoxia, but also a reduction in tumor blood flow and consequently an impairment in the delivery of the chemotherapeutic agents.23–25 However, some research studies suggested that VDAs could otherwise be superior against such large tumors, because tumor vasculature could be already susceptible to occlusion due to elevated interstitial fluid pressure, which are more associated with advanced tumor masses.26 Moreover a recent review study demonstrated that combining VDAs with traditional chemotherapeutic agents has greater potential to eradicate tumor cells.27,28
Herein we aim to combine the VDA agent (CA4) with the topoisomerase I inhibitor (Cpt) chemically for the first time using a suitable hydrophilic linkers of tetraethylene glycol that would permit the formation of an amphiphilic structure that could assemble into micelles, which might in turn improve the water solubility of the resulting molecules and provide a synergistic effect of the combined therapy (Scheme 1).
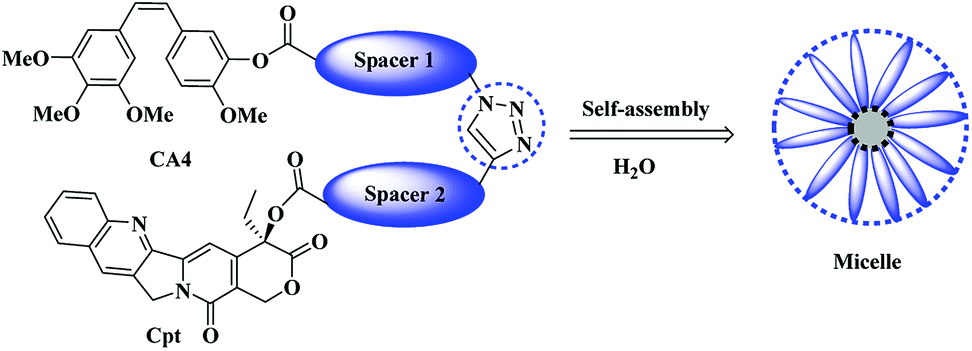 | ||
| Scheme 1 General scheme of the proposed micelle formation. | ||
2. Experimental
2.1. Reagents and instruments
All chemical materials that used in the experiments were of analytical grade and were purchased from commercial suppliers. Tetraethylene glycol, L-ascorbic acid sodium salt 99%, quinoline, propargyl bromide, succinic anhydride 99%, 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC), 2-(3,4,5-trimethoxyphenyl) acetic acid, 3-hydroxy-4-methoxybenzaldehyde and camptothecin were purchased from (Alfa Aesar Company, England). 4-(Dimethylamino) pyridine (DMAP) was purchased from Merck Company. Esterase from porcine liver, pyrene, acetic anhydride, copper, anhydrous copper sulfate were purchased from (Sigma-Aldrich, USA). Sodium azide was purchased from (Riedel-de Haën Company, Germany). Triethylamine and diethyl ether were purchased from (SDFCL Company, India). HeLa cells and 3T3 fibroblasts cell lines were obtained as a kind gift from Dr Johnny Amer (The source was the American Type Culture Collection [ATCC], Manassas, VA, USA). DMEM medium, fetal bovine serum (FBS), L-glutamine and penicillin/streptomycin were purchased from Biological Industries, Beit Haemek. CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) was purchased from Promega (Wisconsin, USA).For flash chromatography, silica gel (Sigma-Aldrich 230–400 mesh) was used. Columns were eluted with positive air pressure. Nuclear Magnetic Resonance (NMR) spectra were recorded with Bruker Avance 400 and 500 MHz spectrometers. Chemical shifts were reported in ppm, and coupling constants were reported in Hz. Infrared spectra were recorded on FTIR Spectrometer (Nicolet iS5, Thermo Fisher Scientific Company, USA). Ultraviolet-visible (UV-Vis) spectra were recorded on a UV/vis (7315 Spectrophotometer, Jenway, UK), using quartz cuvettes. Perkin Elmer luminescence spectrometer was used to calculate the critical micelle concentration (CMC). Atomic force microscope (AFM) imaging was performed by depositing the sample of mica using a tapping mode-AFM (Alquds University) and WSxM software designed by Nanotec Electronica (Madrid, Spain) used for image analysis.29 Particle size analysis was measured by dynamic light scattering (DLS) (NanoBrook Omni, Brookhaven Instruments Corporation, USA) with a fixed scattering angle of 90° at 25 °C. Microplate reader 6000 (Unilab Inc, USA) was used to read the plate for cell viability test. Olympus IX73® inverted microscope operated with Pylon Viewer 64 bit® software was used for bright field and fluorescence imaging for cellular uptake experiment.
2.2. Synthesis
The following compounds were synthesized and characterized according to our previous reported study:10 OH-TEG-OTs (1), OH-TEG-N3 (2), COOH-TEG-N3 (3), CA4 (4) and CA4-TEG-N3 (5).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)). FTIR: 2980, 2128, 1716 cm−1. 1H NMR (500 MHz, CDCl3): δ 6.55 (bs, 1H, COOH), 4.13 (s, 2H, CH2COOH), 4.08 (s, 2H, CH2C
:1)). FTIR: 2980, 2128, 1716 cm−1. 1H NMR (500 MHz, CDCl3): δ 6.55 (bs, 1H, COOH), 4.13 (s, 2H, CH2COOH), 4.08 (s, 2H, CH2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C), 3.80–3.66 (m, 12H, 6CH2O), 2.46 (s, 1H, CCH). 13C NMR (125.7 MHz, CDCl3): δ 175.2, 79.1, 78.3, 71.2, 70.9, 70.8, 70.7, 70.6, 70.2, 69.1, 62.1.:1) to provide pale yellow oil with 78% yield. Rf: 0.32 (DCM/MeOH (20:1)). 1H NMR (500 MHz, CDCl3): δ 8.33 (s, 1H, H7), 8.19 (d, 1H, J = 8.3 Hz, H12), 7.87 (d, 1H, J = 8.3 Hz, H9), 7.76 (t, 1H, J = 6.9 Hz, H11), 7.66 (t, 1H, J = 7.9 Hz, H10), 7.21 (s, 1H, H14), 5.70–5.66 (dd, 2H, J = 17.2 Hz, J = 17.2 Hz, H17), 5.27 (d, 2H, J = 2.8 Hz, H5), 4.31 (d, 2H, J = 7.9 Hz, COCH2O), 4.14 (s, 2H, CH2CC), 3.66–3.49 (m, 12H, 6CH2O), 2.36 (s, 1H, CCH), 2.32–2.15 (m, 2H, 2H19), 0.99 (t, 1H, J = 7.7 Hz, H18). 13C NMR (125.7 MHz, CDCl3): δ 169.7 (CO), 167.2 (C21), 157.3 (C16a), 152.2, 148.8, 146.4, 145.4, 131.2, 130.7, 129.6, 128.4, 128.2, 128.1, 120.3, 95.9 (C2, C3, C6–C16), 78.2, 76.4 (C20), 76.2, 71.0, 70.7, 70.6, 70.5, 70.0, 60.5 (CH2CC), 68.1 (COCH2O), 67.2 (C17), 49.9 (C5), 31.8 (C19), 7.6 (C18).:1) and a yellow oil product was obtained with 58% yield. Rf: 0.15 (DCM/MeOH (9:1)). 1H NMR (500 MHz, CDCl3): δ 8.41 (s, 1H, H7-Cpt), 8.21 (d, 1H, J = 8.5 Hz, H12-Cpt), 7.95 (d, 1H, J = 7.9 Hz, H9-Cpt), 7.84 (t, 1H, J = 7.4 Hz, H11-Cpt), 7.76 (bs, 1H, H-triazol), 7.67 (t, 1H, J = 7.4 Hz, H10-Cpt), 7.19 (s, 1H, H14-Cpt), 7.10 (d, 2H, J = 6.9 Hz, 2CH, Ar-CA4), 6.98 (s, 1H, CH, Ar-CA4), 6.82 (d, 2H, J = 8.3 Hz, 2CH, Ar-CA4), 6.44 (d, 2H, J = 10.1 Hz, CH
C), 3.80–3.66 (m, 12H, 6CH2O), 2.46 (s, 1H, CCH). 13C NMR (125.7 MHz, CDCl3): δ 175.2, 79.1, 78.3, 71.2, 70.9, 70.8, 70.7, 70.6, 70.2, 69.1, 62.1.:1) to provide pale yellow oil with 78% yield. Rf: 0.32 (DCM/MeOH (20:1)). 1H NMR (500 MHz, CDCl3): δ 8.33 (s, 1H, H7), 8.19 (d, 1H, J = 8.3 Hz, H12), 7.87 (d, 1H, J = 8.3 Hz, H9), 7.76 (t, 1H, J = 6.9 Hz, H11), 7.66 (t, 1H, J = 7.9 Hz, H10), 7.21 (s, 1H, H14), 5.70–5.66 (dd, 2H, J = 17.2 Hz, J = 17.2 Hz, H17), 5.27 (d, 2H, J = 2.8 Hz, H5), 4.31 (d, 2H, J = 7.9 Hz, COCH2O), 4.14 (s, 2H, CH2CC), 3.66–3.49 (m, 12H, 6CH2O), 2.36 (s, 1H, CCH), 2.32–2.15 (m, 2H, 2H19), 0.99 (t, 1H, J = 7.7 Hz, H18). 13C NMR (125.7 MHz, CDCl3): δ 169.7 (CO), 167.2 (C21), 157.3 (C16a), 152.2, 148.8, 146.4, 145.4, 131.2, 130.7, 129.6, 128.4, 128.2, 128.1, 120.3, 95.9 (C2, C3, C6–C16), 78.2, 76.4 (C20), 76.2, 71.0, 70.7, 70.6, 70.5, 70.0, 60.5 (CH2CC), 68.1 (COCH2O), 67.2 (C17), 49.9 (C5), 31.8 (C19), 7.6 (C18).:1) and a yellow oil product was obtained with 58% yield. Rf: 0.15 (DCM/MeOH (9:1)). 1H NMR (500 MHz, CDCl3): δ 8.41 (s, 1H, H7-Cpt), 8.21 (d, 1H, J = 8.5 Hz, H12-Cpt), 7.95 (d, 1H, J = 7.9 Hz, H9-Cpt), 7.84 (t, 1H, J = 7.4 Hz, H11-Cpt), 7.76 (bs, 1H, H-triazol), 7.67 (t, 1H, J = 7.4 Hz, H10-Cpt), 7.19 (s, 1H, H14-Cpt), 7.10 (d, 2H, J = 6.9 Hz, 2CH, Ar-CA4), 6.98 (s, 1H, CH, Ar-CA4), 6.82 (d, 2H, J = 8.3 Hz, 2CH, Ar-CA4), 6.44 (d, 2H, J = 10.1 Hz, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), 5.42–5.38 (dd, 2H, J = 17.2 Hz, J = 17.2 Hz, H17-Cpt), 5.22 (d, 2H, J = 2.8 Hz, H5-Cpt), 4.65 (s, 2H, CH2COO), 4.49 (s, 2H, CH2COO), 4.35 (s, 2H, triazol CH2), 3.87–3.83 (m, 5H, CH2 triazol, OCH3), 3.79 (s, 3H, OCH3), 3.75 (s, 6H, 2OCH3), 3.70–3.55 (m, 20H, 10CH2O), 1.22 (m, 2H, 2H19-Cpt), 0.85 (t, 1H, J = 7.6 Hz, H18-Cpt). 13C NMR (125.7 MHz, CDCl3): δ 173.2 (CO), 170.1 (CO), 169.4 (CO), 166.2 (C21), 158.3 (C16a), 155.2, 155.0, 154.3, 154.0, 153.2, 148.6, 146.5, 145.2, 131.5, 130.8, 129.5, 128.6, 128.5, 128.4, 128.2, 127.4, 123.3, 121.3, 120.1, 111.2, 104.4, 96.8, 76.8 (C20), 71.2, 70.6, 70.5, 70.4, 69.3, 68.1 (COCH2O), 67.2 (C17), 60.3, 56.2, 55.3, 50.5 (CH2-triazol), 50.2 (C5), 32.4 (C19), 7.7 (C18).
CH), 5.42–5.38 (dd, 2H, J = 17.2 Hz, J = 17.2 Hz, H17-Cpt), 5.22 (d, 2H, J = 2.8 Hz, H5-Cpt), 4.65 (s, 2H, CH2COO), 4.49 (s, 2H, CH2COO), 4.35 (s, 2H, triazol CH2), 3.87–3.83 (m, 5H, CH2 triazol, OCH3), 3.79 (s, 3H, OCH3), 3.75 (s, 6H, 2OCH3), 3.70–3.55 (m, 20H, 10CH2O), 1.22 (m, 2H, 2H19-Cpt), 0.85 (t, 1H, J = 7.6 Hz, H18-Cpt). 13C NMR (125.7 MHz, CDCl3): δ 173.2 (CO), 170.1 (CO), 169.4 (CO), 166.2 (C21), 158.3 (C16a), 155.2, 155.0, 154.3, 154.0, 153.2, 148.6, 146.5, 145.2, 131.5, 130.8, 129.5, 128.6, 128.5, 128.4, 128.2, 127.4, 123.3, 121.3, 120.1, 111.2, 104.4, 96.8, 76.8 (C20), 71.2, 70.6, 70.5, 70.4, 69.3, 68.1 (COCH2O), 67.2 (C17), 60.3, 56.2, 55.3, 50.5 (CH2-triazol), 50.2 (C5), 32.4 (C19), 7.7 (C18).2.3. Solubility, critical micelle concentration and stability studies
Calibration curves of CPT, CA4 and codrug 9 were constructed as follow::14 ml of 2 M acetic acid was added to 2.99 g of sodium acetate dissolved in water and diluted to 1000 ml with water.Phosphate buffer solution pH 7.4:250.0 ml of 0.2 M potassium dihydrogen phosphate was added to 393.4 ml of 0.1 M sodium hydroxide.
Carbonate buffer solution pH 9.2:0.1 M sodium carbonate was prepared by dissolving 1.06 g anhydrous Na2CO3 in water and diluted to 100 ml and 0.1 M sodium bicarbonate was prepared by dissolving 0.84 g NaHCO3 in water and diluted to 100 ml. Then, 90 ml of diluted sodium bicarbonate were added to 10 ml of diluted sodium carbonate in to 100 ml volumetric flask.
000 rpm for 10 min. After that, exactly 100 μl of the supernatant was taken and diluted. Then, the absorbance was measured at λmax 420 nm and concentrations were obtained using calibration curve with correlation coefficient more than 0.99. The intrinsic solubility represented by steady-state concentrations.2.4. Hydrolysis study by esterase enzyme
In order to study the hydrolysis of the drug by esterase enzyme was conducted as documented previously.32 Briefly, 1 mg of codrug 9 and 1 mg of esterase (10 U ml−1) were dissolved in 5 ml of phosphate buffer (pH 4.7) and gently stirred at 37 °C. An aliquot was withdrawn every 10 min and was replaced with equal volume of fresh medium to mimic the sink conditions. The absorbance of the collected samples was measured by UV spectrophotometer.2.5. Cytotoxicity study
3. Results and discussion
3.1. Synthesis
The synthesis was achieved by using the adequate linkers to provide the correct hydrophobic/hydrophilic balance in order to improve their water solubility, therefore, we utilized two derivatives of tetraethylene glycol one for the Cpt and other for the CA4 since the biocompatible polyethylene glycol (PEG) chain has shown effective improvement in the solubility of hydrophobic anticancer drugs conjugate and the prolong circulation of the formed micelles.34,35The first linker was synthesized through selective monotosylation of tetraethylene glycol followed by nucleophilic substitution with sodium azide to obtain compound 2 and terminated by oxidation reaction utilizing Jones reagent to obtain the bifunctional linker 3. In a final step, an esterification reaction with CA4 to obtain CA4-TEG-N3 5 as shown in Scheme 2.
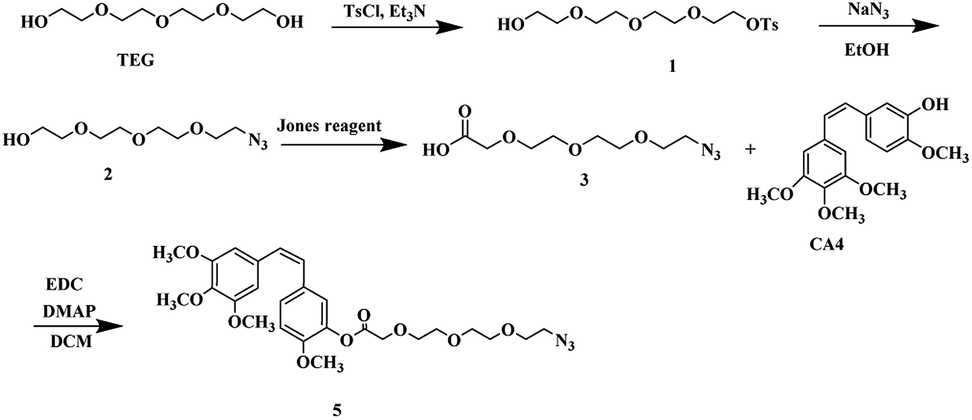 | ||
| Scheme 2 Synthetic scheme of CA4-TEG-N3 5. | ||
Once the first moiety has successfully synthesized, the second linker was prepared by reacting tetraeythylene glycol selectively with propargyl bromide in the presence of sodium hydride in THF. Followed by oxidation reaction to obtain compound 7 that reacts with Cpt through esterification reaction using EDC as a coupling agent and DMAP to obtain Cpt-TEG alkyne 8 as shown in Scheme 3.
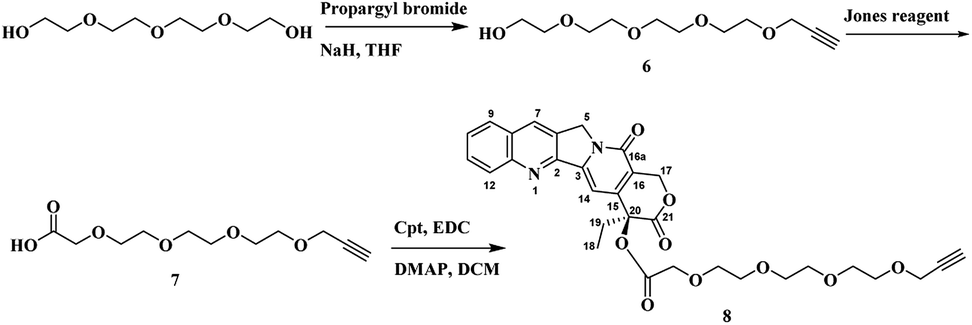 | ||
| Scheme 3 Synthetic scheme of Cpt-TEG-Alkyne 8. | ||
As the two moieties were successfully synthesized CA4-TEG-N3 5 and Cpt-TEG-alkyne 8, they were connected through click reaction, which is occurred in one pot and produced a high yield reaction with a minimum byproduct.36,37 Herein we used anhydrous CuSO4 in the presence of ascorbic acid dissolved in a mixture DCM:H2O to synthesized CA4-TEG-triazole-TEG-Cpt 9 as shown in the Scheme 4. The connection between the two anticancer drugs was through triazole group that are highly enzymatic and chemically stable.38 Moreover, the direct linkage between the CA4, Cpt and linkers was through an ester bond that can be hydrolyzed easily by esterase enzyme that is usually overexpressed in cancer cells with a high intracellular concentration39,40 in order to give a rapid release profile of the anticancer drugs.
 | ||
| Scheme 4 Synthetic scheme of CA4-TEG-triazole-TEG-Cpt 9 (codrug 9). | ||
3.2. Solubility, stability and micelle formation
In order to investigate whether the conjugation of the two low-water-soluble drugs with the hydrophilic linkers could improve the overall aqueous solubility, the shake-flask method was implemented to study the solubility of codrug 9 at 37 °C in sodium phosphate buffer (pH 7.4). The test was performed based on calibration curves constructed by UV-Vis spectrophotometery. The solubility test was performed in triplicates. The results showed an improvement in the total solubility of codrug 9 under the specified conditions with a concentration of 1.8 mM, whereas the solubilities of Cpt and CA4 were 5 μM and 17.2 μM respectively. This enhancement of solubility could be related to the introduced hydrophilic chains that connect between the two anticancer drugs. More interestingly, as the synthesized codrug 9 consists of hydrophobic molecules and hydrophilic chains (amphiphilic structure), this organized structure can self-assembly into diverse supramolecular structure. Moreover, the 1,2,3-traizole group has the potential to enhance the construction of various supramolecular systems as the triazole rings facilitate the π–π interactions and has a polarized C–H bond, which could be used as a H-bond donor.21,41,42 In this direction, we studied the capability of our compound to self-assemble into micelle structure through atomic force microscopy (AFM) and dynamic light scattering (DLS). The image of AFM resembles the formation of micelle supramolecular structure with a spherical shape as shown in Fig. 1A. In addition, the DLS results showed good dispersibility with a PDI of 0.2 and a hydrodynamic diameter of 146 nm as shown in Fig. 1B. This diameter range is optimum to permit a preferential internalization to cancer cells over the normal cells as previously reported.43,44 Moreover, the CMC was estimated using pyrene as a fluorescent probe.31 As the concentration of the compound 9 increased, a red shift of pyrene absorbance was noticed in the excitation spectrum (Fig. 1C). In particular, the absorbance band of pyrene in water was at 333 nm, and it shifted to 336 nm when pyrene was embedded in micelles as the pyrene transfer from the water environment to the hydrophobic core of the micelle. Fig. 1C shows the plot of the intensity ration I336/I333 versus concentration. The CMC value was measured to be 1 × 10−3 mg ml−1 (0.9 mM) as the intersection of the two tangent lines. This value is considered thermodynamically stable enough as a drug delivery vehicle.45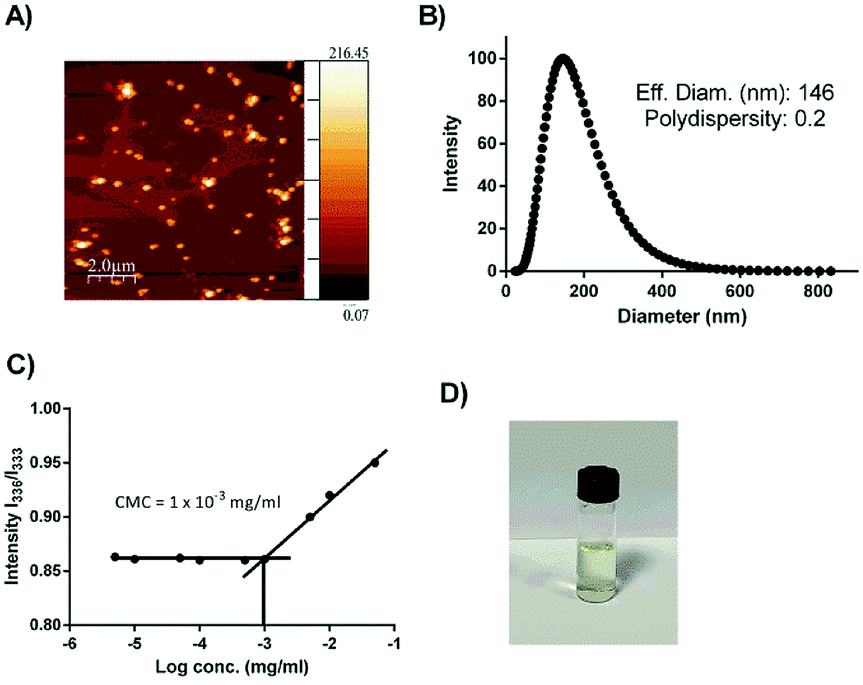 | ||
| Fig. 1 (A) AFM image; (B) DLS result; (C) CMC result of codrug 9 and (D) vial photograph demonstrates the stability of codrug 9 after 1 week. | ||
A stability study was also conducted for one week on the synthesized codrug 9 at three various pHs (4.5, 7.4 and 9.2). The percentage of conversion of the co-drug to their parent drugs were measured for one week based on the prepared calibration curves. The obtain results indicated that the synthesized codrug 9 is totally stable at pH 7.4 during the whole week as shown in Fig. 1D. Regarding pH 4.5, a hydrolysis was observed with 20% of the sample after 24 h and maintained this percentage during the whole week. In the case of pH 9.2, a higher hydrolysis was observed with 50% after 24 h and also maintained this percentage during the whole week as shown in Fig. 2A.
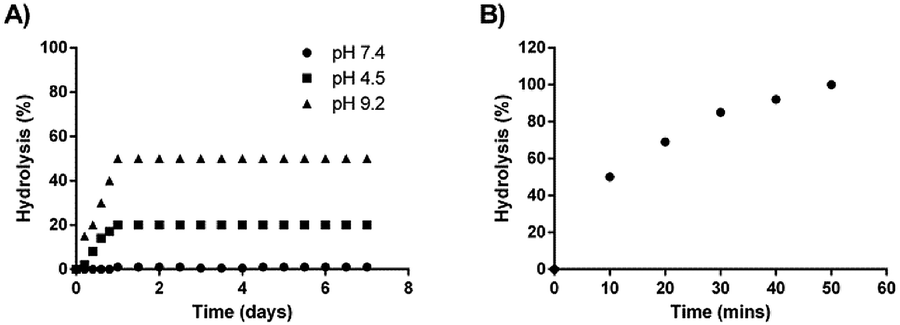 | ||
| Fig. 2 (A) Percentage of hydrolysis at different pHs. (B) Percentage of hydrolysis in the presence of esterase enzyme. | ||
Moreover, the complete hydrolysis of the co-drug was also studied in the presence of esterase enzyme. An incubation study was conducted with porcine liver esterase. It was observed a complete hydrolysis of the co-drug after 50 min of incubation with esterase enzyme, which confirms the total conversion of the co-drug to the two active anticancer drugs after 50 min as shown in Fig. 2B.
3.3. MTS study
The cytotoxicity of codrug 9 was investigated on HeLa cells and was compared with the free CA4 and Cpt alone. The effect of codrug 9 was also investigated on the noncancerous 3T3 fibroblasts. The MTS assay was implemented in order to objectively estimate the cell viability under various concentrations over 48 h treatment interval, which was the minimal time period required for the drug to affect the cells as shown in Fig. 3. The results demonstrated a concentration dependent cytotoxic activity on HeLa cells under all tested concentrations. Interestingly, there was a clear significant improvement in the anticancer activity of the codrug 9 in comparison to the free drugs over the used concentration range which was up to approximately five folds, which might be attributed to a synergistic activity for the combined drugs as they work in two different modes of actions, which allowed a concentration as low as 23.75 nM to exhibit a strong cytotoxic effect. Moreover, the synergistic effect was confirmed by calculating the combination index (CI) values at different lethal doses (LD50, LD75, LD90, LD95) using CompuSyn software developed by Chou and Martin.46 As shown in Table 1, a very low LD50 of the developed codrug 9 (0.29 nM) was obtained in comparison to the CA4 and Cpt with LD50 of 58.3 nM and 50.9 nM, respectively. More interestingly, the CI values at all doses were less than 0.1 which confirm a synergistic effect of the developed co-drug 9. This synergistic activity not only enhances the therapeutic activity but also allows the use of lower doses as confirmed by the calculated dose reduction index (DRI) of each drug as shown in Table 1, which would be expected to decrease the associated side effects. This synergistic effect coincide with the previously reported results of the conjugation of camptothecin and cytarabine through disulfide linkage that showed a rapid cellular uptake with a synergistic anticancer activity in vitro and in vivo studies.47 Importantly, there was no significant cytotoxic activity with 3T3 fibroblasts, which might reflect a preferential activity against cancerous cells.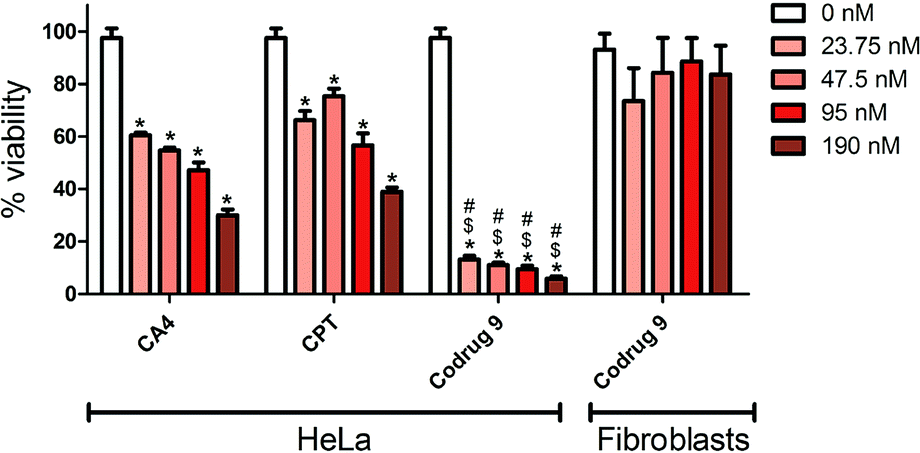 | ||
| Fig. 3 % Viability of HeLa cancer cells and 3T3 fibroblasts determined by MTS assay after 48 h of treatment. *p ≤ 0.05 compared to 0 nM, $p ≤ 0.05 compared to corresponding CA4 concentration, #p ≤ 0.05 compared to corresponding CPT concentration, N = 3. | ||
3.4. Cellular uptake study of FITC loaded micelles
Cellular uptake of the formed micelles into the cancerous cells is an essential step to have an effective anticancer activity. Since Camptothecin and Combretastatin A4 have low fluorescent intensity, FITC was encapsulated in the self-assembled micelles to obtain a better visualization of the internalization.48 The cellular uptake was investigated by inverted fluorescent microscope. Fig. 4 demonstrated the obtained results after the treatment of HeLa cells with FITC loaded micelles at two time intervals (1 h and 2 h). As shown in the Fig. 4, a bright green fluorescence was detected inside the HeLa cells, which indicates an effective and rapid internalization of the developed micelles. Together these findings indicate that the developed micelles have an effective cytotoxicity with rapid internalization by HeLa cancer cells and a good cytocompatibility on 3T3 non-cancerous cells.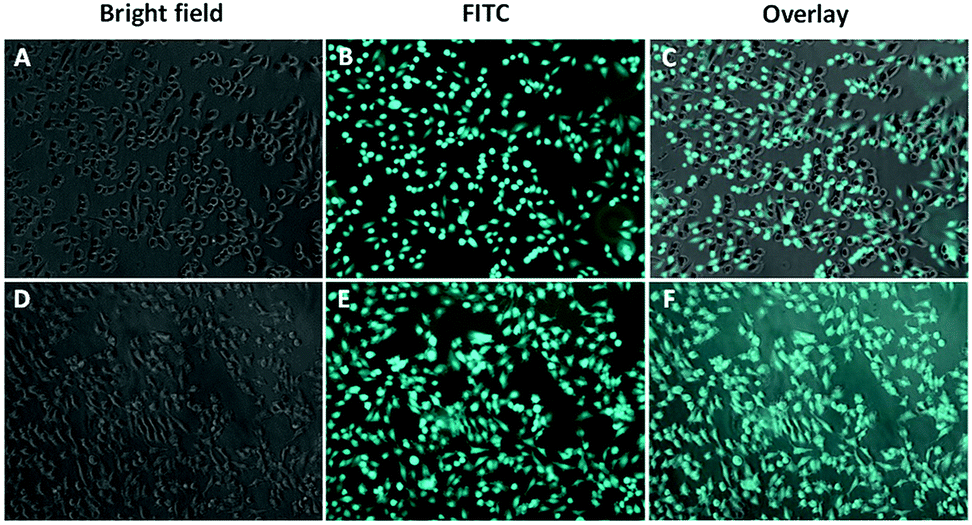 | ||
| Fig. 4 Uptake of FITC-loaded micelles by HeLa cells. The fluorescent FITC signal was detected in HeLa cells treated with FITC-loaded micelles after 1 h (A–C) or 2 h (D–F). | ||
4. Conclusions
In conclusion, codrug 9 was successfully synthesized with a strategy to improve the water solubility of the used anticancer drugs (CA4 and Cpt). The obtained amphiphilic structure has the capability to self-assemble into micelle structure as confirmed by AFM, DLS and CMC determination. The synthesized codrug 9 showed a good stability at the physiological pH, but could completely hydrolyze into the parent drugs in the presence of an esterase enzyme. The MTS study showed a great improvement in the anticancer activity with a strong synergistic effect as confirmed by the calculated CI without cytotoxic effect on 3T3 normal cells. Moreover, the cellular uptake study showed a rapid internalization of the developed micelles within 1 h of the treatment. The developed codrug 9 could provide a novel therapy for cancer and we recommend further in vivo studies to determine the pharmacokinetic behaviour and the anticancer activity in vivo.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the deanship of Scientific Research at An Najah National University under fund identification number ANNU-1718-Sc-001. We acknowledge Dr Mohammad Qneibi and Ms Shorooq Sobuh for facilitating cell imaging in their lab.Notes and references
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-A Cancer Journal for Clinicians, 2018, 68, 7–30 CrossRef PubMed.
- B. Sumer and J. Gao, Nanomedicine, 2008, 3, 137–140 CrossRef PubMed.
- L. Y. Ramirez, S. E. Huestis, T. Y. Yap, S. Zyzanski, D. Drotar and E. Kodish, Pediatr. Blood Cancer, 2009, 52, 497–502 CrossRef PubMed.
- A. K. Ganti, A. Pearce, M. Haas, R. Viney, S.-A. Pearson, P. Haywood, C. Brown and R. Ward, PLoS One, 2017, 12, e0184360 CrossRef PubMed.
- M. A. Raji, Lancet Oncol., 2005, 6, 357 CrossRef PubMed.
- R. Iacovelli, F. Pietrantonio, C. Maggi, F. de Braud and M. Di Bartolomeo, Crit. Rev. Oncol. Hematol., 2016, 98, 24–28 CrossRef PubMed.
- G. Tomasello, M. Ghidini, S. Barni, R. Passalacqua and F. Petrelli, Expert Rev. Clin. Pharmacol., 2017, 10, 649–660 CrossRef CAS PubMed.
- P. Parhi, C. Mohanty and S. K. Sahoo, Drug Discovery Today, 2012, 17, 1044–1052 CrossRef CAS PubMed.
- J. A. Kemp, M. S. Shim, C. Y. Heo and Y. J. Kwon, Adv. Drug Delivery Rev., 2016, 98, 3–18 CrossRef CAS PubMed.
- M. Assali, R. Shawahna, S. Dayyeh, M. Shareef and I.-A. Alhimony, Eur. J. Pharm. Sci., 2018, 122, 179–184 CrossRef CAS PubMed.
- S. Saha, M. Ghosh and S. K. Dutta, RSC Adv., 2017, 7, 53322–53333 RSC.
- X. Zhang, Y. Liu, Y. J. Kim, J. Mac, R. Zhuang and P. Wang, RSC Adv., 2017, 7, 19685–19693 RSC.
- V. Abbot, P. Sharma, S. Dhiman, M. N. Noolvi, H. M. Patel and V. Bhardwaj, RSC Adv., 2017, 7, 28313–28349 RSC.
- G. M. Tozer, C. Kanthou, C. S. Parkins and S. A. Hill, Int. J. Exp. Pathol., 2002, 83, 21–38 CrossRef CAS PubMed.
- G. M. Tozer, C. Kanthou and B. C. Baguley, Nat. Rev. Cancer, 2005, 5, 423–435 CrossRef CAS PubMed.
- P. Hinnen and F. A. L. M. Eskens, Br. J. Cancer, 2007, 96, 1159–1165 CrossRef CAS PubMed.
- R. Dorr, K. Dvorakova, K. Snead, D. Alberts, S. Salmon and G. R. Pettit, Invest. New Drugs, 1996, 14, 131–137 CrossRef CAS PubMed.
- G. J. Rustin, S. M. Galbraith, H. Anderson, M. Stratford, L. K. Folkes, L. Sena, L. Gumbrell and P. M. Price, J. Clin. Oncol., 2003, 21, 2815–2822 CrossRef CAS PubMed.
- N. H. Oberlies and D. J. Kroll, J. Nat. Prod., 2004, 67, 129–135 CrossRef CAS PubMed.
- L. P. Rivory and J. Robert, Pharmacol. Ther., 1995, 68, 269–296 CrossRef CAS PubMed.
- M. Assali, J. J. Cid, M. Pernia-Leal, M. Munoz-Bravo, I. Fernandez, R. E. Wellinger and N. Khiar, ACS Nano, 2013, 7, 2145–2153 CrossRef CAS PubMed.
- S. Sheng, Y. Chen, T. Zhang, M. Ding, Y. Wu, Z. Shen, G. Han and X. Wang, RSC Adv., 2017, 7, 46370–46377 RSC.
- L. Bentzen, S. Keiding, M. R. Horsman, T. Grönroos, S. B. Hansen and J. Overgaard, Acta Oncol., 2009, 41, 304–312 CrossRef.
- R. F. Kallman, G. L. DeNardo and M. J. Stasch, Cancer Res., 1972, 32, 483–490 CAS.
- R. P. Hill, Br. J. Cancer, Suppl., 1980, 4, 230–239 CAS.
- R. K. Jain, Cancer Res., 1987, 47, 3039–3051 CAS.
- D. W. Siemann, D. J. Chaplin and M. R. Horsman, Cancer Invest., 2017, 35, 519–534 CrossRef CAS PubMed.
- H. Liu, Y. Xie, Y. Zhang, Y. Cai, B. Li, H. Mao and R. Yu, RSC Adv., 2016, 6, 113933–113939 RSC.
- I. Horcas, R. Fernández, J. M. Gómez-Rodríguez, J. Colchero, J. Gómez-Herrero and A. M. Baro, Rev. Sci. Instrum., 2007, 78, 013705 CrossRef CAS PubMed.
- M. Assali, M. Joulani, R. Awwad, M. Assad, M. Almasri, N. Kittana and A. N. Zaid, ChemistrySelect, 2016, 1, 1132–1135 CrossRef CAS.
- M. Wilhelm, C. L. Zhao, Y. Wang, R. Xu, M. A. Winnik, J. L. Mura, G. Riess and M. D. Croucher, Macromolecules, 1991, 24, 1033–1040 CrossRef CAS.
- M. Assali, A. N. Zaid, F. Abdallah, M. Almasri and R. Khayyat, Int. J. Nanomed., 2017, 12, 6647–6659 CrossRef CAS PubMed.
- T. C. Chou and P. Talalay, Adv. Enzyme Regul., 1984, 22, 27–55 CrossRef CAS PubMed.
- Y. Guo, P. Zhang, Q. Zhao, K. Wang and Y. Luan, Macromol. Biosci., 2016, 16, 420–431 CrossRef CAS PubMed.
- P. Zhang, W. He, H. Zhang, C. Huang, D. Zhao and Y. Luan, ChemPlusChem, 2016, 81, 1237–1244 CrossRef CAS.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- J. Sheng, N. Ma, Y. Wang, W.-C. Ye and B.-X. Zhao, Drug Des., Dev. Ther., 2015, 2015, 1585–1599 CrossRef PubMed.
- M. P. Mathew, E. Tan, S. Shah, R. Bhattacharya, M. Adam Meledeo, J. Huang, F. A. Espinoza and K. J. Yarema, Bioorg. Med. Chem. Lett., 2012, 22, 6929–6933 CrossRef CAS PubMed.
- C. A. McGoldrick, Y.-L. Jiang, M. Brannon, K. Krishnan and W. L. Stone, BMC Cancer, 2014, 14, 1–13 CrossRef CAS PubMed.
- L. Xu, Y. Li and Y. Li, Asian J. Org. Chem., 2014, 3, 582–602 CrossRef CAS.
- B. Schulze and U. S. Schubert, Chem. Soc. Rev., 2014, 43, 2522 RSC.
- M. P. Desai, V. Labhasetwar, E. Walter, R. J. Levy and G. L. Amidon, Pharm. Res., 1997, 14, 1568–1573 CrossRef CAS.
- A. Prokop and J. M. Davidson, J. Pharm. Sci., 2008, 97, 3518–3590 CrossRef CAS PubMed.
- W. Xu, P. Ling and T. Zhang, J. Drug Delivery, 2013, 2013, 1–15 CrossRef PubMed.
- T. C. Chou, Pharmacol. Rev., 2006, 58, 621–681 CrossRef CAS PubMed.
- W. He, X. Hu, W. Jiang, R. Liu, D. Zhang, J. Zhang, Z. Li and Y. Luan, Adv. Healthcare Mater., 2017, 6, 1700829 CrossRef PubMed.
- W.-M. Li, D.-M. Liu and S.-Y. Chen, J. Mater. Chem., 2011, 21, 12381–12388 RSC.
| This journal is © The Royal Society of Chemistry 2019 |