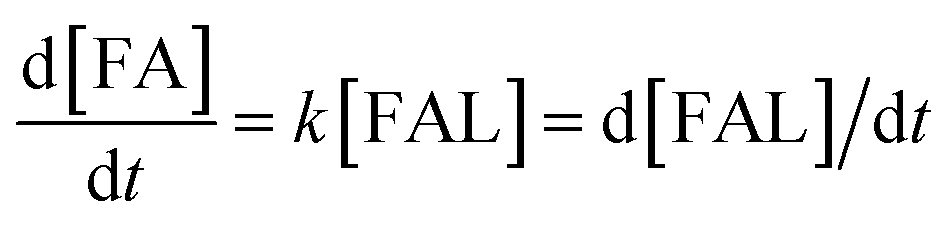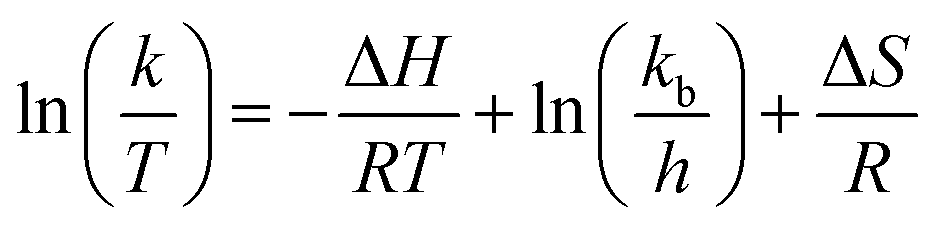DOI:
10.1039/C8RA08616H
(Paper)
RSC Adv., 2019,
9, 3063-3071
Low-temperature catalytic hydrogenation of bio-based furfural and relevant aldehydes using cesium carbonate and hydrosiloxane†
Received
17th October 2018
, Accepted 10th January 2019
First published on 24th January 2019
Abstract
Selective hydrogenation of unsaturated compounds is mainly carried out by using high-pressure hydrogen in the presence of a precious or transition metal catalyst. Here, we describe a benign approach to efficiently catalyze the hydrogenation of furfural (FUR) to furfuryl alcohol (FFA) over commercially available cesium carbonate using nontoxic and cheap polymethylhydrosiloxane (PMHS) as hydrogen source. Good to excellent FFA yields (≥90%) could be obtained at 25–80 °C by appropriate control of the catalyst dosage, reaction time, and the hydride amount. FUR-to-FFA hydrogenation was clarified to follow a pseudo-first order kinetics with low apparent activation energy of 20.6 kJ mol−1. Mechanistic insights manifested that PMHS was redistributed to H3SiMe, which acted as the active silane for the hydrogenation reactions. Importantly, this catalytic system was able to selectively reduce a wide range of aromatic aldehydes to the corresponding alcohols in good yields of 81–99% at 25–80 °C in 2–6 h.
1. Introduction
With dwindling fossil resources, the increasing demand for energy forced the use of renewable resources as alternatives.1 Biomass is the most abundant organic carbon resource that is widely used in the production of biofuels and various platform chemicals.2 Among the value-added molecules, furfural (FUR) is able to be obtained from hemicellulose by acid-catalyzed cascade hydrolysis and dehydration, which can be further converted to a variety of chemicals and liquid fuels,3 such as 2-methylfuran, 2-methyltetrahydrofuran, furfuryl alcohol (FFA), ethyl levulinate, and γ-valerolactone, through different chemical processes involving reduction as the key step.3,4 FFA is particularly attractive because of its many applications in the polymer and fine chemical industries. In addition to the corrosion and solvent resistance, the resulting resin from FFA also has excellent chemical and thermal properties.5 Moreover, FFA can be used to make furan reinforced plastics due to its corrosion resistance,6 which is also an essential intermediate for the manufacture of vitamin C, dispersants, lysine and lubricants.7
In the industry, copper chromite catalysts have been used to hydrogenate FUR to FFA for decades.8 The main disadvantage of this catalytic system is the toxicity of chromium oxides.9 Thus, a number of research interests have been drawn to design eco-friendly catalysts for the conversion of FUR to FFA with high activity and selectivity.10 Precious (e.g., Pd, Pt)11 and transition metals (e.g., Cu, Ni)12 have been explored to be efficient for catalytic hydrogenation of FUR to FFA, although they are either expensive or complicated to prepare, and the use of high-pressure hydrogen gas typically requires a series of pressure-resistant instruments.13 On the other hand, liquid hydrogen sources such as formic acid or alcohols (e.g., isopropanol) have received much attention in recent years.14 However, the use of formic acid as hydrogen donor may lead to the corrosion of the experimental equipment, and the alcohol can further react with the FFA by etherification to reduce the selectivity of FFA.15 Current research efforts involve the development of alternative catalytic systems that do not contain toxic species to avoid the contamination of products.
Hydrosilylation of carbonyl compounds to alcohols is one of important methods that have been widely used in academia and industry.16 This type of conversion process is very attractive because the used hydrosilanes are air- and water-stable hydride source that can be activated under mild conditions by certain catalysts or activators to form silyl ethers with carbonyl groups.17 The resulting silyl ethers can be easily converted to alcohol by additional hydrolysis.18 Among commercially available hydrosilanes, polymethylhydrosiloxane (PMHS) is a very attractive reagent of choice because it is inexpensive, commercially available, non-toxic, biodegradable, and stable to air and moisture.19 In addition, PMHS is of low viscosity and soluble in most organic solvents.20
Herein, selective hydrogenation of FUR to FFA was achieved over a low-cost Cs2CO3 using PMHS as hydride in an operationally simple manner. This catalytic strategy is more practical since Cs2CO3 has unique properties, such as ease to handle, low hygroscopicity, and high compatibility with organic solvents.21 The effects of different experimental parameters, like the type of catalyst and solvent, reaction temperature, reaction time, catalyst dosage, and the hydride amount on the catalytic performance were studied. In addition, the reaction kinetics and mechanism were also investigated.
2. Experimental
2.1 Materials
Furfuryl alcohol, pyridine-2-carboxaldehyde (99%), 4-chlorobenzaldehyde (99%), 2-nitrobenzaldehyde (99%), methanol (99%), 5-methylfurfural (99%), 2-methylthiazole-5-carbaldehyde (99%), salicylaldehyde (99%), 1-naphthaldehyde (99%), furfural, phenylsilane (97%), polymethylhydrosiloxane (PMHS, 99%), triethoxysilane (97%), diphenylsilane (97%), deuterated dimethyl sulfoxide (DMSO-d6, 99.8 atom% D), 1,4-dioxane (99%), potassium carbonate (K2CO3, 99%), cesium carbonate (Cs2CO3, 99%), naphthalene (99%), methanol (MeOH, 99%), triethylsilane (99%), trimethoxysilane (99%) and 1,1,3,3-tetramethyldisiloxane (97%) were purchased from Beijing Innochem Technology Co., Ltd. Tetrahydrofuran (THF, 99%)and 1,1,1,3,5,5,5-heptamethyltrisiloxane (98%) were purchased from TCI (Shanghai) Development Co., Ltd. Lithium carbonate (Li2CO3, 99%), calcium carbonate (CaCO3, 98%), cesium sulfate (Cs2SO4, 98%) and sodium carbonate (Na2CO3, 99%) were purchased from Shanghai Aladdin Industrial Inc. Cesium bicarbonate (CsHCO3, 99%), cesium chloride (CsCl, 98%), acetonitrile (MeCN, 99%), 5-methyltetrahydrofuran (MTHF, 99%), dichloromethane (DCM, 99%), dimethyl sulfoxide (DMSO, 99%) and N,N-dimethylformamide (DMF, 99%) were purchased from Sigma-Aldrich Co. LLC. Other used reagents were of analytical grade without further purification, unless stated otherwise.
2.2 Procedures for catalytic hydrogenation of FUR to FFA
All the reactions were carried out in 15 mL Ace pressure tubes. In a typical procedure, 0.5 mmol FUR, 0.05 mmol Cs2CO3 (16 mg), 2.0 mL DMF and 0.1 g PMHS were added into the tube and sealed. Then, the mixture was transferred into an oil-bath and magnetically stirred at 400 rpm for a specific reaction time. The zero time was established as the tube was placed into the oil bath preheated to 25–80 °C. After the reaction was completed and cooled down to room temperature, 3 mL methanol was added into the reaction system, followed by stirring under ambient conditions for 30 min for alcoholysis of siloxane intermediate. The reaction solution was filtered to remove solid particles, followed by quantitative analysis with gas chromatography (GC) and gas chromatography-mass spectrometry (GC-MS).
2.3 Analytical method
The FFA yield and FUR conversion were quantitatively analyzed by GC (Agilent 7890B) with a HP-5 column (30 m × 0.320 mm × 0.25 μm) and a flame ionization detector. Naphthalene (10 mg) was utilized as internal standard by referring to the standard curves (with R2 > 0.9998) made from commercial samples. Liquid products were identified by GC-MS (Agilent 6890N GC/5973 MS, Santa Clara, CA). The substrate conversion rate (C, mol%) and product yield (Y, mol%) were calculated by following below equations:
2.4 Catalyst recycling study
After each cycle of reactions, the remaining catalyst was separated by centrifugation, washing with DMF and acetone for 4 times, respectively, followed by drying at 80 °C in the atmosphere of N2 for 8 h, which was then directly used for the next run. STEM (scanning transmission electron microscopy) test of the recovered Cs2CO3 was measured by JEOL 2100 TEM/STEM.
3. Results and discussion
3.1 Effect of different catalysts
Initially, different catalysts were screened for hydrogenation of FUR to FFA using PMHS as H-donor at room temperature in DMF. Cs2CO3 was found to show the highest activity, being capable of converting FUR to FFA with 45.5% yield, 55.0% conversion and TOF of 5.6 h−1 at room temperature for 1 h (Table 1, entry 1). In contrast, nearly no reaction took place by using Li2CO3, Cs2SO4, CsCl or CaCO3 as catalyst (entries 2–5). When considering the influence of cations, Cs2CO3, Na2CO3 and K2CO3 were more active in the reaction than other carbonate bases (entries 1, 6 and 7), indicating that the size of the cation played an important role in the reaction. With the increase of the radius of the cation, the ionic property of the salt increased, thus with enhanced solubility.20b Among the ions of alkali and alkaline earth metals, Cs+ has the largest ionic radius and the weakest polarization, which allows the paired carbonate ion to be more freely available to interact with the hydrosilane. In addition to the cation, the effect of the anion on the reaction is not negligible, mainly related to its basicity that has a positive correlation with nucleophilic ability. In this regard, compared with chloridion (entry 4), carbonate with relatively higher nucleophilic ability is more prone to attack PMHS, thus activating the silane to release hydride.20b In view of the superior FFA yield, Cs2CO3 was selected as the optimal catalyst for the reaction.
Table 1 The conversion of FUR to FFA over difference catalystsa
| Entry |
Cat. |
Yield (%) |
Conversion (%) |
TOFb (h−1) |
| Reaction condition: 0.5 mmol FUR, 16 mg catalyst, PMHS (1.47 mmol H−), 2 mL DMF, 25 °C, and 1 h. Turnover frequency (TOF) was evaluated on the basis of (mole of FUR)/(mole of the catalyst × reaction time). |
| 1 |
Cs2CO3 |
45.5 |
55.0 |
5.6 |
| 2 |
Li2CO3 |
0 |
0 |
0 |
| 3 |
Cs2SO4 |
0 |
0 |
0 |
| 4 |
CsCl |
0 |
0 |
0 |
| 5 |
CaCO3 |
0 |
0 |
0 |
| 6 |
K2CO3 |
29.5 |
32.4 |
1.4 |
| 7 |
Na2CO3 |
18.5 |
22.4 |
0.7 |
3.2 Effect of reaction temperature and time
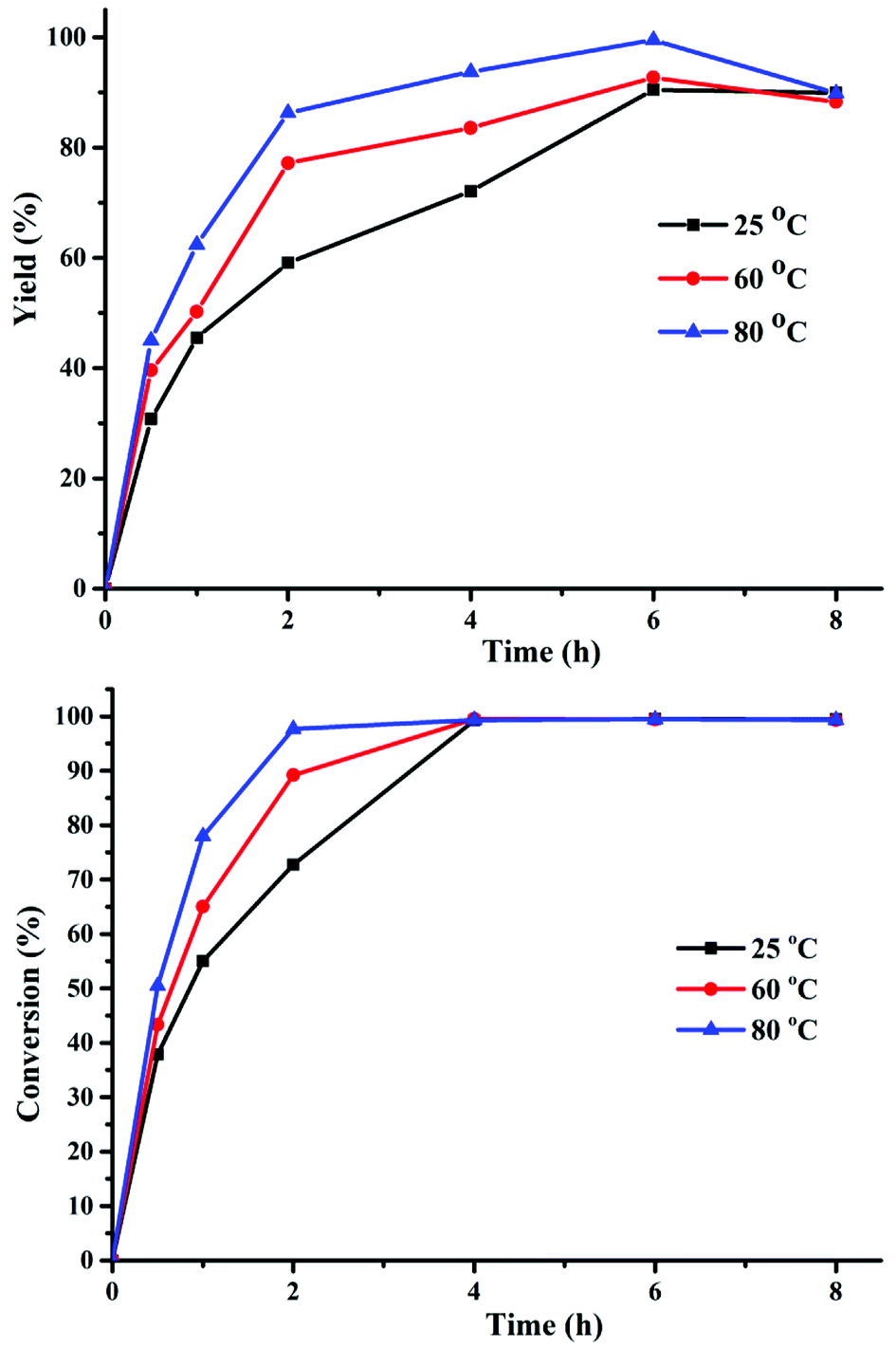
In the presence of Cs2CO3, the effect of reaction temperature and time on the hydrogenation of FUR to FFA was studied. All experiments were conducted at 25, 60, and 80 °C with the reaction time in the range of 0.5 to 8 h, and the results are displayed in Fig. 1. Both FFA yield and FUR conversion increased with the extension of reaction time from 0.5 to 6 h, displaying that a relatively longer reaction time could promote the FFA formation. However, the FFA selectivity decreased as the reaction time was prolonged to 8 h due to the generation of siloxane intermediate (Fig. S1–S3†). Therefore, 6 h was chosen as the optimum reaction time. It was obvious that the reaction efficiency was enhanced as reaction temperature increased from 25 to 80 °C in 6 h. Remarkably, when raising the temperature to 80 °C, the FA yield could reach 99%. Therefore, the optimal temperature of 80 °C and time of 6 h were beneficial to obtain FFA with less intermediates and byproducts being formed.
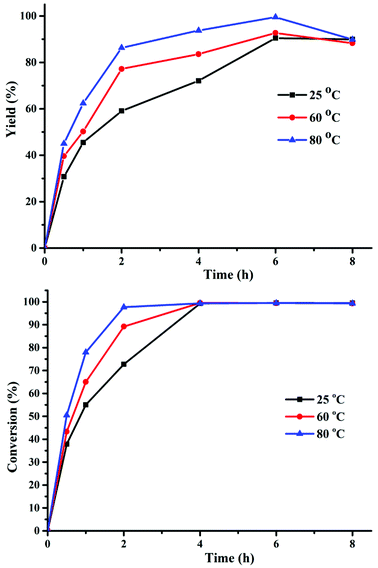 |
| | Fig. 1 The effect of reaction time and temperature on the synthesis of FFA from FUR. Reaction conditions: 0.5 mmol FUR, 16 mg Cs2CO3, PMHS (1.47 mmol H−), 2 mL DMF. | |
3.3 Effect of different solvents
The effect of the used solvents in this chemoselective hydrosilylation reaction was then examined over Cs2CO3. On the one hand, solvent polarity could affect the solvent–reactant interactions.22 As the polarity of the solvent increases, the increased interaction of FUR-solvent may promote the formation of FFA. On the other hand, with the increase of the solvent polarity, the solubility of PMHS into the solvent can also be enhanced, thereby facilitating the interaction of PMHS with the catalyst to release H−. Further interaction of hydride with FUR gives FFA. Highly polar aprotic solvent such as DMF and DMSO displayed high FUR conversion of 99% and satisfactory FFA yield of 99% (Fig. 2). In contrast, the solvents (e.g., THF, DCM, MTHF, CH3CN, 1,4-dioxane) with relatively low polarity exhibited inferior performance with FFA yield of 82%, 70%, 65%, 64%, and 8.2%, respectively. Notably, methanol and water were inactive for the reaction, due to the occurrence of dehydrogenative silylation between PMHS and the protic solvent (Fig. S10†) to release H2 (ref. 23) that cannot be used as active hydrogen source over Cs2CO3.
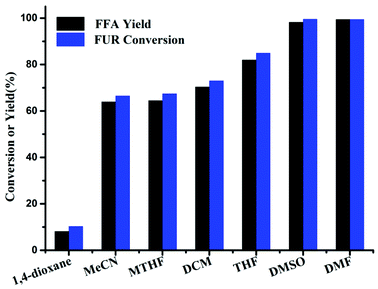 |
| | Fig. 2 Effect of solvents on the synthesis of FFA from FUR. Reaction conditions: 0.5 mmol FUR, 16 mg Cs2CO3, PMHS (1.47 mmol H−), 2 mL solvent, 80 °C, and 6 h. | |
3.4 Effect of different hydrosilanes
The influence of various hydrosilanes on the hydrogenation of FUR to FFA was studied by using Cs2CO3 as catalyst (Table 2). Among the employed simple silanes, phenylsilane and diphenylsilane (Table S1,† entries 6 and 7) revealed relatively higher yields of FFA, which might be due to their relatively poor stability of silicon atoms.24 However, they are usually too dear to be used on a large scale.25 To our astonishment, both the conversion of FUR and the yield of FFA were relatively high (>99%) when using cheap PMHS as the hydrogen source (Table S1,† entries 8). Besides, the polymerized PMHS was easy to form resins to encapsulate the catalyst,26 which would be helpful for the separation of the catalyst from the reaction mixture. From the economic and eco-friendly points of view, PMHS was screened out as the optimal hydrogen source for FUR hydrogenation.
Table 2 Reaction rate constant k at different temperatures
| Temperature (K) |
Reaction rate constant, k (1/min) |
Coefficient of determination (R2) |
| 298 |
0.0141 |
0.9685 |
| 333 |
0.0184 |
0.9474 |
| 353 |
0.0214 |
0.9537 |
3.5 Effect of PMHS dosage
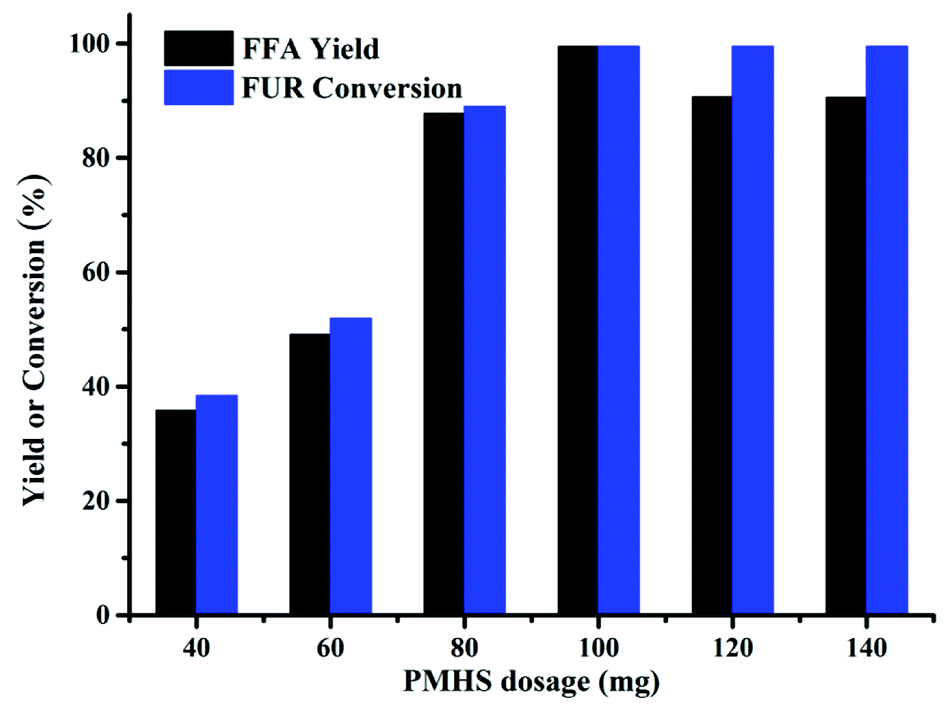
The PMHS dosage was found to have a significant effect on FFA synthesis, as shown in Fig. 3. As the PMHS dosage increased from 0.04 to 0.1 g, the FFA yield increased from 30 to 99% accordingly. Nevertheless, when the PMHS dosage was more than 0.1 g, the FFA yield was decreased despite of constant FUR conversion. The reduction of FFA yield was likely to be the encapsulation of Cs2CO3 by the resins derived from PMHS26 that impeded the contact of FUR to the active sites (carbonate). Therefore, the maximum FFA yield could be acquired by utilizing 0.1 g PMHS at 80 °C for 6 h in DMF.
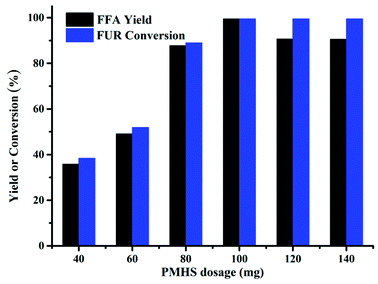 |
| | Fig. 3 Effect of PMHS dosage on the synthesis of FFA from FUR. Reaction conditions: 0.5 mmol FUR, 16 mg Cs2CO3, 2 mL DMF, 80 °C, and 6 h. | |
3.6 Effect of catalyst dosage
The effect of catalyst dosage on the hydrogenation of FUR to FFA under the optimal reaction conditions was further investigated, and the results are shown in the Fig. 4. 99% FUR conversion and 99% FFA yield were obtained in the presence of 16 mg Cs2CO3 at 80 °C for 6 h. When the Cs2CO3 dosage increased from 0 to 16 mg, both FUR conversion and FFA yield increased to 99%. However, when the catalyst dosage was beyond 16 mg, the yield of FFA displayed a markedly reduction ascribed to the adsorption of FFA onto the surface of the superfluous Cs2CO3 solid, or to the physical adsorption of water, thus inhibiting the alcoholysis of the siloxane intermediate to release the product.27 In addition, the fast ineffective depletion of PMHS was most possible to occur by forming pentavalent silicate species (H3Si(Me)–CO3) with carbonate.28 Although the pentavalent silicate species were active for the reduction of carbonyl compounds,29 serious polymerization was to exist when utilizing superfluous Cs2CO3 (e.g., 18 mg), which hence might decrease the catalyst activity.
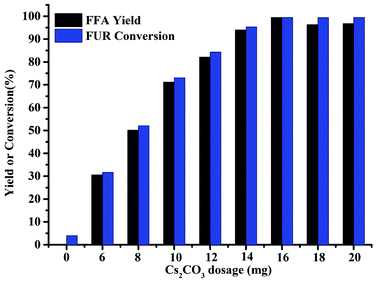 |
| | Fig. 4 Effect of catalyst dosage on the synthesis of FFA from FUR. Reaction conditions: 0.5 mmol FUR, PMHS (1.47 mmol H−), 2 mL DMF, 80 °C, and 6 h. | |
3.7 Catalyst recycling study
Recyclability study for FFA producing from FUR was conducted over Cs2CO3 at 25 °C and 80 °C for 6 h in presence of Cs2CO3 (16 mg), while the catalyst was recycled with decline in FFA yield (Table S2†). STEM image showed the distribution of the evenly dispersed elements, such as Cs, Si, C and O-species of recycled Cs2CO3 (Fig. S4†). From the above data, it was speculated that the reduced catalytic activity could be attributed to interaction of cesium carbonate with hydrosilane. To verify this point, a nuclear magnetic verification experiment was carried out to react cesium carbonate with PhSiH3 and PMHS under the reaction conditions of 25 °C, 6 h in the presence of DMSO-d6. NMR studies (as represented in Fig. S5†) disclosed the formation of silicon formate, which could be converted to value-added chemical – formic acid by hydrolysis.30 Considering the low-cost of Cs2CO3, it would be still promising to co-upgrade carbonate during the reduction process.
3.8 Kinetic study of FUR-to-FFA conversion
To investigate the reaction kinetics of FUR being converted to FFA, the reaction rate constants at various temperatures were determined based on the increased amount of FFA as a function of time (Table 2). It was assumed that the hydrogenation of FUR obeyed first-order kinetics.31 The reaction rate constant (k) was determined by using below equations.| |
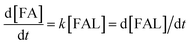 | (1) |
| |
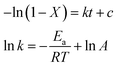 | (2) |
where X denotes the FUR conversion, the −ln(1 − X) versus the reaction time was fitted at different temperatures in Fig. 5, confirming that the reaction followed the pseudo-first order kinetics for all temperatures studied and the rate constants calculated were 0.0092, 0.0191, and 0.0350 min−1 at reaction temperatures of 298, 333, and 353 K, respectively. The apparent activation energy (Ea) of 20.6 kJ mol−1 was determined by the numerical regression assuming valid Arrhenius equation (as shown in eqn (2)), which was much lower than those of previously reported catalysts in the conversion of FUR to FFA (Table 3). Vaidya and Mahajani, using a noble metal-based catalyst (Pt/C) and 2-propanol as solvent, reported a value equal to 28 kJ mol−1, which is a low value considering that the absence of diffusional resistances was verified.32 Sharma et al. reported that Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Zn:Cr:Zr based catalysts for the hydrogenation of FUR to FFA, with apparent activation energy of 102 kJ mol−1.31 Potentially, the final disposal of the metal catalysts might cause serious environmental pollution.36 In this regard, our developed catalytic system meets the economic and eco-friendly design concept.
:Zn:Cr:Zr based catalysts for the hydrogenation of FUR to FFA, with apparent activation energy of 102 kJ mol−1.31 Potentially, the final disposal of the metal catalysts might cause serious environmental pollution.36 In this regard, our developed catalytic system meets the economic and eco-friendly design concept.
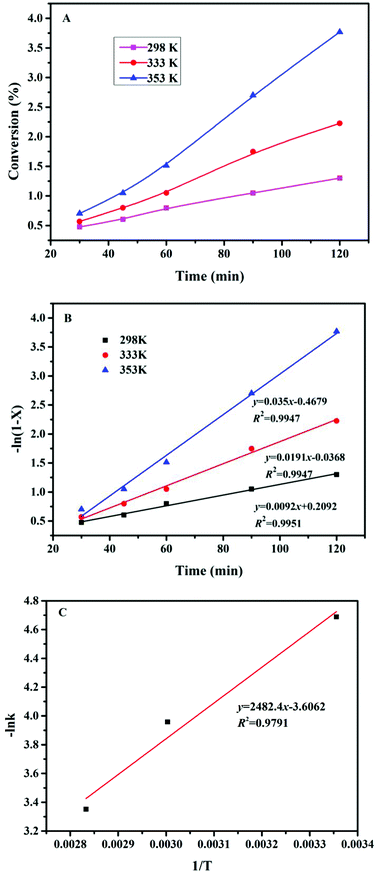 |
| | Fig. 5 (A) Kinetic curves of Cs2CO3 for the hydrogenation of FUR to FFA; (B) linear relationship between time and −ln(1 − X) at different reaction temperatures catalyzed by Cs2CO3; (C) Arrhenius plot of −lnk vs. 1/T catalyzed by Cs2CO3. | |
Table 3 Kinetic parameters of Cs2CO3 and previously reported catalysts for the hydrogenation of FUR to FFAa
| Catalyst |
Activation energy (kJ mol−1) |
References |
| Reaction conditions: 0.05 mmol FUR, 16 mg Cs2CO3, 2 mL DMF, PMHS (1.47 mmol H−). |
| CuMgAl |
127 |
35 |
| Cu:Zn:Cr:Zr |
102 |
31 |
| Au/Al2O3 |
45.0 |
34 |
| Co–Cu/SBA-15 |
38.5 |
33 |
| Pt/C |
28.0 |
32 |
| Cs2CO3 |
20.6 |
This work |
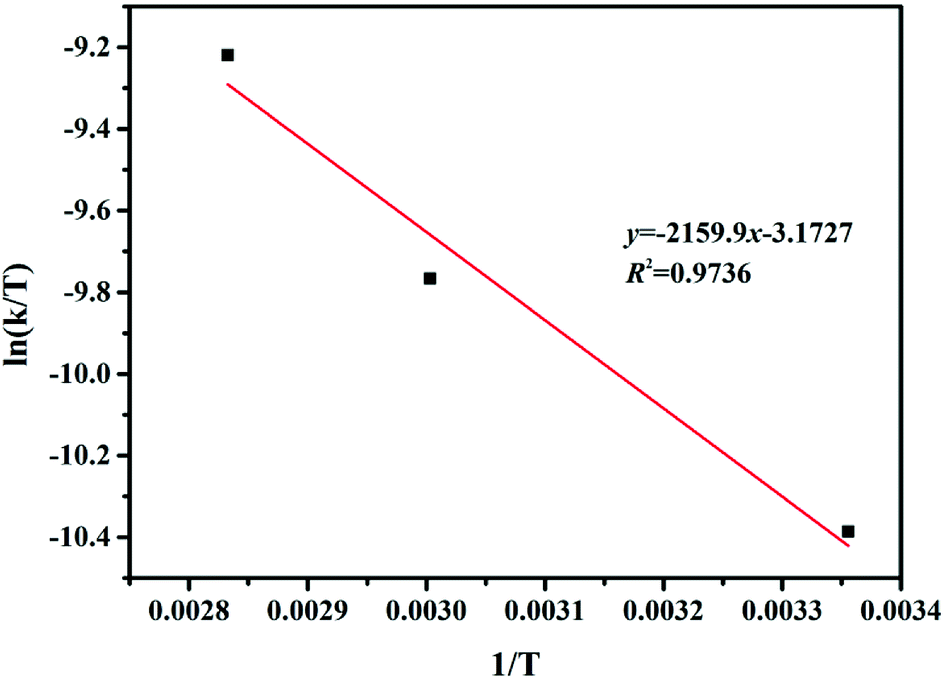
Thermodynamic characteristics of the reaction were also studied based on the results of the kinetic studies. For this purpose, the Eyring–Polanyi equation was used.
| |
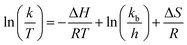 | (3) |
k is the rate constant of temperature
T. Δ
H and Δ
S are the changes of the enthalpy and entropy in the reaction system, respectively.
kb,
h and
R are the Boltzmann, Planck, and gas constants, respectively.
37 Δ
H and Δ
S were calculated from the slope and intercept of the Eyring–Polanyi plot for the reaction (
Fig. 6). The
eqn (3) can model the varies in the Gibbs free energy (Δ
G) at temperature
T. The values of Δ
H, Δ
S and Δ
G for the reaction are shown in
Table 4. The positive values of Δ
H displayed that the hydrogenation of FUR to FFA catalyzed by cesium carbonate is an endothermic reaction, which can be carried out under heating conditions. The negative value of Δ
S displayed that the degree of ordered geometry/alignment of the reactants in the ground state was lower than transient state. The positive value of Δ
G displayed that this reaction was essentially non-spontaneous.
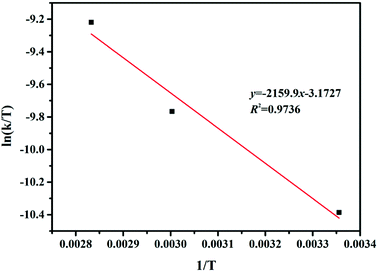 |
| | Fig. 6 Eyring–Polanyi plot between ln(k/T) and 1/T. | |
Table 4 Thermodynamic parameters of the reaction system
| Parameters |
ΔH (kJ mol−1) |
ΔS (kJ mol−1 K−1) |
ΔG (kJ mol−1) |
| 298 K |
333 K |
353 K |
| Values |
18.0 |
−0.223 |
84.5 |
92.3 |
96.7 |
3.9 Reaction pathway for FUR-to-FFA hydrogenation and substrate scope expansion
To definitely illuminate the reaction pathway for converting FUR to FFA, the GC spectra were conducted at 25 °C by changing the reaction time from 0.5 to 6 h (Fig. 7), clearly indicating that the content of FUR was decreased, and the content of FFA increased by prolonging the reaction time. The mechanism of hydrogenation of aldehydes and ketones catalyzed by Cs2CO3 is very similar to the reduction process of amides.38 The interaction of PMHS with carbonate dianion is likely to produce high valency silicate intermediates (Fig. S6 and S7†), which can transfer hydrides to carbonyl groups. During our experiments, a large amount of gas, methyl silane (H3SiMe), was observed immediately after the PMHS was added to the DMF solution of Cs2CO3 mixed with the substrate, which is consistent with previous studies.39 In order to verify the presence of methylsilane, PMHS was directly added into the solution of DMSO-d6 and Cs2CO3. Nuclear magnetic detection of the reaction mixture was performed after 6 h at room temperature (Fig. S8†). Two quartet peaks were observed at δ = 3.4 and 0.19 ppm in 1H NMR spectra, respectively, indicating the formation of MeSiH3 during the reaction process.
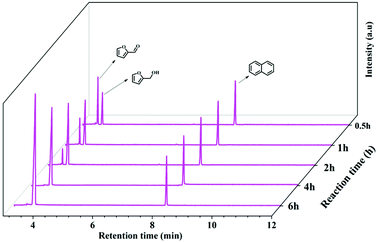 |
| | Fig. 7 GC spectrum at different reaction times for the conversion of FUR to FFA. Reaction condition: 0.5 mmol FUR, 16 mg Cs2CO3, PMHS (1.47 mmol H−), 2 mL DMF, 25 °C. | |
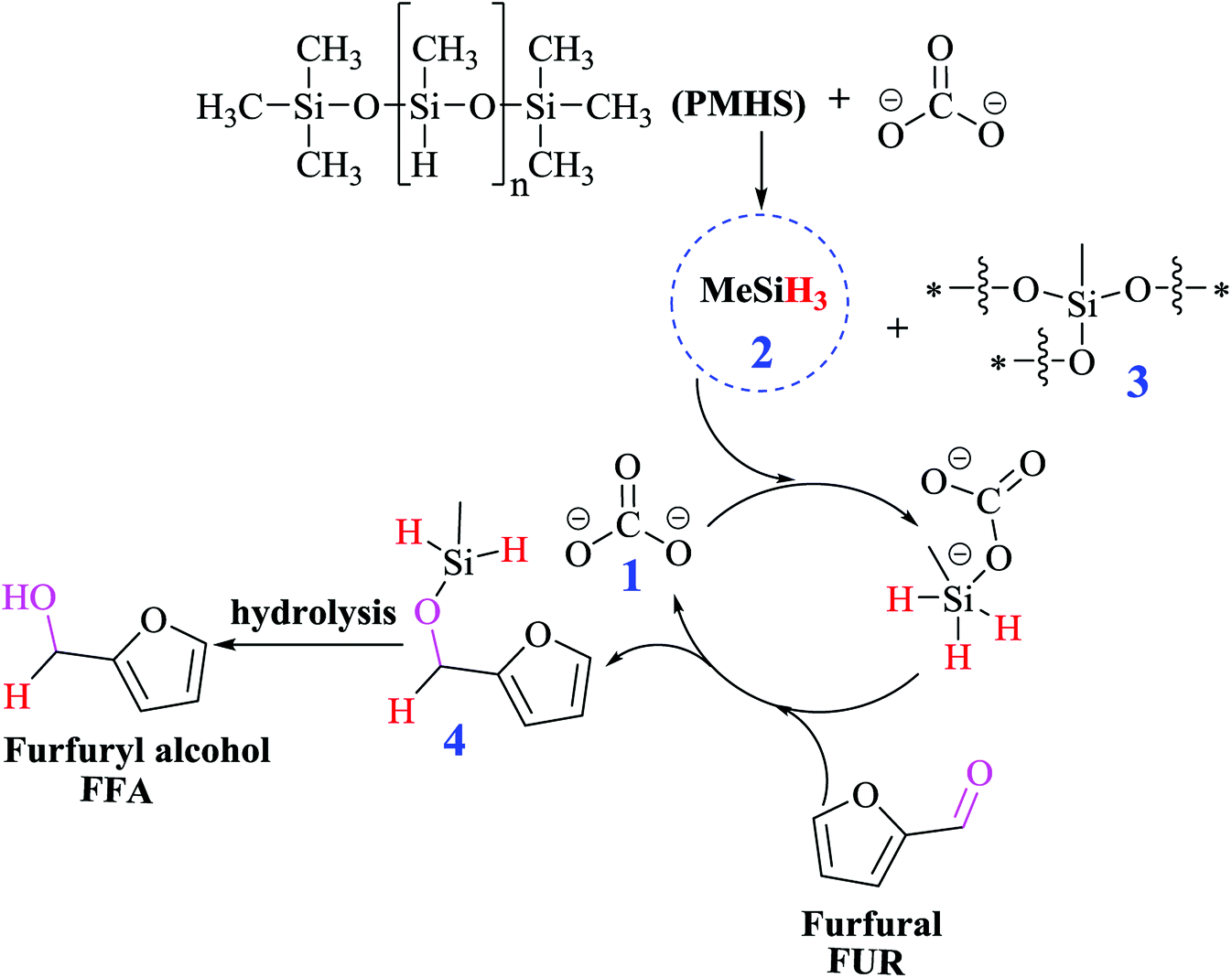
Based on the empirical evidences, the possible reaction mechanism of the hydrogenation of FUR to FFA is shown in Scheme 1. In the presence of Lewis base (carbonate 1), PMHS was first cracked to form small silane (H3SiMe 2), and a series of siloxanes 3. The resulting H3SiMe would form a pentavalent silicate with carbonate (Fig. S6†), followed by reacting with the carbonyl group in FUR to form hexavalent (Fig. S7†), which is a nucleophile in nature40 and can activate aldehyde group by hydride transfer. The release of carbonate 1 to the next catalytic cycle affords silyl ether 4, followed by the hydrolysis of the siloxane intermediate to give FFA.
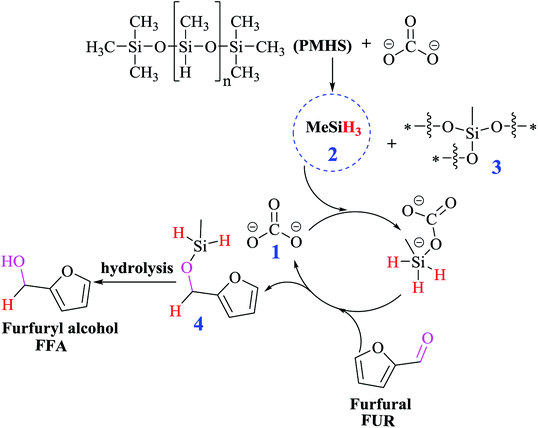 |
| | Scheme 1 Proposed reaction pathway for the conversion of FUR to FFA. | |
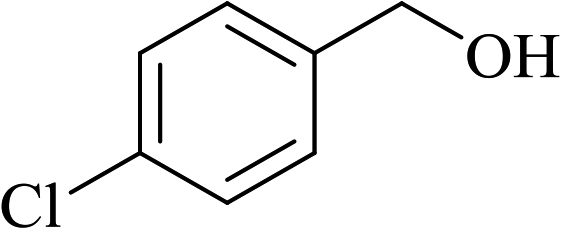
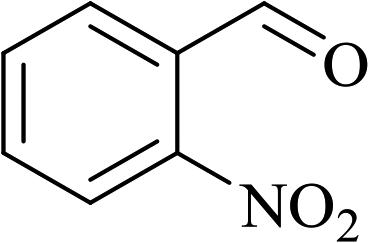
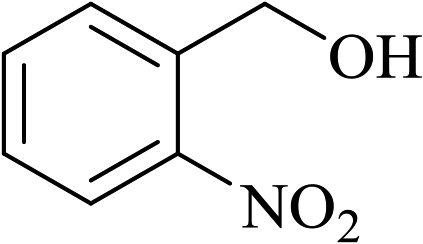
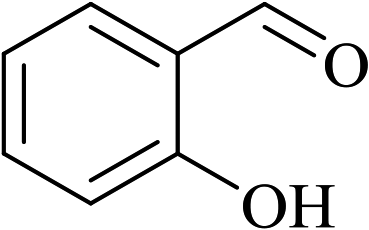
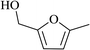
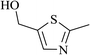
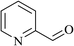
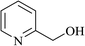
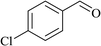
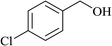
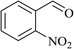
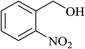
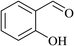
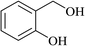
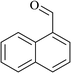
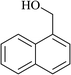
In order to examine the scope of this catalytic system for hydrogenation of FUR to FFA with Cs2CO3 using PMHS as H-donor, different aromatic aldehydes were employed as substrates, and the results are shown in Table 5. The hydrogenation of various aldehydes to corresponding alcohols, as identified by GC-MS (Fig. S9†), could achieved high yields 81–99% under mild reaction conditions. In particular, nitro- and chlorine-groups were tolerated during the reducing process (Table 5, entries 4 and 5).
Table 5 Catalytic hydrogenation of different aldehydes to alcohols with Cs2CO3 and PMHSa
| Entry |
Substrate |
Product |
Temp. (°C) |
Time (h) |
Yield (%) |
| Reaction conditions: 0.5 mmol substrates, 16 mg Cs2CO3, PMHS (1.47 mmol H−), 2 mL DMF. |
| 1 |
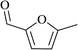 |
 |
80 |
6 |
85 |
| 2 |
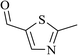 |
 |
80 |
6 |
>99 |
| 3 |
 |
 |
25 |
2 |
>99 |
| 4 |
 |
 |
80 |
6 |
88 |
| 5 |
 |
 |
80 |
6 |
87 |
| 6 |
 |
 |
80 |
6 |
81 |
| 7 |
 |
 |
80 |
6 |
>99 |
4. Conclusions
In summary, an efficient and benign catalytic protocol was developed for selective reduction of FUR to FFA in yields of up to 99% over Cs2CO3 by using eco-friendly and inexpensive PMHS as H-donor. This developed reaction system followed a pseudo-first order reaction with apparent activation energy of 20.6 kJ mol−1, which was much lower than previously reported ones. The mechanistic study illustrated the reaction proceeding via the hydrosilylation process with H3SiMe as the key hydride species formed by the activation of PMHS with carbonate, as clarified by 1H NMR. Moreover, this catalytic strategy was also appropriate for selective hydrogenation of different aromatic aldehydes to corresponding alcohols in good yields.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the financial support of the National Natural Science Foundation of China (21576059 & 21666008), Key Technologies R&D Program of China (2014BAD23B01), Fok Ying-Tong Education Foundation (161030), and Guizhou Science & Technology Foundation ([2018]1037 & [2017]5788).
References
- X. Li, P. Jia and T. Wang, ACS Catal., 2016, 6, 7621–7640 CrossRef CAS.
-
(a) B. Liu and Z. Zhang, ACS Catal., 2015, 6, 326–338 CrossRef;
(b) H. Li, A. Riisager, S. Saravanamurugan, A. Pandey, R. S. Sangwan, S. Yang and R. Luque, ACS Catal., 2018, 8, 148–187 CrossRef CAS;
(c) J. X. Long, W. F. Zhao, Y. F. Xu, H. Li and S. Yong, Catalysts, 2018, 8, 633 CrossRef;
(d) H. Li, W. F. Zhao, W. Dai, J. X. Long, M. Watanabe, S. Meier, S. Saravanamurugan, S. Yang and A. Riisager, Green Chem., 2018, 20, 5327–5335 RSC;
(e) H. Li, Z. Fang, J. Luo and S. Yang, Appl. Catal., B, 2017, 200, 182–191 CrossRef CAS.
- G. Li, W. Jiao, Z. Sun, Y. Zhao, Z. Shi, Y. Yan and Y. Tang, ACS Sustainable Chem. Eng., 2018, 6, 4316–4320 CrossRef CAS.
-
(a) H. Wang, X. Li, X. Lan and T. Wang, ACS Catal., 2018, 8, 2121–2128 CrossRef CAS;
(b) D. Vargas-Hernández, J. M. Rubio-Caballero, J. Santamaría-González, R. Moreno-Tost, J. M. Merida-Robles, M. A. Perez-Cruz and P. Maireles-Torres, J. Mol. Catal. A: Chem., 2014, 383, 106–113 CrossRef;
(c) H. Li, W. Zhao, S. Saravanamurugan, W. Dai, J. He, S. Meier, S. Yang and A. Riisager, Communications Chemistry, 2018, 1, 32 CrossRef;
(d) H. Li, Y. Li, Z. Fang and R. L. Smith Jr, Catal. Today, 2019, 319, 84–92 CrossRef CAS;
(e) H. Li, Z. Fang, R. L. Smith Jr and S. Yang, Prog. Energy Combust. Sci., 2016, 55, 98–194 CrossRef.
- B. J. Barr and S. B. Wallon, J. Appl. Polym. Sci., 1971, 15, 1079–1090 CrossRef.
- B. M. Nagaraja, V. S. Kumar, V. Shasikala, A. H. Padmasri, B. Sreedhar, B. D. Raju and K. R. Rao, Catal. Commun., 2003, 4, 287–293 CrossRef CAS.
-
(a) K. Yan, G. Wu, T. Lafleur and C. Jarvis, Renewable Sustainable Energy Rev., 2014, 38, 663–676 CrossRef CAS;
(b) T. V. Kotbagi, H. R. Gurav, A. S. Nagpure, S. V. Chilukuri and M. G. Bakker, RSC Adv., 2016, 6, 67662–67668 RSC.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS PubMed.
- M. M. Villaverde, N. M. Bertero, T. F. Garetto and A. J. Marchi, Catal. Today, 2013, 213, 87–92 CrossRef CAS.
- Y. Wang, Y. Miao, S. Li, L. Gao and G. Xiao, Mol. Catal., 2017, 436, 128–137 CrossRef CAS.
-
(a) N. Date, V. L. Parola, C. Rode and M. L. Testa, Catalysts, 2018, 8, 252 CrossRef;
(b) M. Hronec and K. Fulajtarová, Catal. Commun., 2012, 24, 100–104 CrossRef CAS.
-
(a) K. Yan, J. Liao, X. Wu and X. Xie, RSC Adv., 2013, 3, 3853–3856 RSC;
(b) S. A. Khromova, M. V. Bykova, O. A. Bulavchenko, D. Y. Ermakov, A. A. Saraev, V. V. Kaichev, R. H. Venderbosch and V. A. Yakovlev, Top. Catal., 2016, 59, 1413–1423 CrossRef CAS.
-
(a) M. S. Li, Y. F. Hao, F. Cárdenas-Lizana and M. A. Keanes, Catal. Commun., 2015, 69, 119–122 CrossRef CAS;
(b) Q. Q. Yuan, D. M. Zhang, L. van Haandel, F. Y. Ye, T. Xue, E. J. M Hensen and Y. J. Guan, J. Mol. Catal. A: Chem., 2015, 406, 58–64 CrossRef CAS;
(c) J. He, H. Li, A. Riisager and S. Yang, ChemCatChem, 2018, 10, 430–438 CrossRef CAS;
(d) S. Srivastava, N. Solanki, P. Mohanty, K. A. Shah, J. K. Parikh and A. K. Dalai, Catal. Lett., 2015, 145, 816–823 CrossRef CAS.
-
(a) D. Scholz, C. Aellig and I. Hermans, ChemSusChem, 2014, 7, 268–275 CrossRef CAS PubMed;
(b) V. Choudhary, S. I. Sandler and D. G. Vlachos, ACS Catal., 2012, 2, 2022–2028 CrossRef CAS;
(c) G. Wu, K. L. More, C. M. Johnston and P. Zelenay, Science, 2011, 332, 443–447 CrossRef CAS PubMed.
- Q. Zhang, S. S. Li, M. M. Zhu, Y. M. Liu, H. Y. He and Y. Cao, Green Chem., 2016, 18, 2507–2513 RSC.
-
(a) A. K. Roy, Adv. Organomet. Chem., 2007, 55, 1–59 CrossRef;
(b) M. Oestreich, J. Hermeke and J. Mohr, Chem. Soc. Rev., 2015, 44, 2202–2220 RSC.
- K. Revunova and G. I. Nikonov, Dalton Trans., 2015, 44, 840–866 RSC.
- A. Muthukumar, N. C. Mamillapalli and G. Sekar, Adv. Synth. Catal., 2016, 358, 643 CrossRef CAS.
- A. Volkov, K. P. Gustafson, C. W. Tai, O. Verho, J. E. Backvall and H. Adolfsson, Angew. Chem., Int. Ed., 2015, 54, 5122–5126 CrossRef CAS PubMed.
-
(a) N. J. Lawrence, M. D. Drew and S. M. Bushell, J. Chem. Soc., Perkin Trans. 1, 1999, 1, 3381–3391 RSC;
(b) G. Kumar, M. A. Muthukumar and G. Sekar, J. Org. Chem., 2017, 33, 4883–4890 Search PubMed.
- W. Xie, M. Zhao and C. Cui, Organometallics, 2013, 32, 7440–7444 CrossRef CAS.
-
(a) K. Fulajtárova, T. Soták, M. Hronec, I. Vávra, E. Dobročka and M. Omastová, Appl. Catal., A, 2015, 502, 78–85 CrossRef;
(b) H. Li and R. L. Smith, Nat. Catal., 2018, 1, 176–177 CrossRef.
-
(a) J. M. Blackwell, K. L. Foster, V. H. Beck and W. E. Piers, J. Org. Chem., 1999, 64, 4887–4892 CrossRef CAS PubMed;
(b) K. Garcés, F. J. Fernández-Alvarez, V. Polo, R. Lalrempuia, J. J. Pérez-Torrente and L. A. Oro, ChemCatChem, 2014, 6, 1691–1697 CrossRef.
- C. Bornschein, S. Werkmeister, K. Junge and M. Beller, New J. Chem., 2013, 37, 2061–2065 RSC.
- D. Addis, S. Das, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 6004–6011 CrossRef CAS PubMed.
- Y. Motoyama, K. Mitsui, T. Ishida and H. Nagashima, J. Am. Chem. Soc., 2005, 127, 13150–13151 CrossRef CAS PubMed.
- W. Wu, W. Zhao, C. Fang, Z. Wang, T. Yang, H. Li and S. Yang, Catal. Commun., 2018, 105, 6–10 CrossRef CAS.
- W. Zhao, W. Wu, H. Li, C. Fang, T. Yang, Z. Wang and S. Yang, Fuel, 2018, 217, 365–369 CrossRef CAS.
- M. Kira, K. Sato and H. Sakurai, J. Org. Chem., 1987, 52, 948–949 CrossRef CAS.
- K. Motokura, D. Kashiwame, A. Miyaji and T. Baba, Org. Lett., 2012, 14, 2642–2645 CrossRef CAS PubMed.
- R. V. Sharma, U. Das, R. Sammynaiken and A. K. Dalai, Appl. Catal., A, 2013, 454, 127–136 CrossRef CAS.
- P. D. Vaidya and V. V. Mahajani, Ind. Eng. Chem. Res., 2003, 42, 3881–3885 CrossRef CAS.
- S. Srivastava, N. Solanki, P. Mohanty, K. A. Shah, J. K. Parikh and A. K. Dalai, Catal. Lett., 2015, 145, 816–823 CrossRef CAS.
- M. Li, Y. Hao, F. Cárdenas-Lizana and M. A. Keane, Catal. Commun., 2015, 69, 119–122 CrossRef CAS.
- M. M. Villaverde, N. M. Bertero, T. F. Garetto and A. J. Marchi, Catal. Today, 2013, 213, 87–92 CrossRef CAS.
- W. Huang, H. Li, B. Zhu, Y. Feng, S. Wang and S. Zhang, Ultrason. Sonochem., 2007, 14, 67–74 CrossRef CAS PubMed.
- H. Pan, H. Li, H. Zhang, A. Wang, D. Jin and S. Yang, Energy Convers. Manage., 2018, 166, 534–544 CrossRef CAS.
- K. Ryoichi, T. Masatoshi and I. Yoshihiko, Tetrahedron Lett., 1998, 39, 1017–1020 CrossRef.
- K. Revunova and G. I. Nikonov, Chem.–Eur. J., 2014, 20, 839–845 CrossRef CAS PubMed.
- S. Rendler and M. Oestreich, Synthesis, 2005, 17, 1727–1747 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08616h |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Song Yang
* and
Song Yang