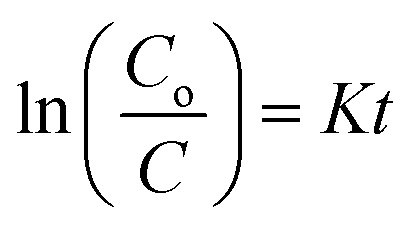DOI:
10.1039/C8RA08208A
(Paper)
RSC Adv., 2019,
9, 3215-3223
Role of MIL-53(Fe)/hydrated–dehydrated MOF catalyst for electrochemical hydrogen evolution reaction (HER) in alkaline medium and photocatalysis†
Received
4th October 2018
, Accepted 15th December 2018
First published on 23rd January 2019
Abstract
The role of breathing behavior in hydrated and dehydrated forms of MIL-53(Fe) is investigated here. The material can be used as an efficient electrocatalyst and photocatalyst for a hydrogen evolution reaction (HER) in an alkaline medium and the same was further tested for the degradation of organic pollutants. The as-synthesized MIL-53(Fe)/hydrated and dehydrated forms were characterized by different analytical techniques to study their structure, morphology, surface analysis, thermal, physical and chemical properties. The breathing behavior of the hydrated and dehydrated forms of MIL-53(Fe) was studied through BET surface analysis. Our results show a low onset potential (−0.155 V and −0.175 V), Tafel slope (71.6 mV per decade, 88.7 mV per decade) and a large exchange current density (1.6 × 10−4 mA cm−2 and 2.5 × 10−4 mA cm−2). Hydrated and dehydrated MIL-53(Fe) degraded an RhB dye solution within 30 minutes thus proving their efficiency as efficient photocatalysts.
1. Introduction
Molecular hydrogen is considered a future energy carrier in the transition from the current hydrocarbon economy and is regarded as a clean energy carrier with the highest gravimetric energy density. Hydrogen as a clean fuel has been produced from renewable energy sources.1 There are diverse effective hydrogen production methods like water electrolysis, photolysis, thermolysis, hydrocarbon steam reforming, biomass pyrolysis, and coal gasification.2–4 Among these methods, electrolysis and electrochemical splitting have been considered as simple eco-friendly methods. The electrochemical hydrogen evolution reaction (HER) is one of the main cathodic reactions in electrochemical energy conversion devices.5 To meet the requirements of a HER, efficient electrocatalysts are imperative owing to the multi electron nature of dihydrogen generation through proton reduction6 and reduction of over potential (η). In general, electrocatalytic materials are found to the best solution for low production costs and reducing the over potential of a hydrogen evolution reaction (HER). Hence, it is an active field of research for the development of new electrode materials in order to reduce the hydrogen evolution over potential at an acceptable current density.7 In the past decades, various catalysts have been synthesized and reported. Among them, a Pt catalyst is the best candidate for a HER reaction, owing to its over potential and stability in an acid medium.8 But its high cost and scarcity prevents its usage for further large scale hydrogen production. Thus, there is an urge for the development of alternative catalysts (e.g., non-noble and metal-free electro catalysts). Compared with metal-free electrocatalysts, porous materials are used as active catalyst materials because of their tunable molecular structures, abundance, and strong tolerance to acidic/alkaline environments.9,10 Metal organic frameworks (MOFs) are a family of crystalline porous solids with coordination networks of metal ions and organic ligands linked together, and have been used as an absorbing class of micro porous crystalline materials due to their intrinsic low density, large accessible pore volumes, tunable textures and well-defined pore size distributions. They have been studied for different applications in gas storage/separation, magnetism, molecular recognition, proton and ion conduction, luminescence, catalysis, and drug carrying/delivery. Also, MOFs behave like semiconductors when exposed to light, thus making it possible for them to be photocatalysts.11–15 MOFs are considered superior materials due to the ease of tuning their pore sizes and shapes from a microporous to a mesoporous scale by changing the connectivity of the inorganic moiety and the nature of the organic linkers. Metal organic frameworks are intensively studied as the coordination networks of tailored nanoporous host materials via the self-assembly of metal ions linked by bridging ligands, which in turn contributes to the bulk volume. Another important criterion is the breathing effect or swelling phenomenon, which leads to an increase or decrease in the cell volume.16 The breathing effect enables the material to accommodate guest molecules. A previous report on MIL-53(Fe) has shown that the anhydrous state exhibited closed pores. The most extraordinary properties of the flexible structure of MOFs rely on their ability to adopt the pore openings to accommodate guest species and different modes (absorption, desorption) of guest molecules;17,18 this breathing effect can produce a dramatic increase and decrease in the cell volume without loss of crystallinity or bond breaking.19 Previous results showed that the material showed a remarkable breathing effect for Cr and Al, wherein removal of water changed the structure from a closed to a large pore material. But it was postulated that this behavior depends on the metal used, and in the case of Fe, the material actually contracts upon dehydration.20 MIL-53(Fe) stands for Material of Institute Lavoisier and the structure of MIL-53(Fe) is built up by the interconnection of an infinite chains of trans corner-sharing octahedral FeO4(OH2) and linear dicarboxylate ions that leads to a 3D solid with lozenge-shaped channels. In particular, the presence of iron offers several advantages compared to other metals, due to its low costs and non-toxicity, which makes MIL-53(Fe) an interesting catalyst. In the case of MIL-53(Fe), it displays a more complex and different behavior in the hydrated form with very narrow pores (vnp) and in the dehydrated form with closed pore structures.21,22 In this report, MIL-53(Fe) is prepared under two different experimental conditions with diverse pore behavior and further tested for its applicability towards a hydrogen evolution reaction (HER) and photocatalytic dye degradation of rhodamine-B. The prepared materials, under two different experimental conditions, are designated as MIL-53(Fe)/hydrated and MIL-53(Fe)/dehydrated samples. The synthesized materials are characterized by X-ray diffraction (XRD), Raman spectroscopy, thermo-gravimetric analysis (TGA), field emission scanning electron microscopy (FE-SEM) and diffuse-reflectance UV-spectroscopy. Further MIL-53(Fe)/hydrated–dehydrated material was subjected to electrochemical testing to investigate the HER performance. Results show that the material rendered a low onset potential, less over potential, a small Tafel slope and a high current density in an alkaline medium, rendering a good electrocatalyst for a hydrogen evolution reaction. Further photocatalytic tests revealed that the material was active towards dye degradation and hence could be used for environmental remedial applications.
2. Experimental
2.1 Materials
All the chemicals were of analytical grade and utilized as received without any further purification. Graphite fine powder, hydrogen peroxide (30 wt%), sodium nitrate (98%), sulphuric acid (99 wt%), potassium permanganate, terephthalic acid, iron(III) chloride hexahydrate, DMF and hydrazine hydrate were purchased from Sigma Aldrich and were used as received without further purification.
2.2. Preparation of metal organic framework MIL-53(Fe)
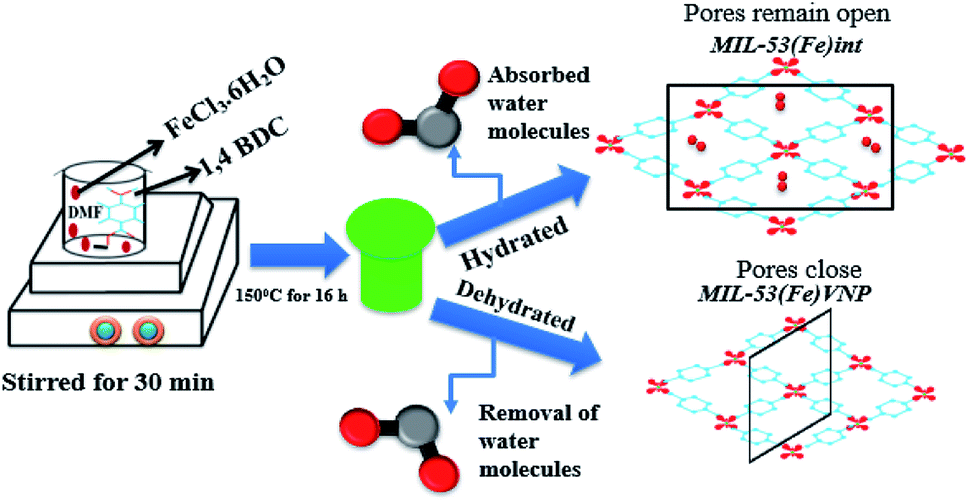
The metal organic framework MIL-53(Fe) was synthesized as per the previously reported method with a slight modification.23 In detail, 10 mmol of 1,4 benzene dicarboxylic acid and 5 mmol of iron(III) chloride were dissolved in 100 ml of dimethyl formamide solution and stirred for 30 min. The solution mixture was further transferred to a Teflon-lined stainless steel autoclave and heated at 150 °C for 16 h. The resulting product was washed with DMF and methanol by centrifugation and dried at 60 °C. To remove the residual unreacted substances, the obtained yellow solid was immersed in DI water for 12 h and calcinated at 60 °C for 12 h to get the hydrated form. Similarly, calcinations were done at 120 °C to get the dehydrated form. The breathing behaviour varied with temperature and solvent process. The pores sizes were enlarged due to the increase in temperature. MIL-53(Al) and MIL-53(Cr) hydrated phases have contracted pores and the dehydrated phases have open pores,20 but in MIL-53(Fe) the hydrated phase has intimidated pores and the dehydrated phase has narrow pores (vnp) due to the reversible breathing behaviour shown in the schematic (Scheme 1).
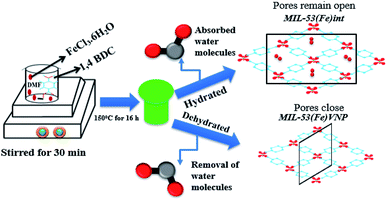 |
| | Scheme 1 A schematic of the synthesis of MIL-53(Fe) metal organic framework composite. | |
2.3 Material characterization
The morphological studies and elemental compositions of the composites were done with FESEM (Hitachi S-4800) and TEM JEOL-2000EX, operated at 120 kV. The crystalline structure of the synthesized composite was identified by using a Rigaku Miniflex powder X-ray diffraction (XRD) (Cu Kα = 1.5406 Å) over a 2θ range from 10–50°. The functional groups were analyzed by Fourier transform infrared spectroscopy (FTIR) (Shimadzu Affinity-1) in the range of 500 cm−1 to 4000 cm−1 and Raman spectroscopy. Thermo gravimetric analyses (TGA) of the MIL-53(Fe) was performed in an air atmosphere with an SDT Q600 (TA Instruments). The optical properties of the sample were measured with diffuse-reflectance UV-vis spectroscopy and photoluminescence properties were recorded using a fluorescence spectrometer (Horiba Jobin Yvon – Fluorolog).
2.4 Preparation of modified glassy carbon electrode (MGCE)
The bare glassy carbon electrode surface (GCE, 3 mm diameter) with a geometric area of 0.071 cm2 was polished with a 0.05 micron alumina slurry, rinsed with DI water, sonicated in ethanol and double distilled water to activate and then dried at room temperature. Before each experiment, the electrodes were first pre-treated by cycling several times from 0.0 V to 0.4 V. The catalyst slurry was prepared by suspending 5 mg of the prepared MIL-53(Fe)/dehydrated, MIL-53(Fe)/hydrated catalyst in 250 μL ethanol. 5 μL of MIL-53(Fe)/hydrated, MIL-53(Fe)/dehydrated and 5 μL of Nafion was dropped on to the clean glassy carbon electrode surface and allowed to dry at room temperature for 1 h.
2.5 Photoelectrochemical studies
The photoelectrochemical properties of the MIL-53(Fe)/dehydrated, MIL-53(Fe)/hydrated catalysts were tested by a CHI 660C electrochemical work station and a 300 W Cermax Xenon short arc lamp with a cut off filter (λ > 420 nm) was used as an irradiation source. A standard three electrode cell with a platinum wire as a counter electrode, Ag/AgCl as a reference electrode and MIL-53(Fe)/dehydrated, MIL-53(Fe)/hydrated coated electrodes as a working electrode were used. A 0.1 M Na2SO4 aqueous solution was used as the electrolyte throughout the experiments. For the preparation of the MIL-53(Fe)/dehydrated and MIL-53(Fe)/hydrated electrodes, a suspension of MIL-53(Fe)/dehydrated or MIL-53(Fe)/hydrated, methanol and Triton X-100 was deposited onto an fluorine doped tin oxide substrate with an area of 0.5 cm2 by the doctor blade method and then dried at 373 K for one hour.
2.6 Photocatalytic dye degradation of rhodamine B
The photocatalytic activity of MIL-53(Fe)/dehydrated and MIL-53(Fe)/hydrated was evaluated with rhodamine B as a dye using a 250 watt LED lamp (400 nm to 800 nm). In a typical experiment, 3 ml of 5 mmol of H2O2 was added to of 20 mg L−1 (20 ppm) of a rhodamine B aqueous solution. To this was added the required amount of the prepared MIL-53(Fe)/hydrated and MIL-53(Fe)/dehydrated catalyst and stirred for 30 min in the dark to establish an adsorption–desorption equilibrium. 2 ml aliquots were withdrawn by syringe at definite time intervals and the photo catalyst was extracted and centrifuged for 10 min. The dye concentration was determined with a UV-vis spectrophotometer by measuring the RhB dye absorption wavelength at 554 nm.
3. Results and discussion
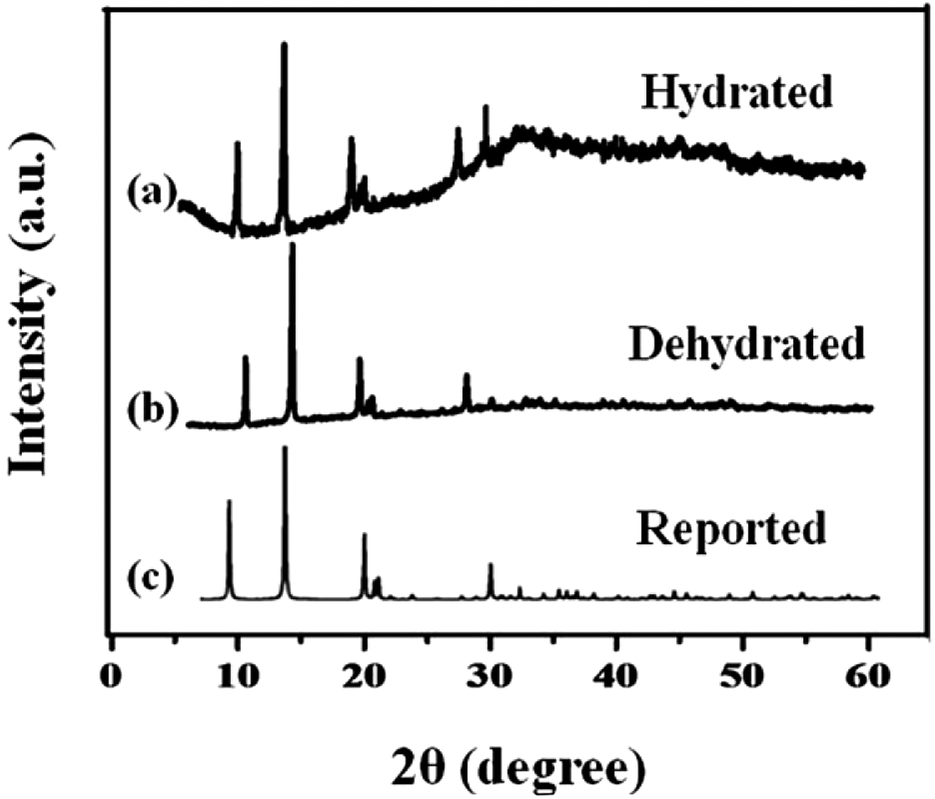
The phase composition and structure of the samples were examined by using X-ray powder diffraction. Fig. 1 shows the XRD pattern of the synthesized MIL-53(Fe) (hydrated & dehydrated) samples and from the figure, it can be observed that the diffraction peaks were seen at 9.08, 12.6, 17.49, 18.39, 25.3, 27.1, which were consistent with the simulated ones.24 The XRD pattern of MIL-53(Fe) indicates high crystallinity and there were no another phases seen in the spectrum. No Fe2O3 or other phases were seen, which indicates the purity of the sample. The peak intensity was not affected by dehydration, which means there was no change in the crystallinity of the materials.
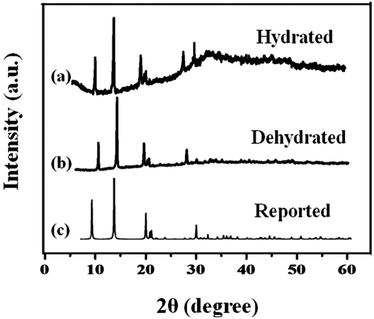 |
| | Fig. 1 X-ray diffraction spectra of MIL-53(Fe) (a) hydrated, (b) dehydrated and (c) simulated peaks. | |
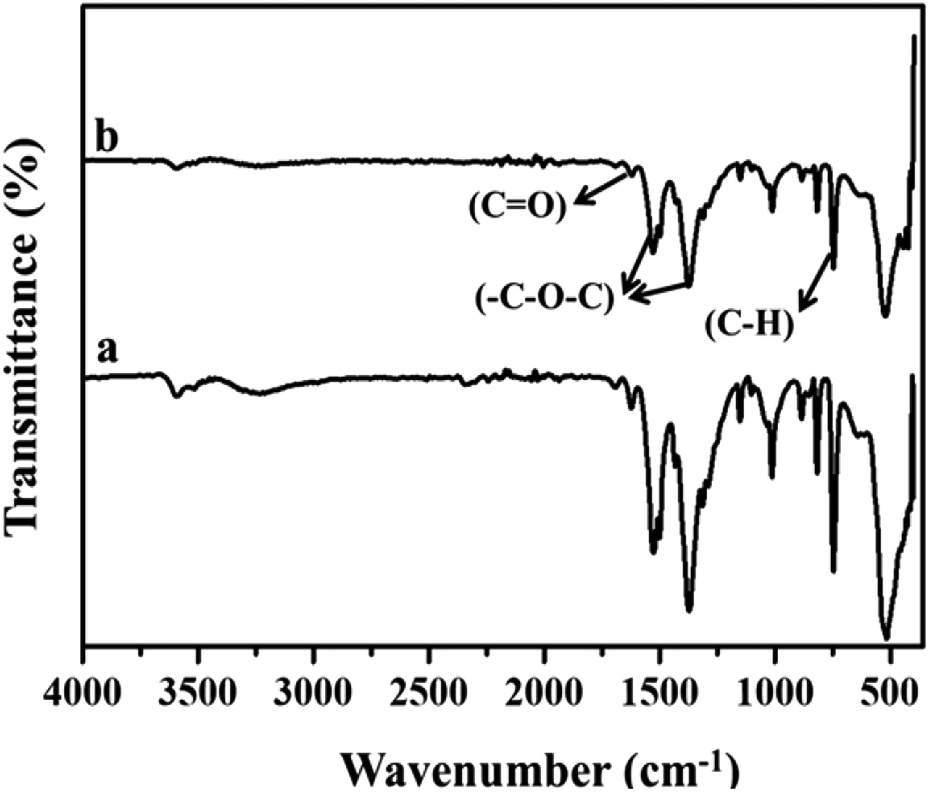
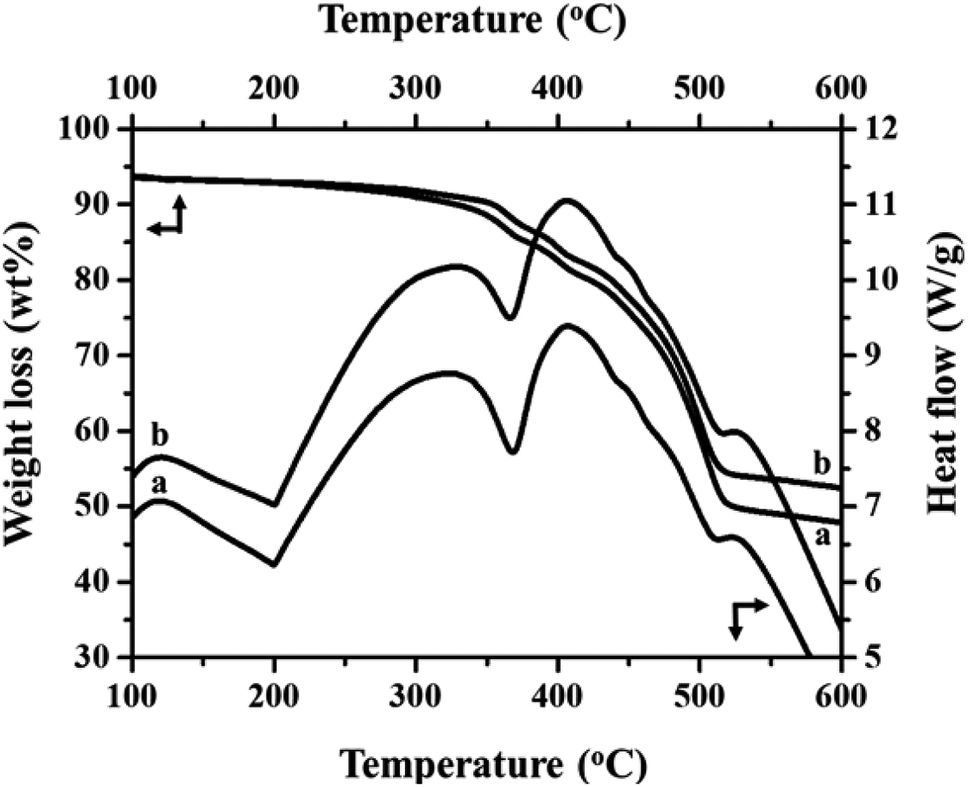
Fig. 2(a) and (b) show the FT-IR spectra of MIL-53(Fe)/hydrated and dehydrated forms. The absorption band peak observed at 3431 cm−1 is due to the stretching vibration of the O–H bonds from the surface absorbed water, which is less intense in the dehydrated form. The absorbance band at 1692 cm−1 indicates the stretching vibration of (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) bonds found in COO−.25 The peaks at 1524 cm−1 and 1380 cm−1 are the symmetric and asymmetric stretching vibrations of C–O pertaining to the carboxylate groups, confirming the presence of the ligand within the sample. The hydrated form shows a band at 863 cm−1 due to δ (OH) modes of the bridging hydroxyl group (Fe–O(H)–Fe)26 and a peak at 747 cm−1 corresponds to the C–H bending vibration of benzene rings.27 The peak at 532 cm−1 corresponds to the vibration peak of ν (Fe–O). Thermogravimetric (TGA) analysis is a useful tool for stability studies of MIL-53(Fe)/dehydrated and hydrated samples (Fig. 3).28 As shown in the figure, the graph illustrates the weight loss percentage as a function of temperature. An initial weight loss of 6.1% and 6.7% was caused by the removal of water and DMF around 100 °C to 200 °C. Major weight losses of 44.4% and 39.6% occurred around 300–400 °C, which might be due to the breaking of terephthalic acid within the pores, which suggests that the terephthalic acid molecules have been completely expelled at 410 °C. The weight loss above 450 °C corresponds to the breakdown of the BDC linker from the framework. The material is stable above 500 °C, and the residual material remaining is 36.46% MIL-53(Fe)/hydrated and 30.97% MIL-53(Fe)/dehydrated, which indicates that the former is more stable than the latter.
O) bonds found in COO−.25 The peaks at 1524 cm−1 and 1380 cm−1 are the symmetric and asymmetric stretching vibrations of C–O pertaining to the carboxylate groups, confirming the presence of the ligand within the sample. The hydrated form shows a band at 863 cm−1 due to δ (OH) modes of the bridging hydroxyl group (Fe–O(H)–Fe)26 and a peak at 747 cm−1 corresponds to the C–H bending vibration of benzene rings.27 The peak at 532 cm−1 corresponds to the vibration peak of ν (Fe–O). Thermogravimetric (TGA) analysis is a useful tool for stability studies of MIL-53(Fe)/dehydrated and hydrated samples (Fig. 3).28 As shown in the figure, the graph illustrates the weight loss percentage as a function of temperature. An initial weight loss of 6.1% and 6.7% was caused by the removal of water and DMF around 100 °C to 200 °C. Major weight losses of 44.4% and 39.6% occurred around 300–400 °C, which might be due to the breaking of terephthalic acid within the pores, which suggests that the terephthalic acid molecules have been completely expelled at 410 °C. The weight loss above 450 °C corresponds to the breakdown of the BDC linker from the framework. The material is stable above 500 °C, and the residual material remaining is 36.46% MIL-53(Fe)/hydrated and 30.97% MIL-53(Fe)/dehydrated, which indicates that the former is more stable than the latter.
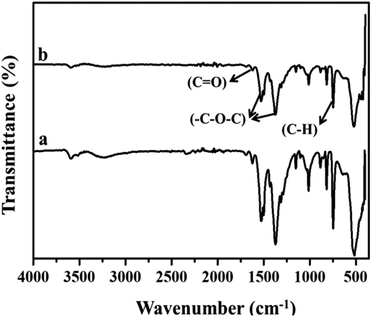 |
| | Fig. 2 FT-IR spectra of MIL-53(Fe) (a) hydrated and (b) dehydrated form. | |
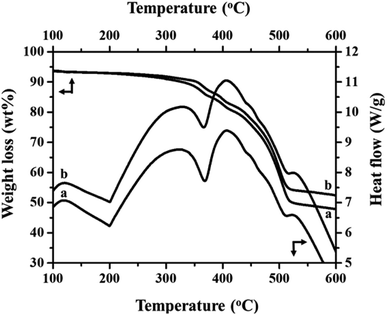 |
| | Fig. 3 TGA/DSC analysis of (a) MIL 53(Fe) dehydrated and (b) hydrated forms. | |
DSC is a useful technique to explain the physiochemical properties of the dehydration and hydration process of the MIL-53(Fe)29 and the respective results are given in Fig. 3 (top-right curves). The DSC curves show three exothermic and two endothermic peaks, which are associated with the ordered thermal decomposition of the MIL-53(Fe) structures. In the DSC curves, there are no exothermic peaks before 300 °C indicating the removal of the residual molecules of the MIL-53(Fe) framework. The peaks at 367 °C and 365 °C are endothermic peaks of the dehydrated and hydrated forms assigned to the thermal decomposition of the MIL-53(Fe) framework.
Fig. S1† shows the UV-Vis diffuse reflectance spectra (DRS) of the MIL-53(Fe)/dehydrated and MIL-53(Fe)/hydrated samples (see ESI, Fig. S1†). The band at 240 nm could be assigned to the ligand-to-metal charge transfer (LMCT) of O(II) → Fe(III) bonding of the carboxylate oxygen to the metal. Furthermore, a short peak at 439 nm is due to the d–d transition of [6A1g = >4A1g + 4Eg (G)] in Fe(III) in the MOFs. The absorbance edge of MIL-53(Fe)/hydrated (322 nm) absorbs at a longer wavelength compared to the MIL-53(Fe)/dehydrated (318 nm), which might be due to the water absorbance.30,31 The optical band gap of the MIL-53(Fe)/dehydrated form is 2.57 eV and is 2.51 eV for the MIL-53(Fe)/hydrated form, as calculated from the Tauq plot. The prominent optical properties in the UV-vis region of MIL-53(Fe) reveal that the catalyst materials could be used efficiently for a photocatalytic purpose, wherein a narrow band gap suggests its good photochemical stability.
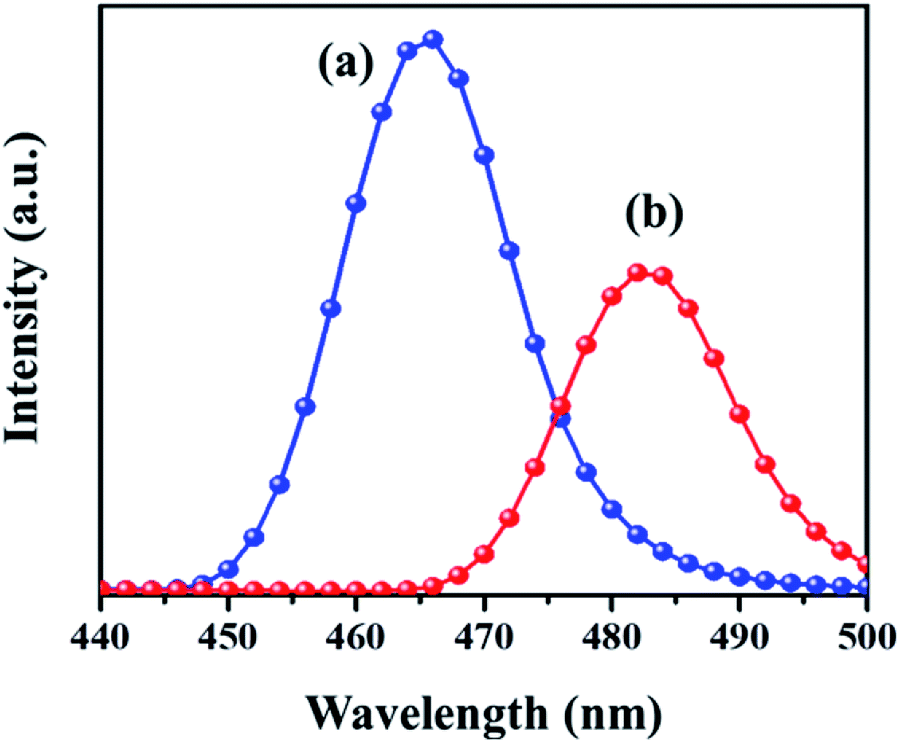
PL analysis helps to understand the photochemical properties and recombination efficiency of the photogenerated electron–hole pair.32 Fig. 4 illustrates the PL spectra of the MIL-53(Fe)/hydrated sample excited at 231 nm and 240 nm. As observed in the figure, its emission spectrum shows a broad peak around 440–500 nm. In the case of the MIL-53(Fe)/dehydrated form, there is a blue shift of the emission observed at 465 nm and for MIL-53(Fe)/hydrated, the peak seen at 483 nm is a red shift emission, and also a lower intensity indicates the rate of recombination of the electron–hole pair due to the photocatalyst dye removal. A dramatic change in the emission spectra under hydration and dehydration conditions for MIL-53(Fe) could be due to the slowdown of the recombination rate for the MIL-53(Fe)/hydrated samples compared to the MIL-53(Fe)/dehydrated sample.
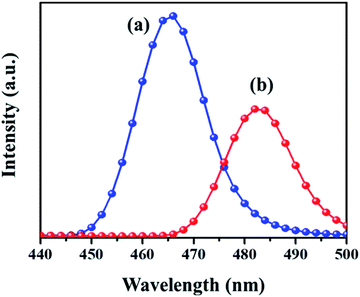 |
| | Fig. 4 Photoluminescence study of (a) dehydrated and (b) hydrated forms. | |
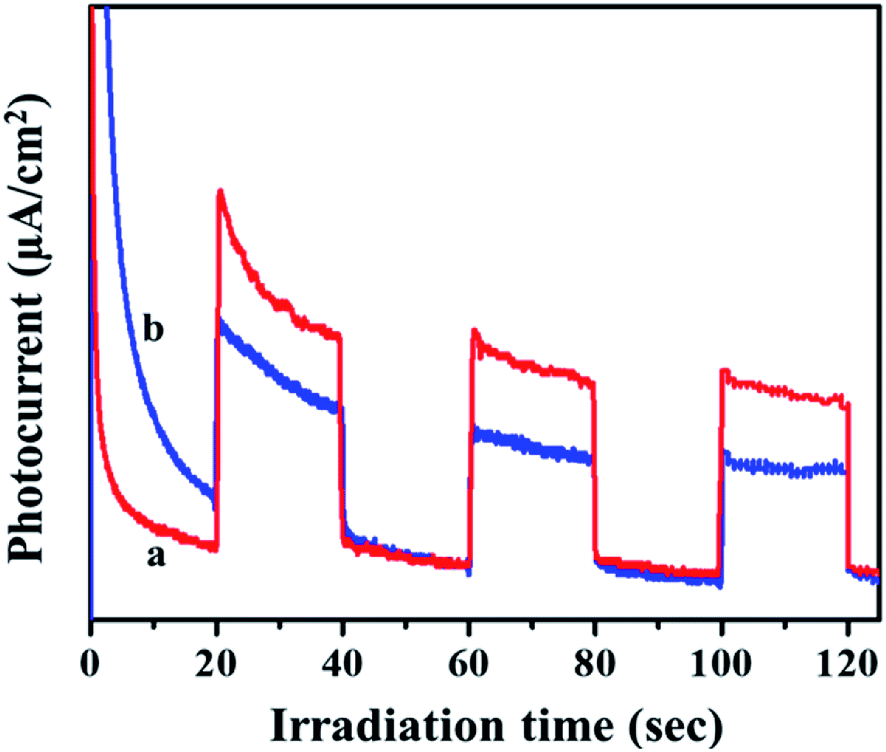
Transient photocurrent measurements are used to demonstrate the process of charge separation and charge transfer through periodic light on and light off regions.33,34 To cater the same, the samples were dispersed and sonicated. A 300 W Xe light outfitted with a cut-off channel was utilized as the illumination source. The plating voltage was 5 V and the time interval was 1 min. The photocurrent was measured with a CHI 660 C electrochemical workstation using a 1.0 M Na2SO4 solution by a two compartment three-electrode framework with Ag/AgCl (3 M NaCl) as a reference electrode, Pt wire as auxiliary electrodes and fluorine doped tin oxide (FTO) glass coated with the prepared catalyst as the working electrode. As seen in Fig. 5, the photo response of the MIL-53(Fe)/hydrated sample has a higher photocurrent compared to the MIL-53(Fe)/dehydrated sample, which indicates that the former has a more efficient charge separation of photo excited electron–hole pairs.
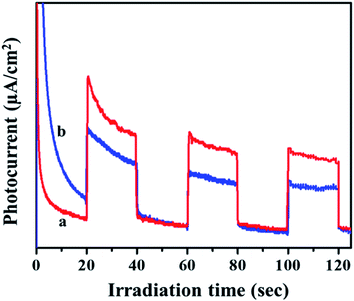 |
| | Fig. 5 Transient photocurrent response of (a) MIL-53(Fe)/hydrated and (b) dehydrated samples. | |
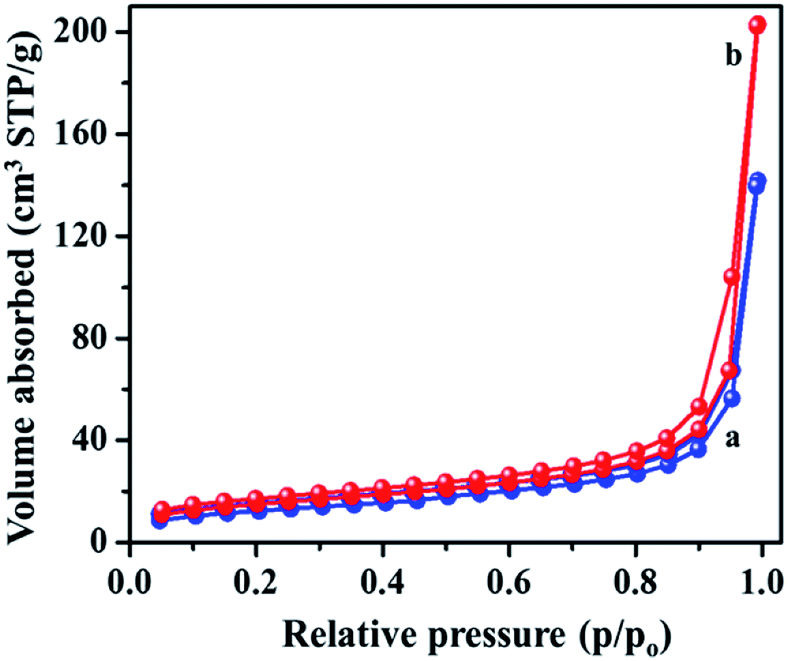
Fig. S2† shows the Raman spectra of the MIL-53(Fe)/dehydrated and MIL-53(Fe)/hydrated samples recorded over a wide range from 1800 cm−1 to 860 cm−1 (see ESI, Fig. S2†). The spectrum is dominated by the vibrational modes of organic linker viz. terephthalic acid (H2bdc) of the metal organic framework material. There was no substantial change in both forms, indicating that the process didn't have much effect on the structure. The specific surface area and pore volume of MIL-53(Fe), both in the dehydrated and hydrated form, were determined by N2 adsorption–desorption isotherms at 77 K (Fig. 6). The curves displayed an intermediate mode between micropores and mesopores. The BET surface area of MIL-53(Fe)/dehydrated and the hydrated sample was calculated to be 43.75 m2 g−1 and 52.813 m2 g−1, respectively, with the pore volumes shown in Table 1. The former sample has a low surface area with very narrow pores (vnp), whereas the hydrated form has a substantially large surface area. The curve shows an intermediate mode between type I and IV isotherms, corresponding to the micropores and mesopores.35 The Barrett–Joyner–Halenda (BJH) mesopore size distribution curve exhibited a pore size centered at about 4.9 nm for MIL-53(Fe)/dehydrated and 5.9 nm for the MIL-53(Fe)/hydrated form. Hence, the results show that the dehydrated form has very narrow pore structures, whereas the hydrated form shows an intermediate pore with an increased pore size. This suggests that the change of the pore volume and surface will have certain effects on the electrocatalytic and photocatalytic activities of MIL-53(Fe).
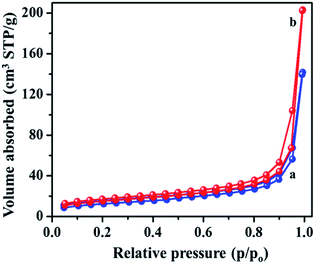 |
| | Fig. 6 BET surface areas calculated from N2 adsorption–desorption isotherms of MIL-53(Fe) (a) dehydrated (b) hydrated. | |
Table 1 N2 BET isotherm – MIL 53(Fe) dehydrated and hydrated
| Sample |
Specific surface area (m2 g−1) |
Pore volume (cm3 g−1) |
Average pore diameter (nm) |
| MIL-53(Fe)/dehydrated |
43.75 |
0.218 |
4.9 |
| MIL-53(Fe)/hydrated |
52.813 |
0.312 |
5.9 |
The surface morphology of the prepared MIL-53(Fe)/dehydrated and hydrated form was further analyzed by FE-SEM (Fig. 7). As seen in the figure, it was observed that the MIL-53(Fe) possess a smooth surface with micro rod-like structures.36
 |
| | Fig. 7 FE-SEM images of (a & b) MIL-53(Fe)/hydrated (c & d) MIL-53(Fe)/dehydrated samples. | |
3.1 HER measurements
Cyclic voltammetry, linear sweep voltammetry, a Tafel plot and electrochemical impedance spectroscopy were recorded at ambient temperature using a CHI Instruments electrochemical workstation. The standard three electrodes were used to determine the electrochemical characteristics of the MIL-53(Fe)/hydrated–dehydrated electrocatalysts, where the working electrode was a modified glassy carbon electrode (GCE) with a surface area of 0.071 cm2 (diameter of the electrodes was 3 mm), Pt wire was used as a counter electrode and Ag/AgCl was used as reference electrodes. All electrochemical performances were investigated in a 1 M NaOH electrolyte. Linear sweep voltammetry was tested at a scan rate of 2 mV s−1. Electrochemical impedance measurements (EIS) were carried out with an amplitude of 0.1 Hz to 105 Hz and the electrochemical double layer capacitance (Cdl) measurement was performed in a potential window where no faradaic process occurs at a varied scan rate of 2 mV s−1, 20 mV s−1, 40 mV s−1, 60 mV s−1, 80 mV s−1, 100 mV s−1. ECSA was calculated from the specific capacitance37 and TOF was calculated from the specific current density.38 The electrolyte was freshly prepared with analytical grade reagents and purified water.
3.2 Polarization test analysis and Tafel kinetic mechanism
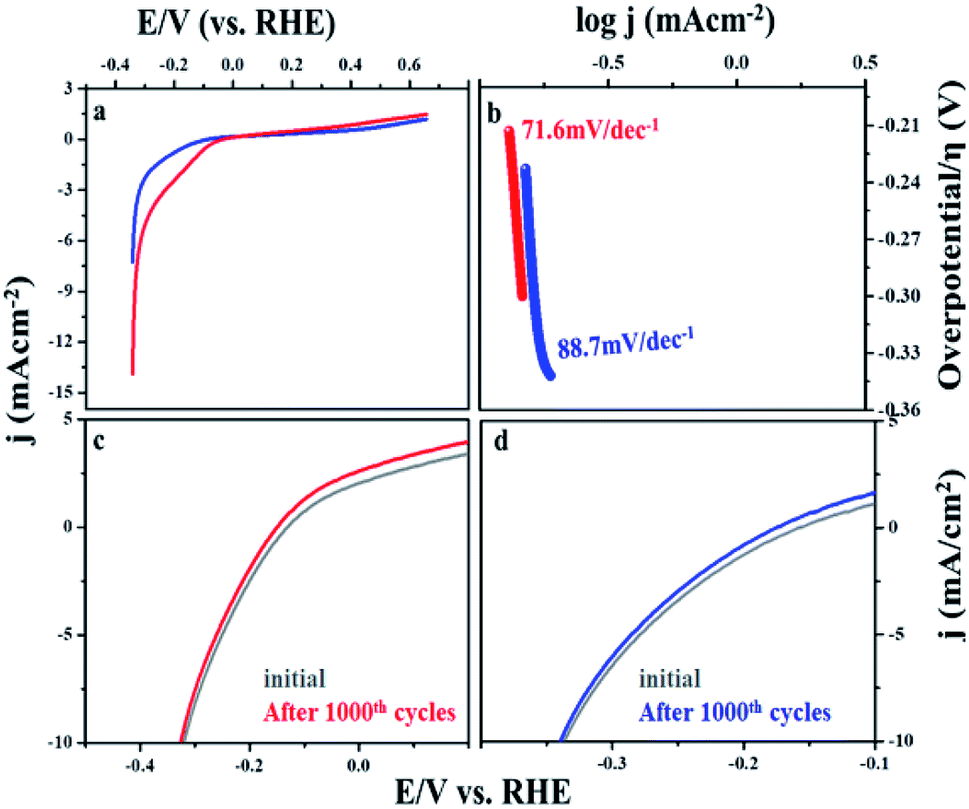
The steady state polarization plots of the MIL-53(Fe)/hydrated–dehydrated samples in 0.1 M NaOH are given Fig. 8. The polarization technique is a systematic and an effective method to investigate the electrochemical activity of the electrocatalysts.39 From the figure, the HER onset potentials of the hydrated and dehydrated samples were observed at −0.155 V and −0.175 V, respectively. The catalytic activity of the MIL-53(Fe)/hydrated–dehydrated samples are slightly different from one another, which might be due to the difference in the electron transfer capability. The exchange current density (Jo) can be accessed by extrapolation of the Tafel curves.40 The value was calculated to be 1.6 × 10−4 mA cm−2 and 2.5 × 10−4 mA cm−2 for the dehydrated and hydrated state, respectively. As compared to MIL-53(Fe)/dehydrated catalysts, MIL-53(Fe)/hydrated catalysts have a higher exchange current density thus showing a faster electrochemical charge transfer through the catalyst–solution interface. An overall result shows a low onset potential with a higher current density at a potential range between −0.6 V to 0.4 V. The enhanced HER activity is further illustrated by comparing with the Tafel slope. The Tafel plots obtained from over potential vs. log current density (η vs. log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) j) are given in Fig. 8b, which could be exploited for quantitative kinetic analysis of HER. The linear regions of the Tafel plots were fitted into the Tafel equation (η = a + b logj), where b is the Tafel slope, j is the current density and a is a constant.41 In general, three principle steps for converting H+ to H2 have been postulated for HER in an alkaline medium, commonly named a Volmer reaction, Heyrovsky reaction and Tafel reaction. A Volmer reaction refers to a proton discharge electrosorption (eqn (1)), an electrochemical desorption is a Heyrovsky reaction (eqn (2)) and the Tafel reaction is a recombination of the two surface absorbed hydrogen atoms (eqn (3)),
j) are given in Fig. 8b, which could be exploited for quantitative kinetic analysis of HER. The linear regions of the Tafel plots were fitted into the Tafel equation (η = a + b logj), where b is the Tafel slope, j is the current density and a is a constant.41 In general, three principle steps for converting H+ to H2 have been postulated for HER in an alkaline medium, commonly named a Volmer reaction, Heyrovsky reaction and Tafel reaction. A Volmer reaction refers to a proton discharge electrosorption (eqn (1)), an electrochemical desorption is a Heyrovsky reaction (eqn (2)) and the Tafel reaction is a recombination of the two surface absorbed hydrogen atoms (eqn (3)),| | |
M + H3O + e− → MHads + H2O (b ∼ 120 mV)
| (1) |
| | |
MHads + MHads → M + H2 (b ∼ 40 mV)
| (2) |
| | |
MHads + H3O + e− → 2M + H2 + H2O (b ∼ 30 mV)
| (3) |
where MHads represents the absorbed hydrogen atom over the surface metal and M represents the catalytically active free sites for HER.42 The Tafel slope of MIL-53(Fe)/hydrated is 71.6 mV per decade and for the MIL-53(Fe)/dehydrated catalyst it is 88.7 mV per decade at an overpotential of 214.4 mV and 233.3 mV, respectively. These Tafel slopes suggest the electrodesorption process as the rate-limiting step following the Volmer–Heyrovsky mechanism. A low Tafel slope indicates that the number of accessible active sites on the catalyst has increased. Thus, the results indicate that the MIL-53(Fe)/hydrated and MIL-53(Fe)/dehydrated catalyst exhibit a low onset potential, high exchange current density, and small Tafel slope, suggesting a good electrocatalytic activity for HER application, as shown in Table 2.
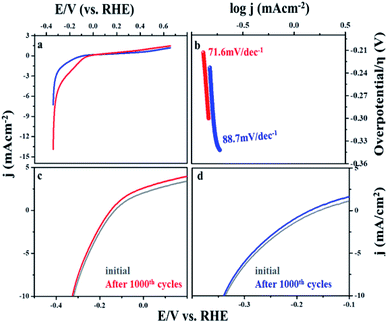 |
| | Fig. 8 (a) HER activity in linear sweep voltammetry (LSV) MIL 53(Fe) dehydrated and hydrated. (b) Tafel polarization peak MIL 53(Fe) dehydrated and hydrated (c and d) cycle stability test for MIL 53(Fe) dehydrated and hydrated form. | |
Table 2 Tafel slope and exchange current density for HER at MIL-53(Fe)/hydrated and MIL-53(Fe)/dehydrated catalyst based electrodes
| S. no |
Catalyst |
−logjo mA cm−2 |
b mV dec−1 |
α |
| 1 |
MIL-53(Fe)/hydrated |
2.5 × 10−4 mA cm−2 |
71.6 |
0.83 |
| 2 |
MIL-53(Fe)/dehydrated |
1.6 × 10−4 mA cm−2 |
88.7 |
0.67 |
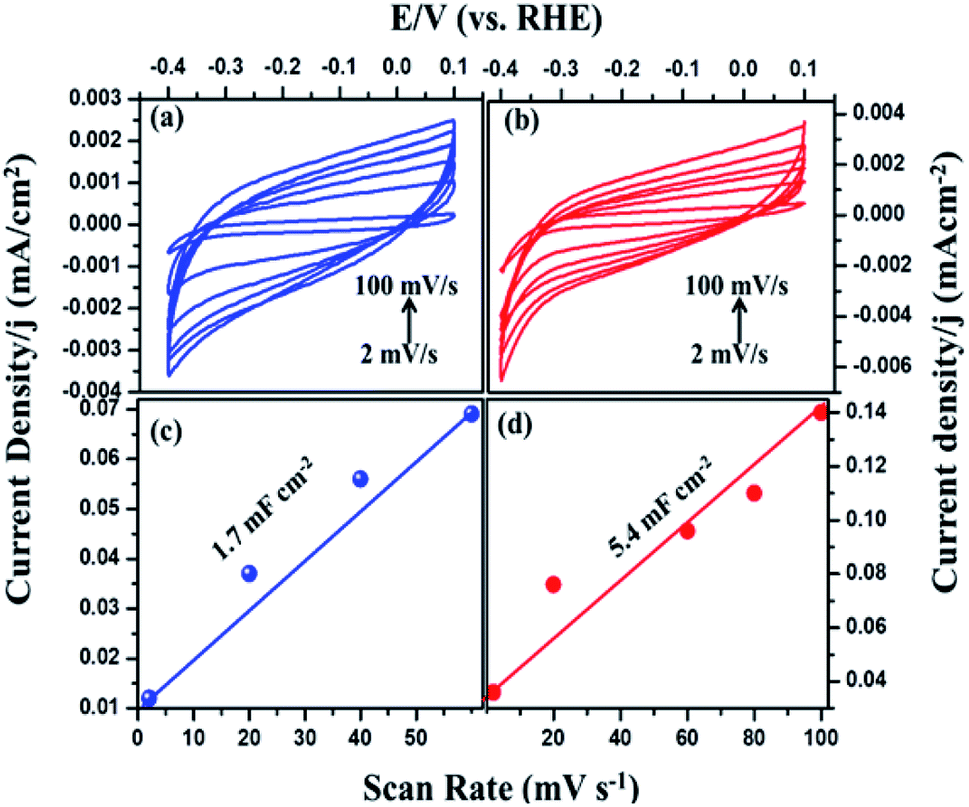
The intrinsic activity of the catalyst materials was studied using the turnover frequency (TOF), which represents the number of hydrogen molecules produced per second per active site of Fig. S3 (see ESI†).43 TOF values of the MIL-53(Fe)/hydrated and dehydrated catalysts were calculated to be 3.0 s−1 and 1.1 s−1 in the case of the MIL-53(Fe)/hydrated form. These values indicate that the MIL-53(Fe)/hydrated catalyst shows better catalytic activity per site compared to the MIL-53(Fe)/dehydrated catalyst. To calculate the TOF, a main factor that needs to be calculated is the measurement of the number of active sites (n) and the electrochemically active surface area (ECSA). The above TOF calculations were made assuming that the surface exposed atoms are the active sites. The estimation of the electrochemically active surface area was conducted by a measurement of the double-layer capacitance in a potential region with no faradaic response following McCory's et al., methodology.44 To better understand the influence of the enhanced HER performance, the electrochemical surface area (ECSA) was evaluated by measuring the double layer capacitance, Cd, as Cdl is proportional to the ECSA.45 The capacitance was obtained from the plots (Fig. 9), which clearly show that the double capacitance layer (Cdl) was higher for MIL-53(Fe)/hydrated (5.4 mF cm−2) compared to the MIL-53(Fe)/dehydrated catalysts (1.7 mF cm−2), indicating the high exposure active site edge in the MIL-53(Fe)/hydrated catalysts in an alkaline medium.
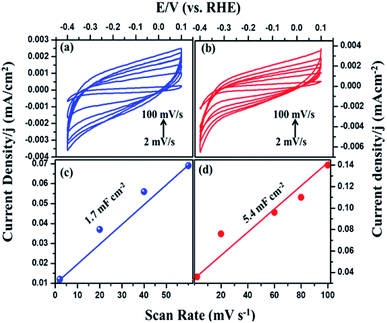 |
| | Fig. 9 Cyclic voltammetry of MIL-53(Fe) (a) dehydrated and (b) hydrated and (c and d) plots showing the extraction of the Cdl for MIL-53(Fe)/dehydrated and hydrated. | |
3.3 Electrochemical impedance spectroscopy
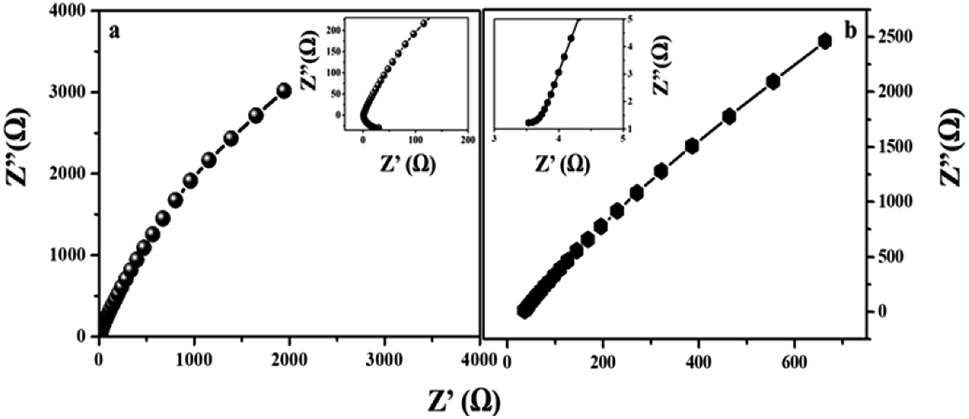
The electrode kinetics under a catalytic HER operating condition was investigated using electrochemical impedance spectroscopy (EIS) to compare the internal resistance of the electrode material (Fig. 10). In the Nyquist plot, the x and y axes symbolize the real and negative imaginary parts of impedance. The intercept at the x-axis gives the solution resistance (Rs) of the cell and the diameter of the semicircle gives the charge transfer resistance (Rct), which is due to the electron conductivity of the material.45
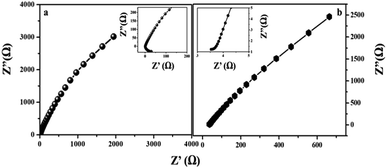 |
| | Fig. 10 Nyquist plots of MIL-53(Fe) (a) hydrated and (b) dehydrated sample electrodes by applying a sine wave with an amplitude of 200 mV and a frequency range from 0.1 Hz to 105 Hz. (Inset. Enlargement of the Nyquist plot of MIL-53(Fe)/hydrated and dehydrated and EIS circuit plot.) | |
After circuit fitting, the charge transfer resistance value of the MIL-53(Fe)/dehydrated sample was calculated to be 18.4 Ω and 1.9 Ω in the case of the MIL-53(Fe)/hydrated form at an applied voltage of 220 mV and an applied frequency from 0.1 Hz to 105 Hz. In the case of the MIL-53(Fe)/hydrated catalyst, a lower resistance is exhibited between the interfaces, thus showing an excellent HER activity in an alkaline electrolyte as compared to the MIL-53(Fe)/dehydrated catalyst.
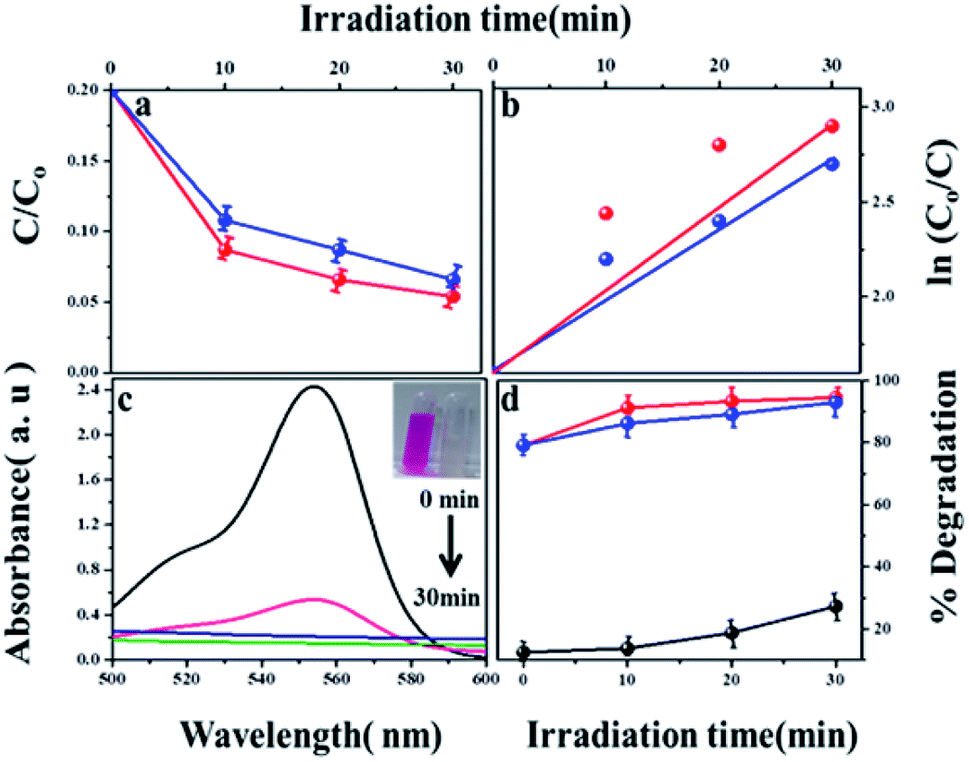
The photocatalytic degradation of refractory organic pollutants in aqueous solutions has importance in various fields such as industry, agriculture and so on. It provides a simple way to examine advanced oxidation processes (AOPs) such as photocatalysis and catalytic activity.46 To further understand the property of the material towards catalysis, MIL-53(Fe) was tested as photocatalysts. Fig. 11(a) shows the variation of the rhodamine-B concentration (C/Co) vs. time for the degradation process using dehydrated and hydrated forms of the synthesized MIL-53(Fe) catalyst. The photons are generally absorbed onto the surface of the photocatalyst. If the surface area of the photocatalyst is enhanced, it leads to an increase in the amount of absorption of photons, which in turn leads to an increase in the recombination rate of electron–hole pairs. The hydrated form of MIL-53(Fe) has more surface area compared to its dehydrated form, thus enabling an increased amount of absorption of organic pollutants, thereby increasing the degradation efficiency. Fig. 11(d) shows the percentage degradation of the dehydrated and hydrated forms of MIL-53(Fe). In the absence of H2O2, the degradation percentage of the dehydrated and hydrated forms of MIL-53 Fe is 47% and 56%, respectively, within 30 min of the process. This is due to the capability of the material to generate the reactive charge carrier. In the presence of H2O2, the degradation efficiency improves by 27.3%, which is due to an increase in the OH radicals. The dehydrated and hydrated forms of MIL-53(Fe) with H2O2 have a degradation efficiency of 93% and 95%, respectively. The variation in the absorbance peak of the hydrated form of MIL-53(Fe) with H2O2 and 20 ppm rhodamine-B, with respect to time, can be seen in Fig. 11(c). After 30 minutes of reaction, it can be seen that the absorption peak has been completely eliminated, which indicates that RhB is degraded under visible light illumination. Fig. 11(b) shows the pseudo-first order kinetics reaction, which describes the relationship between the linear plots of ln(C/Co) vs. irradiation time in Fig. 11(b)
where
K is the rate constant,
t is irradiation time (min
−1),
Co is the concentration of RhB dye and
C is the concentration of RhB at regular time intervals. The rate constant of the MIL-53(Fe)/dehydrated sample is 0.09 min
−1 and the rate constant of MIL-53(Fe)/hydrated is 0.097 min
−1. Thus, the rate constant of the MIL-53(Fe)/hydrated catalyst is slightly higher than the MIL-53(Fe)/dehydrated catalyst.
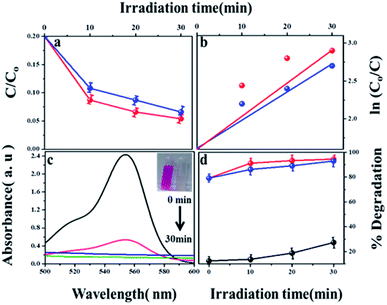 |
| | Fig. 11 (a) Photodegradation of RhB with H2O2 using MIL-53(Fe)/dehydrated and hydrated (b) first order kinetics curve of RhB dye degradation (c) absorption spectrum of RhB with MIL-53(Fe)/hydrated (d) percentage of degradation of H2O2, MIL-53(Fe)/dehydrated and hydrated. | |
4. Conclusions
In summary, MIL-53(Fe)/dehydrated and MIL-53(Fe)/hydrated have been successfully synthesized by a solvothermal method for HER and photocatalytic activity due to their good activity, conductivity, and stability. While MIL-53(Fe)/dehydrated and MIL-53(Fe)/hydrated show a good electrocatalytic activity and photocatalytic activity, the MIL-53(Fe)/hydrated catalyst demonstrates an obviously enhanced catalytic activity for HER with a high exchange current density compared to MIL-53(Fe)/dehydrated. MIL-53(Fe)/hydrated had an exchange current density of 2.5 × 10−4 mA cm−2, and MIL-53(Fe)/dehydrated had an exchange current density of 1.6 × 10−4 mA cm−2. MIL-53(Fe)/hydrated and MIL-53/dehydrated had Tafel slopes of 71.6 mV dec−1, 88.7 mV dec−1, respectively. The Tafel responses of the electrodes show a Volmer–Heyrovsky mechanism during HER. Thus, in result, the breathing behaviour of the MIL-53(Fe) hydrated and dehydrated states showed that the hydrated form has better activity than that of the dehydrated form for the following three possible reasons: (i) surface area and pore volume (ii) photochemical properties and recombination efficiency and (iii) stability. The high surface area and pore volume material has a high conductivity and fast electron transport, effective ion adsorption/desorption and relevant pore architecture for the rapid approach of ions from the electrolyte solution to the catalyst surface. The MIL-53(Fe)-hydrated surface area is 52.813 m2 g−1 and the dehydrated form surface area is 43.75 m2 g−1. The hydrated form has a higher surface area compared to the dehydrated form. This is one of the possible reasons for the better activity of the MIL-53(Fe) – hydrated state than the dehydrated state. A weaker intensity in the PL spectrum indicates a faster recombination rate of a photo-induced electron–hole pair, attributed to the higher photocatalytic activities, thus indicating better activity for the hydrated form. In the hydrous environment, it is more stable than in the anhydrous state because the water molecules provide further stabilization through hydrogen bonding. In the anhydrous state, water molecules are not present in the cavities of the material and hence a network of hydrogen bonded water molecules does not occur to support its stability. This is one of the reason that the hydrated form had better activity than the dehydrated form.
Conflicts of interest
There are no conflicts to declare.
References
- J. N. Armor, Catal. Lett., 2005, 101, 131–145 CrossRef CAS.
- N. H. Afgan, A. Veziroglub and M. G. Carvalho, Int. J. Hydrogen Energy, 2007, 32, 3183–3193 CrossRef CAS.
- A. Contreras, R. Guirado and T. N. Veziroglu, Int. J. Hydrogen Energy, 2007, 32, 4635–4640 CrossRef CAS.
- H. Dong, T. Lei, Y. He, N. Xu, B. Huang and C. T. Liu, Int. J. Hydrogen Energy, 2011, 36, 12112–12120 CrossRef CAS.
- H. Mengci, F. Kong, G. Yin, Z. Lv, X. Sun, H. Shi and B. Gao, RSC Adv., 2018, 8, 14369–14376 RSC.
- L. Wu, X. Guo, Y. Xu, Y. Xiao, J. Qian, Y. Xu, Z. Guan, Y. He and Y. Zeng, RSC Adv., 2017, 7, 32264–32274 RSC.
- A. L. iju Elias and C. hitharanjan Hegde, RSC Adv., 2016, 6, 66204–66214 RSC; Y. Zhao, S. Wang, C. Li, X. Yu, C. Zhu, X. Zhang and Y. Chen, RSC Adv., 2016, 6, 7370–7377 RSC.
- M. A. Amin, N. El-Bagoury, M. H. H. Mahmoud, M. M. Hessien, S. S. Abd El-Rehim, J. Wysock and J. Ryl, RSC Adv., 2017, 7, 3635–3649 RSC.
- G. B. de-Mello, L. Smith, S. J. Rowley-Neale, J. Gruber, S. J. Hutton and C. E. Banks, RSC Adv., 2017, 7, 36208–36213 RSC.
- R. K. Das, Y. Wang, S. V. Vasilyeva, E. Donoghue, I. Pucher, G. Kamenov, H. P. Cheng and A. G. Rinzler, ACS Nano, 2014, 8, 8447–8456 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 974–986 CrossRef CAS PubMed.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- H. Ogihara, M. Fujiia and T. Saji, RSC Adv., 2014, 4, 58660–58663 RSC.
- L. Song, M. Zhao, X. Li, Z. Zhang and L. Qua, RSC Adv., 2016, 6, 70740–70746 RSC.
- T. Tian, L. Ai and J. Jiang, RSC Adv., 2015, 5, 10290–10295 RSC.
- R. Kitaura, K. Seki, G. Akiyama and S. Kitagawa, Angew. Chem., 2003, 42, 428–431 CrossRef CAS PubMed.
- D. Li and K. Kaneko, Chem. Phys. Lett., 2001, 335, 50–56 CrossRef CAS.
- C. Serre, C. Mellot-Draznieks, S. Surble, N. Audebrand, Y. Filinchuk and G. Ferey, Science, 2017, 315, 1828–1831 CrossRef PubMed.
- F. Millange, N. Guillou, R. I. Walton, J. Grene`che, I. Margiolaki and G. Ferey, Chem. Commun., 2008, 39, 4732–4734 RSC.
- P. G. Yot, Z. Boudene, J. Macia, D. Granier, L. Vanduyfhuys, T. Verstraelen, V. Van Speybroeck, T. Devic, C. Serre, G. Férey, N. Stock and G. Maurin, Chem. Commun., 2014, 50, 462 RSC.
- F. Niekiel, J. Lannoeye, H. Reinsch, A. S. Munn, A. Heerwig, I. Zizak, S. Kaskel, R. I. Walton, D. de Vos, P. Llewellyn, A. Lieb, G. Maurin and N. Stock, Inorg. Chem., 2014, 53, 4610 CrossRef CAS PubMed.
- T. Loiseau, C. Serre, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille and G. Férey, Chem.–Eur. J., 2004, 10, 1373–1382 CrossRef CAS PubMed.
- P. Horcajada, C. Serre, G. Maurin, N. A. Ramsahye, F. Balas, M. A. Vallet-Regí, M. Sebban, F. Taulelle and G. R. Ferey, J. Am. Chem. Soc., 2008, 130, 6774–6780 CrossRef CAS PubMed.
- T. R. Whitfeld, X. Wang, L. Liu and A. J. Jacobson, Solid State Sci., 2005, 7, 1096–1103 CrossRef.
- A. Banerjee, R. Gokhale, S. Bhatnagar, J. Jog, M. Bhardwaj, B. Lefez, B. Hannoyer and S. Ogale, J. Mater. Chem., 2012, 22, 19694–19699 RSC.
- C. Costentin, M. Robert and J. M. Saveant, J. Am. Chem. Soc., 2006, 128, 8726–8727 CrossRef CAS PubMed.
- P. Horcajada, C. Serre, G. Maurin, N. A. Ramsahye, F. Balas, M. A. Vallet-Regí, M. Sebban, F. Taulelle and G. r. Ferey, J. Am. Chem. Soc., 2008, 130, 6774–6780 CrossRef CAS PubMed.
- R. El Osta, A. C. Sinclair, N. Guillou, R. I. Walton, F. Vermoortele, M. Maes, D. de Vos and F. Millange, Chem. Mater., 2012, 24, 2781–2791 CrossRef CAS.
- Y. Dosseh, C. Xia and G. A. Simionesco, J. Phys. Chem. B, 2003, 107, 6445–6453 CrossRef.
- G. T. Vuong, M. H. Pham and T. O. Do, Dalton Trans., 2013, 42, 550–557 RSC.
- G. T. Vuong, M. H. Pham and T. O. Do, CrystEngComm, 2013, 15, 9694–9703 RSC.
- M. Long, W. Cai, J. Cai, B. Zhou, X. Chai and Y. Wu, J. Phys. Chem., 2006, 110, 20211–20216 CrossRef CAS.
- Y. Wang, W. Chu, S. Wang, Z. Li, Y. Zeng, S. Yan and Y. Sun, ACS Appl. Mater. Interfaces, 2014, 6, 20197–20204 CrossRef CAS PubMed.
- S. Bordiga, C. Lamberti, G. Ricchiardi, L. Regli, F. Bonino, A. Damin, K. P. Lillerud, M. Bjorgen and A. Zecchina, Chem. Commun., 2014, 20, 2300–2301 Search PubMed.
- A. Tuan, D. Dao, L. Q. Dang, K. T. Nguyen, Q. K. Nguyen, P. T. Dang, H. T. K. Tran, Q. T. Duong, T. V. Nguyen and G. D. Lee, RSC Adv., 2015, 5, 5261–5268 RSC.
- C. Zhang, L. Ai and J. Jiang, Ind. Eng. Chem. Res., 2015, 54, 153–163 CrossRef CAS.
- H. Topsøe, Appl. Catal., 2007, 322, 3–8 CrossRef.
- J. Guo, H. Zhu, S. Zhou, Y. Sun and X. Zhang, J. Mater. Chem. A, 2015, 21, 2993–2999 CAS.
- L. A. Kibler, ChemPhysChem, 2006, 7, 985–991 CrossRef CAS.
- Y.-J. Tang, M.-R. Gao, C.-H. Liu, S.-L. Li, H.-L. Jiang and Y. Q. Lan, Angew. Chem., Int. Ed., 2015, 54, 12928–12932 CrossRef CAS.
- R. Ganesan and J. S. Lee, J. Power Sources, 2006, 217, 157 Search PubMed.
- R. K. Shervedani and A. R. Madram, Int. J. Hydrogen Energy, 2008, 33, 2468–2476 CrossRef.
- X.-Y. Yu, Y. Feng, Y. Jeon, B. Guan, X. W. Lou and U. Paik, Adv. Mater., 2016, 28, 9006–9011 CrossRef CAS.
- F. Song and X. Hu, Nat. Commun., 2014, 5, 4477 CrossRef CAS PubMed.
- Q. Wang, L. Jiao, H. Du, Y. Si, Y. Wang and H. Yuan, J. Mater. Chem., 2012, 22, 21387–21391 RSC.
- L. Qin, Z. Li, Z. Xu, X. Guo and G. Zhang, Appl. Catal., B, 2015, 179, 500–508 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08208a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence d,
Sudhagar Pitchaimuthue,
Soon Kwan Jeongf and
Andrews Nirmala Grace
d,
Sudhagar Pitchaimuthue,
Soon Kwan Jeongf and
Andrews Nirmala Grace