
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAggregation induced emission by pyridinium–pyridinium interactions†
Kaspars Leduskrasts and
Edgars Suna *
*
Latvian Institute of Organic Synthesis, Aizkraukles 21, LV-1006, Riga, Latvia. E-mail: edgars@osi.lv
First published on 11th January 2019
Abstract
Non-covalent intermolecular interactions between pyridinium subunits in a crystal-state are an efficient means to accomplish aggregation induced emission and avoid aggregation caused quenching.
Introduction
Organic luminescent molecules (luminophores) have found widespread applications in the design of optoelectronic devices and sensors.1 In the majority of applications, organic luminophores are either used as thin solid films or they are doped into a polymer matrix (host material). In thin films and in polymer matrices highly aggregated luminophore molecules may experience intermolecular π–π aromatic interactions. The non-covalent π–π interactions usually lead to decay of the excited-state energy of organic luminophores via non-radiative intermolecular energy transfer, thus resulting in decrease of emissive properties in the solid state. This phenomenon is well-known as aggregation caused quenching (ACQ).2 Due to the ACQ, the majority of organic luminophores feature considerably reduced emission or even lack of emission in the solid state, while being highly emissive in diluted solutions.A traditional approach to minimize the detrimental intermolecular π–π stacking relies on a decrease of planarity of organic luminophores. This has been achieved by out-of-plane twisting of aromatic subunits in luminophores as well as by incorporation of steric bulk3 or an anionic moiety4 in proximity to the π-system. Combined with a restriction of intramolecular motions (to minimize the non-radiative dissipation of exciton energy), these approaches have resulted in the design of organic emitters that feature higher emission in the solid-state as compared to solution (known as aggregation induced emission or AIE). First introduced by Tang,5 AIE luminogens (AIEgens) have become a hot topic in materials science during the last decade,6 and they have found ample application in materials science.7 It should be noted, however, that AIEgens may lose the solid state emission if intermolecular non-covalent π–π interactions between emitter molecules can take place.8 Clearly, the search for a new type of interactions that would help to avoid the detrimental intermolecular π–π stacking would facilitate the development of AIEgens with improved emissive properties.
It has been demonstrated that the undesired π–π stacking can be avoided by increasing the distance between interacting π-systems in the solid state.8a We hypothesized that other interactions that are effective at longer distances and are more flexible with respect to mutual spatial orientation9 of the interacting π-systems can be utilized to avoid the undesired π–π stacking. This has led us to propose that interactions between heteroaromatic cationic π-systems can be employed in the design of solid-state luminogens (AIEgens). Interestingly, the pyridinium cation–arene π system interactions have been used by Bull and Fossey to generate luminescent responses in solution.10 In addition, Lu and coworkers reported a fluorescence turn-on in polymer matrices via cation-π interactions.11 However, to the best of our knowledge, interactions between two heteroaromatic cationic π-systems has not been used in the design of AIEgens. Herein we demonstrate that non-covalent intermolecular interactions between two neighboring pyridinium subunits in solid-state is an efficient means to accomplish the solid-state emission (Fig. 1).
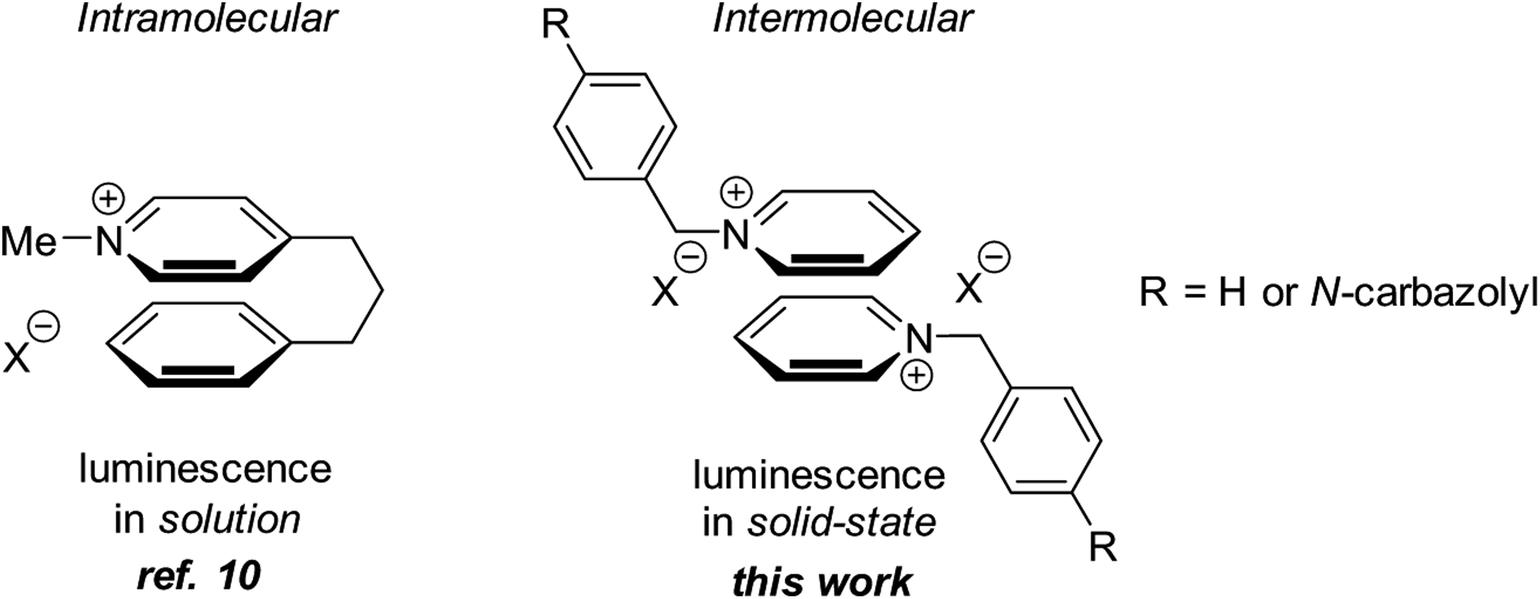 | ||
| Fig. 1 Design of the solid-state AIEgens based on pyridinium–pyridinium interactions. | ||
Results and discussion
Synthesis of quaternary pyridinium salts 1–5
N-Benzyl pyridinium bromide 1 was obtained by alkylation of pyridine with benzyl bromide (Fig. 2). The synthesis of pyridinium carbazoles 2a and 2b involved reduction of aldehyde 6 to benzylic alcohol 7, followed by conversion to benzyl chloride and reaction with pyridine (Fig. 2). The corresponding tosylate 2b was prepared from benzyl alcohol 7 and TsCl in the presence of pyridine as a base. In addition, pyridinium carbazoles 3, 4 possessing substituents in position 4 of the pyridine ring were also synthesized to evaluate influence of steric bulk on the interaction between pyridinium subunits of neighboring molecules in the solid-state (Fig. 2). Finally, 1,4-butylene linker-containing pyridinium carbazole 5 was prepared by double substitution of 1,4-dibromobutane with carbazole and pyridine (Fig. 2). All obtained pyridinium salts 1–5 were crystalline materials and they were characterized by 1H and 13C NMR spectra as well as by HRMS and IR techniques. | ||
| Fig. 2 Synthesis of pyridinium salts 1–5. Reagents and conditions: (a) BnBr, acetone, reflux, 1 h, 94%. (b) NaBH4, THF, MeOH, rt, 16 h, 87%. (c) TsCl, pyridine (for 2a), 4-tBu-pyridine (for 3) or 4-Me-pyridine (for 4), CH2Cl2, rt, 24 h, 57% (2a), 63% (3), 53% (4). (d) SOCl2, CH2Cl2, rt, 30 min, then pyridine, MeCN, 80 °C, 1 h, 70%. (e) Br(CH2)4Br, KOH, DMSO, rt, ultrasound, 30 min, 48%, (f) pyridine, MeCN, 90 °C, 16 h, 69%. | ||
Luminescence data
UV-vis absorption spectra of 1–5 were measured in MeCN solutions (at ca. 10−6 mol L−1 concentration) at room temperature and under ambient atmosphere. All synthesized materials displayed absorption peaks in 235–360 nm range (Fig. 3A), which were attributed to the π–π* transitions. Pyridinium salt 1 showed a narrow absorption band in MeCN solution with a maximum at 285 nm (entry 1, Table 1). Intense solid-state absorbance in the 250–350 nm region with sharply decreasing absorbance intensity above 350 nm was observed for the crystalline salt 1 (see ESI, page S10†). The salt also displayed an emission in MeCN solution (see ESI, page S9†) with photoluminescence quantum yield (PLQY) of 1.9% (entry 1, Table 1). Notably, excitation of the crystalline pyridinium salt 1 at 393 nm resulted in a solid-state emission with PLQY of 5.3% (entry 1, Table 1) and the maximum at 520 nm (see ESI, page S9†). The observed 2.8-fold increase of PLQY in the solid-state as compared to that in the MeCN solution pointed to the AIE properties of pyridinium salt 1. Next, emission properties of pyridinium salts 2–5 with attached carbazole luminophore were evaluated.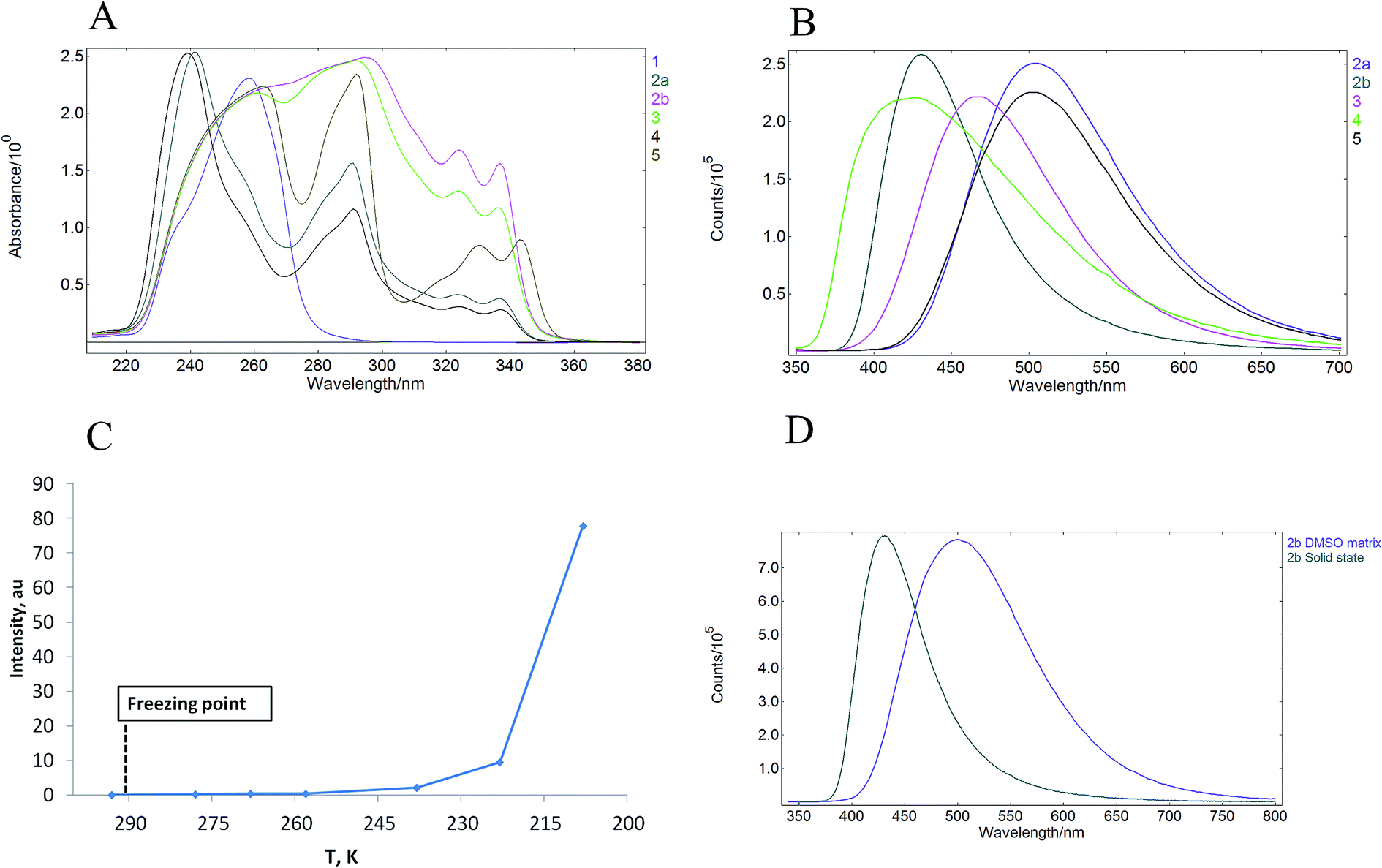 | ||
| Fig. 3 (A) UV-vis spectra of 1–5 in MeCN solutions; (B) solid state emission of 2–5; (C) emission of 2b in DMSO as a function of temperature; (D) normalized intensity of emission of 2b in the solid state and in the DMSO matrix. | ||
| Entry | Compound | λAbs, nm | Solid λEm, nm | Solution φ (%) | Solid φ (%) | αAIE |
|---|---|---|---|---|---|---|
| a Excited at 393 nm.b Excited at 325 nm. | ||||||
| 1 | 1 | 258 | 520a | 1.9 | 5.3 | 2.8 |
| 2 | 2a | 241, 290, 325, 337 | 505b | <0.1 | 17.3 | >173 |
| 3 | 2b | 264, 295, 324, 337 | 432b | <0.1 | 18.7 | >187 |
| 4 | 3 | 260, 294, 325, 337 | 465b | <0.1 | 18.5 | >185 |
| 5 | 4 | 240, 292, 324, 337 | 442b | <0.1 | 9.7 | >97 |
| 6 | 5 | 263, 292, 330, 343 | 503b | <0.1 | 15.9 | >159 |
All pyridinium carbazoles 2–5 were virtually non-emissive in solutions with photoluminescence quantum yields (PLQY) below 0.1%. In sharp contrast, the solid state PLQY for pyridinium salts 2–5 were measured to be in the range from 9.7 to 18.7%. Hence, pyridinium salts featured up to 187 times increase of emission in the solid state as compared to that in MeCN solution (Table 1). These results clearly indicated that pyridinium salts 2–5 possess AIE properties. Interestingly, the solid state emission intensity was not influenced by the structure of the anion as evidenced by comparable solid state PLQY for pyridinium chloride 2a (entry 2, Table 1) and pyridinium tosylate 2b (entry 3). Introduction of bulky t-Bu substituent in the position 4 of pyridinium subunit did not affect the solid state PLQY of 3 (entry 4 vs. entry 3), however the corresponding 4-methyl substituted analogue 4 showed the lowest PLQY values in the carbazole series (entry 5). Finally, AIEgen 5 with a flexible butylene linker between carbazole subunit and pyridinium moiety featured only slightly reduced solid state PLQY as compared to 2b (entry 6 vs. entry 3). This is notable given a higher degree of freedom for intramolecular motions in 5 as compared to 2b due to the long alkyl chain, which may consume the excited-state through non-radiative relaxation. All AIEgens 2–5 showed a broad emission peak in the solid state with no distinctive bands except the maximum in the range of 432–520 nm (Fig. 3B). Importantly, wavelength of the solid-state emission maximum depended on the counter-ion in pyridinium salts 1–5. Higher energy emission peaks in the range of 432–465 nm were observed for AIEgens 2b, 3 and 4, which possess tosylate as the counter ion (Table 1, entries 3–5). In contrast, lower energy emission (503–520 nm) was measured for chloride or bromide counterion-containing AIEgens 1, 2a and 5 (Table 1, entries 1, 2 and 6). Finally, pyridinium carbazoles 2–5 displayed intense solid-state absorbance in the 250–350 nm region with sharply decreasing absorbance intensity above 350 nm. The solid-state absorbance of 2–5 was consistent with that in MeCN solution (see ESI, page S10†).
Photoluminescence behaviour of 2a in various solvent mixtures was investigated to get an additional insight in AIE properties of 2a. Lack of the emission was observed for 2a in acetonitrile (a good solvent). In contrast, enhanced emission was observed in mixtures containing high (90–95% v/v) fraction of Et2O (a poor solvent) in MeCN (see ESI, page S13†). The dynamic light scattering (DLS) measurements indicated that pyridinium salt 2a did not form aggregates in solvent mixtures that contained less than 90% of Et2O in acetonitrile (see ESI, page S13†). Notably, the lack of emission was observed for 2a in these solvent mixtures. In contrast, the formation of aggregates could be observed even by the naked eye if 90–95% (v/v) of Et2O in acetonitrile was used as the solvent. In the latter case, the size of aggregates was larger than 10 μm, thus exceeding limits of DLS measurements. The apparent correlation between the formation of aggregates and luminescence provided an additional support for AIE properties of pyridinium salt 2a.
We have also examined whether AIE properties of pyridinium salts 1–5 originate from restriction of intramolecular rotation (RIR) phenomena.12 To this end we measured a relationship between emission of 2b in DMSO solution and temperature in the range from 293 K to 208 K (Fig. 3C). A solution of 2b in DMSO did not display observable emission at 293 K. Importantly, the emission increase could not be determined after freezing of the DMSO solution of 2b, as well as upon further cooling to 238 K. The emission only started to appear at temperatures below 238 K and the intensity increased considerably at 208 K (Fig. 3C). Lack of the emission for 2b in the DMSO matrix points against the RIR mechanism as the origin of AIE properties of 2b, because considerable enhancement of the emission in solid matrices is usually observed for most of AIEgens that benefit from RIR effect.13 Furthermore, the emission peak for 2b in frozen DMSO matrix showed bathochromic shift as compared to that in the solid state (Fig. 3D). Finally, the solid state emission intensity for crystalline 2b remained unchanged in the temperature range from 293 K to 193 K. These data provide evidence that the luminogens 1–5 show AIE properties in crystalline form.
Single crystal X-ray analysis
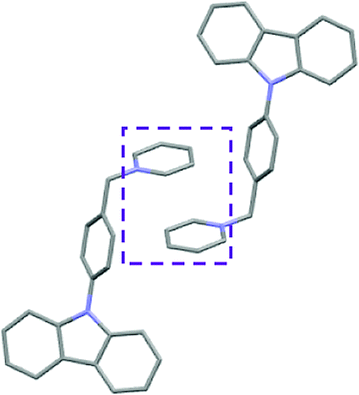
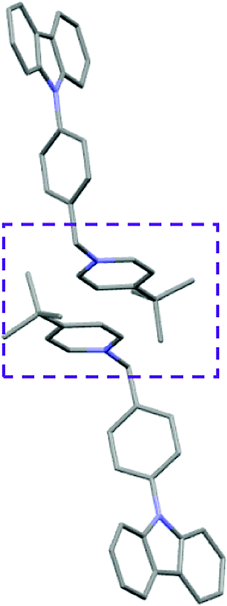
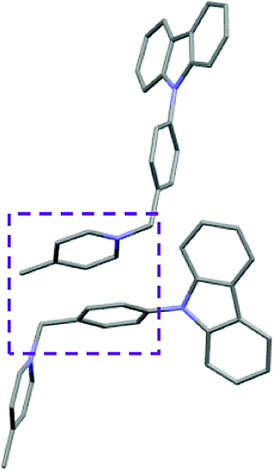
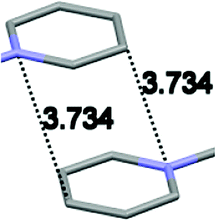
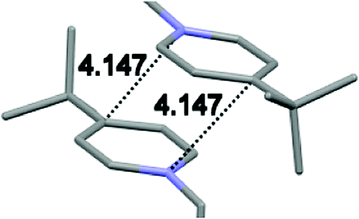
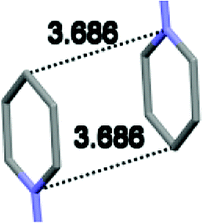
Single crystal X-ray analysis of luminogens 2–5 provided important insight into intermolecular interactions that result in AIE properties (Table 2). Single crystals of pyridinium salts 2–5 suitable for X-ray crystallography could be obtained by vapor diffusion from various solvents systems such as acetone/EtOH, DCM/hexane, EtOAc/MeCN, toluene/DCM or Et2O. In a crystal lattice pyridinium salts 2a, 2b, 3 and 5 feature intermolecular π+–π+ interactions between the charged pyridinium rings. This is evidenced by a strictly parallel off-center orientation of neighboring pyridinium rings with the distance between them spanning a range from 3.686 Å (for 5) to 4.211 Å (for 2a; see Table 2). In contrast, pyridinium salt 4 does not have the π+–π+ interactions between the charged pyridinium rings. Instead, interactions between pyridinium ring and 1,4-disubstituted phenylene moiety can be observed for 4 as evidenced by a relatively close distance (from 3.803 Å to 4.027 Å) between the two nonparallel-oriented π-systems (Table 2).14 It should be noted that pyridinium salt 4 demonstrated lower PLQY as compared to other AIEgens 2a, 2b, 3 and 5 (see Table 1), suggesting that pyridinium–pyridinium interactions are important to achieve greater AIE properties in the crystalline state.15| Compound 2a | Compound 2b | Compound 3 | Compound 4 | Compound 5 |
|---|---|---|---|---|
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
In addition to the intermolecular pyridinium–pyridinium interactions in the crystal lattice of 2b, the aromatic system of tosylate anion is positioned in a non-parallel off-center orientation with respect to pyridinium ring (3.843–4.186 Å distance, see ESI, page S9†). However, the latter interaction apparently does not influence the AIE properties of 2b, as evidenced by similar PLQY values for 2b and the analogous AIEgen 2a, possessing chloride as the counter-ion (see entries 2 and 3, Table 1). The other tosylate-containing AIEgens 3 and 4 do not show the face-to-face interactions between pyridinium cation and π-system of the tosylate anion. Hence, these data demonstrate that the crystal-state AIE effect can be achieved using chloride, bromide and tosylate as the counter-ion for pyridinium. Importantly, nature of counter-ion influences the solid-state emission maximum of AIEgens. Thus, change of the counter-ion in AIEgen 2 from tosylate (2b) to chloride (2a) resulted in a noticeable bathochromic shift of more than 70 nm (Fig. 3B.). The dependence of the solid-state emission maximum on the nature of the counter-ion simplifies the design of AIEgens with the desired emission wavelength.
Conclusions
All synthesized pyridinium salts 1–5 demonstrate AIE properties with up to 187 times emission increase in the crystal-state as compared to solution. Lack of emission for 2b in solution and frozen DMSO matrix (solid amorphous state) speaks against the RIR effect as the origin of the AIE properties for pyridinium salt 2b. Single crystal X-ray analyses provide clear evidence for the presence of non-covalent intermolecular pyridinium–pyridinium and pyridinium–π interactions in the crystal-state of AIEgens 2–5. Consequently, crystal-state AIE effect can be attributed to the intermolecular π+–π+ and π+–π interactions involving pyridinium cations. Hence, non-covalent interactions between two neighboring pyridinium subunits in a crystal-state are an efficient means to accomplish AIE. The nature of counter-ion in pyridinium salts 2–5 does not affect emission efficiency (PLQY) of the crystal-state AIEgens, however, the counter-ion does influence the crystal-state emission maximum of AIEgens. The use of the non-covalent pyridinium–pyridinium or pyridinium–π interactions in the design of AIEgens is alternative and complementary approach to routinely used means to avoid the ACQ effect.3,4 Further studies on the design of the crystal-state AIEgens based on interactions between cationic π systems will be reported in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by Taiwan–Lithuania–Latvia Tripartite Cooperation Fund (Project “New materials and technologies for very-high color rendering and high sunlight spectrum resemblance OLED lighting sources”). We thank Dr A. Mishnev and Dr D. Stepanovs for X-ray crystallographic analysis and P. Dimitrijevs for DLS measurements.References
- (a) D. Volz, J. Photonics Energy, 2016, 6, 020901 CrossRef; (b) X.-D. Wang, O. S. Wolfbeis and R. J. Meier, Chem. Soc. Rev., 2013, 42, 7834–7869 RSC.
- (a) A. S. Klymchenko, Acc. Chem. Res., 2017, 50, 366 CrossRef CAS PubMed; (b) S. A. Jenekhe and J. A. Osaheni, Science, 1994, 265, 765 CrossRef CAS PubMed; (c) Solid-state luminescence enhancement has been also observed: J. Shi, L. E. Aguilar Suarez, S.-J. Yoon, S. Varghese, C. Serpa, S. Y. Park, L. Lüer, D. Roca-Sanjuán, B. Milián-Medina and J. Gierschner, J. Phys. Chem. C, 2017, 121, 23166–23183 CrossRef CAS.
- (a) J. E. Anthony, J. S. Brooks, D. L. Eaton and S. R. Parkin, J. Am. Chem. Soc., 2001, 123, 9482–9483 CrossRef CAS PubMed; (b) Z.-F. Yao, J.-Y. Wang and J. Pei, Cryst. Growth Des., 2018, 18, 7–15 CrossRef CAS.
- J. Wang, X. Gu, P. Zhang, X. Huang, X. Zheng, M. Chen, H. Feng, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, J. Am. Chem. Soc., 2017, 139, 16974–16979 CrossRef CAS PubMed.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhuc and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- (a) T. Butler, W. A. Morris, J. Samonina-Kosicka and C. L. Fraser, ACS Appl. Mater. Interfaces, 2016, 8, 1242 CrossRef CAS PubMed; (b) Z. S. Wang, B. S. Gelfand, P. C. Dong, S. Trudel and T. Baumgartner, J. Mater. Chem. C, 2016, 4, 2936 RSC; (c) O. Shynkaruk, G. He, R. McDonald, M. J. Ferguson and E. Rivard, Chem.–Eur. J., 2016, 22, 248 CrossRef CAS PubMed; (d) M. T. Gabr and F. C. Pigge, RSC Adv., 2015, 5, 90226 RSC; (e) W. A. Morris, T. D. Liu and C. L. Fraser, J. Mater. Chem. C, 2015, 3, 352 RSC; (f) K. R. Ghosh, S. K. Saha and Z. Y. Wang, Polym. Chem., 2014, 5, 5638 RSC; (g) N. B. Shustova, A. F. Cozzolino, S. Reineke, M. Baldo and M. Dinca, J. Am. Chem. Soc., 2013, 135, 13326 CrossRef CAS PubMed; (h) F. Hu, G. X. Zhang, C. Zhan, W. Zhang, Y. L. Yan, Y. S. Zhao, H. B. Fu and D. Q. Zhang, Small, 2015, 11, 1335 CrossRef CAS PubMed; (i) C. Wang, H. Zhang, L. Tian, W. Zhu, Y. Lan, J. Li, H. Wang, G. X. Zhang, D. Q. Zhang, S. L. Yuan and G. T. Li, Sci. China: Chem., 2016, 59, 89 CrossRef CAS.
- (a) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361 RSC; (b) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718 CrossRef CAS PubMed; (c) X. Z. Yan, H. Z. Wang, C. E. Hauke, T. R. Cook, M. Wang, M. L. Saha, Z. X. Zhou, M. M. Zhang, X. P. Li, F. H. Huang and P. J. Stang, J. Am. Chem. Soc., 2015, 137, 15276 CrossRef CAS PubMed; (d) X. Z. Yan, M. Wang, T. R. Cook, M. M. Zhang, M. L. Saha, Z. X. Zhou, X. P. Li, F. H. Huang and P. J. Stang, J. Am. Chem. Soc., 2016, 138, 4580 CrossRef CAS PubMed; (e) A. D. Shao, Y. S. Xie, S. J. Zhu, Z. Q. Guo, S. Q. Zhu, J. Guo, P. Shi, T. D. James, H. Tian and W. H. Zhu, Angew. Chem., Int. Ed., 2015, 54, 7275 CrossRef CAS PubMed; (f) C. Wang, B. J. Xu, M. S. Li, Z. G. Chi, Y. J. Xie, Q. Q. Li and Z. Li, Mater. Horiz., 2016, 3, 220 RSC; (g) Z. L. Xie, C. J. Chen, S. D. Xu, J. Li, Y. Zhang, S. W. Liu, J. R. Xu and Z. G. Chi, Angew. Chem., Int. Ed., 2015, 54, 7181 CrossRef CAS PubMed; (h) Y. T. Gao, G. X. Feng, T. Jiang, C. C. Goh, L. G. Ng, B. Liu, B. Li, L. Yang, J. L. Hua and H. Tian, Adv. Funct. Mater., 2015, 25, 2857 CrossRef CAS; (i) W. Dong, T. Fei, A. Palma-Cando and U. Scherf, Polym. Chem., 2014, 5, 4048 RSC.
- (a) M. Mohan, S. Pangannaya, M. N. Satyanarayan and D. R. Trivedi, ChemistrySelect, 2018, 3, 3803–3813 CrossRef CAS; (b) N. Han, H. Kun, X. Yuanjing, P. Qian, Z. Zujin, H. Rongrong, C. Junwu, S. Shi-Jian, Q. Anjun and B. Z. Tang, Mater. Chem. Front., 2017, 1, 1125–1129 RSC.
- M. S. Marshall, R. P. Steele, K. S. Thanthiriwatte and C. D. Sherrill, J. Phys. Chem. A, 2009, 113, 13628–13632 CrossRef CAS PubMed.
- W. Chen, S. A. Elfeky, Y. Nonne, L. Male, K. Ahmed, C. Amiable, P. Axe, S. Yamada, T. D. James, S. D. Bull and J. S. Fossey, Chem. Commun., 2011, 47, 253–255 RSC.
- J. Zhong, Z. Li, W. Guan and C. Lu, Anal. Chem., 2017, 89, 12472–12479 CrossRef CAS PubMed.
- J. Chen, C. C. W. Law, J. W. Y. Lam, Y. Dong, S. M. F. Lo, I. D. Williams, D. Zhu and B. Z. Tang, Chem. Mater., 2003, 15, 1535–1546 CrossRef CAS.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- Similar interactions between pyridinium cation and aromatic or heteroaromatic π-system have been observed by X-ray analysis: (a) X.-H. Jin, C. Chen, C.-X. Ren, L.-X. Cai and J. Zhang, Chem. Commun., 2014, 15878–15881 RSC; (b) C. Chen, X.-H. Jin, X.-J. Zhou, L.-X. Cai, Y.-J. Zhang and J. Zhang, J. Mater. Chem. C, 2015, 3, 4563–4589 RSC; (c) Y. Gu, Z. Zhao, H. Su, P. Zhang, J. Liu, G. Niu, S. Li, Z. Wang, R. T. K. Kwok, X.-L. Ni, J. Sun, A. Qin, J. W. Y. Lam and B. Z. Tang, Chem. Sci., 2018, 9, 6497–6502 RSC.
- 9-(4-Benzylphenyl)-9H-carbazole 10 (see ESI, page S7†), an analog of 2a,b with benzene ring instead of the pyridinium subunit does not possess AIE properties. It features high solution-state emission (39.9% PLQY) and reduced solid-state emission (35.5% PLQY). For photophysical properties of 10, see ESI.†.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, photophysical properties, X-ray crystallographic data (CIF files), 1H and 13C NMR data. CCDC 1873711–1873715. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ra08771g |
| This journal is © The Royal Society of Chemistry 2019 |