
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical profiling and total quality assessment of Isodon japonica using data-independent acquisition mode combined with superimposed multiple product ion UHPLC-Q-TOF-MS and chemometric analysis†
Yiran Jin‡
ab,
Tingting Tian‡a,
Yinghua Maa,
Ludan Houa,
Shuai Guana,
Wenjing Suna,
Yingfeng Du *a and
Lantong Zhang*a
*a and
Lantong Zhang*a
aDepartment of Pharmaceutical Analysis, School of Pharmacy, Hebei Medical University, 361 East Zhongshan Road, Shijiazhuang, Hebei 050017, P. R. China. E-mail: yingfengdu@hotmail.com; zhanglantong@263.net; Fax: +86-311-86266419; Tel: +86-311-86265625
bThe Second Hospital of Hebei Medical University, Shijiazhuang, Hebei 050000, P. R. China
First published on 11th January 2019
Abstract
In this paper, an analytical strategy combined data acquisition with a practical mining strategy aimed at rapid characterization and quantitation of ent-kaurane diterpenoids in Isodon japonica using ultra high-performance liquid chromatography-triple time-of-flight mass spectrometry (UPLC-Q-TOF-MS/MS). First, an effective self-built filter template based on drug phase I/II metabolic reaction theory and a components library data set were established. Second, the mass spectra of ent-kaurane diterpenoid standards were studied and their mass spectrum cleavage pathways were summarized. Next, the methanol extract of this herb was studied by data-independent acquisition mode (DIA). With the aid of a self-built filter template, the peaks of ent-kaurane diterpenoids were easily picked out and rapidly classified as ent-kaurane diterpenoids from a complex matrix. A total of 24 ent-kaurane diterpenoids were structurally identified. Meanwhile, the self-built filter template provided a convenient and fast method for the structural characterization and Isodon japonica was used to illustrate this approach for the first time. Furthermore, eight major bioactive diterpenoids were simultaneously quantified by a newly developed superimposed multiple product ion (SMPI) with UPLC-Q-TOF-MS/MS method. Principal component analysis (PCA) revealed significant differences in different batches of samples. These combined qualitative and quantitative methods were used to provide a potential approach for the holistic quality evaluation of traditional Chinese medicine (TCM) and its preparations.
1. Introduction
Isodon japonica has been a commonly used medicinal plant in most regions of China, Japan, and Korea for centuries.1 It has been recorded in authoritative literature2 and used as a folk medicine to treat hepatitis, gastritis, mastitis, tonsillitis, liver cancer and breast cancer.3 Ent-Kaurane diterpenoids have been identified as the major bioactive constituents of this plant.4 Pharmacological and clinical studies have revealed that diterpenoids possess multiple features, e.g. anti-angiogenic, anti-inflammatory, antitumor, analgesic, antitubercular, antibacterial and anti-ischemic activities.5–10 To date, most diterpenoids reported from Isodon japonica belonged to ent-kaurane diterpenoids.Generally, efficacies of TCM are based on collective effects of many components even minor or trace material. Hundreds of chemical ingredients are contained in one herbal medicine, so applying a single approach for a comprehensive quality control of TCM is very difficult. Therefore, it is important that ent-kaurane diterpenoids are used as marker components for defining the pharmacodynamic material and for reinforcing the holistic quality control of Isodon japonica.
LC-MS has been considered as a powerful and efficient means for the qualitative and quantitative determination of ingredients.11–13 Though there have been a few reports about analyzing ent-kaurane diterpenoids by LC-MS,14–16 it is difficult to simultaneously analyze many unknown and trace components in complex matrices using conservative LC-MS method. Recently, triple time-of-flight mass spectrometry (Q-TOF-MS) are widely applied for qualitative studies because they have many advantages of plentiful structure-related information, the exact mass and fragmentation patterns in a highly sensitive and convenient manner.17 Currently, the fragmentation behavior and pathways of few ent-kaurane diterpenoids has been studied by TOF-MS.18–20 However, identification and quantitation of ent-kaurane diterpenoids in Isodon japonica or other plants by UPLC-Q-TOF-MS has not been reported.
Available acquisition techniques usually include dependent acquisition (DDA) and data independent acquisition (DIA) in Q-TOF-MS.21 DDA method could automatically acquire data by massive trigger events. In recent years, DIA strategies have been developed, which brings about the acquisition of all ions present at any time in the chromatographic separation in the MS/MS spectrum.22 Meanwhile, identification of the active ingredients of TCM using a LC-MS method was primarily based on characteristic fragment ions combined with manual inspection. However, there are many drawbacks of time consuming and omission due to high background noise. In order to solve this problem, several post-acquisition data mining methods were usually to hunt the possible target compounds. Other than, drug metabolites could be locked quickly based on a well-defined and specified mass defect limits between the target drug and its metabolites. Drug metabolites are generated through the same biochemical mechanisms and thus have analogous mass defects and similar substructures as the parent drug. Similarly, the ingredients in medicinal herbs can be divided into many families, and compounds in the same core structure usually have the same skeleton or substituents.23 Therefore, an appropriate self-built template based on drug phase I/II metabolic reaction theory was established for the fast identification of the composition of the homologues in natural plant extracts by settings a consideration of the reference filter and matching substituents. It is convinced that the unrelated ions could be rapidly and automatically removed from a complex chemical background by an appropriate self-built template range. The eligible compounds were obtained automatically by filtration of complex signals through self-built template for the herbal extracts after DIA scan. Because there were the huge differences in concentration and the different types of components in TCM, it was very difficult to find a method to balance these relationships. In this study, we developed a superimposed multiple product ions (SMPI) LC-Q-TOF method, which has high quantitative resolution with TOF-MS/MS identification to eliminate interferences. According to these considerations, a combination of the above methods was developed and applied to identify and quantify the trace compounds in Isodon japonica. And the data obtained from quantitative analysis were analyzed by principal component analysis (PCA) to estimate the chemical constituents of various geographical locations, plant fractions and collection time.
2. Experiments
2.1. Reagents and materials
Methanol (HPLC grade) and purified water were obtained from TEDIA (TEDIA Scientific Co., Fairfield, USA) and Wahaha (Wahaha Group Co. Ltd, Hangzhou China), respectively.Oridonin was obtained from Zelang Co. Ltd. (Nanjing, Jiangsu, China). Hebeirubesensin K, enmenol, henryin, lasiodonin, glaucocalyxin A, ponicidin and rabdoternin A were separated from different Isodon herbs and donated by Professor Jixia Zhang from Henan Xinxiang Medical University. The contents of these ingredients were above 98% by HPLC. Generally, ent-kaurane diterpenoids are classified into two subtypes: C-20 non-oxygenated subtype (group 1) and C-20 oxygenated subtype (group 2). Group 2 can be further divided into monoepoxy ent-kauranes (group 2–1), diepoxy ent-kauranes (group 2–2) and C-20 oxygenated nonepoxy ring ent-kaurane diterpenoids (group 2–3). Among the 8 references, glaucocalyxin A (14) belonged to group 1; hebeirubescensin K (1), enmenol (4), lasiodonin (6), oridonin (13) and rabdoternin A (24) were included in group 2–1; ponicidin (3) belonged to group 2–2; and henryin (12) belonged to group 2–3.
Thirty-one batches of Isodon japonica were obtained from Chinese various locations and they were from cultivated plantations (Table 1). All plant specimens were identified and conserved in the specimen room of the Pharmacy School (Hebei Medical University).
| Sample | Source | Collecting time | Sample | Source | Collecting time |
|---|---|---|---|---|---|
| S1 | Shexian, Hebei | 2017.08 | S17 | Taihangshan, Shanxi | 2017.08 |
| S2 | Shexian, Hebei | 2017.08 | S18 | Taihangshan, Shanxi | 2017.09 |
| S3 | Shexian, Hebei | 2017.09 | S19 | Huixian, Henan | 2017.08 |
| S4 | Taihangshan, Shanxi | 2017.07 | S20 | Huixian, Henan | 2017.08 |
| S5 | Taihangshan, Shanxi | 2017.08 | S21 | Huixian, Henan | 2017.09 |
| S6 | Taihangshan, Shanxi | 2017.09 | S22 | Zunhua, Hubei | 2017.07 |
| S7 | Huixian, Henan | 2017.08 | S23 | Zunhua, Hubei | 2017.08 |
| S8 | Huixian, Henan | 2017.08 | S24 | Zunhua, Hubei | 2017.09 |
| S9 | Huixian, Henan | 2017.09 | S25 | Zunhua, Hubei | 2017.09 |
| S10 | Zunhua, Hubei | 2017.07 | S26 | Jiutai, Jilin | 2017.08 |
| S11 | Zunhua, Hubei | 2017.08 | S27 | Jiutai, Jilin | 2018.09 |
| S12 | Zunhua, Hubei | 2017.09 | S28 | Jiutai, Jilin | 2017.08 |
| S13 | Shexian, Hebei | 2017.08 | S29 | Jiutai, Jilin | 2017.09 |
| S14 | Shexian, Hebei | 2017.08 | S30 | Anshan, Liaoning | 2017.09 |
| S15 | Shexian, Hebei | 2017.09 | S31 | Anshan, Liaoning | 2017.09 |
| S16 | Taihangshan, Shanxi | 2018.07 |
2.2. Instrumentation and analytical conditions
Chromatographic separation was performed on a Shimadzu (Kyoto, Japan) UPLC instrument including a binary solvent delivery system (LC-30AD), an auto sampler (SIL-30AC), and a column oven (CTO-30A). The column system was used for a Phenomenex Kinetex C18 column (100 mm × 2.1 mm, 2.6 μm) with a column filter and the column temperature was maintained at 25 °C. The mobile phase included water (A) and methanol (B) at a 0.3 mL min−1 flow rate. The followed gradient elution program was 40–50% B from 0 to 10 min, 50–65% B from 10 to 30 min, 65–95% B from 30 to 40 min. The ratio was returned to initial ratio after holding 95% solvent B for 10 min. The injection volume was 2 μL.Mass spectrometry was executed on a Triple TOF™ 5600+ system with Duo-Spray™ ion sources operating in the negative electrospray ionization (ESI) mode. Analyst® TF 1.7 software was used to control the instrument and for data acquisition as well as analysis.
For the qualitative analysis, an MS/MS mass spectrum can be acquired from the fragmentation of a precursor ion by the DIA method i.e. SWATH techniques. The SWATH method (cycle time, 1900 ms) was composed of a TOF MS scan (accumulation time, 250 ms) and a series of product ion scans (accumulation time, 60 ms) in the high-sensitivity mode. Thirty SWATH quadrupole isolation windows were 30 Da wide ranging from 300 to 500 Da. Besides, the CE was set at −35 eV, and the CES was 20 eV in the MS/MS experiments. The nebulizer gas (gas 1), heater gas (gas 2) and curtain gas were set at 55, 55 and 35 psi, respectively. Furthermore, the MS and the MS/MS scans were automatically adjusted and calibrated using calibration delivery system (CDS) (Table 2).
| Analyte | tR (min) | MW | MS (m/z) | MS/MS (m/z) |
|---|---|---|---|---|
| a MW: Molecular weight. | ||||
| Hebeirubescensin K | 5.98 | 366.2042 | 365.2 | 347.1811 + 317.1753 |
| Enmenol | 6.07 | 366.2042 | 365.2 | 347.1864 + 317.1777 |
| Henryin | 7.23 | 392.2199 | 391.2 | 373.2045 + 331.1909 |
| Lasiodonin | 7.40 | 364.1886 | 363.2 | 345.1690 + 333.1696 |
| Oridonin | 7.66 | 364.1886 | 363.2 | 345.1695 + 333.1706 |
| Glaucocalyxin A | 7.85 | 332.1988 | 331.1 | 313.1802 + 285.1850 |
| Ponicidin | 8.43 | 362.1729 | 361.3 | 343.1555 + 297.1495 |
| Rabdoternin A | 9.81 | 364.1886 | 363.2 | 345.1714 + 255.1770 |
In subsequent quantitative analysis, the superimposed multiple product ions (SMPI) LC-Q-TOF transitions of 8 constituents in high-resolution MS data for each sample were acquired by TOF-MS mode. The collision energy (CE) was set at −35 eV, and the collision energy spread (CES) was 0 eV and accumulation time, 100 ms. The other conditions were the same as above. The transitions of 8 constituents in Isodon japonica are listed in ESI Table 2.†
2.3. Preparation of standard solution
Each of eight reference compounds was accurately weighed and dissolved in methanol to make stock solutions (0.415–1.24 mg mL−1). A series of mixed standard solution was also prepared by appropriate dilution with gradient concentrations to build a standard curves. The above solutions were kept in for further experiment.2.4. Preparation of sample solution
All dried samples were sieved through a 40 mesh filter (1 g) precisely weighed and put into a 50 mL capped conical flask. Then, the samples were extracted with 25 mL methanol in an ultrasonic ice-water bath lasting for 50 min. Methanol was added to the sample solution to achieve the original weight. The liquid supernatant solution was filtered (0.22 μm) after centrifugation (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 10 min). The filtrate was kept in a refrigerator at 4 °C before being subjected to qualitative and quantitative analysis.
000 rpm, 10 min). The filtrate was kept in a refrigerator at 4 °C before being subjected to qualitative and quantitative analysis.
2.5. Method validation for quantitative analysis
A sequence of reference solutions was used to determine the linearities of the analytes. The calibration curves were drawn by measuring standard concentration (x) and the peak areas (y) of the analytes. The limits of detection (LOD) and the limit of quantification (LOQ) were measured by injecting a range of dilute solutions (diluted by methanol) with exact concentrations. Under the condition of chromatography, LOD and LOQ were independently determined at S/N ratio of 3 and 10, respectively.Through preparing six replications from the Isodon japonica samples (S1) in one day, the intra-day precisions were obtained. The same step was operated once every day for 3 consecutive days to test inter-day precision. The repeatability was confirmed with six independent sample S1 solutions, and variation was expressed as the relative standard deviation (RSD, %).
The recovery experiment was performed using a known amount of mixed reference solutions with three different concentrations (high, middle and low) added to the known amount of sample S1 and extracted and analyzed using the above method. Triplicate samples were extracted at each concentration. The following formula was used to evaluate the average recoveries: (total detected amount − original amount)/added amount × 100%. To assess the stability of the sample, the sample S1 solutions were kept in the autosampler (4 °C) and detected at different time points (0, 2, 4, 6, 8, 12, 16, and 24 h).
2.6. Peak characterization
The method of identifying ent-kaurane diterpenoids compounds is summarized as follows. First, a self-built chemical library, including compound name, molecular formula, exact molecular weight and structure of the reported compounds based on SciFinder database (http://scifinder.cas.org) was constructed to assist the characterization. Second, the primary and secondary fragment ion and fragmentation behaviors of 8 standards were analyzed to summarize the fragmentation rules. Third, a combination of the Masterview and IDA Explore functions in Peakview 2.1 was used to detect compounds. The characteristics were analyzed by the mass spectrometry fragmentation rules and the in-house chemical library. The error of the elemental composition of the chemical composition is set to 5 ppm, and the error of the secondary fragment ion is set to 10 ppm. Then, the useful parameter ClogP, which was calculated using the program Chemdraw Ultra 14.0, was introduced to distinguish the structural isomers. The compound with the smaller ClogP value commonly has shorter retention time in a reversed phase liquid chromatography system.
2.7. Quantitative data analysis
PCA is an unsupervised pattern recognition technique used for analysis similarities and differences in numerous samples. It can classify and reduce the dimensionality of numerical datasets and can be used to compare and distinguish the quality of medicinal materials24–26 using SPSS 21.0 software (IBM SPSS, USA). The principal component (PC) score plots of the first 2 PCs were generated from the PCA results.3. Results and discussion
3.1. Data post-processing and analytical strategy
Massive data were acquired by DIA method in UPLC-Q-TOF-MS/MS, so multi-data post-processing approaches were inevitably used to analyze and screen the compounds in Isodon japonica samples. Mainly the extracted ion chromatograms (XICS) processing tools in PeakView™ 2.0 software (AB SCIEX, Foster City, CA) furnished the accurate quasi-molecular mass number, isotope pattern fit and mass errors. The errors of the predicted formula (less than ±2 ppm) were searched against the compounds reported in the Isodon genus to obtain tentative authentication. This identification was further confirmed by the probable elementary compositions of the product ions (≤10 ppm) and mentioned cracking patterns using MS/MS spectra. The detail information including retention time, formula, and fragment ions of compounds identified in Isodon japonica was presented in Table 3.| No | tR (min) | Formula | [M − H]− | Error (ppm) | Fragment ions in negative mode | Identification |
|---|---|---|---|---|---|---|
| a Compounds identified by comparison with reference standards. | ||||||
| 1a | 4.89 | C20H30O6 | 365.1965 | −1.3 | 347.1811, 335.1835, 329.1752, 317.1753, 299.1674, 285.1508, 281.1517 | Hebeirubescensin K |
| 2 | 4.97 | C20H26O5 | 345.1706 | −0.3 | 327.1608, 301.1811, 299.1645, 283.1686, 271.1369 | 7β,10α-7-Hydroxy-1,15-dioxokaur-16-en-18-oci acid |
| 3a | 5.57 | C20H26O6 | 361.1650 | −1.9 | 343.1555, 325.1455, 297.1495, 281.1545, 271.1699, 255.1385, 253.2600, 149.0603 | Ponicidin |
| 4a | 5.58 | C20H30O6 | 365.1964 | −1.6 | 347.1864, 335.1844, 329.1742, 317.1777, 299.1664, 281.1530 | Enmenol |
| 5 | 6.61 | C26H42O9 | 497.2757 | 0.3 | 497.2777, 479.2671, 335.2221, 317.2115 | (5β,8α,9β,11β,13α)-11,13,15-Trihydroxykaur-16-en-3-yl beta-D-allopyranoside. |
| 6a | 7.55 | C20H28O6 | 363.1807 | −1.8 | 345.1690, 333.1696, 327.1589, 309.1482, 315.1592, 299.1638297.1497, 281.1532 |
Lasiodonin |
| 7a | 8.90 | C22H32O7 | 407.2070 | −1.2 | 347.1860, 361.1661, 329.1725, 311.1653, 299.1650, 281.1562, 271.1695 | Sodoponin |
| 8 | 8.91 | C20H32O4 | 335.2225 | −0.7 | 317.2126, 299.2180, 289.2180, 271.2061 | Glaucocalyxin F |
| 9 | 9.44 | C20H30O5 | 349.2028 | 0.1 | 331.1919, 313.1821, 303.1968, 295.1758 | Rabdoinflexin B |
| 10 | 9.65 | C20H30O4 | 333.2065 | −1.8 | 315.1955, 297.1844, 289.2166, 287.2009, 271.2057, 149.0614 | Umbrosin A |
| 11 | 10.34 | C20H26O5 | 347.1705 | −0.9 | 327.1596, 317.1782, 309.1491, 301.1818, 283.1703 | 7α,14β -7,14-Dihydroxykaur-16-ene-3,11,15-trione |
| 12a | 10.38 | C22H32O6 | 391.2128 | 0.1 | 373.2045, 331.1909, 313.1805, 295.1679, 287.2007, 269.1897267.1789, 149.0609, 59.0162 |
Henryin |
| 13a | 10.407 | C20H28O6 | 363.1808 | −1.4 | 345.1695, 333.1706, 327.1593, 315.1600, 309.1490, 299.1646, 297.1497, 283.1697, 281.1542, 271.1688, 265.1589, 253.1570, 149.0605 | Oridonin |
| 14a | 10.41 | C20H28O4 | 331.1913 | −0.6 | 269.1906, 149.0598 | Glaucocalyxin A |
| 15 | 10.43 | C20H28O5 | 347.1864 | −0.6 | 347.1857, 329.1776, 311.1653, 301.1775, 299.1651, 285.1848, 283.1718 | 1α,7β,14β-Trihydroxy-7α,20-epoxykaur-16-en-15-one |
| 16 | 11.6 | C20H32O4 | 335.2223 | −1.6 | 317.2116, 307.2272, 289.2161, 271.2100 | 2β,7α,14β-Trihydroxykaur-15-one |
| 17 | 12.15 | C20H32O4 | 333.2068 | −1.0 | 315.1958, 305.2102, 297.1840, 287.2010, 269.1888, 271.2061 | Kamebanin |
| 18 | 13.62 | C20H28O4 | 331.1912 | −0.8 | 313.1832, 285.1862, 269.1914, 149.0616 | 7α,10α,14β-10,14,18-Trihydroxykaura-11,16-dien-15-one |
| 19 | 13.62 | C20H26O6 | 361.1653 | −1.0 | 343.1541, 325.1422, 331.1544, 315.1603, 317.1728, 299.1639, 297.1480 | Macrocalyxin E |
| 20 | 13.63 | C20H26O5 | 345.1704 | −0.8 | 345.1710, 327.1596, 315.1630, 301.1820, 299.1666, 297.1499, 283.1700, 271.1702 | 1α,6β,7α-Trihydroxy-7α,20-epoxykaur-9(11),16-dien-15-one |
| 21 | 13.64 | C22H30O6 | 389.1967 | −0.5 | 347.1852, 345.2071, 329.1752, 327.1962, 311.1642, 301.1818, 285.1858 | 3β,7α,10α14β-7,14-Dihydroxy-12,15-dioxokaur-16-en-3-yl acetate |
| 22 | 14.87 | C20H32O4 | 333.2070 | −0.3 | 315.1961, 289.2197, 287.2017, 271.2048, 257.1901, 149.0601 | Excisanin C |
| 23 | 16.48 | C20H32O4 | 333.2066 | −1.5 | 315.1959, 297.1831 | Glaucocalyxin C |
| 24a | 19.89 | C20H28O6 | 363.1810 | −0.8 | 345.1714, 327.1609, 301.1809, 283.1704, 255.1770 | Rabdoternin A |
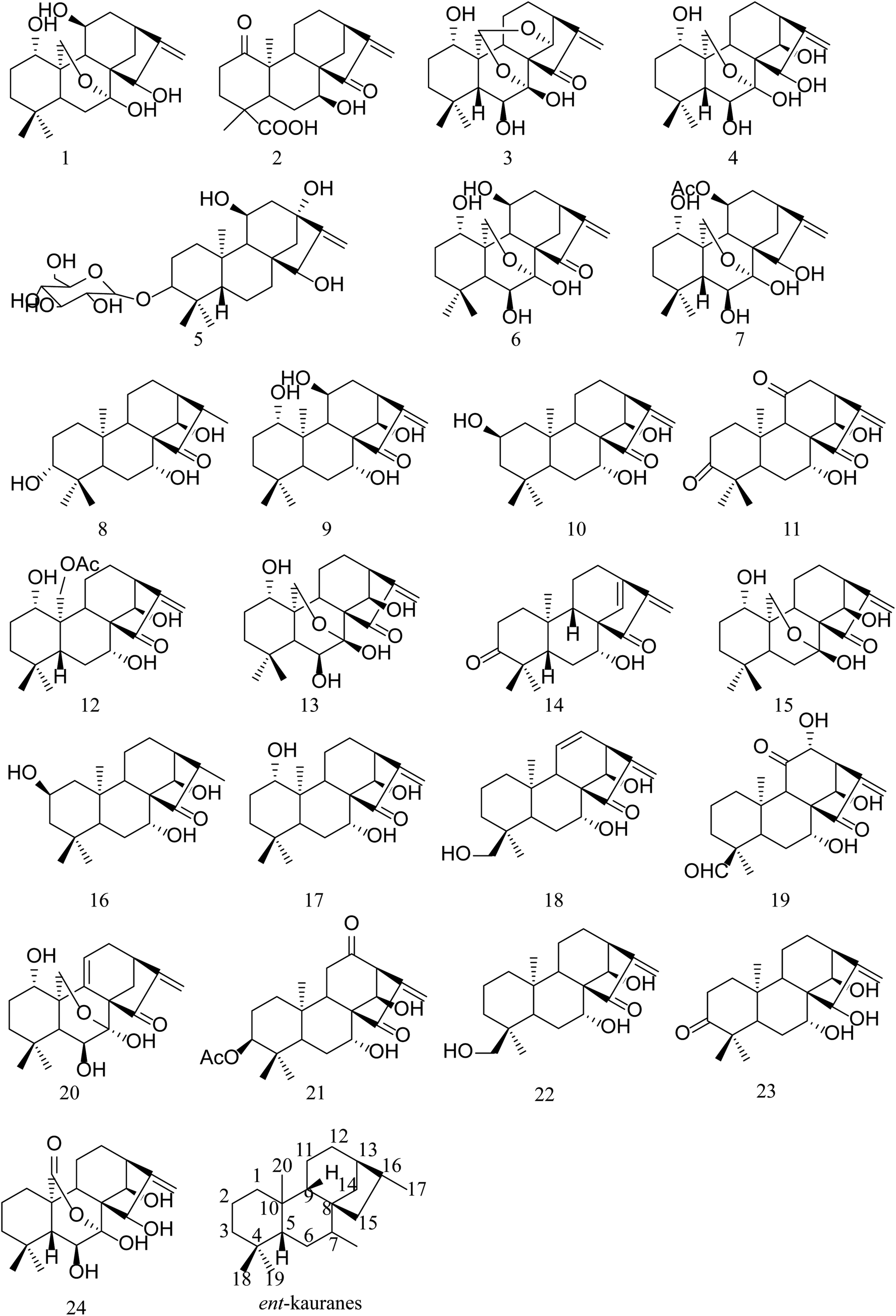 | ||
| Fig. 1 Chemical structures of 24 compounds identified in the Isodon japonica. | ||
First of all, the mass defect deviation was confirmed based on the replacement of several components according to self-built database. The filter chromatogram was set as central formula ± mass defect tolerance range. A central formula was obtained using the average molecular formula of ent-kauranes diterpenoids, and the mass defect tolerance range was equal to the half-width of the mass defect range. The concept of the self-built filter was used first to define the filter reference based on the mother nucleus. According to all the structures of the reported compounds that we summarized, the substituents of ent-kaurane diterpenoids were primarily methyls, hydroxyls, formyls, methoxyls, acetyls, glucosyls and aldehydes. By calculating the mass defects of the multifarious substituents, including desaturation moieties and methyl groups generated the maximum increase in mass defects, except for hydroxyl groups, which led to a decrease. Hence, a maximum number of one glucosyl moiety and three hydroxyls were assigned to acquire a maximum value of mass defects corresponding to C26H42O10. Four hydroxyl groups and one carbonyl group were carefully assigned to acquire the minimum value, matching with C20H26O6.27 Hence, the calculated mass defect range was from 0.1704 to 0.2766 and the filter setting was set as (C23H34O8 ± 53.1 mDa) over the mass range of 300–500 Da. The parameter of the prediction of ent-kauranes diterpenoids were set as follows: the maximum tolerance of mass error was set at 5 ppm; H–C ratio was below 3; DBE (degree of unsaturation) was defined as 5–25; elemental composition ranged from 10 to 50 for carbon; from 10 to 30 for hydrogen; from 2 to 15 for oxygen. All the molecular formulas were assumed to have no nitrogen. As soon as the filtering template was employed in the total ion chromatogram (TIC), the representative fragment ions remained visible but the asymmetrical ions could be eliminated. In the TIC of the Isodon japonica samples, a low abundance of constituent ions were masked by background ions, resulting in mistaken identification of constituent ions of interest during manual detection. To reduce possible complex interference signal ions and characterize the typical ingredients globally and rapidly, the filtering technique was employed to obtain characteristic chromatographic peaks. In Fig. 2, it is obvious that the self-built filtering technique can significantly reduce the noise level. Next, the observed ions were calculated using the formula finder and communicated with ChemSpider v.1.1 (AB SCIEX, Foster City, CA) to predict unknown chemical compounds.
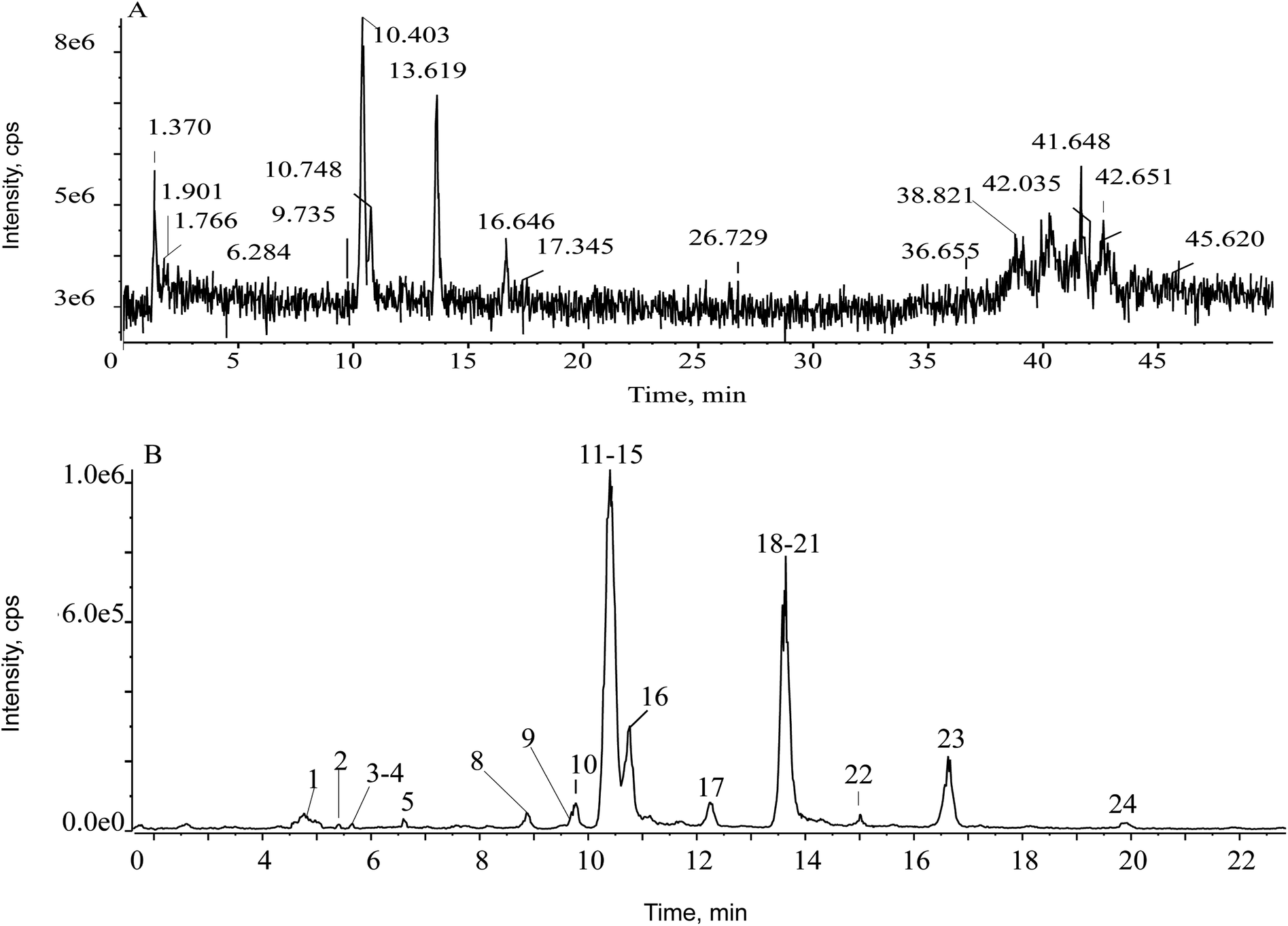 | ||
| Fig. 2 Total-ion chromatograms (TIC) of the extract of S1: (A) the unfiltered TIC; (B) the corresponding filtered chromatogram. | ||
3.2. Optimization of sample extraction method
To acquire optimized extraction ability, we compared two extraction methods including ice-water ultrasonic extraction and reflux extraction. The amounts of the major chemical components extracted by ice-water ultrasonic method were higher than these by reflux method, while supersonic extraction has the advantages of less solvent required and shorter extraction time. Thereafter, different extraction solvents, extraction solvent volume and extraction time were investigated. By comparing the sum numbers and areas of characteristic peaks in each chromatogram, the sample extraction method is defined.3.3. Quantitative analysis method development
To acquire good separation and an ideal distribution, a mixed solution of eight ent-kaurane diterpenoids standards and the crude extract (S1) were used to optimize and evaluate the chromatographic conditions. Various chromatographic columns were screened, including a Thermo Hypersll C18 column (100 mm × 2.1 mm, 3.0 μm particle size) and a Phenomenex Kinetex C18 column (100 mm × 2.1 mm, 2.6 μm particle size). The Phenomenex Kinetex C18 column showed sharper peaks and less peak tailing than the Thermo columns. Different mobile phases (acetonitrile–water, methanol–water), flow rates (0.2, 0.3, 0.4 mL min−1), and gradient programs were compared to ensure the best instrument conditions for suitable chromatographic performance for qualitative analysis. After various tests, the mobile phase was set at a flow rate of 0.3 mL min−1 to achieve the best peak resolution, better peak shapes and a stable baseline.Both negative and positive ionization modes were investigated to assess the mass spectrometric conditions. The negative mode was chosen because the ionization of ent-kaurane diterpenoids had higher sensitivity, more stable and repeatable ions, and lower background noise. So, the negative ionization mode was selected for the following experiments. Moreover, for optimization of (SMPI) conditions for quantitative analysis, the mass spectrometry cleavage patterns of investigated eight known reference standards were definitively detected in LC-Q-TOF-MS/MS.
3.4. Identification of ent-kaurane diterpenoids in Isodon japonica
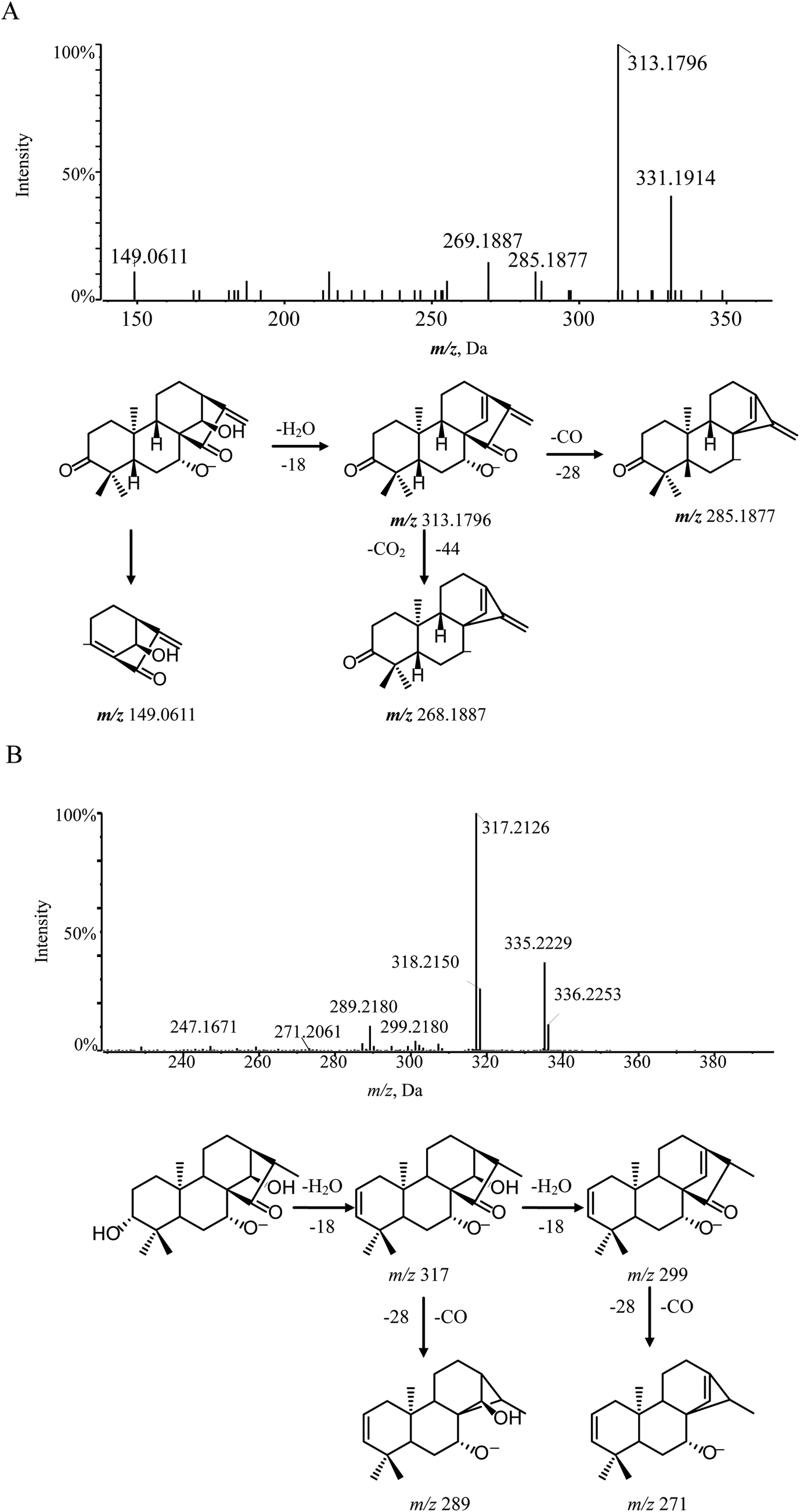 | ||
| Fig. 3 MS/MS spectra and cleavage pathways of glaucocalyxin A (A) and compound 8 (B). | ||
According to the mass fragmentation behavior of glaucocalyxin A (14), the cleavage patterns of group 1 were summarized. The distinctive ions were observed by the consecutive loss of H2O and CO. For example, compounds 8 and 16 were eluted in the chromatogram at tR 8.91 min and 11.6 min, respectively, which are isomers with the same molecular formula, C20H32O4. Compounds 8 and 16 may have similar skeletons, as their MS/MS spectra are consistent with glaucocalyxin A (14). The MS/MS spectra of compound 8 and 16 showed three distinctive ions at m/z 317 ([M − H–H2O]−) and 299 ([M − H–H2O]−), which demonstrated that the hydroxyl group was easily lost. The other daughter ions at m/z 289 and 271 were produced by the loss of CO from the fragment ions at m/z 317 and 299. Based on the above MS/MS data, compounds 8 (Fig. 3B) and 16 were tentatively assigned as glaucocalyxin F and 1α, 6β, 7α-trihydroxy-7α, 20-epoxykaur-9 (11), 16-dien-15-one. Moreover, compounds 2, 5, 9, 10, 11, 17, 18, 19, 21, 22, 23 were tentatively identified by comparison with the fragmentation patterns (Fig. S1†).
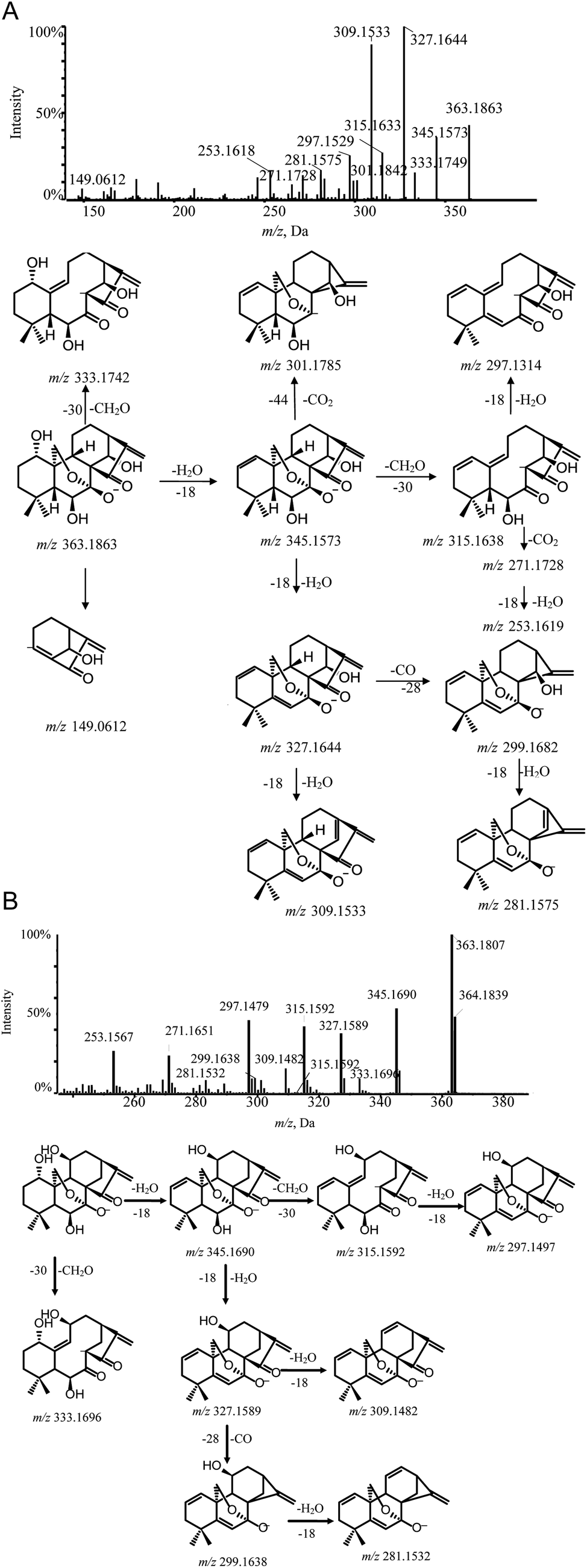 | ||
| Fig. 4 MS/MS spectra and cleavage pathways of oridonin (A) and lasiodonin (B). | ||
Lasiodonin (6) also belonged to group 2–1 and its deprotonated ion [M − H]− ion was m/z 363.1807. Lasiodonin and oridonin are a pair of isomers. They also possessed similar fragmentation pathways and a distinctive m/z 333.1696 fragment ion which indicated that a neutral fragment CH2O was lost from the deprotonated ion at m/z 363. Subsequent product ions were observed through successive losses of H2O from the deprotonated molecule ion m/z 363.1807, yielding the typical product ions at m/z 345.1690, 327.1589, 309.1482. Moreover, the [M − H–CH2O–2H2O]− (m/z 315.1680), [M − H–CO–3H2O]− (m/z 281.1532) and [M − H–CO–2H2O]− (m/z 299.1638) were also detected (Fig. 4B).
Hebeirubescensin K (1) demonstrated an precise mass of [M − H]− ion at m/z 365.1965 corresponding to the molecular formula C20H30O6. Enmenol (4) eluted in the chromatogram at retention time (tR) 5.58 min and gave a [M − H]− ion at m/z 365.1964, which showed that compounds 1 and 4 were a pair of isomers. The diagnostic ion at m/z 335 produced by the loss of a CH2O residue (30 Da) indicated the presence of the 7, 20-epoxy unit in the structure of compound 1 and compound 4. The MS/MS spectrum of compound 1 and compound 4 also contained abundant fragment ions such as [M − H–H2O]− (m/z 347), [M − H–2H2O]− (m/z 329), [M − H–CH2O–H2O]− (m/z 317), [M − H–CH2O–2H2O]− (m/z 299) and [M − H–CH2O–3H2O]− (m/z 281). The cleavage pathway of hebeirubescensin K (1) and enmenol (4) and the MS/MS spectra are shown in Fig. S2-A and S2-B,† respectively.
Rabdoternin A (24) also belonged to the 7, 20-epoxy-ent-kaurne-type diterpenoids. It produced fragment ions at m/z 345.1714 and 327.1609 via the continuous loss of H2O from the precursor ion (m/z 363.1810). Furthermore, the diagnostic product ions ([M − H–H2O–CO2]−) (m/z 301.1809) and [M − H–2H2O–CO2]− (m/z 283.1704) were yielded by the continuous cracking of the C-7–O bond and the C-20–C-10 bond to remove the neutral fragment CO2. The cleavage pathway of rabdoternin A (24) and the MS/MS spectrum of rabdoternin A are shown in Fig. S2-C.†
As above, other unknown 7, 20-epoxy-ent-kauranes, including 7, 15, and 20 were tentatively identified by comparison with the fragmentation patterns of standards (Fig. S3†).
Ponicidin (3) is a representative of group 2–2. Its MS/MS spectrum showed a remarkable deprotonated parent ion at m/z 361.1673 and subsequently lost a molecule of H2O at C-1 to form a dominant ion at m/z 343.1571. Furthermore, the characteristic fragment ion at m/z 299.1663 was observed and interpreted as the cracking of the C-7–O bond, the C-20–C-10 bond, and the C-14–O bond, accompanied by H-20 transferring to C-14 via the four-member transition state between H-20, C-20, O, and C-14 to eliminate the neutral fragment CO2. This cleavage was considered a major fragmentation pathway for C-20 diepoxy ent-kauranes. The typical product ions at m/z 271.1714 and m/z 253.1579 were detected in the MS/MS spectrum by the successive elimination of CO and H2O from the [M − H–H2O–CO2]− (m/z 299.1663) molecular ion. Moreover, other distinctive minor ions, such as [M − H–2H2O]−, [M − H–2H2O–CO]− and [M − H–2H2O–CO2]− at m/z 325.1456, m/z 297.1508 and m/z 281.1562, were also detected. The fragmentation pathway of ponicidin (3) and the MS/MS spectra are shown in Fig. 5.
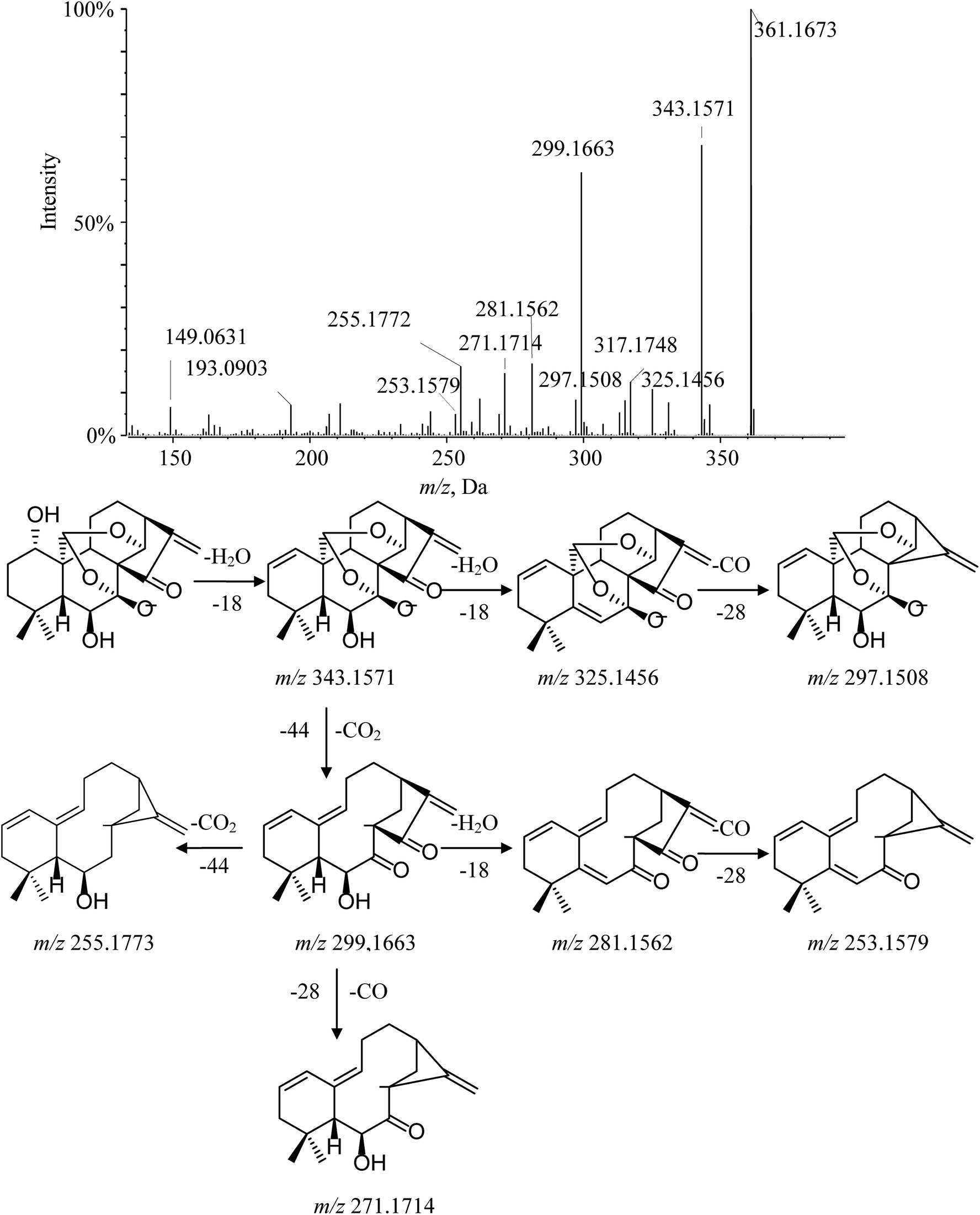 | ||
| Fig. 5 MS/MS spectrum and cleavage pathway of ponicidin. | ||
Henryin (12) is a C-20 oxygenated nonepoxy diterpenoid (group 2–3); a dominant [M − H]− (m/z 391.2134) was detected in the MS/MS spectrum. The diagnostic product ion at m/z 331.1912 was observed by the loss of 60 Da, which demonstrated that a small molecular CH3COOH was lost from the deprotonated ion at m/z 391.2134. Characteristic fragment ions 287.1996 and 269.1908 were generated by the successive losses of CO2 and H2O. Furthermore, the typical fragment ions at m/z 373.2034 ([M − H2O–H]−), 313.1817 ([M − H–H2O–AcOH]−), 295.1721 ([M − H–2H2O–AcOH]−) and 267.1799 ([M − H–2H2O–AcOH–CO]−), which have a similar cleavage behavior to the C-20 non-oxygenated ent-kauranes diterpenoid, were also observed in high-resolution mass spectrometry (HRMS) data. The fragmentation pathway of henryin (12) and the MS/MS spectra are shown in Fig. 6.
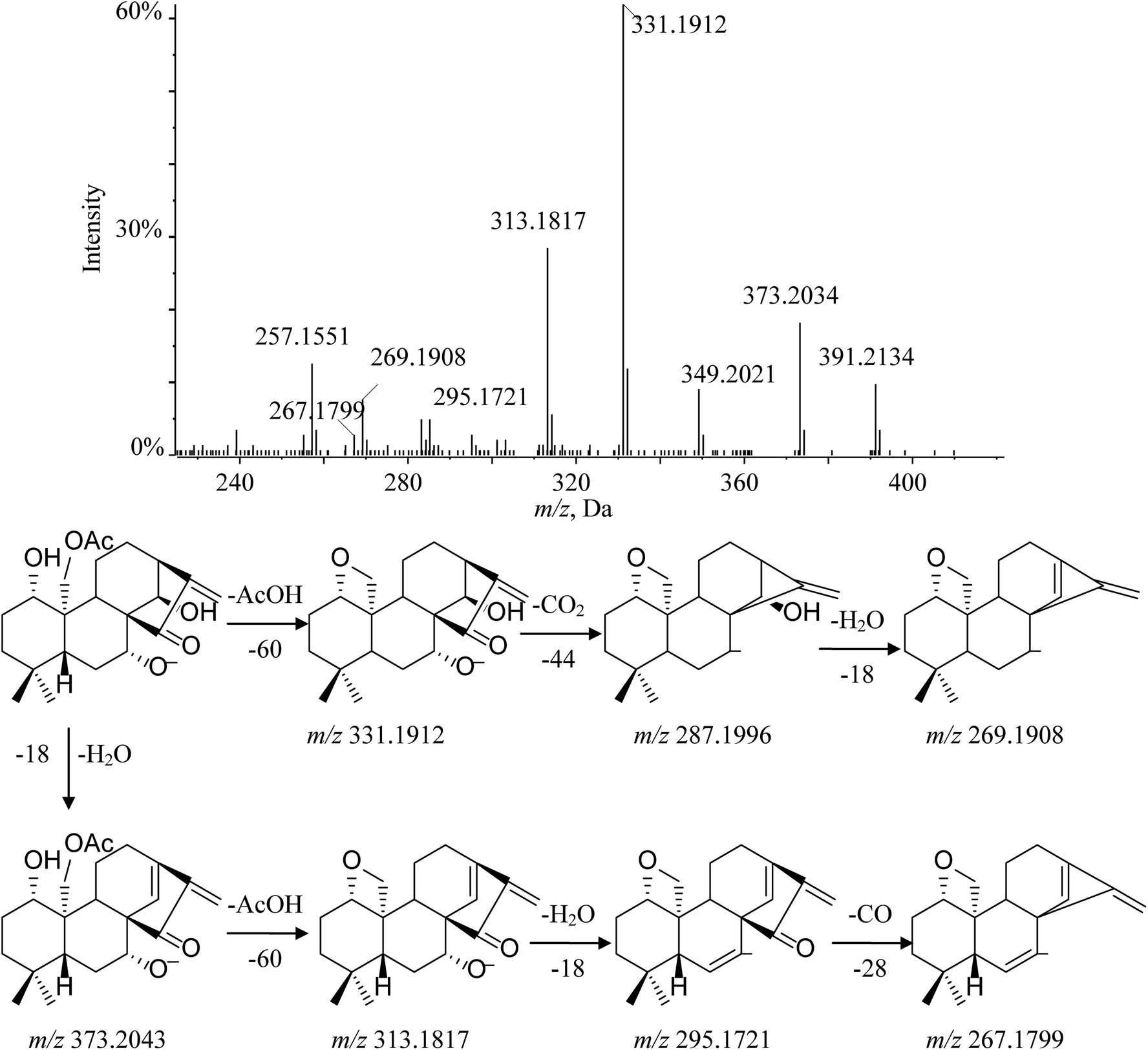 | ||
| Fig. 6 MS/MS spectrum and cleavage pathway of henryin. | ||
3.5. Quantification of eight major bioactive ent-kaurane diterpenoids and chemometric analysis
The established approach was subsequently used for analysis eight ent-kaurane diterpenoids in 31 batch samples of Isodon japonica. We performed the full validation including limit of detection, limit of quantification linearity, precision and accuracy. These detailed results are summarized in Tables 4 and 5. Up to now, there are only a few literatures on the determination active ingredients content in this herb, but they were limited in high performance liquid chromatography (HPLC) to determine of one or two diterpenoids.28,29 As known, quantitative analysis of one or several active components does not represent its quality because the curative effects of herbal medicines result from the integrated effects of a number of multiple compounds. Compared with the previous method in the literatures, the results manifested that the method was rapid, simple, specific and reliable. The quantitative analyses were carried out with the help of external standard methods. The individual and total amounts of the analytes were summarized in Table 6. The results showed successful application of this suggested method for the quantification of the major ent-kaurane diterpenoids in different Isodon japonica samples (Fig. 7). As seen in Table 6, the contents of the 8 major bioactive compounds varied to some degree. All samples examined were rich in glaucocalyxin A (14), and henryin (12) was found to be the predominant component, with a much higher content than the other compounds.| compounds | Calibration curve | r2 | Liner range (μg mL−1) | LOQ (ng mL−1) | LOD (ng mL−1) |
|---|---|---|---|---|---|
| Hebeirubescensin K | y = 2.61 × 105x + 9.41 × 105 | 0.9991 | 0.03467–3.4670 | 1.450 | 0.480 |
| Enmenol | y = 2.58 × 106 − 3.92 × 103 | 0.9992 | 0.004233–0.4233 | 1.100 | 0.550 |
| Henryin | y = 1.69 × 105 + 3.28 × 105 | 0.9985 | 0.22833–22.830 | 0.293 | 0.117 |
| Lasiodonin | y = 4.02 × 105x − 28.9 | 0.9995 | 0.0048438–0.48438 | 0.832 | 0.555 |
| Oridonin | y = 6.64 × 104x + 9.29 × 103 | 0.9999 | 0.3830–38.30 | 1.663 | 0.554 |
| Glaucocalyxin A | y = 3.26 × 105x + 4.16 × 103 | 0.9993 | 0.68–68.00 | 1.435 | 0.287 |
| Ponicidin | y = 1. 92 × 104x − 1.15 × 103 | 0.9983 | 0.06867–6.867 | 3.554 | 1.185 |
| Rabdoternin A | y = 1.02 × 106x + 1.38 × 103 | 0.9987 | 0.08125–8.125 | 0.677 | 0.271 |
| Analyte | Precision (%) | Repeatability (%) (n = 6) | Accuracy (%) | |||
|---|---|---|---|---|---|---|
| Intra-day (n = 6) | Inter-day (n = 3) | Concentrations | Recovery (n = 3) | RSD (n = 3) | ||
| Hebeirubescensin K | 3.5 | 3.0 | 1.8 | Low | 98.87 | 2.9 |
| Middle | 98.12 | 2.8 | ||||
| High | 99.35 | 3.1 | ||||
| Enmenol | 2.1 | 1.3 | 1.2 | Low | 99.43 | 3.5 |
| Middle | 99.25 | 3.1 | ||||
| High | 99.51 | 2.7 | ||||
| Henryin | 1.1 | 1.7 | 3.4 | Low | 93.45 | 1.5 |
| Middle | 94.26 | 2.4 | ||||
| High | 93.57 | 2.6 | ||||
| Lasiodonin | 2.5 | 2.3 | 4.8 | Low | 96.75 | 4.2 |
| Middle | 95.83 | 3.1 | ||||
| High | 94.62 | 2.7 | ||||
| Oridonin | 1.2 | 2.7 | 2.6 | Low | 102.52 | 5.6 |
| Middle | 95.86 | 4.1 | ||||
| High | 96.27 | 3.7 | ||||
| Glaucocalyxin A | 1.6 | 3.3 | 1.3 | Low | 101.81 | 3.2 |
| Middle | 98.54 | 2.9 | ||||
| High | 98.24 | 2.7 | ||||
| Ponicidin | 1.2 | 4.5 | 1.8 | Low | 99.25 | 2.9 |
| Middle | 98.75 | 3.4 | ||||
| High | 98.64 | 3.7 | ||||
| Rabdoternin A | 2.4 | 4.9 | 3.9 | Low | 103.32 | 2.1 |
| Middle | 101.04 | 2.6 | ||||
| High | 97.54 | 2.4 | ||||
| Samples | Analystes (mg g−1, mean) | Total | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Hebeirubescensin K | Enmenol | Henryin | Lasiodonin | Oridonin | Glaucocalyxin A | Ponicidin | Rabdoternin A | ||
| a N.D.: not detected. | |||||||||
| S1 | 4.21 | 0.76 | 30.07 | N.D. | 76.30 | 1207.58 | 150.53 | 10.61 | 1480.07 |
| S2 | 4.65 | 0.81 | 34.19 | N.D. | 77.39 | 1226.37 | 164.82 | 10.83 | 1519.06 |
| S3 | 4.14 | 0.71 | 29.76 | N.D. | 69.29 | 1210.72 | 145.12 | 10.31 | 1470.05 |
| S4 | 5.11 | 0.97 | 439.90 | N.D. | 12.01 | 1056.05 | 40.39 | 1.13 | 1555.57 |
| S5 | 5.18 | 0.93 | 440.70 | N.D. | 13.82 | 1079.74 | 44.83 | 1.22 | 1586.43 |
| S6 | 4.91 | 0.77 | 435.20 | N.D. | 10.27 | 952.91 | 39.42 | 1.05 | 1444.53 |
| S7 | 1.64 | 0.33 | 12.95 | 0.41 | N.D. | 550.73 | 2.39 | N.D. | 568.46 |
| S8 | 1.56 | 0.34 | 11.91 | 0.51 | N.D. | 593.70 | 2.95 | N.D. | 610.97 |
| S9 | 1.44 | 0.29 | 9.07 | 0.37 | N.D. | 427.30 | 2.23 | N.D. | 440.71 |
| S10 | 1.82 | 0.25 | 327.90 | 0.29 | N.D. | 796.056 | 2.29 | N.D. | 1128.61 |
| S11 | 1.83 | 0.26 | 394.50 | 0.32 | N.D. | 857.89 | 3.02 | N.D. | 1257.82 |
| S12 | 1.58 | 0.23 | 321.20 | 0.28 | N.D. | 762.05 | 1.88 | N.D. | 1087.22 |
| S13 | 2.01 | 0.43 | 50.62 | N.D. | 16.86 | 1086.41 | 35.41 | 1.40 | 1193.15 |
| S14 | 2.13 | 0.45 | 52.88 | N.D. | 17.89 | 1104.59 | 37.73 | 1.45 | 1217.11 |
| S15 | 1.91 | 0.42 | 47.97 | N.D. | 16.61 | 1084.80 | 34.92 | 1.39 | 1188.02 |
| S16 | 2.74 | 0.47 | 172.10 | N.D. | 3.71 | 934.80 | 18.96 | 0.66 | 1133.44 |
| S17 | 2.47 | 0.49 | 186.10 | N.D. | 3.66 | 1019.16 | 23.28 | 0.76 | 1235.91 |
| S18 | 1.64 | 0.45 | 155.90 | N.D. | 1.89 | 924.99 | 17.99 | 0.64 | 1103.50 |
| S19 | 1.73 | 0.32 | 184.10 | 0.31 | N.D. | 854.21 | 2.33 | N.D. | 1042.99 |
| S20 | 1.70 | 0.31 | 188.20 | 0.31 | N.D. | 839.94 | 2.77 | N.D. | 1033.23 |
| S21 | 1.83 | 0.30 | 155.70 | 0.27 | N.D. | 799.23 | 1.95 | N.D. | 959.28 |
| S22 | 1.64 | 0.25 | 120.60 | 0.12 | N.D. | 460.08 | 1.80 | N.D. | 584.49 |
| S23 | 1.33 | 0.23 | 178.10 | 0.14 | N.D. | 716.26 | 1.89 | N.D. | 897.97 |
| S24 | 1.35 | 0.26 | 179.90 | 0.11 | N.D. | 738.20 | 2.27 | N.D. | 922.09 |
| S25 | 1.23 | 0.22 | 171.30 | 0.08 | N.D. | 593.72 | 1.83 | N.D. | 768.39 |
| S26 | N.D. | N.D. | 110.75 | N.D. | N.D. | 817.24 | N.D. | N.D. | 927.99 |
| S27 | N.D. | N.D. | 11.65 | N.D. | N.D. | 856.27 | N.D. | N.D. | 867.92 |
| S28 | N.D. | 0.04 | 204.39 | 0.13 | N.D. | 631.68 | 6.86 | N.D. | 843.095 |
| S29 | N.D. | 0.34 | 32.46 | 0.01 | N.D. | 745.26 | 12.42 | N.D. | 790.49 |
| S30 | N.D. | 0.04 | 191.81 | N.D. | N.D. | 871.30 | 4.37 | 0.13 | 1067.65 |
| S31 | N.D. | 0.39 | 85.76 | N.D. | N.D. | 776.50 | 3.27 | 0.07 | 865.99 |
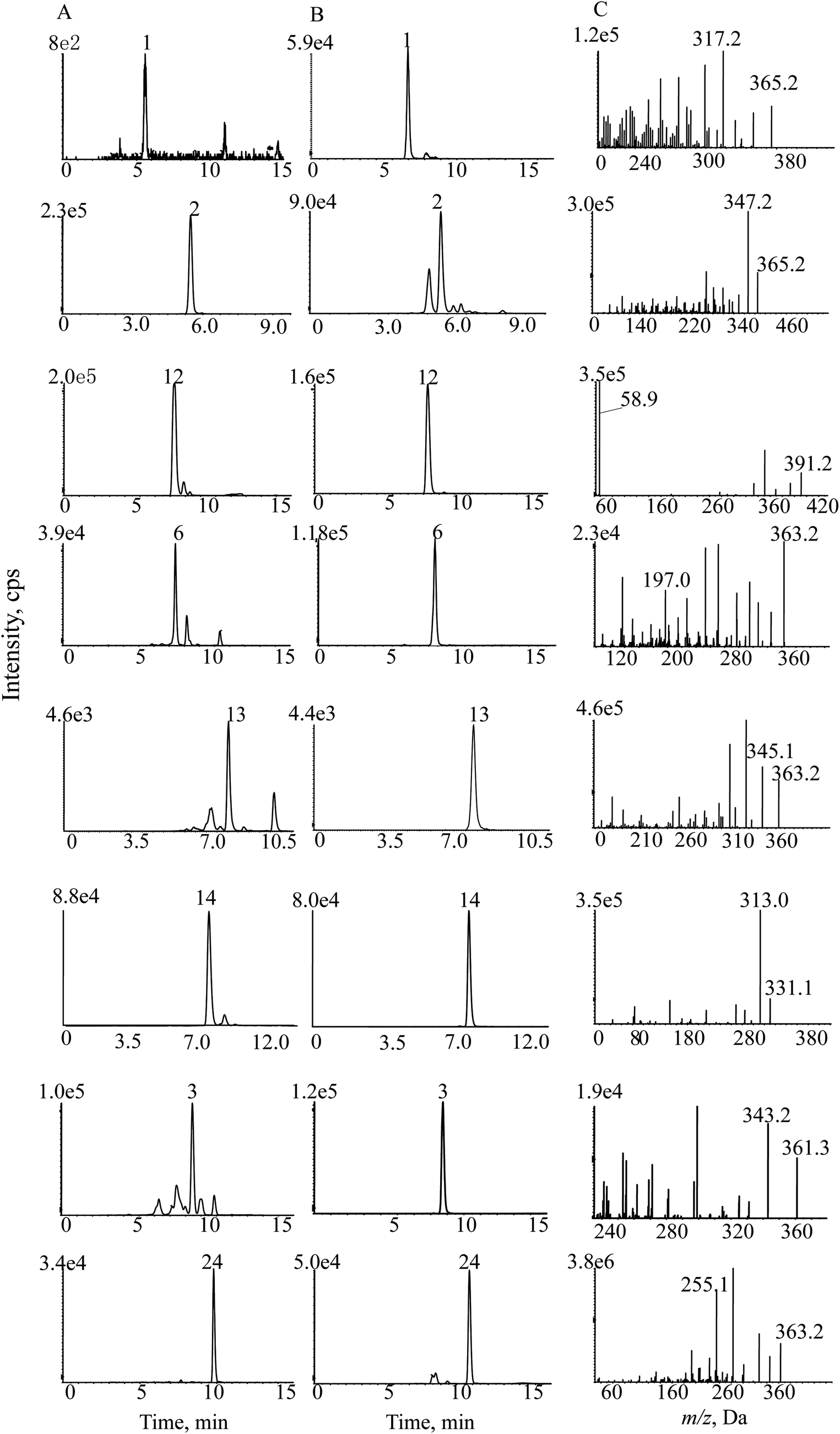 | ||
| Fig. 7 SMPI chromatograms of Isodon japonica. Samples (A), reference substances (B) and (C) SMPI transitions of 8 standards. | ||
All quantitative data were analyzed using PCA. The contents of eight diterpenoids were set as variables, and different batches of samples were set as observations. The first two principal components (PC1 and PC2), with more than 80.83% of the total variance, were extracted for analysis. PC1 accounted for 61.38% of the variance and PC2 accounted for 19.45% of the variance. As shown in Table S1,† PC1 included strong contributions from ponicidin, oridonin and rabdoternin A (3, 13 and 24); PC2 was directly correlated with henryin (12). A two-component PCA score scatter plot (Fig. 8) exhibited that samples were clearly divided into two clusters: one for samples originating from north China, including samples S1–S6 and S13–S18 in domain A; and the other for samples originating from central China and northeast China, including samples S7–S12 and S19–S31 in domain B, which indicated that PC1 (ponicidin, oridonin and rabdoternin A) was important for discrimination. Therefore, the total amount of tested diterpenoids was distinctively different between various areas; samples from Hebei and Shanxi belong to north China, Hubei belongs to central China and Jilin and Liaoning Provinces to northeast China. The content of total diterpenoids in samples from north China was higher than for herbs in central China and northeast China. The results indicate that place of origin has an impact on product quality, which suggests that the quality of this herb could be assured if locality is standardized. Therefore, we developed a generally effective and applicable approach to analyze ent-kaurane diterpenoids in Isodon japonica samples based on the self-built method. More importantly, this strategy could be extensively used to identify the bioactive structural analogues from other herbal preparations.
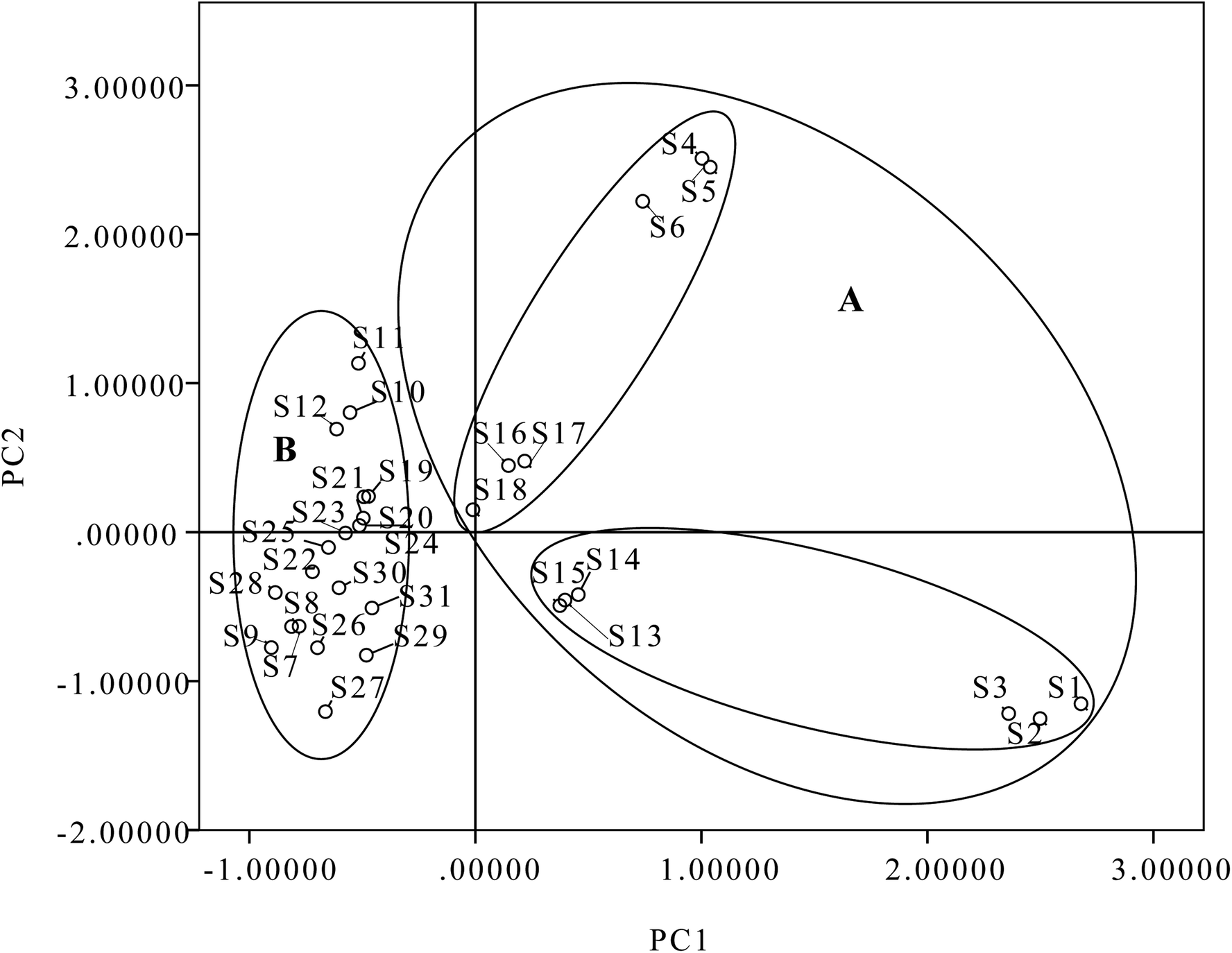 | ||
| Fig. 8 Scatter plot obtained by PCA of 31 batches of samples on the first two principal components: (A) north China; (B) central China and northeast China. | ||
4. Conclusion
A major challenge in TCM analysis is to ensure rapid and accurate holistic quality control. In the present study, a practical and effective self-built filtration method was developed to identify trace ent-kaurane diterpenoids in Isodon japonica extract for the first time. This study has demonstrated the unprecedented advantage in identification for a certain class of components in TCM using this filtration technology. Moreover, all samples were quantified in the SMPI mode by HPLC-MS/MS, and PCA analysis was performed for discrimination based on the contents of the eight major components. According to the qualitative and quantitative analysis results, the chemical profiles of all samples demonstrated a high level of similarity but there were differences in contents of ent-kaurane diterpenoids from different sources. The proposed methods may play an important role in overall quality control of medicinal herbs and will be helpful in further pharmacological studies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Our work received several financial supports including the National Natural Science Foundation of China (No. 81402894), the Hebei Education Department Key Project (No. ZD2017244) and the Hebei Province Food and Drug Administration Projects (No. ZD2015021, No. ZD2015029).References
- T. T. Cui, L. J. Zhao, Y. Liu and W. Jiang, Toxicological studies on Isodon japonica var. glaucocalyx, Guide. China. Med., 2010, vol. 8, pp. 246–249 Search PubMed.
- State Administration of Traditional Chinese Medicine, Chinese materia medica, Shanghai Scientific and Technical Publishers, Shanghai, 1999, p. 6166. Chinese Search PubMed.
- X. D. Shen, B. Wang, C. Y. Liu and J. Zhang, Study of the chemical ingredients of Isodon japonica var. glaucocalyx, Chin. Tradit. Herb. Drugs, 2009, 40, 1883–1885 CAS.
- Z. M. Jin, W. Sha and X. Y. Hu, Study on bacteriostasis of the extracts of Isodon japonica (Burm. f.) Hara var. glaucocalyx (Maxim.) Hara, Guangxi Sci., 2007, 14, 160–162 CAS.
- Y. R. Jin, Y. F. Du, X. W. Shi and P. W. Liu, Simultaneous quantification of 19 diterpenoids in Isodon amethystoides by high-performance liquid chromatography-electrospray ionization tandem mass spectrometry, J. Pharm. Biomed. Anal., 2010, 53, 403–411 CrossRef CAS.
- W. H. Huang, Y. Y. Liang, J. J. Wang, G. Q. Li, G. C. Wang, Y. L. Li and H. Y. Chung, Anti-angiogenic activity and mechanism of kaurane diterpenoids from Wedelia Chinensis, Phytomedicine, 2016, 23, 283–292 CrossRef CAS PubMed.
- L. Yang, Y. B. Zhang, L. F. Chen, N. H. Chen, Z. N. Wu, S. Q. Jiang, L. Jiang, G. Q. Li, Y. L. Li and G. C. Wang, New labdane diterpenoids from Croton laui and their anti-inflammatory activities, Bioorg. Med. Chem. Lett., 2016, 26, 4687–4691 CrossRef CAS.
- C. S. Niu, Y. Li, S. G. Ma, L. Li, J. Qu and S. S. Yu, Analgesic diterpenoids from the twids of Pieris Formosa, Tetrahedron, 2016, 72, 44–49 CrossRef CAS.
- Z. G. Liu, Z. L. Li, D. H. Li, N. Li, J. Bai, F. Zhao, D. L. Meng and H. M. Hua, ent-Abietane-type diterpenoids from the roots of Euphorbia ebracteolata with their inhibitory activities on LPS-induced NO production in RAW 264.7 macrophages, Bioorg. Med. Chem. Lett., 2016, 26, 1–5 CrossRef CAS PubMed.
- N. Tiwari, J. Thakur, D. Saikia and M. M. Gupta, Antitubercular diterpenoids from Vitex trifolia, Phytomedicine, 2013, 20, 605–610 CrossRef CAS PubMed.
- J. Li, Y. H. Wang, J. Troy, T. J. Smillie and I. A. Khan, Identification of phenolic compounds from Scutellaria lateriflora by liquid chromatography with ultravioletphotodiode array and electrospray ionization tandem mass spectrometry, J. Pharm. Biomed. Anal., 2012, 63, 120–127 CrossRef CAS PubMed.
- S. Song, X. P. Zheng, W. D. Liu, R. F. Du, Z. M. Feng, P. C. Zhang and L. F. Bi, Rapid identification of unstable acyl glucoside flavonoids of Oxytropis racemosa Turcz by high-performance liquid chromatography-diode arraydetection-electrospray ionisation/multi-stage mass spectrometry, Phytochem. Anal., 2013, 24, 155–161 CrossRef CAS.
- J. Yang, X. D. Wen, B. X. Jia, Q. Mao, Q. Wang and M. X. Lai, Quality evaluation of Potentilla discolor by high performance liquid chromatography coupled with diode array detection and electrospray ionisation tandem mass spectrometry, Phytochem. Anal., 2011, 22, 547–554 CrossRef CAS.
- X. J. Yin, X. Y. Huang, H. Peng and J. J. Chen, LC-MS guided isolation of ent-kaurane diterpenoids from Nouelia insignis, Fitoterapia, 2016, 111, 42–48 CrossRef PubMed.
- L. Z. Lin, Q. Gao, C. Cui, H. F. Zhao, L. W. Fu, L. M. Chen, B. Yang, W. Luo and M. M. Zhao, Isolation and identification of ent-kaurane-type diterpenoids from Rabdosia serra (MAXIM.) HARA leaf and their inhibitory activities against HepG-2, MCF-7, and HL-60 cell lines, Food Chem., 2012, 131, 1009–1014 CrossRef CAS.
- X. R. Hou, L. L. Wan, Z. J. Zhan, C. P. Li and W. G. Shan, Analysis and determination of deterpenoids in unprocessed and processed Euphorbia lathyris seeds by HPLC-ESI-MS, J. Pharm. Anal., 2011, 1, 197–202 CrossRef CAS PubMed.
- Y. Zhou, X. Liu, J. Yang, Q. B. Han, J. Z. Song, S. L. Li, C. F. Qiao, L. S. Ding and H. X. Xu, Analysis of caged xanthones from the resin of Garcinia hanburyi using ultra-performance liquid chromatography/electrospray ionization quadrupole time-of-flight tandem mass spectrometry, Anal. Chim. Acta, 2008, 629, 104–118 CrossRef CAS.
- T. T. Tian, Y. R. Jin, Y. H. Ma, W. W. Xie, H. J. Xu, K. R. Zhang, L. T. Zhang and Y. F. Du, Identification of metabolites of oridonin in rats with a single run on UPLC-Triple-TOF-MS/MS system based on multiple mass defect filter data acquisition and multiple data processing techniques, J. Chromatogr. B, 2015, 1006, 80–92 CrossRef CAS PubMed.
- Y. Yang, X. J. Yin, H. M. Guo, R. L. Wang, R. Song, Y. Tian and Z. J. Zhang, Identification and comparative analysis of the major chemical constituents in the extracts of single Fuzi herb and Fuzi-Gan cao herb-pair by UFLC-IT-TOF/MS, Chin. J. Nat. Med., 2014, 12, 0542–0553 CAS.
- Y. Zhou, G. Xu, F. F. K. Choi, L. S. Ping, Q. B. Han, J. Z. Song, C. F. Qiao, Q. S. Zhao and H. X. Xu, Qualitative and quantitative analysis of diterpenoids in Salvia species by liquid chromatography coupled with electrospray ionization quadrupole time-of-flight tandem mass spectrometry, J. Chromatogr. A, 2009, 1216, 4847–4858 CrossRef CAS.
- Y. P. Sun, L. Li, M. Liao, M. Su, C. C. Wan, L. T. Zhang and H. L. Zhang, A systematic data acquisition and mining strategy for chemical profiling of Aster tataricus rhizoma (Ziwan) by UHPLC-Q-TOF-MS and the corresponding anti-depressive activity screening, J. Pharm. Biomed. Anal., 2018, 154, 216–226 CrossRef CAS PubMed.
- W. W. Xie, Y. R. Jin, L. D. Hou, Y. H. Ma, H. J. Xu, K. R. Zhang, L. T. Zhang and Y. F. Du, A practical strategy for the characterization of ponicidin metabolites in vivo and in vitro by UHPLC-Q-TOF-MS based on nontargeted SWATH data acquisition, J. Pharm. Biomed. Anal., 2017, 145, 865–878 CrossRef CAS PubMed.
- T. Xie, Y. Liang, H. P. Hao, J. A, L. Xie, P. Gong, C. Dai, L. S. Liu, A. Kang, X. Zheng and G. J. Wang, Rapid identification of ophiopogonins and ophiopogonones in Ophiopogon japonicas extract with a practical technique of mass defect filtering based on high resolution mass spectrometry, J. Chromatogr. A, 2012, 1227, 234–244 CrossRef CAS.
- F. Zhou, J. Y. Peng, Y. J. Zhao, W. S. Huang, Y. R. Jiang, M. Q. Li, X. D. Wu and B. Y. Lu, Varietal classification and antioxidant activity prediction of Osmanthus fragrans Lour. Flowers using UPLC-PDA/QTOF-MS and multivariable analysis, Food Chem., 2017, 217, 490–497 CrossRef CAS PubMed.
- F. X. Zhang, M. Li, L. R. Qiao, Z. H. Yao, C. Li, X. Y. Shen, Y. Wang, K. Yu, X. S. Yao and Y. Dai, Rapid characterization of Ziziphi Spinosae Semen by UPLC/Qtof MS with novel informatics platform and its application in evaluation of two seeds from Ziziphus species, J. Pharm. Biomed. Anal., 2016, 122, 59–80 CrossRef CAS PubMed.
- L. Guo, S. L. Zeng, Y. Zhang, P. Li and E. H. Liu, Comparative analysis of steroidal saponins in four Dioscoreae herbs by high performance liquid chromatography coupled with mass spectrometry, J. Pharm. Biomed. Anal., 2016, 117, 91–98 CrossRef CAS PubMed.
- Z. B. Xiang, X. Y. Liu and X. H. Li, Chemical constituents of Isodon Japonica var. glaucocalyx, Pharm. Chem. J., 2015, 48, 804–814 CrossRef CAS.
- X. D. He, Z. J. Zhang, Z. A. He and J. X. Zhang, Simultaneous determination of glaucocalyxin A and glaucocalyxin B in Isodon japonica by HPLC, Phys. Test. Chem. Anal., Part B., 2012, 48, 1395–1397 CAS.
- Y. T. Zhang, D. X. Sha, M. Sha and C. L. Yuan, Determination of glaucocalyxin A in the leaves of plectranthus japonicus (Burm.) kojdz. var. glaucocalyx (Maxim.) koidz. by HPLC, China J. Chin. Mater. Med., 1991, 16, 679–680 CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08732f |
| ‡ The authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |