
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanism and modeling of hexavalent chromium interaction with a typical black soil: the importance of the relationship between adsorption and reduction†
Jia Zhanga,
Huilin Yinab,
Samuel Barniea,
Minghai Wei *ab and
Honghan Chena
*ab and
Honghan Chena
aBeijing Key Laboratory of Water Resources & Environmental Engineering, China University of Geosciences, Beijing 100083, China. E-mail: wmh87@163.com
bChinese Academy for Environmental Planning, China
First published on 14th February 2019
Abstract
Black soils have a significant retention effect on the migration of Cr(VI) towards groundwater, and Cr(VI) adsorption and reduction are both involved in this process. However, the adsorption and reduction of Cr(VI) were always investigated separately in previous studies resulting in an unclear relationship between them. In this study, the adsorption and reduction kinetic processes of Cr(VI) by a typical black soil were separately investigated under different initial Cr(VI) concentrations (40–400 mg L−1) and pH conditions (3.5–7.0) by the means of desorption treatment, and the equilibrium relationship between aqueous and adsorbed Cr(VI) was innovatively established based on the kinetic data. It was found that under pH 5.7 the adsorbed Cr(VI) content on soil particles was linearly correlated with the remaining Cr(VI) concentration in solution with time (R2 = 0.98), and the reduction rate of Cr(VI) in the reaction system was linearly correlated with the adsorbed Cr(VI) content on soil particles with time (R2 = 0.99). With pH decreasing from 7.0 to 3.5, the partition of Cr(VI) between solid and aqueous phases turned out to be of a non-linear nature, which can be fitted better by the Freundlich model. The retention of Cr(VI) by black soil was determined to follow the “adsorption–reduction” mechanism, where the Cr(VI) was first rapidly adsorbed onto the soil particles by a reversible adsorption reaction, and then the adsorbed Cr(VI) was gradually reduced into Cr(III). A two-step kinetic model was developed accordingly, and the experimental data were fitted much better by the two-step adsorption–reduction kinetic model (R2 = 0.89 on average) compared with the traditional first-order and second-order kinetic models (R2 = 0.66 and 0.76 on average respectively). This paper highlights the novel two step kinetic model developed based on the proposed “adsorption–reduction” mechanism of Cr(VI) retention by a typical black soil.
1. Introduction
Hexavalent chromium Cr(VI) is an important industrial material, which is widely utilized in alloy, electroplating, leather tanning, dyeing and wood preservation.1 According to the Toxic Release Inventory from USEPA, 52![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 tons of Cr has been released into the environment by 1762 industrial facilities.2 In China, the electroplating industry alone can generate 400 million tons of wastewater and 50 thousand tons of solid waste per year,3 and the instances of wastewater discharging into the environment directly or by the means of seepage pits have led to serious Cr(VI) contamination of soil and groundwater.4,5 As known, Cr(VI) is extremely toxic and carcinogenic to plants and human beings,6,7 which is considered as one of the four top priority soil heavy metal contaminants of concern by United State Department of Defense.8–10
600 tons of Cr has been released into the environment by 1762 industrial facilities.2 In China, the electroplating industry alone can generate 400 million tons of wastewater and 50 thousand tons of solid waste per year,3 and the instances of wastewater discharging into the environment directly or by the means of seepage pits have led to serious Cr(VI) contamination of soil and groundwater.4,5 As known, Cr(VI) is extremely toxic and carcinogenic to plants and human beings,6,7 which is considered as one of the four top priority soil heavy metal contaminants of concern by United State Department of Defense.8–10
The soil layer has been found to have significant retention effect on the migration of Cr(VI) towards groundwater, and many investigations have focused on the topic of Cr(VI) adsorption by soils.11,12 It has been extensively reported that Cr(VI) anions (Cr2O72−, HCrO4− and CrO42−) tend to be bound to positively charged soil particles by forming inner-sphere (specific adsorption) or outer-sphere (non-specific adsorption) complexes,8 and the adsorption of Cr(VI) by soil is mainly influenced by pH condition, electrolyte species, inorganic minerals (e.g. iron oxides, aluminum oxides, manganese oxides, and clay) and organic matters (e.g. humus).2,13,14 In addition, substantial attentions were focused on the kinetic and thermodynamic aspects of Cr(VI) adsorption by soil in previous studies. Notably, the adsorption of Cr(VI) by soils was quite different from the other heavy metal cations, such as Pb2+, Cu2+ and Cd2+. The adsorption of heavy metal cations by soils can generally reach equilibrium state within hours, however, the adsorption of Cr(VI) by soils always go through a slow adsorption process (even still proceeding after 180 days),15 which always led to a poor fitting performance of the kinetic data by traditional first-order or second-order kinetic models.
Cr(VI) is of strong oxidation property especially under acidic condition, and can be reduced into Cr(III) gradually by inorganic or organic electron donors in the soil environment.16,17 Cr(III) mainly exists in cations or colloidal precipitate, such as Cr3+, CrOH2+, Cr(OH)2+, and Cr(OH)3, and can be readily adsorbed or precipitate on soil particles. Additionally, Cr(III) is less harmful compared with Cr(VI), which even act as an essential trace element for human bodies, and thus reducing Cr(VI) into less mobile and toxic Cr(III) is regarded as one of the most effective strategies for Cr(VI) contaminated soil and groundwater remediation.18–21
The natural reduction of Cr(VI) on soil particles is mainly influenced by pH condition, redox potential, and inorganic and organic electron donors.22,23 For Cr(VI) adsorption, pH condition mainly determines the soil particle variable charge character according to zero point of charge (ZPC), but for Cr(VI) reduction, pH condition mainly determines the proton concentration, which is a necessary element participating in this reaction. Additionally, soil organic matters (SOM) are considered to act as the main electron donors for Cr(VI) reduction in soil environment,24,25 but the reduction rate of Cr(VI) by SOM is much slower compared with inorganic electron donors, such as Fe(II) and S(II), and that is why the real equilibrium state of Cr(VI) adsorption by soil is almost impossible to be reached within a short time except for that Cr(VI) is exhausted, which may be account for the slow adsorption process of Cr(VI) by soils.
As can be seen, the retention of Cr(VI) by soil includes adsorption and reduction, and both of them are of great significance for Cr(VI) retention in the soil. The adsorption determines the macroscopic retention effect of Cr(VI) migration, while the reduction determines the toxicity (environmental risk) and stability (leaching potential) of Cr.8 These two processes interplay with each other, neither of which should be ignored. However, the adsorption and reduction of Cr(VI) were always investigated separately in previous studies resulting from the unclear relationship between them.26–28 Therefore, it is urgent to establish a quantitative relationship between adsorption and reduction of Cr(VI) in soil, which is the key point to reveal the underlying mechanism of Cr(VI) retention by soils, and this is of vital importance for the prediction and assessment of Cr(VI) migration/transformation and environmental risk in soils and sediments.
In this study, the adsorption and reduction processes of Cr(VI) by a typical black soil were investigated respectively under different initial Cr(VI) concentration (40–400 mg L−1) and pH (3.5–7.0) conditions by the means of desorption treatment. The main purposes of this study were to (1) reveal the partition characteristic of Cr(VI) adsorption from aqueous phase to soil solid phase, (2) establish the quantitative relationship between adsorption and reduction of Cr(VI) in the typical black soil, and (3) simulate the experimental data by the kinetic model developed according to the proposed “adsorption–reduction” mechanism of Cr(VI) retention by the typical black soil.
2. Materials and methods
2.1 Materials
The stock solution of Cr(VI) and NaCl were prepared by the chemicals of K2Cr2O7 and NaCl respectively, and both of these chemicals were obtained from Sinopharm Chemical Reagent Co., Ltd with analytical purity (AR). The working solution was prepared by diluting certain volumes of Cr(VI) and NaCl stock solutions with deionized water just before it was to be used. The final concentration of Cr(VI) in the working solutions ranged from 40 to 400 mg L−1, and the concentration of NaCl was 0.01 M, which acted as the background electrolyte.The used black soil sample was obtained from an undisturbed area located at the northeast of China (43°18′36′′N, 128°23′26′′E), where is located at one of the three major black soil regions worldwide (the other two are the great plains of Ukraine and the Mississippi River basin of America, respectively). This black soil sample can be classified as Mollisol soil according to USDA soil classification system, and it is the typical black soil in the region of Northeast China. The location, landform, climate and vegetation can be found in our previous published paper.29 The surface soil within the depth of 30 cm was sampled after the topsoil containing substantial plant roots had been removed. The soil sample was air-dried and ground, and then was passed through a 100-mesh sieve.
2.2 Characterization of the black soil sample
The pH of the black soil sample was determined by mixing soil with 1 M KCl solution in the solid to liquid ratio of 1:2.5 (w:v), and the pH value was measured after stirring the mixture for 5 min and standing for 3 h. The zero point of charge (ZPC) was determined by the method described elsewhere.30 The organic matter content of the soil sample was determined by the Walkley–Black procedure.31 The chemical composition of the soil sample was determined by X-ray Fluorescence spectroscopy (XRF). The mineral composition of the soil sample was characterized by X-ray diffraction (XRD) on a machine of Rigaku D/MAX-rA.
The pHKCl of the soil sample was determined to be 4.6, and the content of organic matter was determined to be 11.64%. The SiO2 was determined to be the main constituent of the soil sample (44.36%), and the contents of Al2O3 and Fe2O3 were determined to be 16.52% and 6.79% respectively. The other inorganic compositions contents were all under 3%.
2.3 Sorption experiment
In order to reveal the underlying mechanism of Cr(VI) retention by the soil sample under various conditions, the sorption experiment was conducted under different initial Cr(VI) concentrations and pH values. In the experiment under different initial Cr(VI) concentrations, 2 g black soil samples were added into a series of 40 mL amber bottles, and 20 mL working solutions with Cr(VI) concentrations of 40, 100, 250 and 400 mg L−1 were added into the bottles as well. The working solutions contained 0.01 M NaCl as background electrolyte, and the initial pH was adjusted to 5.7 by adding 0.1 M HCl and NaOH solutions. All the above experiments were performed in triplicate, and a blank group (without Cr(VI)) and a control group (without soil sample) were set for each time step. The bottles were put into the shaker incubator, and shaken at 175 rpm at 25 °C. At given time intervals, a batch of bottles was taken out from the shaker incubator, and then centrifuged at 3000 rpm for 10 min. The supernatant was filtrated with 0.45 μm membrane, and the Cr(VI) and Cr(T) concentration in the filtrate were determined by the methods of UV spectrophotometry (SHIMADZU UV-1800) and ICP-OES (SPECBlUE) respectively. The Cr(III) concentration in solution was determined by the difference between the concentrations of Cr(T) and Cr(VI). The pH value and total organic carbon (TOC) in solution were measured simultaneously.In the experiment under different initial pH values, the same amount of soil samples were added into a series of amber bottles as well, and then 20 mL working solutions with initial pH of 3.5, 5.7 and 7.0 were added into the bottles. The initial Cr(VI) concentration of the working solutions was 250 mg L−1. The other experiment conditions and procedures were same with those conducted under different initial Cr(VI) concentrations.
In order to differentiate the adsorption and reduction processes of Cr(VI) in the reaction system, the amount of Cr(VI) adsorbed on the surface of soil particles must be determined. Considering that the specific and non-specific adsorbed Cr(VI) may both exist on the surface of soil particles at the same time, and the background electrolyte solution has almost no desorption effect on the specific adsorbed Cr(VI), NaOH solution was used for the desorption of both specific and non-specific adsorbed Cr(VI) on soil particles in this experiment, which is considered to be able to desorb the specific adsorbed Cr(VI) by ligand exchange. The soil sample after reacting with Cr(VI) were collected by centrifuging (the centrifugation procedure mentioned above), and 20 mL 0.5 M NaOH solution was added in the bottle.32 The mixture was shaken in the shaker incubator at 175 rpm at 25 °C for 12 h, and then centrifuged at 3000 rpm for 10 min. The supernatant was filtrated with 0.45 μm membrane, and the concentrations of Cr(VI) and Cr(T) were determined by the same methods mentioned above. Considering that the color of dissolved humus in the alkaline solution may influence the determination of Cr(VI) concentration by the method of UV spectrophotometry, the blank control of deionized water was replaced by the same dosage of alkaline solution, and the color-developing agent of 1,5-diphenylcarbazide was replaced by the same dosage of deionized water. The above procedures were repeated three times until the concentration of Cr(VI) in alkaline solution turned to be very low, and then the Cr(VI) concentrations from the three times of determination were added up to calculate the Cr(VI) content on the soil sample.
2.4 FTIR characterization of humic acid extracted from the black soil
Humic acid (HA) was extracted from the soil samples with 0.5 M NaOH solution for 12 h, and the suspensions were centrifuged at 4500 rpm for 15 min to obtain the supernatant. The pH of the supernatant was adjusted to 1 by adding 6 M HCl to make HA precipitated, and the suspensions were centrifuged at 4500 rpm for 15 min to obtain the HA samples. The HA samples were freeze-dried, and characterized by an IR spectrometer (Bruker LUMOS, Germany) at room temperature. The samples were uniformly mixed with dried KBr powder at mass ratio of 1:200. Each spectrum was obtained after 64 scans with 2 cm−1 resolution.
3. Results and discussion
3.1 Cr(VI) retention processes
 | ||
| Fig. 1 Concentration variation of Cr(VI) and Cr(T) in solution with time under different initial Cr(VI) concentration conditions. Initial Cr(VI) concentration are 400, 250, 100, and 40 mg L−1 respectively; initial pH is 5.7; background electrolyte is 0.01 M NaCl; 2 g soil sample is contained in 20 mL solutions. Error bars are SEM (n = 3). | ||
The pH in all reaction systems increased with time as shown in Fig. S2,† and the variation amplitudes decreased with initial Cr(VI) concentration resulting from the buffering effect of Cr(VI) anions. The TOC concentration in solution increased with time as well resulting from soil organic matter (SOM) dissolution (Fig. S3†), however, the variation amplitudes increased with initial Cr(VI) concentration, which is in contrast with the pH trend. Notably, the lower equilibrium pH condition resulting from the higher initial Cr(VI) concentration ought to be unfavorable for the dissolution of SOM (such as humic acid), therefore, the abnormal occurrence of the higher TOC concentration under lower pH condition might be induced by the oxidation of SOM by Cr(VI), where lower molecular weight SOM with higher solubility might be produced by SOM oxidative decomposition. Additionally, a positive correlation was found between the concentrations of TOC and Cr(III) in solution (Fig. S4†), which can be used to interpret the higher Cr(III) concentration in solution under higher initial Cr(VI) concentration condition resulting from the high solubility of dissolved organic matter (DOM) and Cr(III) complex.
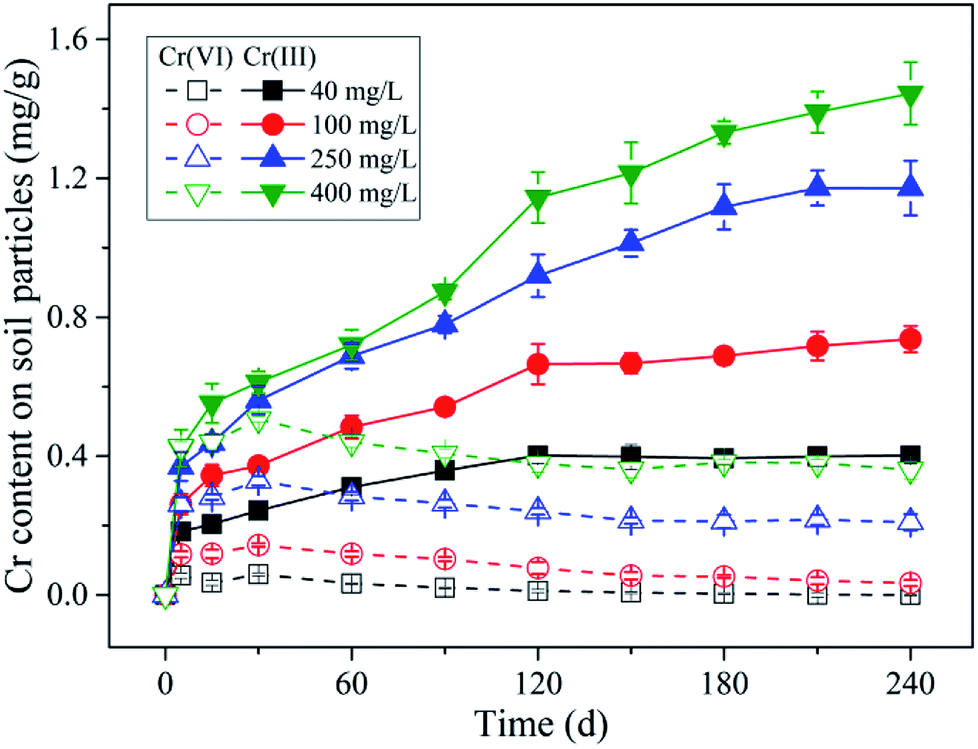 | ||
| Fig. 2 Content variation of Cr(VI) and Cr(III) on soil particles with time under different initial Cr(VI) concentration conditions. Initial Cr(VI) concentration are 400, 250, 100, and 40 mg L−1 respectively; initial pH is 5.7; background electrolyte is 0.01 M NaCl; 2 g soil sample is contained in 20 mL solutions. Error bars are SEM (n = 3). | ||
3.2 pH effect on Cr(VI) retention processes
 | ||
| Fig. 3 pH effect on concentration variation of Cr(VI) and Cr(T) in solution with time. Initial pH are 3.5, 5.7, and 7.0 respectively; initial Cr(VI) concentration is 250 mg L−1; background electrolyte is 0.01 M NaCl; 2 g soil sample is contained in 20 mL solutions. Error bars are SEM (n = 3). | ||
Throughout the experiment, the pH kept almost constant with a slight increasing, resulting from the proton consumption by Cr(VI) reduction (Fig. S5†). Meanwhile, the TOC in solution decreased with pH decreasing (Fig. S6†), due to the poor solubility of SOM under strong acidic condition (such as humic acid). It was worthy to note that the Cr(III) concentration in solution increased with pH increasing, however, in theory Cr(III) cations were supposed to be much more soluble under acidic conditions than neutral conditions. Considering that the TOC concentration under pH 7.0 (110 mg C L−1) was much higher than that under pH 3.5 (51 mg C L−1), and the positive correlation between the concentrations of TOC and Cr(III) in solution under different pH conditions as well (Fig. S7†), it can be further confirmed that the occurrence of high Cr(III) concentration in solution under neutral condition was mainly related to the formation of complex between Cr(III) cations and DOM.
 | ||
| Fig. 4 pH effect on content variation of Cr(VI) and Cr(III) on soil particles with time. Initial pH are 3.5, 5.7, and 7.0 respectively; initial Cr(VI) concentration is 250 mg L−1; background electrolyte is 0.01 M NaCl; 2 g soil sample is contained in 20 mL solutions. Error bars are SEM (n = 3). | ||
3.3 Adsorption–reduction mechanism of Cr(VI) retention
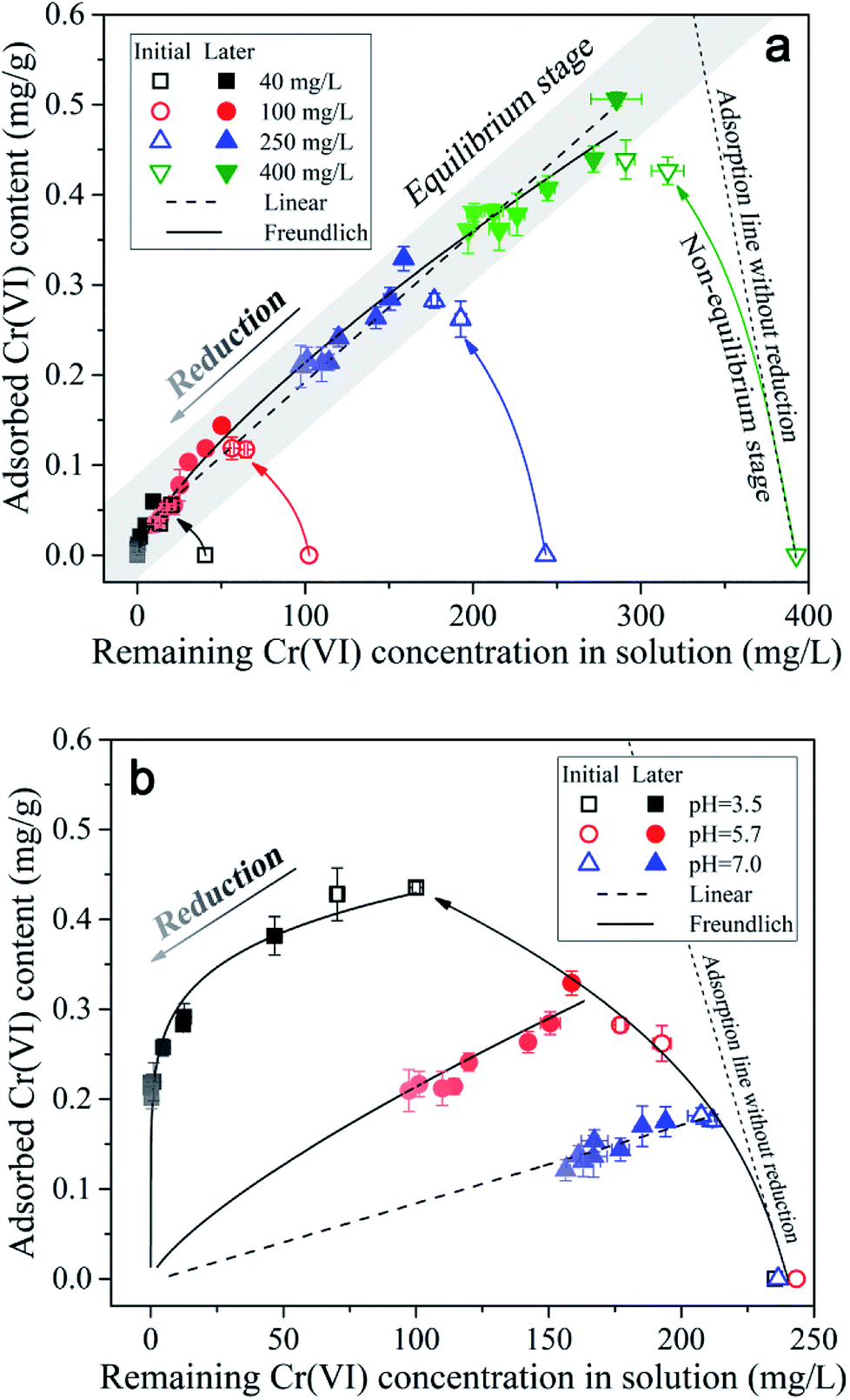 | ||
| Fig. 5 The partition of Cr(VI) between solid and aqueous phases in soil environment under (a) different initial concentration and (b) different initial pH conditions. The labels of initial and later mean the initial stage (0–30 d) and later stage (30–240 d) respectively, and the open symbols and solid symbols represent the data points from initial and later stage respectively. The color shade of the symbols from dark to light represents the data points from 30 d to 240 d, during which the Cr(VI) was progressively reduced into Cr(III) in the reaction system. | ||
As indicated in Fig. 5a, under different initial Cr(VI) concentration conditions, the adsorbed Cr(VI) was found to be positively correlated with the remaining Cr(VI) in solution in the same linear zone (the grey band), which implied that a near-linear partition rule was in charge of the Cr(VI) adsorption process. With time, the data points of different initial Cr(VI) concentration conditions all kept moving towards the original point in the diagram as indicated by the color shade from dark to light, and this was mainly induced by the Cr(VI) losing in the reaction system resulting from Cr(VI) reduction. Notably, throughout the reduction process of Cr(VI), the adsorbed Cr(VI) content on soil particles kept almost a constant ratio (partition coefficient) in respect to the remaining Cr(VI) concentration in solution, which indicated that the adsorption rate of Cr(VI) was much more higher than the reduction rate.
Additionally, it can be found in Fig. 5a that the open symbols representing the data points from initial stage (0–30 d) were a little deviated from the solid symbols representing the data points from later stage (30–240 d), especially for the symbols of higher initial Cr(VI) concentrations. As mentioned above, the content of adsorbed Cr(VI) on solid phase increased within 30 d as shown in Fig. 2, which was not consistent with the decreasing trend of Cr(VI) concentration in solution. This indicated that the equilibrium state of Cr(VI) adsorption from aqueous phase to solid phase has not been reached in 30 d, and thus this period can be called the non-equilibrium stage. The short dash line (adsorption line) in the figure reflected the non-equilibrium stage of Cr(VI) adsorption without Cr(VI) reduction in theory, where the increasing of adsorbed Cr(VI) content on solid phase was accompanied by the linear decrease of remaining Cr(VI) concentration in solution. However, in this experiment a part of Cr(VI) was reduced into Cr(III) in the non-equilibrium stage, therefore, the real adsorption line was a little deviated from the theoretical line without reduction.
The data points from equilibrium stage were selected to be fitted by the linear and Freundlich isotherm models as shown in Fig. 5a, and the equations and fitting parameters are listed in Table S1.† It was found that both of the two models had a good performance in the data fitting, and the Freundlich model (R2 = 0.99) fitted the data a little better compared with the linear model (R2 = 0.98). As indicated, the adsorption of Cr(VI) followed a near-linear partition rule within the initial Cr(VI) concentration of 400 mg L−1, and this implied that the adsorption capacity of Cr(VI) by the soil under pH 5.7 had not been reached yet. The maximum Cr(VI) adsorption quantity of 0.51 mg g−1 was reached under the initial Cr(VI) concentration of 400 mg L−1 by 30 d, and the partition coefficient (Kd) was determined to be 0.0017 L g−1 according to the fitting results of the linear model. Notably, the intercept of the linear model was not zero (0.027 mg g−1), and this was mainly induced by the non-linear character under low remaining Cr(VI) concentrations in solution ranging from 0 to 50 mg L−1, where the local partition coefficient was much higher than that under higher Cr(VI) concentrations.
The partitioning of Cr(VI) between aqueous and solid phases under different pH conditions is shown in Fig. 5b. It can be seen that with pH decreasing the data points can be fitted by the non-linear Freundlich model better compared with the linear model (the fitting parameters can be found in the Table S2†), and the reason may be that the maximum adsorption capacity was almost reached under lower pH conditions resulting from the saturation of complexation sites. Notably, as the repulsion force between Cr(VI) anions and negatively charged soil particles decreased with pH decreasing from 7.0 to 3.5, the maximum adsorption capacity was expected to be increased with pH decreasing. However, when the remaining Cr(VI) concentration in solution was greater than 323 mg L−1, the adsorption amount of Cr(VI) under pH 3.5 was not larger than that under pH 5.7 according to the extrapolation results of Freundlich model. Considering that the soil particle was negatively charged under the experimental pH condition (ZPC = 2.49), according to the electrical double layer theory, the diffusion layer will be compressed with pH decreasing from 7.0 to 3.5, which will decrease the ion exchangeable capacity of the soil particles, and this may account for the decreasing of Cr(VI) adsorption capacity under pH 3.5.
In addition, it can be seen in Fig. 5b that the data points from initial stage (0–30 d) under pH 3.5 and 7.0 were not deviated from the line of isotherm adsorption model like that under pH 5.7, and this indicated that the Cr(VI) adsorption equilibrium state had been reached within 5 d under pH 3.5 and 7.0. This might be mainly determined by the relationship between adsorption rate and reduction rate of Cr(VI). Under pH 3.5, the repulsion force between Cr(VI) anions and negatively charged soil particles was much lower compared with pH 5.7, as a result, the Cr(VI) adsorption rate was far higher than its reduction rate, resulting in the fast accumulation of Cr(VI) on soil particles. While, under pH 7.0, the proton concentration was far lower compared with that under pH 5.7, as a result, the Cr(VI) reduction rate was much lower than its adsorption rate, leading to the fast accumulation of Cr(VI) on soil particles as well. Under pH 5.7, the adsorption rate and reduction rate were comparable, therefore, the non-equilibrium stage will last longer.
In respect to the reducing agent for Cr(VI) reduction, Fe(II), S(II) and SOM were considered as the main electron donors in soil environment. However, the iron element in the soil mainly existed in trivalent form according to the XRD characterization result (Fig. S8†), and the content of sulfur element in the soil only accounted for 0.163% (w:w) according to the element analysis result. Consequently, the SOM was considered as the main electron donors for Cr(VI) reduction, which accounted for 11.64% of the black soil mass. In order to illustrate the changes of humus in soil induced by Cr(VI) reduction, the humic acid (HA) fraction, which is the majority and most active component of humus in soil for Cr(VI) reduction, was extracted from the soil samples after reacting with Cr(VI) for 240 d at different initial Cr(VI) concentration and different pH conditions, and then FTIR spectroscopy was utilized to determine the functional groups variation (shown in Fig. S15†). As indicated, the absorbance bands can be found at the bands of 1720, 1639, 1384, 1230 and 1031 cm−1, which can be attributed to carboxyl, carbonyl, methyl, phenol and hydroxyl respectively.36–43 Because the methyl has been determined to be relatively resistant to Cr(VI) oxidation,44,45 the ratios of absorbances at other functional groups bands to the absorbance of methyl were used to indicate the variation of carboxyl, carbonyl, phenol and hydroxyl, and the results are shown in Fig. S16 and S17.† As indicated, compared with the blank control group (without Cr(VI)), the ratio of carboxyl, carbonyl, phenol and hydroxyl to methyl decreased significantly with pH deceasing and initial Cr(VI) concentration increasing, especially phenol and carboxyl, which have been determined to act as the main electron donor and complexation site for Cr(VI) respectively in our previous studies.44
Considering that the maximum TOC concentration in solution was about 110 mg C L−1, which only accounted for less than 2% of the mass of SOM, therefore, the reduction of Cr(VI) in solution by DOM can be neglected. This implied that the reduction of Cr(VI) mainly occurred on the soil particles, meaning that the retention of Cr(VI) by black soil more tended to follow the adsorption–reduction mechanism.
Additionally, if the retention of Cr(VI) by black soil followed the adsorption–reduction mechanism, the Cr(VI) reduction rate in the reaction system should be related to the content of adsorbed Cr(VI) on soil particles. In order to verify this speculation, a correlation analysis was conducted between the adsorbed Cr(VI) amount on soil particles and Cr(VI) reduction rate in the reaction system, and the results were shown in Fig. S9.† As indicated, a strong positive correlation (R2 = 0.99) was found between them, where the Cr(VI) reduction rate proportionally increased with adsorbed Cr(VI) content increasing, resulting from the increasing initial Cr(VI) concentration, and this further confirmed that the Cr(VI) retention by the black soil followed the adsorption–reduction mechanism.
In order to verify whether the adsorbed Cr(VI) content also positively correlated with Cr(VI) reduction rate under various pH conditions, the corresponding correlation analysis was conducted, and the results were shown in Fig. S10.† As indicated, however, a poor linear correlation (R2 = 0.85) was found between them, where a relatively high reduction rate was achieved with a relatively low adsorbed Cr(VI) content under pH 3.5. This might be mainly induced by the much higher proton concentration in the reaction system under pH 3.5 compared with that under higher pH conditions, because the proton participated in the reduction of Cr(VI) as well. On the other hand, with pH decreasing from 7.0 to 3.5, the anion species of Cr(VI) changed from CrO42− to HCrO4− as shown in Fig. S11,† and the corresponding standard electrode potential increased from −0.13 (CrO42−/Cr3+) to 1.33 V (HCrO42−/Cr3+) resulting in the increasing of Cr(VI) oxidizability.
Additionally, under the experimental condition of this study, the reduced Cr(III) content on soil particles can reached as high as 2.16 mg g−1, while the maximum adsorbed Cr(VI) content on soil particles was only 0.38 mg g−1. This implied that the Cr(VI) reduction played a much more important role compared with Cr(VI) adsorption in Cr(VI) retention by the black soil. However, the adsorption is equally important in this process, because it directly determines the environmental risk and leaching potential of Cr(VI) in soils.
3.4 Modeling of Cr(VI) retention
According to the analysis above, the retention of Cr(VI) by black soil was determined to follow the adsorption–reduction mechanism, and this process can be described by the following two-step interface reactions as has been conceptualized by Laidler:46Step one: adsorption of Cr(VI) from aqueous phase to solid phase
| soil + Cr(VI) ⇌ soil–Cr(VI) | (1) |
Step two: reduction of adsorbed Cr(VI) into Cr(III)
| soil–Cr(VI) + H+ + e− → soil–Cr(III) + H2O | (2) |
Accordingly, a two-step adsorption–reduction kinetic model for Cr(VI) retention by black soil was developed, the development processes of which can be found in the ESI,† and the model can be described by the following differential equations:
 | (3) |
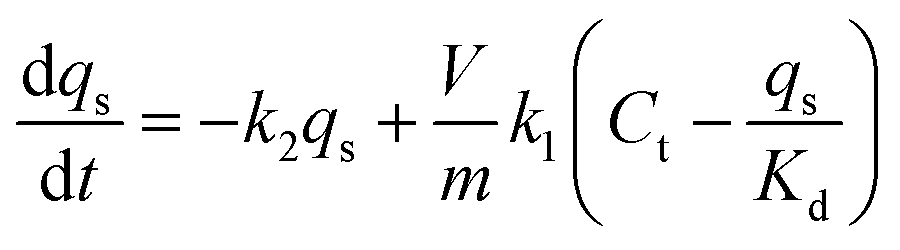 | (4) |
 | (5) |
The kinetic experimental data of Cr(VI) retention by the black soil under different initial Cr(VI) concentrations and pH conditions were simulated using the 1stOpt software by optimizing the parameters of k1, k2 and Kd from eqn (3)–(5), and the fitting results and optimized parameters are respectively shown in Fig. 6 and Table 1. As indicated in Fig. 6, the experimental data can be fitted better by the two-step kinetic model under different initial Cr(VI) concentrations and pH conditions compared with the traditional first or second order kinetic models (the fitting results by first and second order kinetic models are shown in Fig. S12 and Table S3†).
 | ||
| Fig. 6 The time evolution of Cr(VI) concentration in solution, adsorbed Cr(VI) content and reduced Cr(III) content under different initial Cr(VI) concentration and pH conditions. The curves are the fitting results of the two-step adsorption–reduction kinetic model. (a–d) represent the fitting results of initial Cr(VI) concentration ranging from 40 to 400 mg L−1 under pH 5.7. (e) and (f) represent the fitting results of initial pH 3.5 and 7.0 under initial Cr(VI) concentration of 250 mg L−1. | ||
| Condition | Cr(VI) concentration (mg L−1) | pH | Kd (L g−1) | k1 (d−1) | k2 (d−1) | R2 |
|---|---|---|---|---|---|---|
| a | 40 | 5.7 | 0.0060 | 50.6394 | 0.1138 | 0.85 |
| b | 100 | 5.7 | 0.0044 | 52.8358 | 0.0626 | 0.91 |
| c | 250 | 5.7 | 0.0018 | 54.5005 | 0.0994 | 0.85 |
| d | 400 | 5.7 | 0.0018 | 55.2086 | 0.0708 | 0.83 |
| e | 250 | 3.5 | 0.0100 | 12.4845 | 0.0660 | 0.95 |
| f | 250 | 7.0 | 0.0009 | 59.9781 | 0.1638 | 0.94 |
As can be seen in Table 1, the optimized Kd of condition (c) and (d) were both 0.0018 L g−1, which was consistent with the result (0.0017 L g−1) of linear correlation between adsorbed Cr(VI) content and remaining Cr(VI) concentration in solution under different initial Cr(VI) concentrations, and the optimized Kd of condition (f) was 0.0009 L g−1, which was also consistent with the correlation result (0.0009 L g−1). However, the optimized Kd of condition (a) and (b) were found to be a little larger than that of condition (c) and (d), and this was mainly induced by the non-linear partition characteristic under low remaining Cr(VI) concentrations in solution ranging from 0 to 50 mg L−1 as mentioned before.
It can be found that the optimized k1 was much higher than the optimized k2, and this indicated that the Cr(VI) adsorption rate was much higher than its reduction rate, which further confirmed that the reduction of Cr(VI) was the rate limiting step of Cr(VI) retention by the black soil. Additionally, the optimized k2 value of reduction rate constant was found to be much higher than that in theory (0.0138 d−1) obtained from the correlation slope between Cr(VI) reduction rate and adsorbed Cr(VI) content, and this might be mainly induced by the fast reduction of Cr(VI) within 5 d. As various electron donors may exist in the soil environment, and the one with higher reducibility will be consumed by Cr(VI) preferentially, resulting in the stepwise feature of Cr(VI) reduction. While the reduction rate constant in theory was determined only by the correlation in the range from 5 to 120 d, therefore, the kinetic model with multiple reduction constants associated with the reducibility of different electron donors might be of significance in the following studies.
In addition, the majority of the optimized k1 values ranged from 50 to 60 d−1 except for that under condition (e), and this might be mainly induced by the disproportionate relationship between the adsorbed Cr(VI) content and remaining Cr(VI) concentration in solution, which led to the poor fitting result under this reaction condition. It can be seen in Fig. 6e that a significant amount of adsorbed Cr(VI) still persisted on the soil particles, when the remaining concentration of Cr(VI) in solution has been very low. This indicated a specific adsorption of Cr(VI) that was isolated from the electron donors might have occurred on the soil particles, and it cannot be simulated well by the current two-step kinetic model, the underlying mechanism of which still deserved to be further investigate.
4. Conclusions
In this study, the adsorption and reduction of Cr(VI) retention by a typical black soil were investigated respectively by the batch experiment. A near-linear partition law was found between the adsorbed Cr(VI) content and remaining Cr(VI) concentration in solution under pH 5.7, which can be well fitted by both of the linear (R2 = 0.98) and Freundlich model (R2 = 0.99). With pH decreasing, the partition showed a strong non-linear feature, which can be described by the Freundlich model better. Additionally, the adsorbed Cr(VI) was considered as the intermediate state for Cr(VI) reduction, and the reduction rate was positively correlated with the adsorbed Cr(VI) content. Therefore, the retention of Cr(VI) by black soil was determined to follow the adsorption–reduction mechanism, and the two-step kinetic model was proposed accordingly. The two-step kinetic model has a much better fitting performance compared with traditional first and second order kinetic models, and this further implied the reliability of the proposed adsorption–reduction mechanism of Cr(VI) retention by black soil. The findings of this study will contribute to the understanding and modeling of the Cr(VI) migration and transformation in soil environment. However, the influences of Cr(VI) non-linear partition feature under different pH conditions, various electron donors with different reducibility, and Cr(VI) specific adsorption isolated with electron donors on the adsorption–reduction kinetic model still need to be further studied. Additionally, the release mechanism of Cr(III) may be another topic that deserves to be further investigated as well.Conflicts of interest
There are no competing interests to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (41672239), China Geological Survey (1212011121173), and National Science and Technology Major Project (2016ZX05040-002-003).References
- B. Dhal, H. N. Thatoi, N. N. Das and B. D. Pandey, J. Hazard. Mater., 2013, 250–251, 272–291 CrossRef CAS PubMed.
- G. Choppala, N. Bolan and B. Seshadri, J. Hazard. Mater., 2013, 261, 718–724 CrossRef CAS PubMed.
- J. Liu, X. H. Zhang, H. Tran, D. Q. Wang and Y. N. Zhu, Environ. Sci. Pollut. Res., 2011, 18, 1623–1632 CrossRef CAS PubMed.
- K. Dermentzis, A. Christoforidis, E. Valsamidou, A. Lazaridou and N. Kokkinos, Global NEST J., 2011, 13, 412–418 Search PubMed.
- C.-L. Hsu, S.-L. Wang and Y.-M. Tzou, Environ. Sci. Technol., 2007, 41, 7907–7914 CrossRef CAS PubMed.
- E. Tassi, M. Grifoni, F. Bardelli, G. Aquilanti, S. La Felice, A. Iadecola, P. Lattanzi and G. Petruzzelli, Environ. Sci.: Processes Impacts, 2018, 20, 965–976 RSC.
- I. Aharchaou, J. S. Py, S. Cambier, J. L. Loizeau, G. Cornelis, P. Rousselle, E. Battaglia and V. Dal, Environ. Toxicol. Chem., 2017, 37, 983–992 CrossRef PubMed.
- P. M. Jardine, M. A. Stewart, M. O. Barnett, N. T. Basta, S. C. Brooks, S. Fendorf and T. L. Mehlhorn, Environ. Sci. Technol., 2013, 47, 11241–11248 CrossRef CAS PubMed.
- B. Yousaf, G. Liu, Q. Abbas, R. Wang, H. Ullah, M. M. Mian and A. Rashid, RSC Adv., 2018, 8, 25983–25996 RSC.
- M. M. Mian, G. Liu, B. Yousaf, B. Fu, H. Ullah, M. U. Ali, Q. Abbas, M. A. M. Munir and L. Ruijia, Chemosphere, 2018, 208, 712–721 CrossRef CAS PubMed.
- X. Zhang, J. Tong, B. X. Hu and W. Wei, Environ. Sci. Pollut. Res., 2018, 25, 459–468 CrossRef CAS PubMed.
- T. Mpouras, M. Chrysochoou and D. Dermatas, J. Contam. Hydrol., 2017, 197, 29–38 CrossRef CAS PubMed.
- J. Jiang, Y. Wang, R. Xu and C. Yang, Environ. Earth Sci., 2011, 66, 1155–1162 CrossRef.
- H. B. Bradl, J. Colloid Interface Sci., 2004, 277, 1–18 CrossRef CAS PubMed.
- S. Butera, S. Trapp, T. F. Astrup and T. H. Christensen, J. Hazard. Mater., 2015, 298, 361–367 CrossRef CAS PubMed.
- G. Choppala, N. Bolan, A. Kunhikrishnan, W. Skinner and B. Seshadri, Environ. Sci. Pollut. Res., 2015, 22, 8969–8978 CrossRef CAS PubMed.
- J. Zhang, H. L. Yin, B. Samuel, F. Liu and H. H. Chen, RSC Adv., 2018, 8, 3522–3529 RSC.
- M. Otero, L. Cutillas-Barreiro, J. C. Novoa-Munoz, M. Arias-Estevez, M. J. Fernandez-Sanjurjo, E. Alvarez-Rodriguez and A. Nunez-Delgado, Solid Earth, 2015, 6, 373–382 CrossRef.
- G. Choppala, A. Kunhikrishnan, B. Seshadri, J. H. Park, R. Bush and N. Bolan, J. Geochem. Explor., 2018, 184, 255–260 CrossRef CAS.
- S. S. Fan, Y. Wang, Y. Li, J. Tang, Z. Wang, J. Tang, X. D. Li and K. Hu, RSC Adv., 2017, 7, 7576–7590 RSC.
- A. H. Whitaker, J. Pena, M. Amor and O. W. Duckworth, Environ. Sci.: Processes Impacts, 2018, 20, 1056–1068 RSC.
- L. Leita, A. Margon, A. Pastrello, I. Arcon, M. Contin and D. Mosetti, Environ. Pollut., 2009, 157, 1862–1866 CrossRef CAS PubMed.
- J. Zhang, L. Chen, H. Yin, S. Jin, F. Liu and H. Chen, Environ. Pollut., 2017, 225, 86–92 CrossRef CAS PubMed.
- M. K. Banks, A. P. Schwab and C. Henderson, Chemosphere, 2006, 62, 255–264 CrossRef CAS PubMed.
- P. M. Jardine, S. E. Fendorf, M. A. Mayes, I. L. Larsen, S. C. Brooks and W. B. Bailey, Environ. Sci. Technol., 1999, 33, 2939–2944 CrossRef CAS.
- M. T. Fernández-Pazos, B. Garrido-Rodriguez, J. C. Nóvoa-Muñoz, M. Arias-Estévez, M. J. Fernández-Sanjurjo, A. Núñez-Delgado and E. Álvarez, Water, Air, Soil Pollut., 2012, 224, 1366 CrossRef.
- A. A. Khan, M. Muthukrishnan and B. K. Guha, J. Hazard. Mater., 2010, 174, 444–454 CrossRef CAS PubMed.
- J. Jiang, R. Xu, Y. Wang and A. Zhao, Chemosphere, 2008, 71, 1469–1475 CrossRef CAS PubMed.
- J. Zhang, H. Yin, H. Wang, L. Xu, B. Samuel, F. Liu and H. Chen, Environ. Sci. Pollut. Res., 2018, 25, 16913–16921 CrossRef CAS PubMed.
- S. Lu, W. Tan, F. Liu and X. Feng, Acta Pedol. Sin., 2006, 43, 756–763 CAS.
- S. Lettens, V. B. De, P. Quataert, W. B. Van, B. Muys and O. J. Van, Eurasian J. Soil Sci., 2010, 58, 1244–1253 CrossRef.
- K. Pyrzynska, TrAC, Trends Anal. Chem., 2012, 32, 100–112 CrossRef CAS.
- G. Arslan, S. Edebal
![[i with combining dot above]](https://www.rsc.org/images/entities/char_0069_0307.gif) and E. PehlVan, Desalination, 2010, 255, 117–123 CrossRef CAS.
and E. PehlVan, Desalination, 2010, 255, 117–123 CrossRef CAS. - S. Barnie, J. Zhang, H. Wang, H. Yin and H. Chen, Chemosphere, 2018, 212, 209–218 CrossRef CAS PubMed.
- P. Janos, V. Hula, P. Bradnova, V. Pilarova and J. Sedlbauer, Chemosphere, 2009, 75, 732–738 CrossRef CAS PubMed.
- P. E. Fanning and M. A. Vannice, Carbon, 1993, 31, 721–730 CrossRef CAS.
- N. H. Hsu, S. L. Wang, Y. C. Lin, G. D. Sheng and J. F. Lee, Environ. Sci. Technol., 2009, 43, 8801–8806 CrossRef CAS PubMed.
- J. M. O'Reilly and R. A. Mosher, Carbon, 1983, 21, 47–51 CrossRef.
- C. Sellitti, J. L. Koenig and H. Ishida, Carbon, 1990, 28, 221–228 CrossRef CAS.
- T. T. Zhao, W. Z. Ge, Y. X. Nie, Y. X. Wang, F. G. Zeng and Y. Qiao, Fuel Process. Technol., 2016, 150, 71–77 CrossRef CAS.
- W. Chen, N. Habibul, X. Y. Liu, G. P. Sheng and H. Q. Yu, Environ. Sci. Technol., 2015, 49, 2052–2058 CrossRef CAS PubMed.
- Z. Filip, K. Haider, H. Beutelspacher and J. Martin, Geoderma, 1974, 11, 37–52 CrossRef CAS.
- S. Paim, L. Linhares, A. Mangrich and J. Martin, Biol. Fertil. Soils, 1990, 10, 72–76 CAS.
- J. Zhang, H. Yin, L. Chen, F. Liu and H. Chen, Environ. Pollut., 2018, 237, 740–746 CrossRef CAS PubMed.
- J. Zhang, H. Yin, H. Wang, L. Xu, B. Samuel, J. Chang, F. Liu and H. Chen, Sci. Total Environ., 2019, 651, 2975–2984 CrossRef CAS PubMed.
- K. J. Laidler, Pure Appl. Chem., 1996, 68, 149–192 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra08154a |
| This journal is © The Royal Society of Chemistry 2019 |