
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAu@PdAg core–shell nanotubes as advanced electrocatalysts for methanol electrooxidation in alkaline media†
Wenke Yanga,
Qing Zhanga,
Cheng Peng *ab,
Eyu Wua,
Shaowei Chen*b,
Yanyun Mac,
Jie Houa,
Yuexiao Hea,
Bangkai Zhanga and
Lifei Denga
*ab,
Eyu Wua,
Shaowei Chen*b,
Yanyun Mac,
Jie Houa,
Yuexiao Hea,
Bangkai Zhanga and
Lifei Denga
aCollege of Materials Science and Engineering, Huaqiao University, Xiamen 361021, PR China. E-mail: chengpeng@hqu.edu.cn; Fax: +86 592 6162225; Tel: +86 592 6162225
bDepartment of Chemistry and Biochemistry, University of California, 1156 High Street, Santa Cruz, California 95064, USA. E-mail: shaowei@ucsc.edu; Tel: +1 831 4595841
cInstitute of Functional Nano & Soft Materials (FUNSOM), Jiangsu Key Laboratory for Carbon-Based Functional Materials & Devices, Soochow University, 199 Ren'ai Road, Suzhou, 215123, Jiangsu, PR China
First published on 8th January 2019
Abstract
Developing active and cost-effective electrocatalysts for methanol electrooxidation is crucial to the commercialization of direct methanol fuel cells (DMFCs). In this study, Au@PdAg core–shell nanotubes are synthesized in an aqueous solution by sequential galvanic displacement between Ag nanowires and AuCl4− and PdCl42−. High-resolution transmission electron microscopy studies demonstrate that the obtained Au@PdAg nanotubes consist of a Au-rich interior that is encapsulated with a three-dimensionally dendritic, porous PdAg alloy shell, forming a core–sheath nanostructure. Electrochemical studies indicate that the as-prepared Au@PdAg nanotubes exhibit apparent electrocatalytic activity and stability towards methanol electrooxidation in alkaline media. This remarkable high performance can be attributed to their large specific surface area and unique porous morphology.
1. Introduction
Direct methanol fuel cells (DMFCs) have drawn a surge of interest over recent decades due to their high power density, high energy-conversion efficiency, and zero or low emission of pollutants.1–4 Pt-based nanostructured materials have been used as the leading catalysts for methanol electrooxidation in DMFCs.5–9 However, there are two major disadvantages with Pt-based anodic catalysts. Firstly, the catalyst surface can be easily poisoned by the strong adsorption of CO-like intermediates during the oxidation of methanol fuel, which greatly lowers the catalytic performance.10,11 Secondly, the high costs and limited supply of Pt seriously hinder its widespread applications in fuel cells. As a viable alternative, Pd-based catalysts have been attracting much attention in recent years due to their high abundance, good electrocatalytic activities and great resistance to CO poisoning for methanol oxidation in alkaline environment.12–15In recent research, it has been shown that the fabrication of multimetallic Pd-based alloy nanostructures is an attractive way to improve the electrocatalytic performance, due largely to the synergistic interactions between the components.16,17 This also addresses the cost and stability issues by the incorporation of economical and robust metal components into the catalysts.18–20 In addition, it is well-known that the electrocatalytic performance highly depends on the shape as well as surface structure, size and composition of the catalysts.21–23 Accordingly, the preparation of architecturally controlled multimetallic Pd-based alloy electrocatalysts is of great significance for further improvement of their electrocatalytic activity. In the past decades, there have been a number of studies on the synthesis of Pd-based electrocatalysts with well-defined morphologies, such as concave polyhedral structures, nanosheets, nanorings, right bipyramids, and nanoboxes.24–28 Among these, one-dimensional tubular structures with a nanometer-sized wall are particularly attractive, not only because of the high surface area, but also for the reduced vulnerability than small nanoparticles to dissolution, Ostwald ripening, and aggregation.29,30 Meanwhile, extensive research has demonstrated that multimetallic nanocatalysts can significantly improve the catalytic activity resulting from the synergistic interactions between the different metallic components. For example, Guo et al. reported the preparation of PtPd bimetallic nanotubes with a petal-like surface, which exhibited apparent activity and stability toward ethanol electrooxidation.31 Han and co-workers reported the synthesis of PtPdAg ternary alloy nanotubes with a nanoporous framework, which exhibited superior electrocatalytic activity and stability over commercial Pd/C catalyst toward ethanol electrooxidation.32 As such, nanotubes that consist of multiple metallic components are promising platforms for enhancing the electrocatalytic performance of Pd-based electrocatalysts.33,34
Despite extensive efforts in the synthesis of Pd-based nanocatalysts with well-defined morphologies, it remains a synthetic challenge to integrate a well-defined composition and architecture into a given multimetallic Pd-based nanostructure, due to the difficulty of controlling the reduction kinetics of multiple metal precursors of different redox potentials. Therefore, it is essential to develop an efficient and straightforward synthetic route to prepare multimetallic Pd-based nanotubes with a desirable structure to further optimize their electrocatalytic performance. Galvanic displacement, as a fast and simple route for the preparation of nanoalloy catalysts, has been used to synthesize an excellent electrocatalyst with high mass activities and good long-term durability.35 However, to the best of our knowledge, Au@PdAg core–shell nanotubes with a nanoporous framework have not been reported before by using this method.
In this work, trimetallic Pd-based core–shell nanotubes with a nanoporous framework, Au@PdAg core–shell nanotubes (Au@PdAg-NTs), were synthesized by a facile and cost-effective method based on sequential galvanic displacement reactions between Ag nanowires and AuCl4− and PdCl42−. The morphology, crystal structure and composition of the obtained samples were characterized by scanning electron microscopy (SEM), high resolution transmission electron microscopy (HRTEM), and X-ray diffraction (XRD) measurements. Moreover, electrochemical tests were carried out to analyze the electrocatalytic activity towards methanol oxidation in alkaline media. The results show that Au@PdAg-NTs might serve as promising electrocatalysts for high-performance alkaline DMFCs.
2. Experimental section
2.1 Materials
Ethylene glycol ((CH2OH)2), sodium chloride (NaCl), poly(vinyl pyrrolidone) (PVP, K-30), silver nitrate (AgNO3), chloroauric acid (HAuCl4), sodium tetrachloropalladate (Na2PdCl4), ethanol (CH3CH2OH), methanol (CH3OH), and potassium hydroxide (KOH) were all purchased from Sinopharm Chemical Reagents Co., Ltd. Pd/C commercial catalyst (10 wt%) and Nafion solution (1 wt% in ethanol) were obtained from Alfa Aesar and DuPont, respectively. All chemicals were used as received without any treatment. Water was supplied by a water purifier system (18.2 MΩ cm). All chemicals were of analytical grade.2.2 Synthesis of Au@PdAg core–shell nanotubes
In a typical procedure, 2.5 mL of ethylene glycol and 0.11 mL of a NaCl solution (20 mM in ethylene glycol) were refluxed at 160 °C for 1 h under vigorous stirring. After that, 2.23 mL of a AgNO3 solution (0.1 M in ethylene glycol) and 2.23 mL of a PVP solution (0.3 M in ethylene glycol) were injected into the above solution at a rate of 20 mL h−1, simultaneously. The reaction mixture continued to be refluxed at 160 °C for another 45 min, resulting in an opaque gray solution, signifying the formation of Ag nanowires.36 After the mixture solution was cooled down to 5 °C, 40 mL of pure water and 100 mL of a freshly prepared HAuCl4 solution (0.294 mM in pure water) were added dropwise into suspension, which was under stirring at 5 °C for 12 h. Then, 100 mL of a freshly prepared Na2PdCl4 solution (0.588 mM in pure water) was added dropwise into the above suspension, and stirred for another 12 h at room temperature. During the entire synthetic process, the reaction solution was under magnetic stirring and the flask was capped with a glass stopper except for the addition of reagent. The final solution was centrifuged at a rate of 3000 rpm for 10 min to remove the supernatant. The product was washed with a saturated NaCl solution three times to remove the AgCl byproduct that might have settled on the samples. Finally, the products, Au@PdAg-NTs, were centrifuged and washed with H2O until the supernatant was clear and dispersed in H2O (1 mg mL−1) for further use.2.3 Materials characterization
XRD (Bruker D8 advance X-ray diffractometer, Cu-Kα radiation, λ = 0.154059 nm at 40 kV and 40 mA, 2θ range of 25–90°, scan rate 10° min−1) and field-emission scanning electron microscopy (FESEM, HITACHI S-4800) studies were carried out to examine the structure and morphology of Au@PdAg-NTs. High-resolution transmission electron microscopy (HRTEM), high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and energy dispersive X-ray spectroscopy (EDS) measurements were performed with a FEI Tecnai G2 F20 S-TIWN transmission electron microscope operated at 200 kV.2.4 Electrochemical tests
Electrochemical measurements were conducted with an electrochemical workstation (CHI 660D, CH Instruments Inc.) using a three-electrode configuration. The working electrode was prepared by pipetting a 30 μL aliquot of the catalyst ink (prepared by ultrasonically mixing 20 μL of the electrocatalyst and 10 μL of the Nafion solution) on the surface of a cleaned glassy carbon electrode, which was dried at room temperature. The electrodes prepared from Au@PdAg-NTs and Pd/C commercial catalysts were denoted as Au@PdAg-NTs/GC and Pd/C/GC, respectively. The counter electrode was a Pt foil. Hg/HgO (1 M KOH) was used as the reference electrode, which was calibrated against a reversible hydrogen electrode (RHE), and all potentials reported in this paper were referred to the RHE. Cyclic voltammetric (CV) measurements were performed in a 1 M KOH solution with or without 1 M methanol at room temperature in the potential range of −0.9 to +0.4 V at the potential sweep rate of 50 mV s−1. Linear sweep voltammetric (LSV) studies were conducted in a 1 M KOH + 1 M CH3OH solution between −0.9 to +0.4 V at different temperatures at a potential scan rate of 50 mV s−1. Chronoamperometric (CA) analysis was conducted at −0.25 V in 1 M KOH + 1 M CH3OH at room temperature for 1000 s.3. Results and discussion
3.1 Structural characterization
The structure and the composition of Au@PdAg-NTs were first examined by SEM and TEM measurements. As depicted in Fig. S1,† the original Ag nanowires exhibited a diameter of about 100 nm, length of about 3 μm and a smooth surface. Yet, after galvanic replacement reaction with AuCl4− and PdCl42−, the resultant Au@PdAg-NTs show an obvious hollow tubular structure with a rough and porous wall, with a diameter of 160 to 180 nm, length of 1 to 2 μm, and an average wall thickness of approximately 10 nm, as manifested in SEM (Fig. 1a) and TEM (Fig. 1b) studies.37–39 | ||
| Fig. 1 (a) SEM and (b and c) TEM images of as-synthesized Au@PdAg-NTs. Inset to panel (b) is the electron diffraction pattern of the Au@PdAg-NTs. (d) HAADF-STEM image and (e) the corresponding EDS cross-sectional composition line profiles of Au@PdAg-NT. (f) and (g) High-magnification HRTEM images of the corresponding red regions in (c). (h) STEM image and (i–l) the corresponding elemental maps of a section of Au@PdAg-NT. | ||
The electron diffraction patterns (inset to Fig. 1b) suggest good crystallinity of Au@PdAg-NTs. In fact, the HRTEM image in Fig. 1c shows well-defined crystalline lattice fringes in Au@PdAg-NTs. Upon closer inspection, there appear to be two major types of lattice fringes, as exemplified by the dashed boxes marked as “f” and “g”: one with an interplanar spacing of 0.235 nm (Fig. 1f) that is consistent with that of the (111) planes of fcc Au (0.235 nm),40 and the other with an interplanar spacing of 0.230 nm and 0.199 nm (Fig. 1g), which are between the lattice spacing of (111) and (002) planes of pure Ag and pure Pd,40,41 indicating the formation of PdAg alloy. The formation of PdAg alloy can also be confirmed by XPS analysis (Fig. S3†). This suggests that the sample mostly consisted of a gold-rich interior and a PdAg alloy dendritic shell.42 Consistent results of the elemental distribution were obtained in HAADF-STEM measurements (Fig. 1d), where linear scans of the cross-sectional profiles (Fig. 1e) and elemental mapping measurements (Fig. 1h–l) showed that the wall of Au@PdAg-NTs was rich in Au in the interior and PdAg alloy on the surface.
The crystalline phase of the obtained sample was then analyzed by powder XRD measurements. From Fig. 2, it can be seen that Au@PdAg-NTs exhibited broad XRD patterns, likely due to the nanocrystalline nature of the material, with four dominant diffraction peaks at ca. 38.5°, 44.6°, 65.1° and 78.1°, which are again in the intermediate between those of pure Pd (JCPDS card no. 65-6174, pink) and Ag (JCPDS card no. 65-8428, in green)/Au (JCPDS card no. 65-8601, blue), suggesting the formation of PdAg and/or PdAu alloy. It should also be noted that the ratio of the peak intensity of the (111) and (200) diffractions, I(111)/I(200), was estimated to be 3.76 for Au@PdAg-NTs, much larger than that (2.22) of bulk Pd (JCPDS no. 65-6174), suggesting preferential orientation along the (111) direction in Au@PdAg-NTs. In addition, in EDS analysis (Fig. S2†), Pd, Au and Ag can be readily identified in Au@PdAg-NTs, in good agreement with the above XRD results. From the EDS data, it can also be found that the atomic ratio of the Pd, Au and Ag is about 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :17:56.
:17:56.
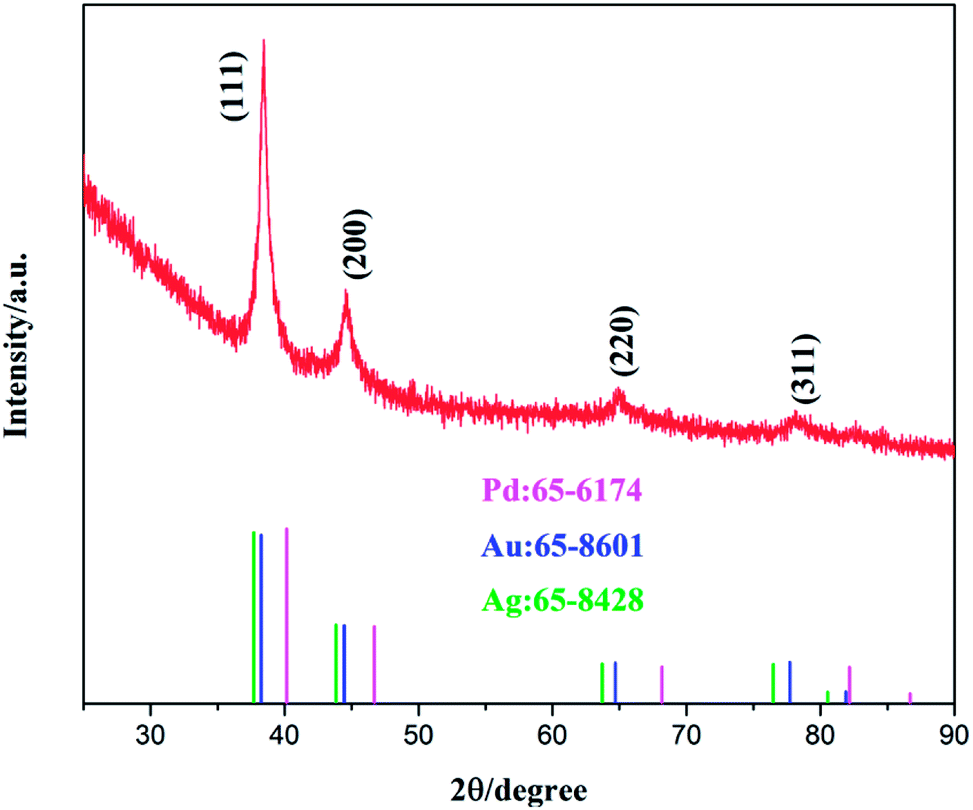 | ||
| Fig. 2 XRD patterns of the as-synthesized Au@PdAg-NTs and the standard patterns of Pd (#65-6174), Au (#65-8601) and Ag (#65-8428). | ||
3.2 Formation mechanism
The formation mechanism of Au@PdAg-NTs is proposed in Fig. 3. At the initial stage of the reaction when HAuCl4 was added into the suspension of Ag nanowires, galvanic replacement occurred preferentially at surface defect sites with a high surface energy (such as steps, stacking faults, etc.), where Ag was oxidized and dissolved into the solution, resulting in the formation of small pits in the Ag nanowires. This might be facilitated by the adsorption of released Cl− ions on the Ag surface, AuCl4− + 3Ag → Au + 3AgCl + Cl−.43 Because of Ag corrosion, the electrons would quickly migrate to the surface of the Ag nanowires and be captured by AuCl4− to generate Au atoms via galvanic reduction. Because of the good match between Ag and Au in terms of crystal structures (fcc) and lattice constants (4.086 vs. 4.078 Å for Ag and Au, respectively),44 the newly formed Au atoms were most likely deposited epitaxially on the surface of the Ag nanowires, which is helpful for preserving the morphology of the Ag nanowire template, leading to the formation of a thin and incomplete layer of Au atoms on the surface of Ag nanowire. Meanwhile, because of higher activity of the fresh surfaces of the etch pits, the interior Ag atoms became gradually dissolved into the solution forming a nanotube nanostructure. Nevertheless, alloying will not occur between Au and the underlying Ag due to slow interdiffusion between them at low temperature and the high stability of Au nanoparticles (the bonding energy is 226.2 kJ mol−1 for Au–Au, and 162.9 kJ mol−1 for Ag–Ag).45,46 Consequently, a porous incomplete Au shell was formed on the Ag template surface along with continuous etching of the silver core and deposition of Au.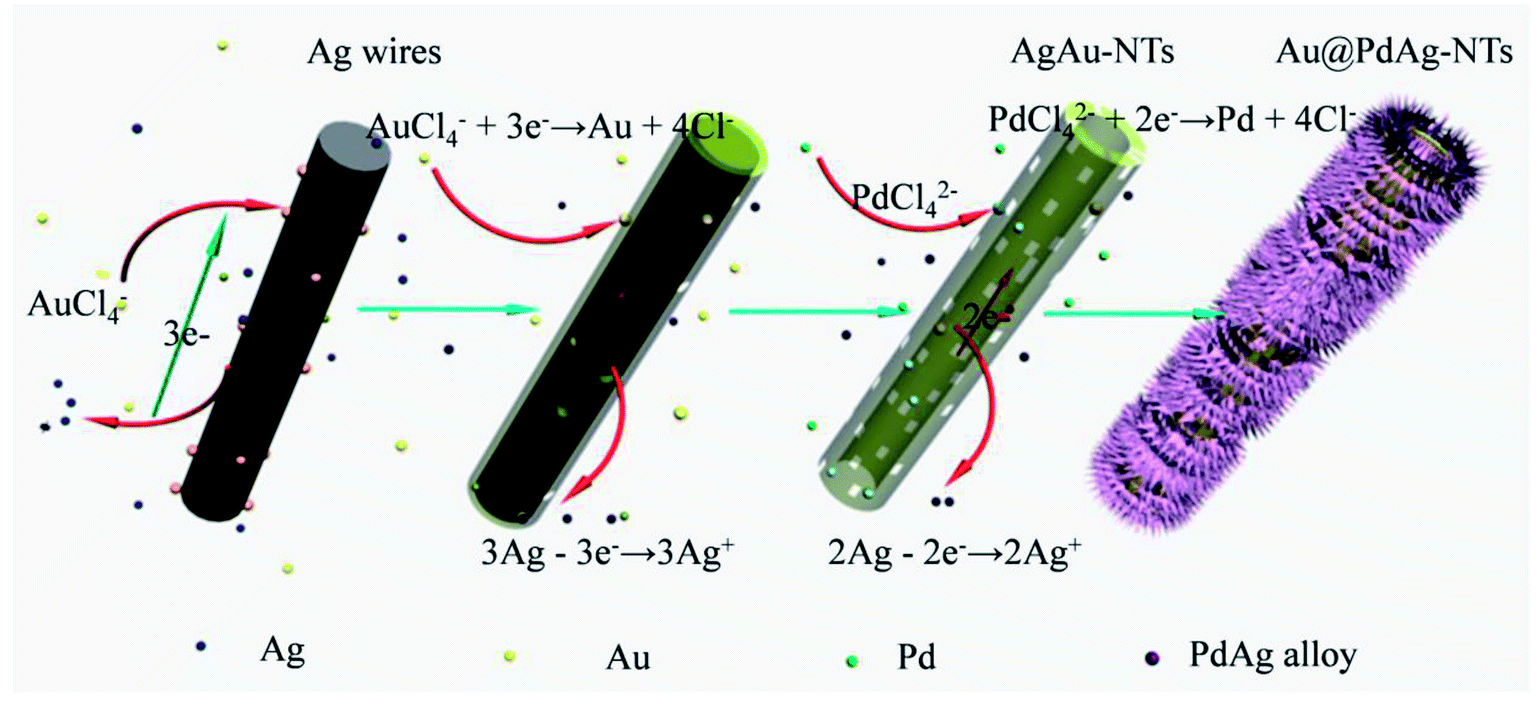 | ||
| Fig. 3 Schematic illustration of the formation process of Au@PdAg-NTs by sequential galvanic replacement reactions between Ag nanowires and AuCl4− and PdCl42−. | ||
In the second step, after AuCl4− was consumed, PdCl42− was added into the above suspension. The galvanic replacement only occurred between the remaining Ag and PdCl42− because the standard reduction potential decreases in the order of AuCl4−/Au > PdCl42−/Pd > AgCl/Ag. As a result, Ag atoms will be continuously oxidized and dissolved into the solution, while the newly formed Pd atoms were poorly epitaxially deposited on the surfaces of the Ag–Au nanotubes due to the relatively large mismatch of the lattice constants between Pd and Ag or Au (∼4.8%),47 forming a rough surface. Because the bonding energies of Pd–Pd (100 kJ mol−1) is lower than that of Pd–Ag (137 kJ mol−1),44 solid–solid inter-diffusion occurred over the surface of the Ag template, resulting in the formation of a conformal and homogeneous PdAg alloy shell.
As the reaction proceeded, the interior Ag of the Ag template gradually disappeared or diffuse, resulting in the formation of tube walls that are rich of Au in the interior. Additionally, based on the stoichiometric relationship, the galvanic replacement of two or three Ag atoms by one Pd or Au atom led to the formation of lattice vacancies, and hence a significant increase of the total surface energy.48 These lattice vacancies coalesced to form pinholes at specific spots in the wall of the tubes by an Ostwald ripening process in order to decrease the surface energy, leading to the formation of nanoporous Au core and PdAg alloy dendritic shell by surface diffusion.49 Eventually, a Au@PdAg-NTs nanostructure comprised of a Au-rich interior and three-dimensional PdAg alloy dendritic shell was formed.
3.3 Electrochemical studies
Fig. 4a shows the CV curves of Au@PdAg-NTs and commercial Pd/C modified electrodes in a N2-saturated 1.0 M KOH solution at the potential scan rate of 50 mV s−1. For both samples, nonzero voltammetric currents started to appear at ca. +0.2 V in the anodic scan, due to the formation of a PdOx layer, and the broad cathodic peak at around −0.2 V can be attributed to the reduction of PdOx. It can be seen that the peak potential of PdOx reduction is about 0.03 V more positive for the Au@PdAg-NTs than for the commercial Pd/C, suggesting reduced oxophilicity of the former. This may facilitate the complete oxidation of intermediate CO formed during methanol electrooxidation at the anode of DMFCs.50 In addition, the hydrogen adsorption/desorption (Had/Hdes) peaks for the commercial Pd/C catalyst are better-defined than those of Au@PdAg-NTs, indicating that for Pd-containing catalyst the adsorption of hydrogen cannot be distinguished from its desorption and the Pd surface was significantly modified due to the incorporation of Au and Ag (the double-layer charging current for the Au@PdAg-NTs catalyst was obviously smaller than that of Pd/C, likely due to contributions from the carbon support in the latter).51 Interestingly, the voltammetric results of Au@PdAg-NTs did not show the typical electrochemical features of Ag or Au in alkaline media, suggesting no separate Ag or Au domains were formed/accessible on the surface of Au@PdAg-NTs and the stability of Ag and Au in alkaline media for the as-prepared sample was enhanced.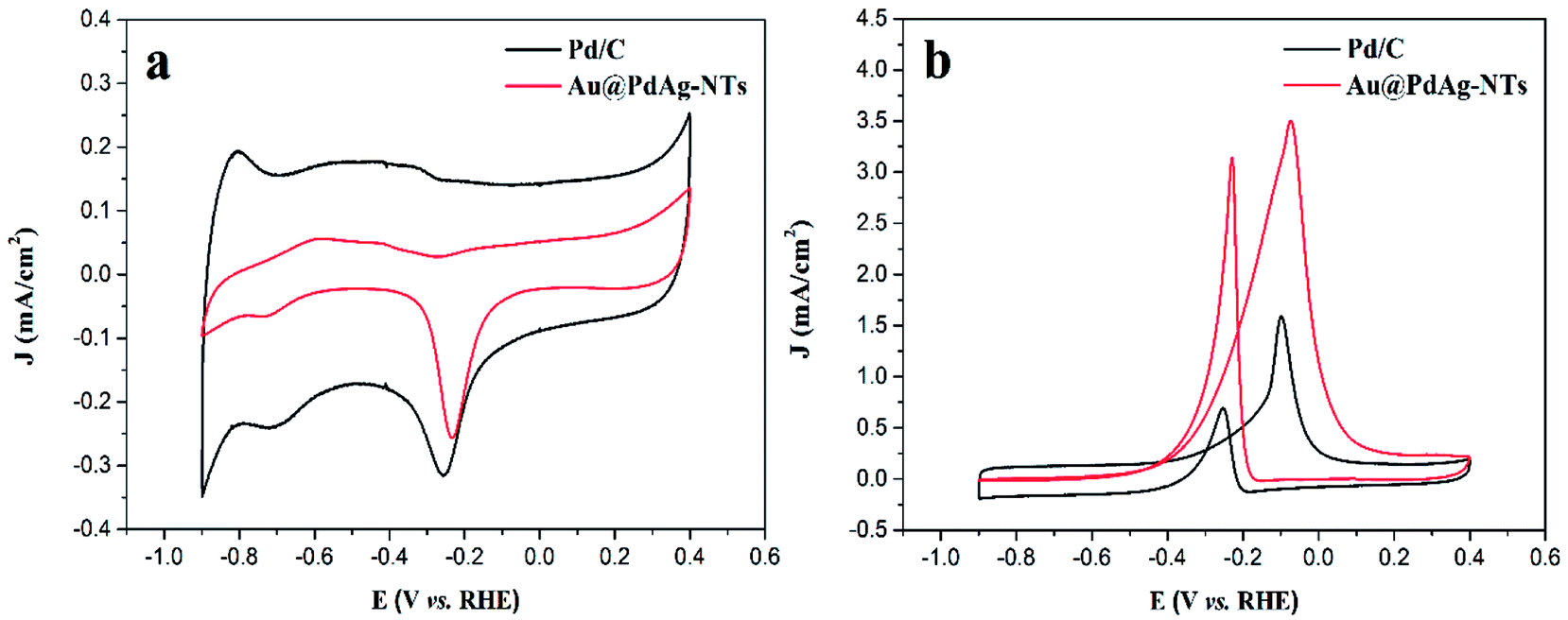 | ||
| Fig. 4 Cyclic voltammograms of the Au@PdAg-NTs/GC and Pd/C/GC electrodes in 1.0 M KOH solution (a) without and (b) with 1.0 M CH3OH at a scan rate of 50 mV s−1. | ||
The steady-state cyclic voltammograms of Au@PdAg-NTs/GC and Pd/C/GC electrodes in 1.0 M CH3OH + 1.0 M KOH are shown in Fig. 4b. Note that the voltammetric currents have been normalized to the effective electrochemical surface areas (ECSA) estimated from the reduction charge of PdOx according to the CV curves in Fig. 4a.52 It is evident from Fig. 4b that Au@PdAg-NTs exhibited two anodic current peaks, similar to results observed in ethanol electrooxidation on Pd-based electrocatalysts in alkaline media and methanol electrooxidation on Pt-based electrocatalysts in acid media.33,53 It is apparent that the electrocatalytic activity of the Au@PdAg-NTs/GC electrode is significantly higher than that of Pd/C/GC electrode, as manifested by the forward anodic peak current density that is 3.50 mA cm−2 for the former, 2.2 times higher that for the latter (1.59 mA cm−2). Furthermore, the onset potential (Eonset = −0.40 V) for methanol oxidation at the Au@PdAg-NTs/GC electrode is 100 mV more negative than that (−0.30 V) at the Pd/C/GC electrode. This onset potential is also more negative than those of the relevant Pd-based catalysts reported in the literature,54–56 again, implying a higher intrinsic electrocatalytic activity for methanol oxidation with the Au@PdAg-NTs.57,58
In order to gain further insights into the superior electrocatalytic performance of Au@PdAg-NTs, LSV tests were conducted within the temperature range of 20 to 40 °C (Fig. 5a and b). One can see that for both Pd/C and Au@PdAg-NTs, the anodic currents for methanol oxidation increased with increasing temperature. Fig. 5c and d show the corresponding Arrhenius plots of the voltammetric currents at −0.02 V vs. temperature. It can be seen that both Pd/C and Au@PdAg-NTs exhibited a linear profile, with an obviously higher current density for Au@PdAg-NTs than for Pd/C. In addition, from the linear regression, the apparent activation energy (Ea) was estimated to be 41.5 and 28.7 kJ mol−1 for Pd/C and Au@PdAg-NTs, respectively.59 The markedly lower activation energy of Au@PdAg-NTs is again consistent with the higher electrocatalytic activity towards methanol oxidation than Pd/C, as was observed above in Fig. 4.
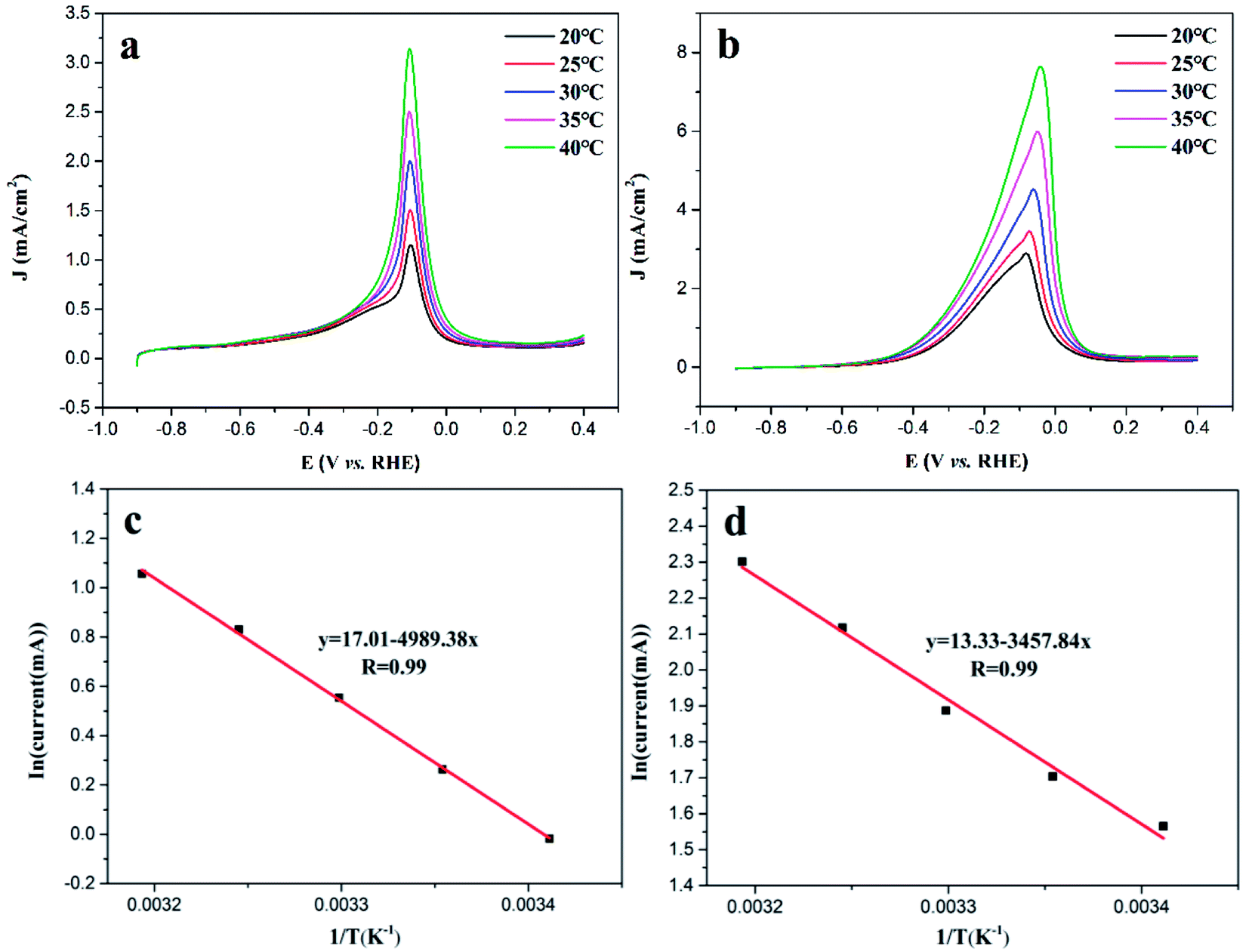 | ||
| Fig. 5 LSV curves of (a) commercial Pd/C/GC and (b) Au@PdAg-NTs/GC electrodes in 1 M KOH + 1 M CH3OH solutions at different temperatures. Potential scan rate 50 mV s−1. Arrhenius plots of the voltammetric currents at −0.02 V (vs. RHE) with temperature for (c) commercial Pd/C and (d) Au@PdAg-NTs. | ||
Electrochemical stability is another key issue for electrocatalysts in DMFCs. To evaluate the stability of the catalysts under continuous operating conditions, CA measurements were performed (Fig. 6). It can be seen that both the initial and steady-state current densities for methanol oxidation on Au@PdAg-NTs are markedly higher than those on commercial Pd/C, further demonstrating a significantly enhanced electrocatalytic activity of Au@PdAg-NTs. This result also coincides with the above CV and LSV data. Furthermore, Au@PdAg-NTs exhibited much slower current attenuation in comparison to Pd/C, signifying enhanced stability in the electrooxidation of methanol. For instance, at 1000 s, the current density (Jt=1000 s) was 0.275 mA cm−2 for Au@PdAg-NTs, more than three times higher than that (0.075 mA cm−2) for Pd/C.
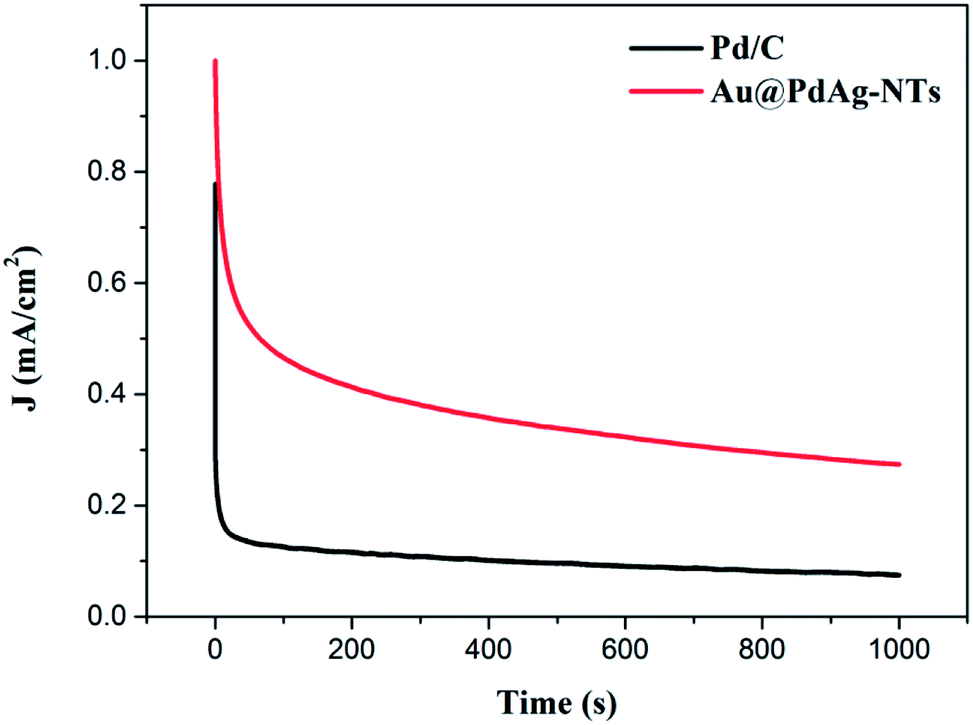 | ||
| Fig. 6 Chronoamperometric curves of the Au@PdAg-NTs/GC and Pd/C/GC electrodes in 1.0 M KOH+1 M CH3OH solution at the electrode potential of −0.15 V (vs. RHE). | ||
These results are summarized in Table S1,† where the much enhanced catalytic activity of Au@PdAg-NTs, as compared to that of Pd/C, may be attributed to the following factors. First, the nanotubular structure of Au@PdAg-NTs can not only facilitate electron transport, but also improve mass transport and catalyst utilization.60 Furthermore, Au@PdAg-NTs can be connected to each other to form a three-dimensional hollow framework in which efficient electrical pathways are established. In addition, the porous wall and hollow interior in Au@PdAg-NTs can promote mass transport. These unique characteristics are important for the efficient electrochemical oxidation of methanol at the interface. Second, the micrometer-sized length of the Au@PdAg-NTs makes it less vulnerable to dissolution, Ostwald ripening and aggregation than commonly used catalysts that are of small size and spherical shape, which will be helpful for the electrocatalytic stability of the catalysts in methanol electrooxidation. Additionally, the dendritic surface structure of Au@PdAg-NTs can not only provide sufficient accessible active sites but also prevent undesirable agglomeration usually encountered with nanoparticle electrocatalysts. Finally, alloying Pd and Ag may change the electronic properties of Pd and enhance the reactivity of Pd for the poisonous intermediates formed in the process of methanol electrooxidation, leading to improved poison tolerance of the Au@PdAg-NTs catalysts.61,62
4. Conclusions
Au@PdAg-NTs with a Au-rich interior and a PdAg alloy dendritic shell were synthesized in aqueous solution by sequential galvanic displacement reactions between Ag nanowires and AuCl4− and PdCl42−. The obtained samples showed an excellent electrocatalytic performance toward methanol oxidation as compared to commercial Pd/C, due to their large specific surface area and unique porous tubular morphology, with a significantly negative shift of the onset potential and markedly enhanced anodic peak current density. This facile and cost-effective strategy may be exploited as a generic method to prepare multimetallic nanostructures for catalysis of diverse important reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Scientific Research Fund of Huaqiao University (No. 10Y0195*), Fundamental Research Funds for the Central Universities (No. JB-ZR1138), the Project Sponsored by the Scientific Research Foundation for Returned Overseas Chinese Scholars, State Education Ministry and the Natural Science Foundation of Fujian Province (Grant No. 2017J01016). We also acknowledge the National Natural Science Foundation of China (NSFC) (21203130), Collaborative Innovation Center of Suzhou Nano Science & Technology, the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD) and the Fund for Innovative Research Teams of Jiangsu Higher Education Institutions.References
- G. Acres, Recent advances in fuel cell technology and its applications, J. Power Sources, 2001, 100, 60–66 CrossRef CAS.
- E. Antolini, Catalysts for direct ethanol fuel cells, J. Power Sources, 2007, 170, 1–12 CrossRef CAS.
- C. Lamy, A. Lima, V. LeRhun, F. Delime, C. Coutanceau and J. Leger, Recent advances in the development of direct alcohol fuel cells (DAFC), J. Power Sources, 2002, 105, 283–296 CrossRef CAS.
- F. Vigier, S. Rousseau, C. Coutanceau, J. Leger and C. Lamy, Electrocatalysis for the direct alcohol fuel cell, Top. Catal., 2006, 40, 111–121 CrossRef CAS.
- A. Chen and P. Holt-Hindle, Platinum-Based Nanostructured Materials: Synthesis, Properties, and Applications, Chem. Rev., 2010, 110, 3767–3804 CrossRef CAS PubMed.
- W. Chen, J. Kim, S. H. Sun and S. W. Chen, Phys. Chem. Electro-oxidation of formic acid catalyzed by FePt nanoparticles, Chem. Phys., 2006, 8, 2779–2786 CAS.
- W. Chen, J. Kim, L. Xu, S. H. Sun and S. W. Chen, Langmuir-Blodgett thin films of Fe20Pt80 nanoparticles for the electrocatalytic oxidation of formic acid, J. Phys. Chem. C, 2007, 111, 13452–13459 CrossRef CAS.
- D. Chu and S. Gilman, Methanol Electro-oxidation on Unsupported Pt-Ru Alloys at Different Temperatures, J. Electrochem. Soc., 1996, 143, 1685–1690 CrossRef CAS.
- W. Chen, J. Kim, S. H. Sun and S. W. Chen, Electrocatalytic Reduction of Oxygen by FePt Alloy Nanoparticles, J. Phys. Chem. C, 2008, 112, 3891–3898 CrossRef CAS.
- A. Tripkovic, K. Popovic, B. Grgur, B. Blizanac, P. Ross and N. Markovic, Methanol electrooxidation on supported Pt and PtRu catalysts in acid and alkaline solutions, Electrochim. Acta, 2002, 47, 3707–3714 CrossRef CAS.
- Y. Chen, A. Miki, S. Ye, H. Sakai and M. Osawa, Formate, an Active Intermediate for Direct Oxidation of Methanol on Pt Electrode, J. Am. Chem. Soc., 2003, 125, 3680–3681 CrossRef CAS PubMed.
- M. Arenz, V. Stamenkovic, P. Ross and N. Markovic, Surface (electro-)chemistry on Pt(1 1 1) modified by a Pseudomorphic Pd monolayer, Surf. Sci., 2004, 573, 57–66 CrossRef CAS.
- C. Xu, L. Cheng, P. Shen and Y. Liu, Methanol and ethanol electrooxidation on Pt and Pd supported on carbon microspheres in alkaline media, Electrochem. Commun., 2007, 9, 997–1001 CrossRef CAS.
- K. Jukk, N. Alexeyeva, P. Ritslaid, J. Kozlova, V. Sammelselg and K. Tammeveski, Electroreduction of oxygen on sputter-deposited Pd nanolayers on multi-walled carbon nanotubes, Electrocatalysis, 2013, 4, 42–48 CrossRef CAS.
- W. Li, X. Zhao and A. Manthiram, Room-temperature synthesis of Pd/C cathode catalysts with superior performance for direct methanol fuel cells, J. Mater. Chem. A, 2014, 2, 3468–3476 RSC.
- W. Huang, X. Kang, C. Xu, J. Zhou, J. Deng, Y. Li and S. Cheng, 2D PdAg alloy nanodendrites for enhanced ethanol electroxidation, Adv. Mater., 2018, 1706962 CrossRef PubMed.
- L. Zhang, Q. Chang, H. Chen and M. Shao, Recent advances in palladium-based electrocatalysts for fuel cell reactions and hydrogen evolution reaction, Nano Energy, 2016, 29, 198–219 CrossRef CAS.
- F. Maroun, F. Ozanam, O. Magnussen and R. Behm, The Role of Atomic Ensembles in the Reactivity of Bimetallic Electrocatalysts, Science, 2001, 293, 1811–1814 CrossRef CAS PubMed.
- A. Ruban, B. Hammer, P. Stoltze, H. Skriver and J. Nørskov, Surface electronic structure and reactivity of transition and noble metals, J. Mol. Catal. A: Chem., 1997, 115, 421–429 CrossRef CAS.
- F. Cheng, X. Dai, H. Wang, S. Jiang, M. Zhang and C. Xu, Electrochim. Acta, 2010, 55, 2295–2298 CrossRef CAS.
- J. Chen, B. Lim, E. Lee and Y. Xia, Shape-controlled synthesis of platinum nanocrystals for catalytic and electrocatalytic applications, Nano Today, 2009, 4, 81–95 CrossRef CAS.
- Y. Xiong and Y. Xia, Shape-controlled synthesis of metal nanostructures: the case of silver, Adv. Mater., 2007, 19, 3385–3391 CrossRef CAS.
- S. Habas, H. Lee, V. Radmilovic, G. Somorjai and P. Yang, Shaping binary metal nanocrystals through epitaxial seeded growth, Nat. Mater., 2007, 6, 692–697 CrossRef CAS PubMed.
- Q. Chen, Y. Jia, S. Xie and Z. Xie, Well-faceted noble-metal nanocrystals with nonconvex polyhedral shapes, Chem. Soc. Rev., 2016, 45, 3207–3220 RSC.
- C. Hu, X. Mu, J. Fan, H. Ma, X. Zhao, G. Chen, Z. Zhou and N. Zheng, Interfacial Effects in PdAg Bimetallic Nanosheets for Selective Dehydrogenation of Formic Acid, ChemNanoMat, 2016, 2, 28–32 CrossRef CAS.
- M. Liu, Y. Lu and W. Chen, Electrocatalysts: PdAg Nanorings Supported on Graphene Nanosheets: Highly Methanol-Tolerant Cathode Electrocatalyst for Alkaline Fuel Cells, Adv. Funct. Mater., 2013, 23, 1289–1296 CrossRef CAS.
- X. Xia, S. Choi, J. Herron, N. Lu, J. Scaranto, H. Peng, J. Wang, M. Mavrikakis, M. Kim and Y. Xia, Quantitative Analysis of the Coverage Density of Br– Ions on Pd{100} Facets and Its Role in Controlling the Shape of Pd Nanocrystals, J. Am. Chem. Soc., 2013, 135, 15706–15709 CrossRef CAS PubMed.
- C. Hu, H. Cheng, Y. Zhao, Y. Hu, Y. Liu, L. Dai and L. Qu, Newly-Designed Complex Ternary Pt/PdCu Nanoboxes Anchored on Three-Dimensional Graphene Framework for Highly Efficient Ethanol Oxidation, Adv. Mater., 2012, 24, 5493–5498 CrossRef CAS PubMed.
- C. Cui, J. Yu, H. Li, M. Gao, H. Liang and S. Yu, Remarkable enhancement of electrocatalytic activity by tuning the interface of Pd-Au bimetallic nanoparticle tubes, ACS Nano, 2011, 5, 4211–4218 CrossRef CAS PubMed.
- S. Yu, U. Welp, L. Hua, A. Rydh, W. Kwok and H. Wang, Fabrication of Palladium Nanotubes and Their Application in Hydrogen Sensing, Chem. Mater., 2005, 17, 3445–3450 CrossRef CAS.
- S. Guo, S. Dong and E. Wang, Pt/Pd bimetallic nanotubes with petal-like surfaces for enhanced catalytic activity and stability towards ethanol electrooxidation, Energy Environ. Sci., 2010, 3, 1307–1310 RSC.
- B. Choi, Y. Lee, S. Kang, J. Hong, J. Kim, I. Park and S. Han, Multimetallic Alloy Nanotubules with Nanoporous Framework, ACS Nano, 2012, 6, 5659–5667 CrossRef CAS PubMed.
- C. Cui, H. Li, J. Yu, M. Gao and S. Yu, Ternary heterostructured nanoparticle tubes: a dual catalyst and its synergistic enhancement effects for O2/H2O2 reduction, Angew. Chem., Int. Ed., 2010, 49, 9149–9152 CrossRef CAS PubMed.
- Z. Chen, M. Waje, W. Li and Y. Yan, Supportless Pt and PtPd Nanotubules as Electrocatalysts for Oxygen-Reduction Reactions, Angew. Chem., Int. Ed., 2007, 46, 4060–4063 CrossRef CAS PubMed.
- S. M. Alia, Y. S. Yan and B. S. Pivovar, Galvanic Displacement as a route to highly active and durable extended surface electrocatalysts, Catal. Sci. Technol., 2014, 4, 3589–3600 RSC.
- Y. Sun, B. Mayers, T. Herricks and Y. N. Xia, Polyol synthesis of uniform silver nanowires: a plausible growth mechanism and the supporting evidence, Nano Lett., 2003, 3(7), 955–960 CrossRef CAS.
- L. Shi, A. Wang, T. Zhang, B. Zhang, D. Su, H. Li and Y. Song, One-Step Synthesis of Au-Pd Alloy Nanodendrites and Their Catalytic Activity, J. Phys. Chem. C, 2013, 117, 12526–12536 CrossRef CAS.
- B. Lim, M. Jiang, P. Camargo, E. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Pd–Pt bimetallic nanodendrites with high activity for oxygen reduction, Science, 2009, 324, 1302–1305 CrossRef CAS PubMed.
- Y. Lee, M. Kim, Y. Kim, Y. S. Kang, J. Lee and S. Han, Synthesis and electrocatalytic activity of Au-Pd alloy nanodendrites for ethanol oxidation, J. Phys. Chem. C, 2010, 114, 7689–7693 CrossRef CAS.
- W. He, X. Wu, J. Liu, X. Hu, K. Zhang, S. Hou, W. Zhou and S. Xie, Design of AgM Bimetallic Alloy Nanostructures (M = Au, Pd, Pt) with Tunable Morphology and Peroxidase-Like Activity, Chem. Mater., 2010, 22, 2988–2994 CrossRef CAS.
- A. K Singh, Y. Hwang and D. Kim, Heterogeneous PdAg alloy catalyst for selective methylation of aromatic amines with formic acid under an additive-free and solvothermal one-pot condition, NPG Asia Mater., 2015, 7, e222 CrossRef.
- J. Huang, S. Vongehr, S. Tang, H. Lu and X. Meng, Highly Catalytic Pd–Ag Bimetallic Dendrites, J. Phys. Chem. C, 2010, 114, 15005–15010 CrossRef CAS.
- R. Newman and K. Sieradzki, Metallic corrosion, Science, 1994, 263, 1708–1709 CrossRef CAS PubMed.
- X. Xia, Y. Wang, A. Ruditskiy and Y. Xia, 25th Anniversary Article: Galvanic Replacement: A Simple and Versatile Route to Hollow Nanostructures with Tunable and Well-Controlled Properties, Adv. Mater., 2013, 25, 6313–6333 CrossRef CAS PubMed.
- Y. G. Sun, Z. Tao, J. Chen, T. Herricks and Y. N. Xia, Ag Nanowires Coated with Ag/Pd Alloy Sheaths and Their Use as Substrates for Reversible Absorption and Desorption of Hydrogen, J. Am. Chem. Soc., 2004, 126, 5940–5941 CrossRef CAS PubMed.
- B. Goris, L. Polavarapu, S. Bals, G. Van Tendeloo and L. M. Liz Marzan, Monitoring Galvanic Replacement Through Three-Dimensional Morphological and Chemical Mapping, Nano Lett., 2014, 14, 3220–3226 CrossRef CAS PubMed.
- K. D. Gilroy, A. Sundar, P. Farzinpour, R. A. Hughes and S. Neretina, Mechanistic study of substrate-based galvanic replacement reactions, Nano Res., 2014, 7, 365–379 CrossRef CAS.
- Y. G. Sun, B. T. Mayers and Y. N. Xia, Template-Engaged Replacement Reaction: A One-Step Approach to the Large-Scale Synthesis of Metal Nanostructures with Hollow Interiors, Nano Lett., 2002, 2, 481–485 CrossRef CAS.
- A. R. Roosen and W. C. Carter, Simulations of microstructural evolution: anisotropic growth and coarsening, Phys. A, 1998, 261, 232–247 CrossRef CAS.
- S. Wang, S. Jiang, T. White, J. Guo and X. Wang, Electrocatalytic Activity and Interconnectivity of Pt Nanoparticles on Multiwalled Carbon Nanotubules for Fuel Cells, J. Phys. Chem. C, 2009, 113, 18935–18945 CrossRef CAS.
- Y. Lu and W. Chen, PdAg Alloy Nanowires: Facile One-Step Synthesis and High Electrocatalytic Activity for Formic Acid Oxidation, ACS Catal., 2012, 2, 84–90 CrossRef CAS.
- R. Pattabiraman, Electrochemical investigations on carbon supported palladium catalysts, Appl. Catal., A, 1997, 153, 9–20 CrossRef CAS.
- S. Guo, S. Dong and E. Wang, Constructing Carbon Nanotube/Pt Nanoparticle Hybrids Using an Imidazolium-Salt-Based Ionic Liquid as a Linker, Adv. Mater., 2010, 22, 1269–1272 CrossRef CAS PubMed.
- Z. Yin, M. Chi, Q. Zhu, D. Ma, J. Sun and X. Bao, Supported bimetallic PdAu nanoparticles with superior electrocatalytic activity towards methanol oxidation, J. Mater. Chem. A, 2013, 1, 9157–9163 RSC.
- L. M. Luo, R. H. Zhang, D. Chen, Q. Y. Hu, X. Zhang, C. Y. Yang and Z. W. Zhou, Hydrothermal synthesis of PdAu nanocatalysts with variable atom ratio for methanol oxidation, Electrochim. Acta, 2018, 259, 284–292 CrossRef CAS.
- S. Yan and Sh. Zhang, Investigation of methanol electrooxidation on Au/C catalyst in alkaline medium, Int. J. Hydrogen Energy, 2011, 36, 13392–13397 CrossRef CAS.
- T. Vidaković, M. Christov and K. Sundmacher, The use of CO stripping for in situ fuel cell catalyst characterization, Electrochim. Acta, 2007, 52, 5606–5613 CrossRef.
- J. Zeng, A simple eco-friendly solution phase reduction method for the synthesis of polyhedra platinum nanoparticles with high catalytic activity for methanol electrooxidation, J. Mater. Chem., 2012, 22, 3170–3176 RSC.
- J. Cohen, D. Volpe and H. D. Abruña, Interfacial rheology of stable and weakly aggregated two-dimensional suspensions, Phys. Chem. Chem. Phys., 2007, 9, 49–77 RSC.
- X. Wang, W. Li, Z. Chen, M. Waje and Y. Yan, Durability investigation of carbon nanotube as catalyst support for proton exchange membrane fuel cell, J. Power Sources, 2006, 158, 154–159 CrossRef CAS.
- Z. Liang, T. Zhao, J. Xu and L. Zhu, Mechanism study of the ethanol oxidation reaction on palladium in alkaline media, Electrochim. Acta, 2009, 54, 2203–2208 CrossRef CAS.
- B. Hammer and J. Norskov, Theoretical Surface Science and Catalysis – Calculations and Concepts, Adv. Catal., 2000, 45, 71–129 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08781d |
| This journal is © The Royal Society of Chemistry 2019 |