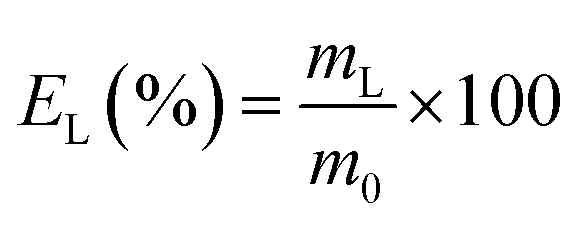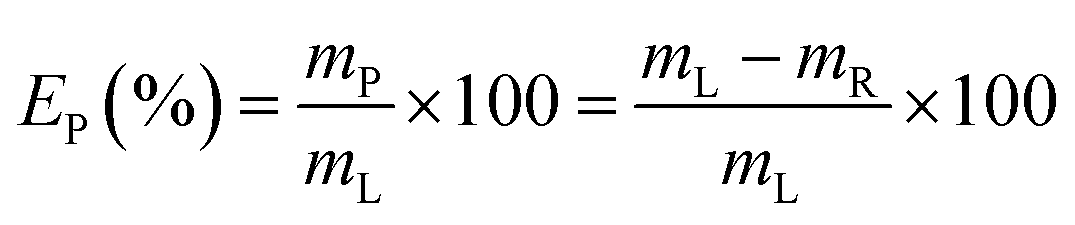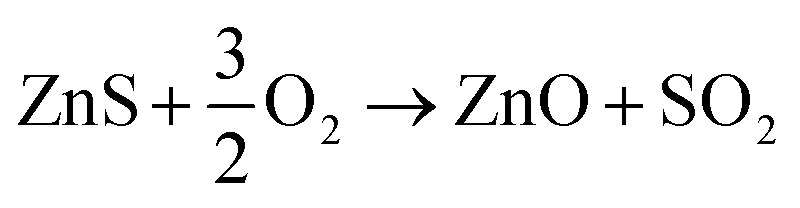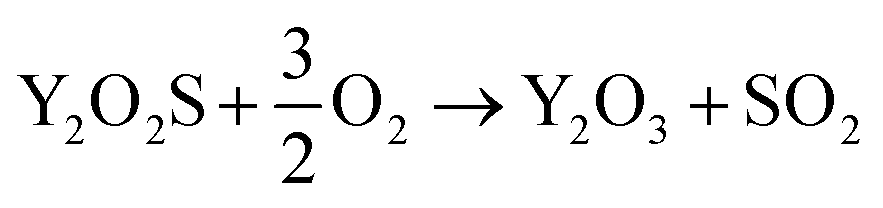DOI:
10.1039/C8RA08158A
(Paper)
RSC Adv., 2019,
9, 1378-1386
Integrated process for the recovery of yttrium and europium from CRT phosphor waste†
Received
2nd October 2018
, Accepted 21st December 2018
First published on 11th January 2019
Abstract
An integrated process flow sheet for the recovery of yttrium and europium from waste cathode-ray tube (CRT) phosphors was developed. This flow sheet is based on a sequence of roasting, leaching with organic acids and precipitation steps. Zinc was efficiently removed from the roasted CRT phosphors by leaching with acetic acid, giving access to the rare earth content. Yttrium and europium were quantitatively leached from the residue by a 1 mol L−1 methanesulphonic acid (MSA) solution. Precipitation with oxalic acid gave a mixed Y/Eu oxalate of high purity (>99 wt%). Co-precipitation of zinc was less than 2 wt%.
Introduction
Cathode-ray tubes (CRTs) have widely been used in the past in television screens and computer monitors.1–3 This technology is currently declining, due to the replacement of CRT-based screens by LCDs, LCD/LEDs and plasma discharge displays.4 A large number of CRTs are thus being currently discharged and need to be properly treated. It is reported that in Europe 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–150000 tons of end-of-life CRT screens are collected every year.5 One of the main components of a CRT is the panel glass, which is covered by a mixture of luminescent powders for producing images by excitation with an electron beam (electroluminescence). The blue and green phosphors are based on zinc sulphide (ZnS:Ag for the blue phosphor and ZnS:Cu for the green phosphor), while the red phosphor in the later generations of CRTs is Y2O2S:Eu3+.4 This phosphor is rich in the rare-earth elements (REEs) yttrium and europium and is a potential secondary resource for these critical raw materials. As reported by Tian et al. (2016), CRT phosphor powder accounts for about 7 g in a CRT screen and it contains about 30% Zn, 4% Al, 16% Y, 1% Eu and 17% S.1 Yttrium and europium concentration values are much higher when compared to their content in primary ores.6 This makes their recovery from the selected waste stream of potential interest. Although many papers have focused on the recovery of REEs from fluorescent lamp phosphor waste,7–13 much fewer studies on REEs recycling from CRTs are available.4 The treatment of CRT phosphor waste is challenging and several issues still need to be addressed. In hydrometallurgical processes for recycling of CRT phosphors, the waste phosphor powder mixture is leached with strong acids, such as H2SO4.14–19 Due to the presence of sulphides, the leaching with strong acids causes the release of toxic H2S gas, unless an oxidising agent is used.4 Dexpert-Ghys et al. proposed a two-step process to recover Y2O2S:Eu3+: an alkaline attack with NaOH/NaClO, aimed at the selective dissolution of ZnS, followed by thermal treatment at 1300–1400 °C to convert the rare-earth oxysulphide Y2O2S:Eu3+ into the rare-earth oxide Y2O3:Eu3+.20 An alternative approach was proposed by Pan et al., in which roasting of the phosphors with NH4Cl was followed by leaching of YCl3 and EuCl3 with water.21 A recovery process was proposed by Önal and Binnemans, which is based on sulphating roasting in the presence of ZnSO4.22 In this way, Y2O2S:Eu3+ was transformed into rare-earth sulphates that could be afterwards selectively leached by water. Recently, subcritical water extraction (SWE) was proven to be an effective method for the leaching of yttrium and europium from waste CRT phosphor; quantitative dissolution was achieved by using 0.75 mol L−1 H2SO4 within 30 minutes at 125 °C, with high selectivity over zinc and lead.23
000–150000 tons of end-of-life CRT screens are collected every year.5 One of the main components of a CRT is the panel glass, which is covered by a mixture of luminescent powders for producing images by excitation with an electron beam (electroluminescence). The blue and green phosphors are based on zinc sulphide (ZnS:Ag for the blue phosphor and ZnS:Cu for the green phosphor), while the red phosphor in the later generations of CRTs is Y2O2S:Eu3+.4 This phosphor is rich in the rare-earth elements (REEs) yttrium and europium and is a potential secondary resource for these critical raw materials. As reported by Tian et al. (2016), CRT phosphor powder accounts for about 7 g in a CRT screen and it contains about 30% Zn, 4% Al, 16% Y, 1% Eu and 17% S.1 Yttrium and europium concentration values are much higher when compared to their content in primary ores.6 This makes their recovery from the selected waste stream of potential interest. Although many papers have focused on the recovery of REEs from fluorescent lamp phosphor waste,7–13 much fewer studies on REEs recycling from CRTs are available.4 The treatment of CRT phosphor waste is challenging and several issues still need to be addressed. In hydrometallurgical processes for recycling of CRT phosphors, the waste phosphor powder mixture is leached with strong acids, such as H2SO4.14–19 Due to the presence of sulphides, the leaching with strong acids causes the release of toxic H2S gas, unless an oxidising agent is used.4 Dexpert-Ghys et al. proposed a two-step process to recover Y2O2S:Eu3+: an alkaline attack with NaOH/NaClO, aimed at the selective dissolution of ZnS, followed by thermal treatment at 1300–1400 °C to convert the rare-earth oxysulphide Y2O2S:Eu3+ into the rare-earth oxide Y2O3:Eu3+.20 An alternative approach was proposed by Pan et al., in which roasting of the phosphors with NH4Cl was followed by leaching of YCl3 and EuCl3 with water.21 A recovery process was proposed by Önal and Binnemans, which is based on sulphating roasting in the presence of ZnSO4.22 In this way, Y2O2S:Eu3+ was transformed into rare-earth sulphates that could be afterwards selectively leached by water. Recently, subcritical water extraction (SWE) was proven to be an effective method for the leaching of yttrium and europium from waste CRT phosphor; quantitative dissolution was achieved by using 0.75 mol L−1 H2SO4 within 30 minutes at 125 °C, with high selectivity over zinc and lead.23
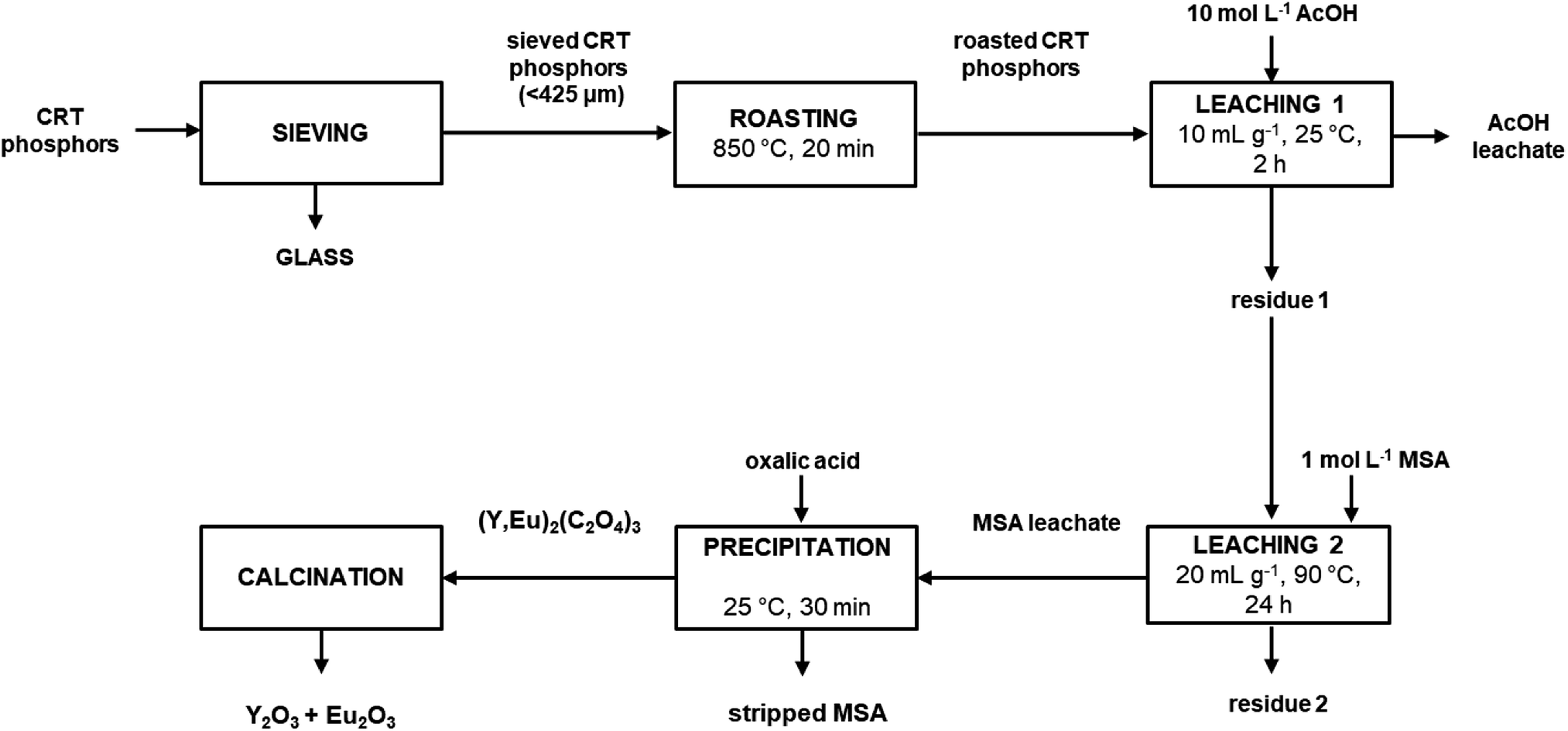
In this paper, an alternative approach is proposed to recover yttrium and europium from waste CRT powder, which overcomes the issues related to the release of toxic H2S gas during leaching by making use of an integrated process. The first step of the recovery process is a roasting step at relatively high temperature (850 °C) to oxidise the zinc sulphide to zinc oxide. In the second step, the oxidised material is selectively leached with acetic acid (CH3COOH, AcOH), giving access to the rare earth-rich fraction of the waste powder. Yttrium and europium are recovered from the leaching residue by a second leaching step with methanesulphonic acid (CH3SO3H, MSA), a biodegradable lixiviant with low toxicity.24 Finally, precipitation with oxalic acid gives a mixed Y/Eu oxalate which after calcination yielded the corresponding mixed oxide.
Experimental
Chemicals and materials
The CRT phosphor powder was obtained from Relight srl (Rho, Italy). In their treatment plant, waste cathode-ray tube televisions and screens are processed in a dedicated section through a disassembly step aimed at separating the several components, such as metallic fractions, plastic casings, cables, printed circuit boards and the cathode-ray tube itself.25 The CRT is then cut in two parts, i.e. the cone and the screen glass. The fluorescent powder, which covers the screen glass, is carefully removed by brushing and under aspiration. Lithium metaborate (reagent grade, >99 wt%) was obtained from Scharlau S.L, lithium tetraborate (78 wt%) from XRF Scientific and lithium bromide (250 g L−1 solution) from ICPH. Zinc oxide (>99%), sodium chloride (>99%) and zinc acetate dihydrate (>99.5%) were obtained from Panreac. Calibration of the alkaline fusion analysis was carried out with 88b Dolomitic Limestone Standard Reference Material from NIST, DC60104 Clay and DC 73303 Rock Certified Reference materials from NCS, No. 156 Slag from Czechslovak Reference Material, BCS-CRM 381 and 382 Basic Slags from British Chemical Standard Certified Reference Material. Hydrofluoric acid (48 wt%) was obtained from Scharlau S.L and Fisher Scientific, nitric acid (65 wt%) from Sigma-Aldrich and Chem Lab, hydrochloric acid (37 wt%) from Honeywell/Fluka and VWR Chemicals. Acetic acid (100 wt%) was purchased from VWR Chemicals, methanesulphonic acid (99.5%) from Carl Roth and oxalic acid (≥99.9%) from Merck. Boric acid >95% and ≥99.5% were obtained from Panreac and Merck, respectively. Triton X-100 was obtained from Acros Organics and the silicone solution in isopropanol from SERVA Electrophoresis GmbH. The standard solutions (1000 μg mL−1 Ga, Dy, Zn, Y, Eu, Sc, La in 3–5 wt% nitric acid) were obtained from ChemLab. Water was always of ultrapure quality. All chemicals were used as received without any further purification.
Analytical techniques
For the analysis of Al, Ca, Fe, K, Mg, Na and Si, the initial CRT phosphor waste was dissolved by alkaline fusion with lithium metaborate.26 The melt was dissolved in diluted nitric acid, based on ISO 14869-2.27 For the analysis of Y, Eu, Zn, S, Sr and Pb, microwave-assisted acid digestion based on the 3052 EPA method was used.28 A representative amount of sample was digested in 2 mL of concentrated nitric acid, 6 mL of concentrated hydrochloric acid and 1 mL of hydrofluoric acid (48 wt%). A second digestion procedure with boric acid was performed to permit the complexation of fluoride and thus protect the quartz plasma torch of the analytical instrument. Both solutions were analysed by Inductively Coupled Plasma-Optical Emission Spectrometry (ICP-OES) using an axial Agilent VISTA-MPX spray chamber.29 The mineralogical composition of the CRT powder was determined by X-ray diffraction analysis (XRD) with a Bruker D8 Advance X-ray diffractometer with a CuKα source and a scintillation detector from 15° to 90° 2θ using a step width of 0.030° and 1 second per step. Phase identification was performed with EVA software. In addition, the CRT powder was analysed by Scanning Electron Microscopy (SEM) using a Jeol SM-5910LV apparatus. The images were taken in high vacuum mode with 20 kV voltage and a solid-state backscatter detector (SSD-BSD). An Oxford Instruments INCAx-act Energy Dispersive X-ray detector (EDS) was used for chemical semi-quantitative analysis. The sample was embedded under vacuum conditions in epoxy resin, pre-polished with abrasive powder and coated with a thin layer of graphite. The zinc mineralogy of the roasted CRT phosphors was determined as follows: separation of sulphide from the sample matrix was accomplished by suspending the sample in concentrated HCl by vigorous agitation. The sample was distilled under acidic conditions at 100 °C using an argon stream to avoid oxidation of sulphides. H2S gas was released from the sample and collected in gas scrubbing bottles containing zinc(II) and a strong acetate buffer. Sulphide was precipitated as zinc sulphide (EPA Method 9030B). The zinc sulphide precipitate was quantified by titration as reported in the EPA Method 9034. The quantification of zinc oxide and zinc silicate was performed by XRD using ZnO and Zn2SiO4 reference standards. The calibration curve was carried out with standards of 12.5, 25 and 50 wt% of ZnO. To complete the standards, NaCl was used. The areas of two peaks free of interferences (2θ = 36.2° and 67.9°) were measured in each diffractogram and two curves were created with the areas obtained in the three standards in each peak. The measured areas were matched with the curves and then the concentration of ZnO was calculated with the average of the two peaks. Zn2SiO4 was used for the XRD quantification. To complete the standards, ZnO and NaCl were used. Then, the following standards were prepared: 10 wt% Zn2SiO4/40 wt% ZnO/50 wt% NaCl, 15 wt% Zn2SiO4/35 wt% ZnO/50 wt% NaCl, 30 wt% Zn2SiO4/30 wt% ZnO/40 wt% NaCl. The area of one peak free of interferences (2θ = 38.7°) was measured in each diffractogram and one curve was created with the areas obtained for the three standards. The areas measured in the samples were matched with the curves and then the concentration of Zn2SiO4 was calculated. The balance to 100% was assigned to other zinc compounds. Yttrium and europium content in the roasted CRT samples was determined by borate fusion by using a LENEO Fusion Instrument. A sample of 100 mg was mixed in a platinum crucible with 2.5 g of lithium tetraborate, Li2B4O7 (flux); 20 drops of lithium bromide solution (250 g L−1) were then added (non-wetting agent). The crucible was placed in the furnace and heated at 1050 °C for 6 minutes. Afterwards, the molten mixture was poured into a HNO3 5 vol% solution while stirring. The zinc content was determined by microwave digestion, using the Speedwave Xpert Microwave System. A 100 mg sample was contacted with a mixture of 6 mL of HCl (37 wt%), 2 mL of HNO3 (65 wt%) and 5 mL of HF (48 wt%). This method resulted to be particularly effective for the dissolution of silicates. Yttrium and europium could not be determined through this method due to the formation of insoluble rare-earth fluorides which occurred under the investigated experimental conditions. Differently from the procedure used for the characterization of the initial phosphors, where a lower amount of HF was used for the digestion of the sample (1 mL), in the case of the roasted CRT phosphors a larger volume of HF (5 mL) was employed. This led to a decrease of the total nitric acid concentration in the mixture which affected the rare earths solubility. It is reported that rare earth fluorides solubility monotonously increases with the nitric acid concentration and upon attaining a certain maximum again decreases.30 Samples were, afterwards, analysed by total reflection X-ray fluorescence spectrometry (TXRF) with a Bruker S2 Picofox TXRF spectrometer equipped with a molybdenum source.31 Polypropylene microtubes were filled with a certain amount of sample and an appropriate internal standard was added. The internal standard was selected in such a way that the X-ray fluorescence energy of the standard was as close as possible to the element to be determined in order to reduce the effects caused by secondary X-rays absorption and minimising at the same time peaks overlap. Samples were finally diluted to 1 mL with a Triton X-100 solution (5 wt%).32 After shaking the samples on a vibrating plate (IKA MS 3 basic), a small droplet (1 μL) was put on a quartz carrier, previously treated with a silicone–isopropanol solution to avoid spreading of the droplet. The quartz carriers were then dried for 30 minutes at 60 °C prior to analysis. Each sample was measured for 200 s (at 50 kV voltage and 600 μA current). TXRF was also employed for determining metal concentration in the acetic acid and MSA leachate.
The purity of the final mixed oxide was determined by ICP-OES on a Perkin Elmer Optima 8300 equipped with a Scott spray chamber assembly. Calibration solutions were prepared containing 1–50 ppm Zn, Y and Eu in 2 vol% HNO3; La and Sc were selected as internal standards.
Sieving and particle size distribution analysis
The particle size distribution was measured to test the separation of the glass particles from the CRT fluorescent powder. An industrial circular sieve shaker FTI-0550 (Filtra Vibración, S.L., Spain) was used to obtain the different fractions of the CRT phosphor. The sieve shaker has a material inlet duct and discharge ducts, so it can operate continuously. Chemical analysis of each fraction was performed to determine the concentration of Y, Eu, Zn, S, Na and K, which are the main elements present in the CRT powder. Yttrium and europium concentration were used to calculate the percentage of Y2O2S:Eu3+ in each fraction, while the zinc content is related to the ZnS amount. Finally, the Na and K content was determined because they are representative components of the glass and can be used to determine the amount of glass in each fraction. In this way, the distribution of the three main compounds could be assessed for each fraction.
Roasting
Preliminary roasting tests were carried out in a muffle furnace by placing 10 g of CRT in a 90 × 50 mm porcelain tray. Optimisation tests were performed in an electric rotary furnace HGTE-160 (COEL S.A., Spain), which operates in continuous mode and is equipped with a rotation and lifting control system that allows the slope of the furnace and the rotation speed to be varied. The electric rotary furnace was required during the scaling up to increase the air–solid contact with the sample to ensure homogeneous roasting. The rotary furnace was heated up to the working temperature. Rotation and slope were adjusted to set the residence time. Temperatures from 800 °C to 1000 °C were tested with a residence time varying between 5 and 40 minutes. A flow of 1.6 m3 h−1 of air was injected into the furnace to provide the oxygen required for the roasting reaction. When the operation temperature was reached, the CRT powder was fed at constant flow (about 0.3 kg h−1). The optimisation of the roasting process was based on the conversion of the compounds containing Zn, Y and Eu into their respective oxides and minimising the formation of non-desirable compounds such as zinc silicates or yttrium silicates. Since it is crucial for further treatment to minimise the amount of zinc silicate in the final roasted product, the mineralogy of the zinc-containing phases in the roasted CRT phosphor was determined using XRD analysis.
Leaching
The leaching tests were performed in glass vials located on a mechanical shaker (TMS-200 Turbo Thermoshaker), unless otherwise specified. The roasted CRT phosphors were contacted with acetic acid and several parameters were varied in order to determine the optimal operative conditions. The agitation was kept constant at 2500 rpm in all the experiments. The leachate was separated from the solid residue through centrifugation (5300 rpm, 30 minutes, Thermo Scientific Heraeus Labofuge 200 centrifuge) and analysed by TXRF to determine the leaching efficiency EL (%), which was calculated according to eqn (1):| |
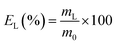 | (1) |
where mL is the mass of the dissolved metal in the leachate (mg) and m0 is the mass of metal in the initial sample (mg). The leaching residue was further treated to recover yttrium and europium. To this aim, MSA was selected as leaching agent. Leaching tests were performed as a function of the lixiviant concentration, by keeping the temperature (T = 90 °C), the liquid-to-solid ratio (L/S = 20 mL g−1) and the contact time (t = 24 h) constant. Unless otherwise specified, each experiment was performed in duplicate.
Y and Eu recovery from the leachate
Yttrium and europium recovery from the leachate was investigated through precipitation with oxalic acid. Precipitation tests were performed in glass vials located on a mechanical shaker, at T = 25 °C and for 30 minutes. The solid residue was separated from the leachate through centrifugation (5300 rpm, 30 minutes) and the supernatant was analysed to determine the precipitation efficiency EP (%), which was calculated according to eqn (2):| |
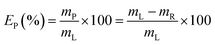 | (2) |
where mP is the mass of metal precipitated (mg) and mR is the remaining mass of metal in the leachate after precipitation (mg). Similarly to the leaching tests, each precipitation test was performed in duplicate. The obtained mixed oxalate was dried in a muffle furnace (t = 12 h, T = 50 °C) and then calcined (t = 5 h, T = 950 °C) in order to get the corresponding oxide. After dissolution in a 6 mol L−1 HCl solution, the purity of the oxide was determined by ICP-OES analysis.
Results and discussion
Characterisation, sieving and particle size distribution analysis
Table 1 shows the results obtained for the characterisation of the initial CRT phosphor powder while Fig. S1† includes the XRD pattern, showing that the main compounds in the waste powder are zinc sulphide (ZnS) and yttrium oxysulphide (Y2O2S). Yttrium is the most relevant rare earth present in the initial CRT phosphor powder (15.5 wt%) together with some europium (0.96 wt%). The distribution of the elements in the CRT phosphor waste can be seen in Fig. S2,† obtained by SEM/EDS. A section of the CRT phosphor micrograph was enlarged and four representative spots were analysed (Fig. 1). On the basis of the distribution of the elements by SEM/EDS and the results by XRD, the main phases present in the sample were identified and a semi-quantitative analysis was carried out by SEM/EDS. With the elemental analysis obtained (mass percentage normalised to 100 of each element in the particle) and the atomic mass of the element, a mineral formula was calculated. The particles studied were consistent with ZnS, glass, Y2O2S and aluminium (Table 2).
Table 1 Composition of the CRT phosphor powder
| Element |
Concentration, wt% |
| Zn |
30.6 |
| S |
16.5 |
| Y |
15.5 |
| Si |
7.18 |
| Al |
1.95 |
| Pb |
1.73 |
| K |
1.62 |
| Na |
1.47 |
| Ca |
1.05 |
| Eu |
0.96 |
| Sr |
0.62 |
| Mg |
0.37 |
 |
| | Fig. 1 Micrograph of the CRT phosphor powder (1000× magnification) showing the particles analysed. | |
Table 2 Elemental analysis with EDS of the indicated positions in the SEM micrograph in Fig. 1 and approximate composition of the CRT phosphor powder
| |
Elements detected with EDS |
Mineralogical phase |
Concentration, wt% |
| Position 1 |
Zn, S |
ZnS |
46 |
| Position 2 |
K, Ca, O, Na, Mg, Al, Si, Sr, Ba |
Glass |
25 |
| Position 3 |
O, Eu, Y, S |
Y2O2S:Eu3+ |
23 |
| Position 4 |
Al, Y, S, Cl |
Metallic Al |
1.5 |
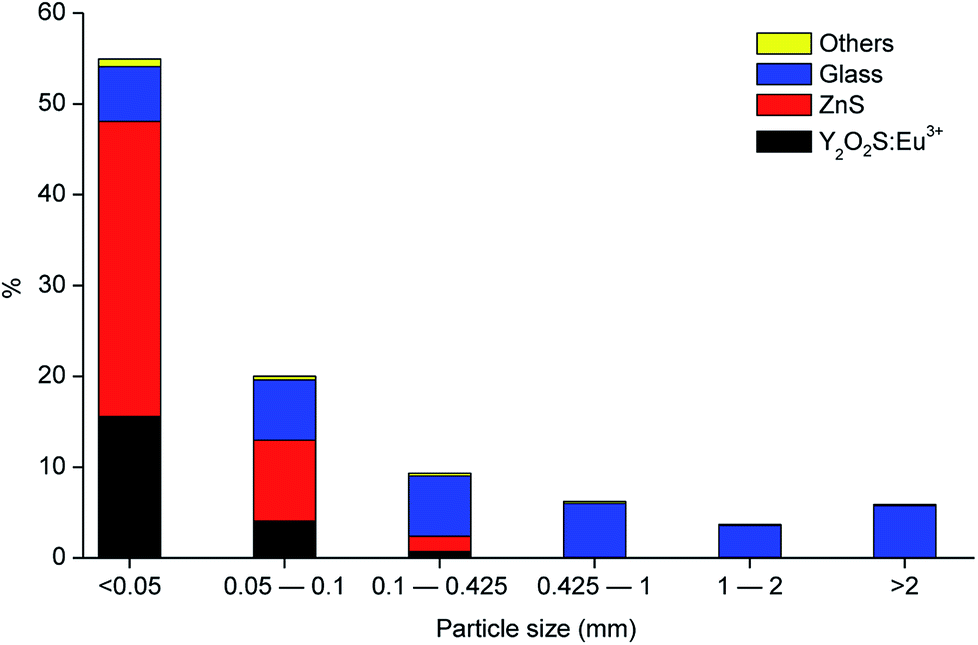
Fig. 2 shows the different fractions of the CRT phosphor powder obtained after sieving. The particle size distribution study showed that 75 wt% of the CRT phosphor powder is related to the fractions with particle size smaller than 100 μm (55 wt% belongs to the fraction <50 μm and 20 wt% to the fraction 50–100 μm) and that the fractions with particle size larger than 425 μm are mostly constituted by glass. Fig. 3 shows the gravimetric distribution of each fraction sieved, expressed as percentage of the total amount of sample, where each colour represents an individual compound. Chemical analysis of all the sieved fractions allowed calculating the representative element for each fraction (Na and K for glass, Zn for zinc sulphide and Y and Eu for Y2O2S:Eu3+). Fractions smaller than 425 μm were all mixed to obtain the selected sample for further tests and then analysed. The results are shown in Table 3.
 |
| | Fig. 2 Appearance of the different sieved fractions of the CRT phosphor powder. | |
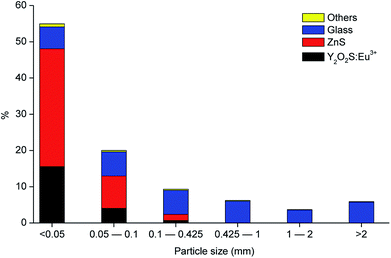 |
| | Fig. 3 Particle size distribution of the CRT phosphor powder, including the composition in each fraction (referred to the total amount of sample). | |
Table 3 Composition of the <425 μm CRT phosphor powder fraction
| Element |
Concentration, wt% |
Element |
Concentration, wt% |
| Zn |
32.8 |
Eu |
1.37 |
| Y |
19.1 |
Na |
1.28 |
| S |
16.6 |
Sr |
1.14 |
| Pb |
2.08 |
Ca |
0.30 |
| Al |
1.92 |
Cd |
0.22 |
| K |
1.55 |
Fe |
0.15 |
Roasting
Preliminary tests showed that the glass contained in the CRT powder produces zinc and yttrium silicates during the roasting process. Considering this circumstance, the amount of non-desirable compound produced during the roasting step was reduced by separating the glass particles from the CRT fluorescent powder. The CRT fraction smaller than 425 μm was subjected to a roasting step aimed at converting ZnS into ZnO, thus avoiding H2S release during the following leaching step. During roasting, the following reactions take place (eqn (3) and (4)).| |
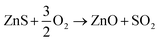 | (3) |
| |
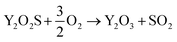 | (4) |
ZnO and Zn2SiO4 concentrations were measured by quantitative XRD and their Zn content was calculated and compared to the total amount of Zn in the CRT sample. Fig. S3 and S4† show the diffractograms of a CRT sample roasted at 850 °C and 1000 °C, respectively. Characteristic peaks of ZnO (2θ = 36.2° and 67.9°) obtained for the roasted sample at 850 °C showed a higher intensity than those obtained at 1000 °C, which is a proof of the decrease in ZnO when the roasting temperature increased. On the contrary, Zn2SiO4 concentration (characteristic peak at 2θ = 38.7°) increased with temperature. The optimum conditions for roasting are those that maximise the ZnO concentration and minimise the Zn2SiO4 formation, along with a complete sulphide decomposition. Table 4 reports the ZnS, ZnO and Zn2SiO4 amount with respect to the total Zn value. The balance to 100% was assigned to other non-identified zinc compounds. It was found that samples roasted at 850 °C had a negligible amount of ZnS and 75 wt% of the zinc was present in the form of ZnO. When the roasting temperature was increased, the Zn2SiO4 content also increased and the fraction of zinc present as ZnO decreased to about 60 wt% at 900 °C and to about 50 wt% at 1000 °C. Roasting at a lower temperature (800 °C) gave worse results for ZnS decomposition, so that this condition was not considered to be acceptable. The residence time was optimised by determining the sulphide concentration in the roasted material and the amount of Zn obtained as ZnO as a function of the time, varied between 0 and 40 minutes.
Table 4 Distribution of zinc over the different zinc compounds present in the roasted CRT, as function of temperature and residence time
| |
Roasting temperature |
| 1000 °C |
900 °C |
850 °C |
800 °C |
| Residence time, min |
10.25 |
11.5 |
27.0 |
36.0 |
8.25 |
15.0 |
20.0 |
10.75 |
15.5 |
20.75 |
33.0 |
9.25 |
16.0 |
19.75 |
| wt% Zn as ZnS |
0.5 |
0.3 |
0.6 |
0.2 |
1.5 |
0.3 |
0.2 |
1.2 |
0.6 |
0.5 |
0.3 |
3.6 |
3.4 |
3.3 |
| wt% Zn as ZnO |
51.6 |
51.0 |
49.0 |
48.6 |
60.4 |
59.9 |
59.2 |
74.3 |
73.1 |
72.6 |
70.8 |
79.6 |
79.3 |
79.0 |
| wt% Zn as Zn2SiO4 |
20.3 |
20.0 |
20.1 |
19.4 |
10.5 |
10.6 |
10.6 |
8.4 |
8.3 |
8.3 |
8.3 |
8.3 |
8.4 |
8.5 |
| wt% Zn in other forms |
27.6 |
28.7 |
30.3 |
31.8 |
27.7 |
29.3 |
30.0 |
16.1 |
18.1 |
18.6 |
20.6 |
8.5 |
8.9 |
9.2 |
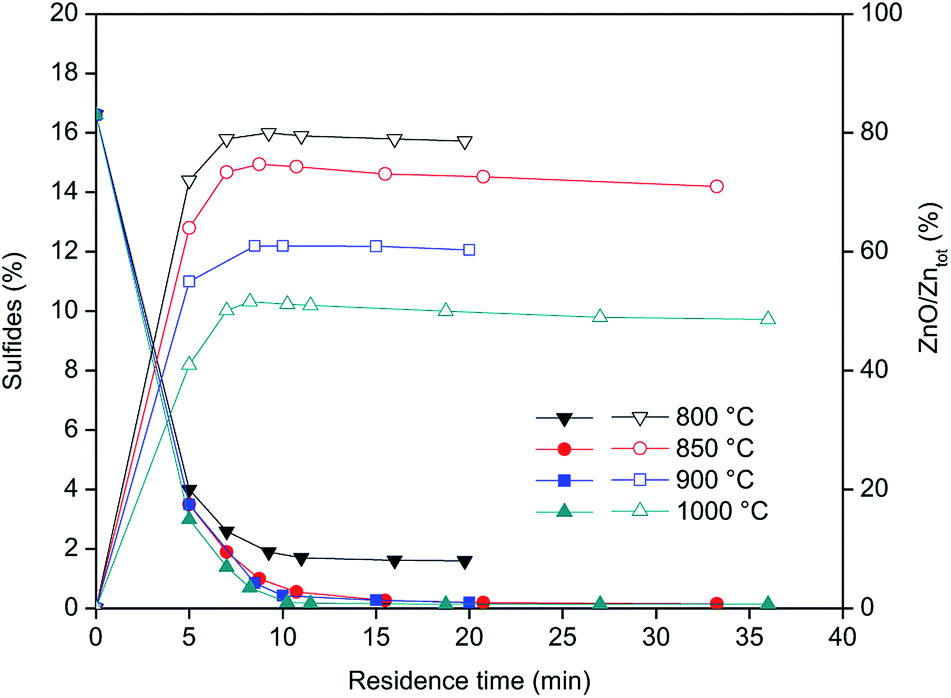
Fig. 4 shows the concentration of sulfides (left Y-axis) and Zn as ZnO (right Y-axis) during the roasting of the CRT powder at different temperatures. ZnS is expressed as percentage of sulphide present in the roasted samples, while ZnO represents the percentage of Zn present in the form of ZnO versus the total zinc content. The results obtained showed that roasting of CRT powders can be conducted successfully in a rotary furnace and that ZnS can be fully decomposed at temperatures above 850 °C. Temperatures higher than 850 °C significantly increase the formation of non-desirable Zn2SiO4, which complicates further zinc extraction prior to recovery of yttrium and europium. The final concentration of Zn2SiO4 is mainly a function of the roasting temperature, so it is preferred to carry out the roasting at the lowest possible temperature that allows decomposition of all the sulphides present in the CRT powder. The optimum conditions for CRT powder roasting were 850 °C with a residence time of 10 to 20 minutes, because when the roasting time is increased up to 33 minutes, the amount of zinc present in the form of ZnO decreases.
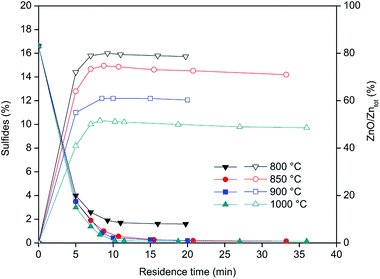 |
| | Fig. 4 Sulphides concentration (left axis, closed symbols) and zinc content as ZnO with respect to the total zinc (right axis, open symbols) as a function of the roasting residence time. | |
Leaching
The roasted phosphor samples were subjected to a leaching step with acetic acid to remove zinc. This leaching agent was selected due to its capability of dissolving zincite (ZnO) and for its green properties.33–35 Preliminary tests were performed on samples roasted at T = 1000 °C under non-optimised conditions, by varying the acetic acid concentration, temperature and liquid-to-solid ratio (L/S). It was found that the leaching efficiency decreased with increasing roasting time (ESI, Table S1†). This is due to the formation of zinc silicates (such as willemite, Zn2SiO4), which are insoluble in acetic acid. It was also found that temperature, liquid-to-solid ratio and contact time have a negligible influence on the leaching yields (ESI, Tables S2–S4†). Variation of the acetic acid concentration showed that the maximum leaching efficiency was achieved for an acetic acid concentration of 10 mol L−1 (ESI, Table S5†). This leaching step with 10 mol L−1 acetic acid is an example of a solvometallurgical process since solvent concentration is higher than 50 vol%.36 The advantages of a solvometallurgical process compared to a hydrometallurgical one are, among others, the potential high selectivity and a reduced water consumption.
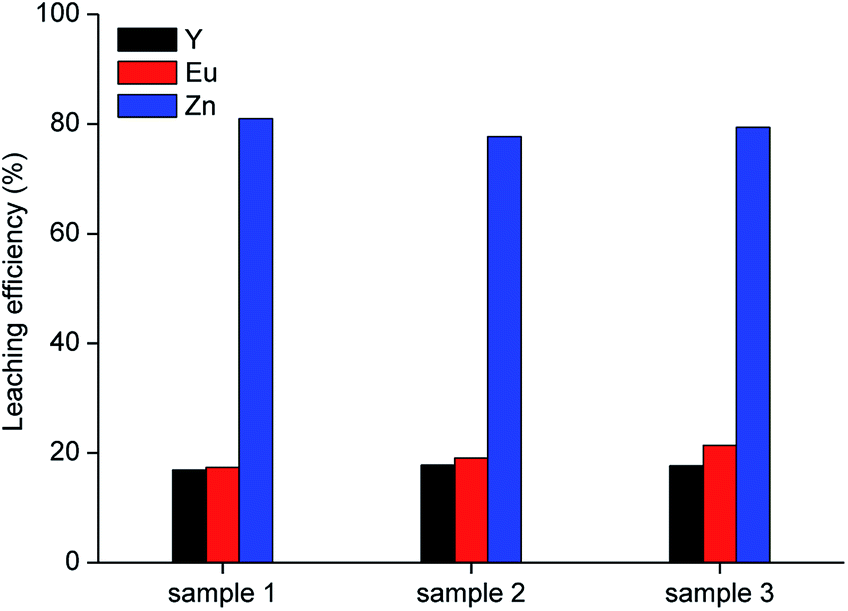
Further leaching tests were then performed on the samples obtained under the optimised roasting temperature (850 °C). In particular, three samples were selected, where more than 70 wt% of the zinc was present as ZnO and the amount of Zn2SiO4 was less than 9 wt%. Y, Eu and Zn content in these samples is shown in Table 5.
Table 5 Chemical composition (wt%) of the selected roasted CRT phosphors
| |
Sample 1 |
Sample 2 |
Sample 3 |
| 850 °C, 20.75 min |
850 °C, 15.5 min |
850 °C, 10.75 min |
| Zn |
30.2 |
33.0 |
31.3 |
| Y |
17.0 |
16.7 |
16.9 |
| Eu |
1.2 |
1.0 |
1.0 |
Leaching was performed with 10 mol L−1 acetic acid in water at room temperature, for 2 h, by employing a liquid-to-solid ratio of 10 mL g−1, which represent the best conditions obtained from the preliminary leaching tests. Results are shown in Fig. 5. It was observed that about 80 wt% of zinc can be leached from the roasted samples with the selected lixiviant. This yield can be reasonably linked to the fraction of zinc present in the phosphors as ZnO, which is soluble in carboxylic acids.37
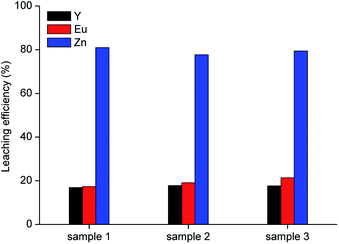 |
| | Fig. 5 Leaching tests with 10 mol L−1 acetic acid on three samples roasted under the optimised conditions (T = 25 °C, t = 2 h, L/S = 10 mL g−1). | |
Since sample 1 showed the lowest ZnS concentration (0.5 wt%), it was selected for further investigation. A sample of 10 g was leached with 10 mol L−1 acetic acid under the optimised conditions (T = 25 °C, t = 2 h, L/S = 10 mL g−1). The residue was separated from the leachate by vacuum filtration using a folded filter paper (grade 601, cellulose, particle retention 5 to 13 μm), followed by washing with water and was then dried at 105 °C until constant mass. The mass loss was found to be about 43 wt%. The metal concentration in the acetic acid leachate and the composition of the residue are reported in Table 6.
Table 6 Metal concentration in the leachate after leaching with 10 mol L−1 acetic acid and composition of the residue (T = 25 °C, t = 2 h, L/S = 10 mL g−1)
| |
Zn |
Y |
Eu |
| Leachate concentration, mg L−1 |
24264 |
2847 |
219 |
| Residue composition, wt% |
4.8 |
24.8 |
1.4 |
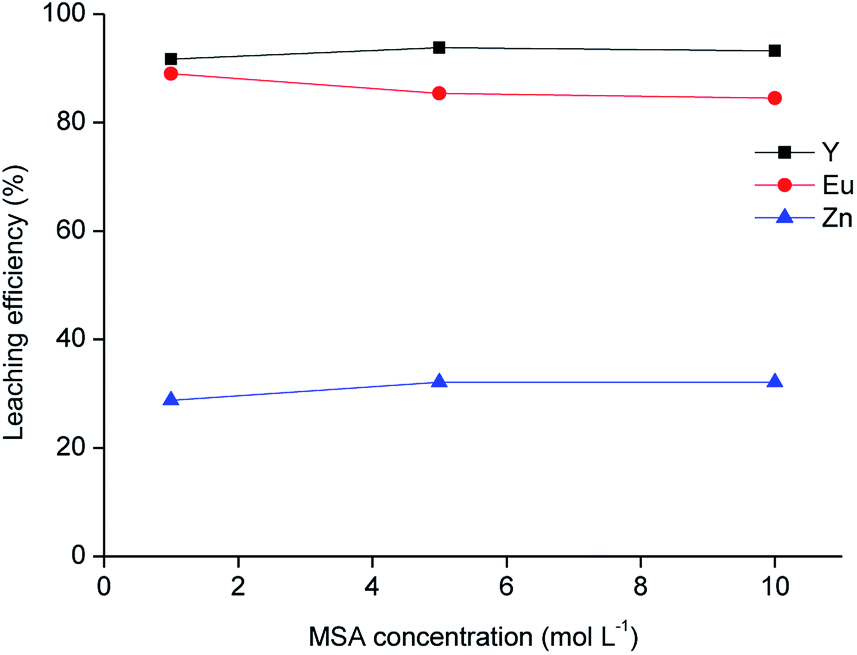
The residue obtained from acetic acid leaching was subsequently submitted to leaching with MSA. MSA was selected as an acidic lixiviant because of its characteristics of thermal stability, low toxicity and biodegradability. Due to these characteristics, MSA is labelled as a green acid.38 MSA is a strong acid (pKa = −1.8).39 One of the main advantages compared to sulphuric acid is the higher solubility of rare-earth methanesulphonates in aqueous solutions.40 The leaching tests with different dilutions of MSA in water performed on the solid residue as a function of the leaching agent concentration are shown in Fig. 6, while the metal concentration in the MSA phase is reported in Table 7. About 90 wt% of Y and Eu was dissolved with the selected lixiviant and the dissolution was already effective at a MSA concentration of 1 mol L−1. The residue obtained from the leaching test with 1 mol L−1 MSA was separated from the solid residue by centrifugation, washed with water and then dried at 105 °C until constant mass. The mass loss after leaching was about 49 wt%.
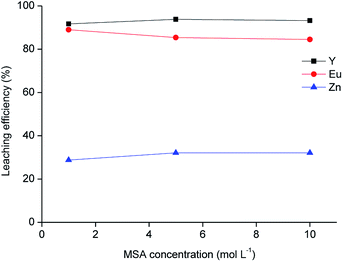 |
| | Fig. 6 Leaching of the residue as a function of methanesulphonic acid concentration (T = 90 °C, t = 24 h, L/S = 20 mL g−1). | |
Table 7 Metal concentration in the leachate (mg L−1) after leaching with different concentrations of methanesulphonic acid (MSA) (T = 90 °C, t = 24 h, L/S = 20 mL g−1)
| MSA concentration, mol L−1 |
Zn |
Y |
Eu |
| 1 |
704 |
11473 |
615 |
| 5 |
780 |
11779 |
594 |
| 10 |
729 |
11573 |
578 |
Recovery of yttrium and europium from the leachate
Yttrium and europium were recovered from the 1 mol L−1 MSA leachate by precipitation with oxalic acid. The precipitation reaction is reported in eqn (5):| | |
2(Y3+, Eu3+) + 3H2C2O4 → (Y, Eu)2(C2O4)3(s) + 6H+
| (5) |
It was found that by adding twice the stoichiometric amount (with respect to yttrium and europium) of solid oxalic acid, quantitative precipitation of both rare earths was achieved. About 2 wt% of zinc present in the MSA leachate was co-precipitated. To obtain a sufficient amount of mixed oxalate for the subsequent calcination step, the whole process was repeated once more starting from 10 g of roasted sample and by treating the entire amount of residue coming from the first leaching step with 1 mol L−1 MSA. The MSA leachate was then subjected to a precipitation step with solid oxalic acid. The obtained mixed oxalate was separated from the leachate by filtration while washing with water, dried at 50 °C for 12 h and then calcined at 950 °C for 5 h, according to a literature procedure (Fig. 7).9 After dissolution in 6 mol L−1 HCl, the mixed oxide was analysed by ICP-OES to determine its purity which was found to be >99.0 wt%. The recovery process flow sheet of the whole process is shown in Fig. 8. It was found that about 75 wt% yttrium and europium could be recovered as a mixed oxide from the waste CRT phosphors powder. Further research is needed to investigate their recovery from the acetic acid leachate so that the global recovery efficiency can be improved.
 |
| | Fig. 7 Mixed yttrium/europium oxide obtained via the recovery process described in present work. | |
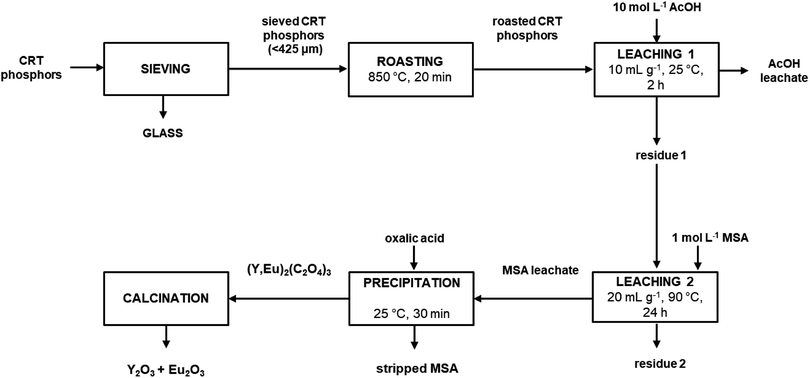 |
| | Fig. 8 Process flow sheet. | |
Conclusions
An integrated process flow sheet comprising roasting, leaching, precipitation as a mixed oxalate and calcination was developed to recover yttrium and europium from waste CRT phosphors. The roasting step allowed an efficient conversion of the zinc sulphide present in the initial phosphor powder into zinc oxide, thus facilitating further processing of the waste. Compared to common hydrometallurgical processes, where leaching is normally performed on non-treated material, in our process the release of toxic H2S gas is avoided. SO2 generated during the roasting step can be used in the production of H2SO4. The recovery of the rare earths was then performed by employing a sequence of leaching steps with organic acids (acetic acid and methanesulphonic acid). Both acids are labelled as ‘green solvents’ because of their low toxicity and low environmental impact. Acetic acid proved to be selective towards dissolution of zinc, but still about 20 wt% yttrium and europium were unavoidably co-dissolved, thus lowering the global recovery yield. The stripped methanesulphonic acid could be reused in consecutive leaching steps; in this way, reagent consumption could be reduced. The obtained product was a mixed yttrium europium oxide with a purity of more than 99.0 wt%. This oxide can be further purified by solvent extraction to obtain pure yttrium and europium oxides for the relevant applications.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work has received funding from the European Union's Horizon 2020 Research and Innovation Programme under Grant Agreement No 680629 (REMAGHIC: New Recovery Processes to produce Rare Earth-Magnesium Alloys of High Performance and Low Cost) (project website: http://www.remaghic-project.eu). The authors acknowledge Relight srl (Rho, Italy) for providing the CRT phosphor powder.
Notes and references
- X. Tian, X. Yin, Y. Gong, Y. Wu, Z. Tan and P. Xu, J. Cleaner Prod., 2016, 135, 1210–1217 CrossRef CAS.
- N. Singh, J. Li and X. Zeng, J. Cleaner Prod., 2016, 133, 188–200 CrossRef CAS.
- C. H. Lee and C. S. His, Environ. Sci. Technol., 2002, 36, 69–75 CrossRef CAS PubMed.
- K. Binnemans, P. T. Jones, B. Blanpain, T. Van Gerven, Y. Yang, A. Walton and M. Buchert, J. Cleaner Prod., 2013, 51, 1–22 CrossRef CAS.
- N. Singh, J. Wang and J. Li, Procedia Environ. Sci., 2016, 31, 465–474 CrossRef.
- C. K. Gupta and N. Krishnamurthy, Extractive Metallurgy of Rare Earths, CRC Press, Elsevier, 2005 Search PubMed.
- K. Binnemans and P. T. Jones, J. Rare Earths, 2014, 32, 195–200 CrossRef CAS.
- S. Van Loy, K. Binnemans and T. Van Gerven, J. Cleaner Prod., 2017, 156, 226–234 CrossRef CAS.
- D. Dupont and K. Binnemans, Green Chem., 2015, 17, 856–868 RSC.
- C. Tunsu, M. Petranikova, M. Gergoric, C. Ekberg and T. Retegan, Hydrometallurgy, 2015, 156, 239–258 CrossRef CAS.
- C. Tunsu, M. Petranikova, C. Ekberg and T. Retegan, Sep. Purif. Technol., 2016, 161, 172–186 CrossRef CAS.
- N. M. Ippolito, V. Innocenzi, I. De Michelis and F. Vegliò, J. Cleaner Prod., 2017, 153, 287–298 CrossRef CAS.
- L. Gijsemans, F. Forte, B. Onghena and K. Binnemans, RSC Adv., 2018, 8, 26349–26355 RSC.
- L. V. Resende and C. Morais, Miner. Eng., 2010, 23, 277–280 CrossRef CAS.
- V. Innocenzi, I. De Michelis, F. Ferella, F. Beolchini, B. Kopacek and F. Vegliò, Waste Manage., 2013, 33, 2364–2371 CrossRef CAS PubMed.
- V. Innocenzi, I. De Michelis, F. Ferella and F. Vegliò, Waste Manage., 2013, 33, 2390–2396 CrossRef CAS PubMed.
- L. V. Resende and C. Morais, Miner. Eng., 2015, 70, 217–221 CrossRef CAS.
- X. Yin, Y. Wu, X. Tian, J. Yu, Y. N. Zhang and T. Zu, ACS Sustainable Chem. Eng., 2016, 4, 7080–7089 CrossRef CAS.
- X. Tian, X. Yin, Y. Gong, Y. Wu, Z. Tan and P. Xu, J. Cleaner Prod., 2016, 135, 1210–1217 CrossRef CAS.
- J. Dexpert-Ghys, S. Regnier, S. Canac, T. Beaudette, P. Guillot, B. Caillier, R. Mauricot, J. Navarro and S. Sekhri, J. Lumin., 2009, 129, 1968–1972 CrossRef CAS.
- X. Pan, L. Peng, W. Chen, J. Wang and Z. Chen, Appl. Mech. Mater., 2013, 295–298, 1840–1845 Search PubMed.
- M. A. R. Önal and K. Binnemans, Hydrometallurgy, 2019, 183, 60–70 CrossRef.
- E. Y. Lin, A. Rahmawati, J. H. Ko and J. C. Liu, Sep. Purif. Technol., 2018, 192, 166–175 CrossRef CAS.
- M. D. Gernon, M. Wu, T. Buszta and P. Janney, Green Chem., 1999, 1, 127–140 RSC.
- https://www.relightitalia.it/en/our-services/treatment, accessed July 17th, 2018.
- CEN/TR 15018 Characterization of waste — Digestion of waste samples using alkali-fusion techniques.
- ISO 14869-2 Soil quality — Dissolution for the determination of total element content — Part 2: Dissolution by alkaline fusion.
- EPA 3052 Microwave assisted acid digestion of siliceous and organically based matrices.
- EN ISO 11885 Water quality. Determination of selected elements by inductively coupled plasma optical emission spectrometry.
- G. Dadabaeva and D. D. Ikrami, Dokl. Akad. Nauk Tadzh. SSR, 1976, 19, 31–33 CAS.
- S. Riaño, M. Regadío, K. Binnemans and T. Vander Hoogerstraete, Spectrochim. Acta, Part B, 2016, 124, 109–115 CrossRef.
- M. Regadío, S. Riaño, K. Binnemans and T. Vander Hoogerstraete, Anal. Chem., 2017, 89, 4595–4603 CrossRef PubMed.
- R. K. Henderson, C. Jimenez-Gonzalez, D. J. C. Constable and A. D. Curzons, Green Chem., 2011, 13, 854–862 RSC.
- R. K. Henderson, A. P. Hill, A. M. Redman and H. F. Sneddon, Green Chem., 2015, 17, 945–949 RSC.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
- K. Binnemans and P. T. Jones, J. Sustain. Metall., 2017, 3, 570–600 CrossRef.
- J. M. Steer and A. J. Griffiths, Hydrometallurgy, 2013, 140, 34–41 CrossRef CAS.
- M. Gernon, M. Wu, T. Buszta and P. Janney, Green Chem., 1999, 1, 127–140 RSC.
- F. C. Walsh and C. Ponce de León, Surf. Coat. Technol., 2014, 259, 676–697 CrossRef CAS.
- BASF SE, Lutropur® – the friendly acid. The purest form of MSA methanesulfonic acid made, 2011 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: XRD and micrograph of the CRT phosphor powder, preliminary leaching tests with acetic acid. See DOI: 10.1039/c8ra08158a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Lourdes Yurramendib,
José Luis Aldanab,
Bieke Onghena
a,
Lourdes Yurramendib,
José Luis Aldanab,
Bieke Onghena