
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceScope of organometallic compounds based on transition metal-arene systems as anticancer agents: starting from the classical paradigm to targeting multiple strategies
Mehvash Zaki
 *a,
Suboot Hairat
b and
Elham S. Aazam
a
*a,
Suboot Hairat
b and
Elham S. Aazam
a
aDepartment of Chemistry, King Abdulaziz University, Jeddah, Saudia Arabia. E-mail: mehvashzaki@gmail.com; Tel: +91 8979086156, +966 561835672
bDepartment of Biotechnology, Wachemo University, Hossana, Ethiopia
First published on 24th January 2019
Abstract
The advent of the clinically approved drug cisplatin started a new era in the design of metallodrugs for cancer chemotherapy. However, to date, there has not been much success in this field due to the persistence of some side effects and multi-drug resistance of cancer cells. In recent years, there has been increasing interest in the design of metal chemotherapeutics using organometallic complexes due to their good stability and unique properties in comparison to normal coordination complexes. Their intermediate properties between that of traditional inorganic and organic materials provide researchers with a new platform for the development of more promising cancer therapeutics. Classical metal-based drugs exert their therapeutic potential by targeting only DNA, but in the case of organometallic complexes, their molecular target is quite distinct to avoid drug resistance by cancer cells. Some organometallic drugs act by targeting a protein or inhibition of enzymes such as thioredoxin reductase (TrRx), while some target mitochondria and endoplasmic reticulum. In this review, we mainly discuss organometallic complexes of Ru, Ti, Au, Fe and Os and their mechanisms of action and how new approaches improve their therapeutic potential towards various cancer phenotypes. Herein, we discuss the role of structure-reactivity relationships in enhancing the anticancer potential of drugs for the benefit of humans both in vitro and in vivo. Besides, we also include in vivo tumor models that mimic human physiology to accelerate the development of more efficient clinical organometallic chemotherapeutics.
 Mehvash Zaki | Dr Mehvash Zaki is an Assistant Professor in the Chemistry Department, King Abdulaziz University, and prior to that she was a Research Associate from the Council of Scientific and Industrial Research (CSIR), Government of India. She received her Ph.D. degree in Chemistry from Aligarh Muslim University, India in 2014. During her Ph.D. tenure, she was awarded the Maulana Azad National Fellowship (MANF) from the University Grant Commission, Government of India. She has published several research articles and reviews in peer reviewed journals of high international repute and participated in national and international conferences. Her research work mainly includes the molecular design and synthesis of metal-based cancer chemotherapeutic drug candidates and their in vitro interaction with DNA/serum proteins, in addition to nuclease, protease and Topoisomerase (I and II) inhibition studies. |
 Suboot Hairat | Dr Suboot Hairat is currently working as an Assistant Professor of Biotechnology, Wachemo University, Ethiopia. He completed his Ph.D. in Plant Molecular Biology from the South Campus, University of Delhi, India and has vast research experience in his field. His research focuses on the “Evaluation of members of the triticeae family for thermotolerance, and transcriptome analysis and functional characterization of Lipid Transfer Proteins in Bread wheat (Triticumaestivum)”. He has published many research articles pertinent to his specialization area in the peer reviewed journals of international repute and participated in conferences/symposium. He also worked as a Research Associate in the renowned research institute National Institute of Plant Genomic Research (NIPGR), New Delhi, India. Later he become an Associate Scientist in the Central Instrumentation Facility, University of Delhi. His research interests include Plant Molecular Biology and Biotechnology, i.e. understanding the molecular mechanisms of thermotolerance in plants, but recently he became interested in recognizing the effect of metal-based drugs on the treatment of cancer. |
 Elham S. Aazam | Prof. Elham S. Aazam is a Professor in the Department of Chemistry, King Abdulaziz University, Jeddah, Saudi Arabia. She completed Ph.D. in Chemistry (2002) from the University of Sussex, Brighton, United Kingdom and joined the Department of Chemistry, King Abdulaziz University, Jeddah, Saudi Arabia as an Assistant Professor in 2004. She has also presented her research work and delivered invited lectures in various national and international conferences/symposia. She has also supervised many research theses and successfully completed a number of independent research projects. She was awarded the Summer Research Scholarship (British council) at Sussex University in 2004 and visited the IbnuSina Institute in Malaysia in 2006. Later she also visited as a guest Assistant professor in the Chemistry department, University of Sussex in UK. Additionally, she is a member of the Royal Society of Chemistry (RSC), American Chemical Society (ACS), Saudi Chemical Society and others. Her research focuses on the synthesis and X-ray crystal structure determination of Metallocenes, Schiff bases of Coumarins, Titanium complexes, Aluminum and Zinc organometallics and their application in catalysis. She is also interested in the photocatalytic properties of nano-materials. |
Introduction
Current emphasis towards the development organometallic chemotherapeutics has attracted many researchers in the search for new cancer therapeutic agents with improved activity and less toxicity.1–4 For centuries, organometallic compounds have been reviewed as catalysts, but are now studied in the exploration of new potential anticancer drugs after the landmark investigation on titanocene by Kopf and Kopf-Maier.5,6 Subsequently, several titanocene-based organometallic complexes entered clinical trials and their mechanism of action was found to be different to that of the clinically approved drug cisplatin.7,8 Since then, scientists have focused more on the development of organometallic transition metal antitumor drug candidates since these complexes show high kinetic stability and reactivity due to the presence of a metal–carbon bond and π-bound arenes. Due to the presence of the transition metal-arene system, the hydrophilicity and hydrophobicity of these complexes can be easily controlled, resulting in better cellular uptake inside tumor cells compared to normal cells.9,10 Also, these complexes show much better selectivity towards cancer cells, which have a slightly lower pH than that of normal cells, as seen with the [Ru(η6-p-cymene)Cl2(pta)] complex, resulting in pH-dependent DNA damage.11 Similarly attaching a ferrocene moiety in place of the phenyl ring in tamoxifen led to the formation of ferrocifen, which showed improved activity in breast cancer cells and will enter a clinical trial soon.12,13 Therefore the use of organometallic complexes provides much scope for rational drug design with better cellular uptake, minimum unwanted side effects and different modes of action inside tumor cells. These molecules are less toxic and show different modes of action than cisplatin, and directly interact with the biological target DNA and undergo ligand hydrolysis.14–18 The kinetic properties of organometallic complexes such as ligand exchange depend upon various factors such as ligand substitution, redox reactions, kinetic stability, low oxidation state of the metal centre and lipophilic character. Modeled after the action of cisplatin, the pharmacokinetic activity of sandwiched and half-sandwiched organometallic compounds of ruthenium, titanium, carbene complexes of gold and ferrocenyl derivatives of iron can be varied by substituting different groups on the phenyl/cyclopentadienyl ring. Another class of organometallics includes metallocenes and metal carbonyls, which display enhanced biological activities due to their high stability and the release of CO at the selective target.19–21 It is known that the covalently grafted organometallic unit is basically inert to ligand substitution; however, it increases the activity of compounds through the modification of their pharmacokinetic profile or acts as a structural mimic.22 Additionally, many organometallic complexes are highly reactive due to the presence of labile biomolecule ligands, such as nucleobases.23–29 This type of complex acts as a prodrug, where as they enter the body, they undergo ligand substitution at the target site. Currently, the design of organometallic compounds for cancer chemotherapy is flourishing due to their lower toxicity, controlled ligand substitution and alternative mechanism of action compared to that of the classic metal-based drugs.30Besides, organometallic complexes show different modes of action compared to classic metal-based drugs. These complexes show enhanced activity towards cancer cells compared to normal coordination complexes due to the following reasons: (a) direct binding with the target site, (b) indirect interaction through non-covalent interactions at the target, (c) activity mainly due to the metal-arene system, where the metal acts as a carrier in vivo, (d) production of reactive oxygen species (ROS),31 (e) organometallic complexes are photoactive and act as photosensitizer,32 (f) selectivity and efficiency towards multiple targets such as thioredoxin reductase (TrRx), mitochondria, endoplasmic reticulum, DNA, proteins and enzymes, (g) controlled ligand substitution and redox reactions and (h) kinetic stability and relative lipophilicity and the presence of a metal in a low oxidation state.
The idea of introducing various functionalities in metal-based drugs to better target cancer cells is not a new concept.33–41 For many centuries, the use of metallodrugs for cancer chemotherapy has been the focus of many scientists. Currently, with the use of modern technology and better understanding of cancer mechanisms, the strategies for the development of new multifunctional chemotherapeutic drugs have accelerated with less toxicity and different mechanisms of action. Although random screening is still a useful weapon in drug discovery, presently, scientists rationally design drugs via the implementation of various pharmacologically active ligands, modification of functional groups and targeting different sites. The main step in drug design is the identification of the site of action, which may be DNA, proteins, enzymes and any organelle in cancer cells. Once the target site of action has been chosen, another challenge for scientists working in the field of metal-based drugs is to design drugs that will directly act on this target without causing side effects to healthy cells. However, it is difficult to determine whether the synthesized complexes need to be inert to redox activity or these reactions need to be controlled under biological conditions to reach the target site. After detailed studies in this field, many scientists concluded that metal- and/or ligand-centered redox reactions provide a unique platform for drug design with mechanisms totally different from that of organic drugs.
In this review we mainly focus on the interactions of organometallic metal-based drugs with various target sites, including DNA, proteins, endoplasmic reticulum (ER) and enzymes. Despite the fact that the traditional clinical platinum antitumor drugs have DNA as their major target, nowadays DNA is not considered as the favored target in the development of antitumor drugs. This is mainly because DNA is present in all types of cells, and thus drugs targeting DNA cause high toxicity and adverse side effects on normal healthy cells besides their action on cancer cells. However, it has been found that chemotherapeutic drugs that target a protein or enzyme are not always successful since cells readily become resistant to such drugs and utilize alternative metabolic pathways. Therefore, before designing drugs, it should be considered that they must target multiple sites to avoid resistance by cancer cells. Accordingly, strategies for targeting multiple sites and the search for new therapeutic targets are becoming increasingly important in the development of chemotherapeutic drugs. Currently, scientists are continuously working to determine new target sites for metallodrugs and much progress has been made on target-site validation. Therefore, this suggests that quest for the design of chemotherapeutic drugs and progress in identifying effective targeting sites are still ongoing in the field of medicinal inorganic chemistry.42
From a chemical perspective, the comparatively robust nature of the organometallic scaffold present in metal complexes represents an ideal template for rational drug design. This has helped researchers to determine the mechanism of the anticancer activity of drugs and identify the mechanisms by which drug activity can be optimized.43,44 In organometallic complexes, their metal–carbon (M–C) bonds endow these coordination compounds with special properties. Additionally, they have high a trans effect and trans influence, where the lability of bonds to other ligands (M–L) in the complex can be greatly influenced by the presence of M–C bonds. On the other hand, π-bonded aromatic arene and cyclopentadienyl ligands can act both as electron donors and π-acceptors. Therefore, these ligands can modify the donor/acceptor behavior (and reactivity) of other ligands in the complex.45 Accordingly, in this review, we outline the recent developments in organometallic complexes as antitumor agents together with their general mechanisms of action and the approaches used to improve their anticancer efficacy. In particular, detailed analysis of the systematic variations of complexes based on a single scaffold has increased the understanding of how each part of a molecule contributes to the function of complexes as antitumor agents.
Organometallic ruthenium complexes targeting human serum albumin (HSA), mitochondria, endoplasmic reticulum (ER), DNA and enzymes
Organometallic compounds display distinct chemical properties compared to other metal complexes due to the presence of metal–carbon bonds. These complexes show a high trans effect, which enhances the lability of other ligands in the complex, thereby resulting in a spectrum of antitumor activity. Specifically, P. J. Dyson and P. J. Sadler et al. found that two ruthenium(II)-arene compounds, namely [Ru(η6-p-cymene)Cl(en)], where, en = ethylenediamine (termed RAED-C)46,47 and [Ru(η6-p-cymene)Cl2(pta)], where, pta = 1,3,5-triaza-7-phosphaadamantane (termed RAPTA-C) (Fig. 1), showed interesting in vivo profiles for cancer cell growth.48 It has been observed that RAED-C reduces the growth of primary tumors; whereas, RAPTA-C strongly affects solid tumor metastases.49–56 This is attributed to the differential preferential binding site of each molecule to chromatin, where the RAED-C complex binds with DNA, while RAPTA-C binds with the histone core and exerts cytotoxicity.57 Arguably, the RAPTA-C complex has been found to be the most successful ruthenium organometallic chemotherapeutic agent in the literature. Besides binding to DNA/histone proteins, Ru(II) organometallics preferentially inhibit the activity of some enzymes such as thioredoxin reductase and cathepsin B, which are mainly involved in cancer progression.58 Generally, their mechanism of action is thought to involve hydrolysis of the Ru–X bond, generating an active Ru–OH2 species, which is responsible for the cytotoxic activity of these complexes. However, the rate of hydrolysis is very important, where, if the complexes hydrolyze too fast they may not reach the target site, and thus a key factor for this type of complex. It has been well established in the literature that the primary cellular target for Ru(II) organometallics is DNA, which is similar to other metal-based drugs. Therefore, the factors affecting DNA binding such as rate and extent of binding and non-covalent interactions such as hydrogen bonding and DNA intercalation are crucial for their antitumor activity.59 Consequently, improvement of the selectivity and broadening of the therapeutic action of ruthenium organometallics have prompt researchers to develop more effective antitumor chemotherapeutics. In recent years, a plethora of new strategies have emerged to increase the efficiency of ruthenium organometallics via ligand modulation to access diverse compounds endowed with a wide range of functionality, thereby providing a new platform for the development of prospective new organometallic chemotherapeutics.60 These strategies involve the targeting of human serum albumin (HSA), mitochondria, endoplasmic reticulum (ER), DNA and enzymes. Some complexes target HSA to reach the cancer cells. HSA is a carrier protein found in the blood in high quantities, and it has been observed that this protein accumulates in cancer cells. Thus, if a complex interacts with this protein then it will easily reach the target cancer cells. Similarly, mitochondria and endoplasmic reticulum are involved in the production of reactive oxygen species (ROS), and if drugs disrupt their proper functioning then cancer cells will undergo cell death due to the high production of ROS. In addition, DNA and different enzymes (TrRx and Topo) are well-known targets for classical drugs. Thioredoxin reductase (TrRx) is one of the enzymes present in cancer cells, which is involved in the removal of ROS. This enzyme is present in a very low concentration in cancer cells; thus, if the function of this enzyme is inhibited, then cancer cells will undergo cell arrest or apoptosis due to the accumulation of ROS. Thus, all these strategies are very important in the design of chemotherapeutic drugs.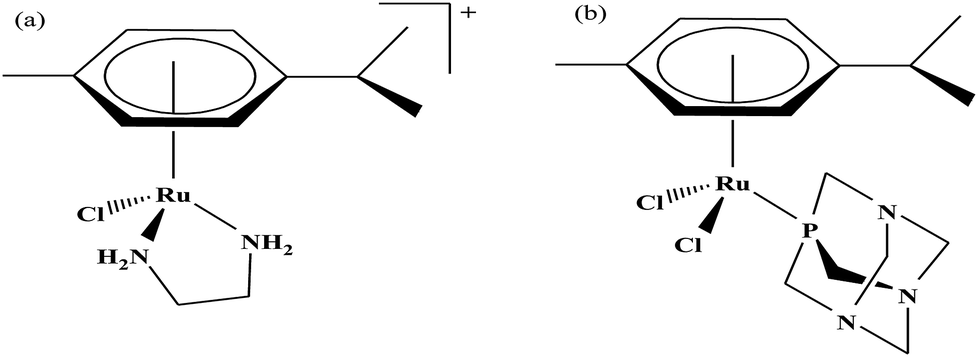 | ||
| Fig. 1 Chemical structures of (a) RAED-C and (b) RAPTA-C. | ||
In this review, we only discuss selected examples of ruthenium complexes showing promising antitumor activities. The use of half-sandwich organometallic complexes provides a new strategy in the development of new chemotherapeutics agents with enhanced in vitro and in vivo activities since these complexes are highly stable and selective towards low pH levels, which are commonly found in solid tumors.61 Recently P. Heffeter and W. Kandioller et al.62 tested poorly soluble metal-based half-sandwich complexes of the type [Ru(η6-p-cymene)(NQ)Cl], [Ru(η6-p-cymene)(PT)Cl] and [Ru(η6-p-cymene)(CHM)Cl] where, NQ = 2-hydroxy-3-methylnaphthalene-1,4-dione, PT = 3-hydroxy-2-methyl-4H-pyran-4-thione, and CHM = 2-(4-chlorophenyl)-3-hydroxy-4H-chromen-4-one, with the aim of controlled, pH-triggered release of the active metallodrugs (Fig. 2). The aqueous solubility of these complexes could be enhanced via their conjugation to hydrophilic polymers such as poly(organo)-phosphazene macromolecules. The synthesized conjugates and the complexes were well-characterized and compared for their antiproliferative activity on various human carcinomas. It was observed that the conjugation of the free organometallics to phosphazenes significantly diminished their in vitro activity compared to that of the free complexes. However, the conjugates showed excellent antitumor profiles in vivo and there was considerable tumor shrinkage after administration of the first dose of loaded polymers. This effect was much more pronounced in the case of the naphthoquinone-based Ru(II) conjugate [Ru(η6-p-cymene)(NQ)Cl], where, specifically, one mouse experienced more than 100% survival time. Generally, the conjugation of organometallics to poly(organo)phosphazenes for drug delivery has given researchers new impetus in the treatment of cancer. This strategy not only stabilizes reactive organometallics, but also tremendously reduces adverse side effects such as the deformation of organs in vivo.
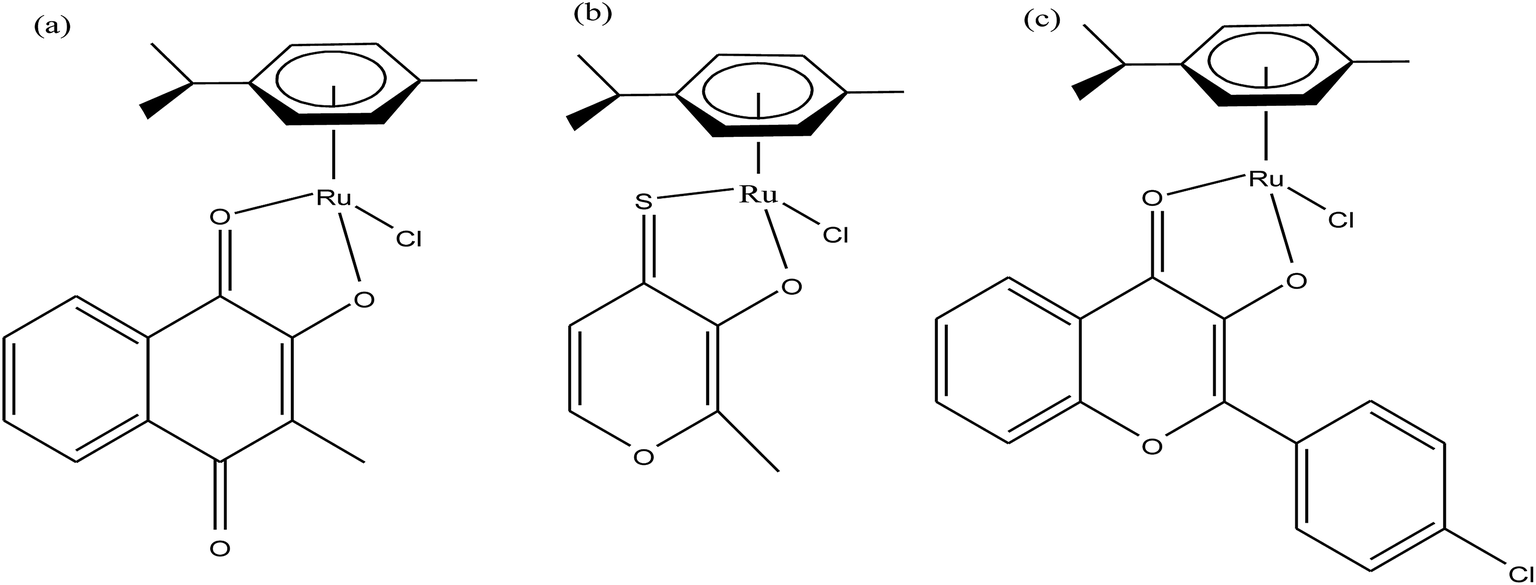 | ||
| Fig. 2 Chemical structures of complexes (a) [Ru(η6-p-cymene)(NQ)Cl], (b) [Ru(η6-p-cymene)(PT)Cl] and (c) [Ru(η6-p-cymene)(CHM)Cl] synthesized for macromolecular conjugation. | ||
It is estimated that the addition of a half-sandwich metal center increases the antitumor activity of compounds, but in some cases moderate activity has been observed, as reported by G. S. Smith et al.63 Here, we suggest that modification in the arene ring system and cyclopentadienyl ligands increases the hydrophilicity, which may be helpful to increase the antitumor efficacy. G. S. Smith et al. synthesized the di- and trinuclear ruthenium complexes [Ru2(η6-p-cymene)2(DIXD)Cl2] and [Ru3(η6-p-cymene)3(BTPE)Cl6] (where, DIXD = diisonicotinic acid 1,4-xylylene diester, BTPE = benzene-1,3,5-tricarboxylic acid tripyridin-4-ylmethyl ester) of polypyridyl ester ligands (Fig. 3) and evaluated their antitumor activity on the A2780 (cisplatin-sensitive) and A2780cisR (cisplatin-resistant) human ovarian cancer cell lines. The results revealed that the dinuclear ruthenium complex did not show promising results, but the trinuclear ruthenium complex exhibited good antiproliferative activity, which is mainly attributed to its increased number of aryl ester groups and type of metal moiety in the complex. It was observed that the trinuclear complex [Ru3(η6-p-cymene)3(BTPE)Cl6] was less active on the A2780cisR cell line with an IC50 value of 84.4 μM compared to the A2780 cell line with an IC50 value of 53.0 μM. However the complex [Ru3(η6-p-cymene)3(BTPE)Cl6] was not as active as cisplatin on both cancer cell lines (IC50 = 1.5 μM for A2780 and 25 μM for A2780cisR cells). Also, the toxicity studies for the trinuclear complex [Ru3(η6-p-cymene)3(BTPE)Cl6] on a non-tumorous human embryonic kidney (HEK) cell line showed that it was less toxic compared to the traditional drug cisplatin. Thus, it can be concluded that antitumor activity increases with an increase in the size and the number of metal centers, but unfortunately in this case the activity was moderate. However, there is a much scope to increase the pharmacological activity of these complexes through modification of the ligand structure and the metal moieties such as the use of arene ligands.
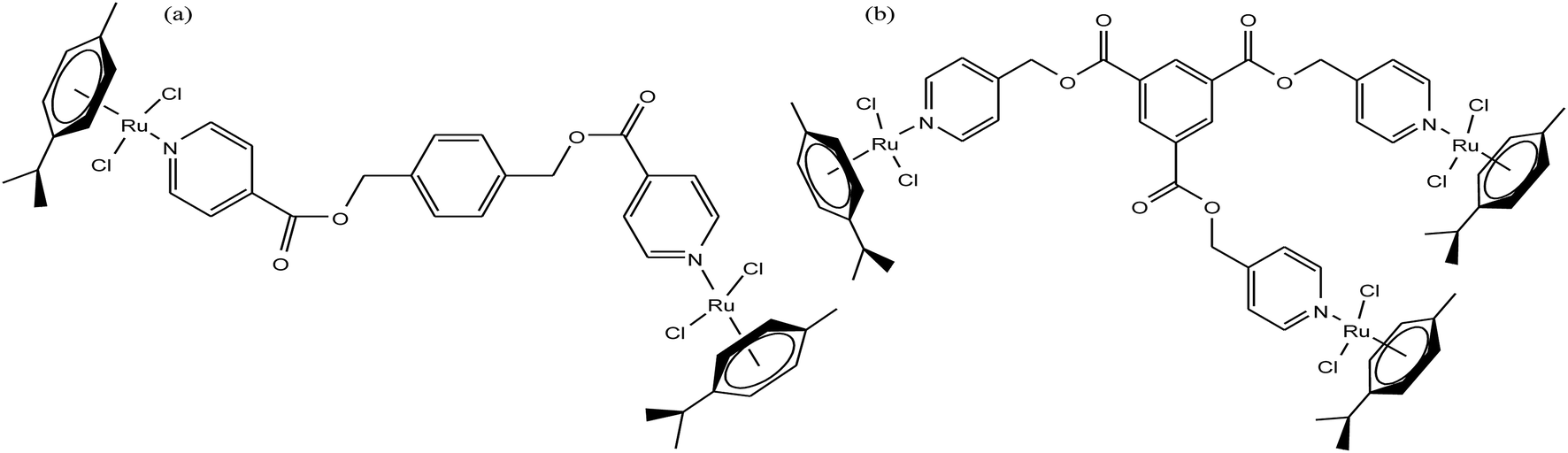 | ||
| Fig. 3 Chemical structure of (a) dinuclear [Ru2(η6-p-cymene)2(DIXD)Cl2] and (b) trinuclear [Ru3(η6-p-cymene)3(BTPE)Cl6] ruthenium complexes of polypyridyl ester. | ||
Half-sandwich complexes, often called ‘piano-stool’ complexes, provide a new platform for the design of chemotherapeutic candidates. Their thermodynamic and kinetic parameters can be easily controlled via modification of their aromatic ring system and by increasing the size of the coordinated arene moiety, which increases their activity in human ovarian cancer cell lines. Besides, changing the chelating ligand in these metal arene complexes also have a good impact on their nucleobase selectivity and the kinetics of the drug molecules.64 Due to the scarcity of the literature on the biological properties of arene ruthenium complexes bearing aroylhydrazones, R. Ramesh et al.65 combined an arene ruthenium unit with a benzhydrazone ligand to generate a series of organometallic compounds (Fig. 4) with promising antitumor activities. All the complexes were thoroughly screened for their cytotoxicity against human cervical cancer cells (HeLa), human breast cancer cells (MDA-MB-231) and human liver carcinoma cells (Hep G2) under in vitro conditions. It was inferred that three complexes, [Ru(η6-C6H6)(Cl)(L3)], [Ru(η6-p-cymene)(Cl)(L1)] and [Ru(η6-p-cymene)(Cl)(L3)] (where, L1 and L3 are indole-3-carboxaldehyde benzhydrazone ligands), were highly active against all the tested cell lines with very low IC50 values compared with that of the classic drug cisplatin. Moreover, the in vitro cytotoxic activity studies of these complexes on healthy mouse embryonic fibroblast cells (NIH 3T3) confirmed their high selectivity towards cancer cells. For the [Ru(η6-C6H6)(Cl)(L3)] and [Ru(η6-p-cymene)(Cl)(L3)] complexes, their high activity is correlated with the electron-donating methoxy substituent at the phenyl ring of the ligand, which increases the lipophilic behavior and allows easy permeation through the cell membrane. Further evaluation using fluorescence staining and flow cytometry techniques revealed that the [Ru(η6-C6H6)(Cl)(L3)] and [Ru(η6-p-cymene)(Cl)(L3)] complexes induced mitochondria-mediated apoptosis in MDA-MB-231 cancer cells (Fig. 5), suggesting their chemopreventive and chemotherapeutic potential for human cancers.
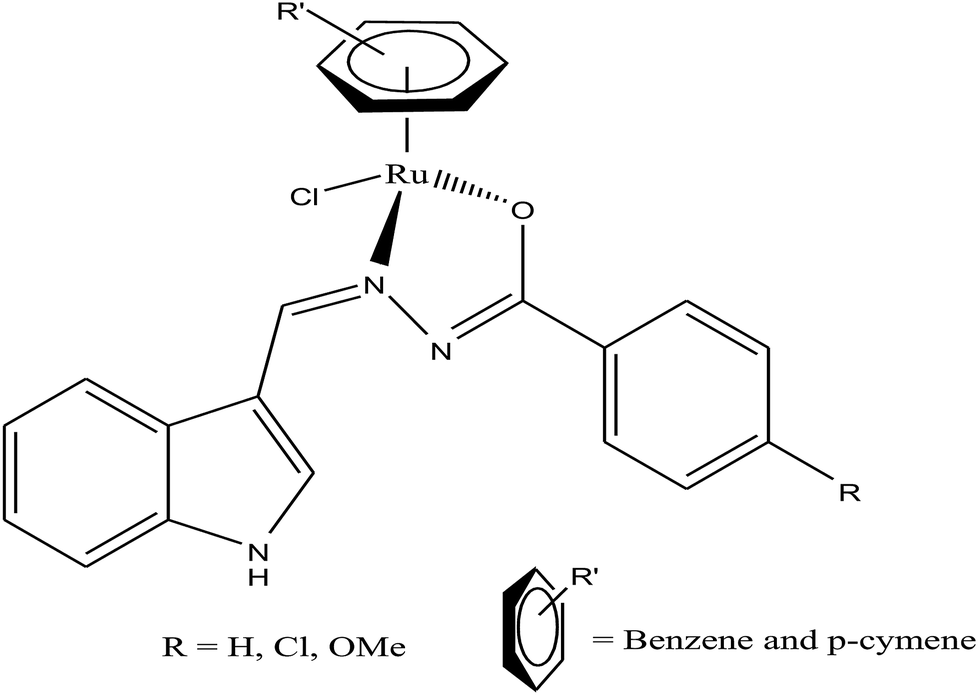 | ||
| Fig. 4 Chemical structure of ruthenium(II) arene benzhydrazone complexes. | ||
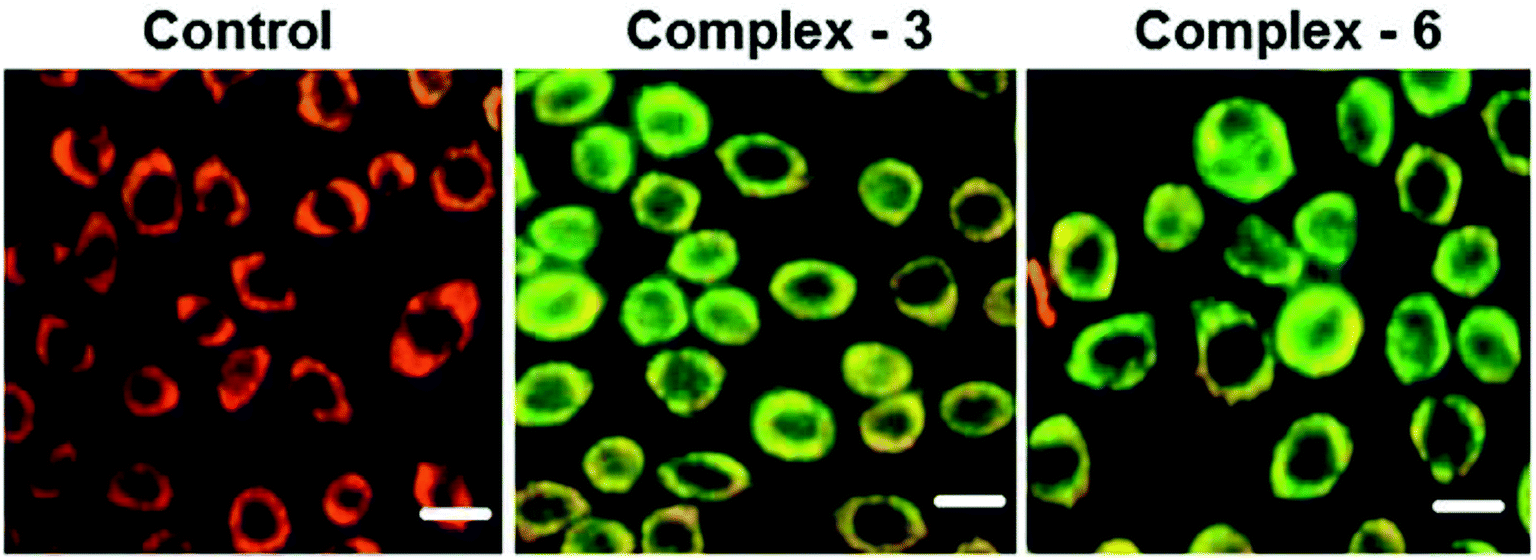 | ||
| Fig. 5 MDA-MB-231 cells were treated with complexes [Ru(η6-C6H6)(Cl)(L3)] and [Ru(η6-p-cymene)(Cl)(L3) for 24 h. The scale bar is 20 mm. This figure was reproduced from ref. 65 with permission from the Royal Society of Chemistry. | ||
In continuation of their work, R. Ramesh et al.66 also reported another series of organometallic ruthenium(II) arene benzhydrazone complexes, in which the ruthenium unit was combined with an anthraquinone moiety and the benzhydrazone ligand (Fig. 6). The in vitro cytotoxicity of all the complexes against HeLa, MDA-MB-231, Hep G2 and NIH 3T3 revealed that they are significantly active due to the presence of extended π conjugation and chelation of Ru(II) ions. However, the [Ru(η6-p-cymene)(Cl)(L3)] complex showed higher efficacy with very low IC50 values compared to cisplatin due to the substitution of the electron-donating methoxy group in the benzhydrazone ligand. This can be explained by the fact that the methoxy group increases the lipophilic character while the p-cymene moiety imparts hydrophobicity, thereby enhancing the cellular accumulation and increasing the antitumor activity. Besides, it was observed that the complexes arrested the proliferation of MDA-MB-231 cells to a much greater extent than HeLa and Hep-G2 cells. Additionally, morphological changes were investigated for the [Ru(η6-p-cymene)(Cl)(L1)] and [Ru(η6-p-cymene)(Cl)(L3)] complexes (where, L1 and L3 are 9-anthraldehyde benzhydrazone derivatives) using various biochemical apoptosis assays, and the results suggested that both complexes induced apoptosis in MDA-MB-231 cancer cells. Also, hemolytic activity studies showed that there was only negligible red hemoglobin release, implying that the complexes are negligibly toxic and are safe to normal cells. Overall, it can be concluded that ruthenium-arene-based benzhydrazone complexes are promising antitumor agents that further warrant in vivo studies.
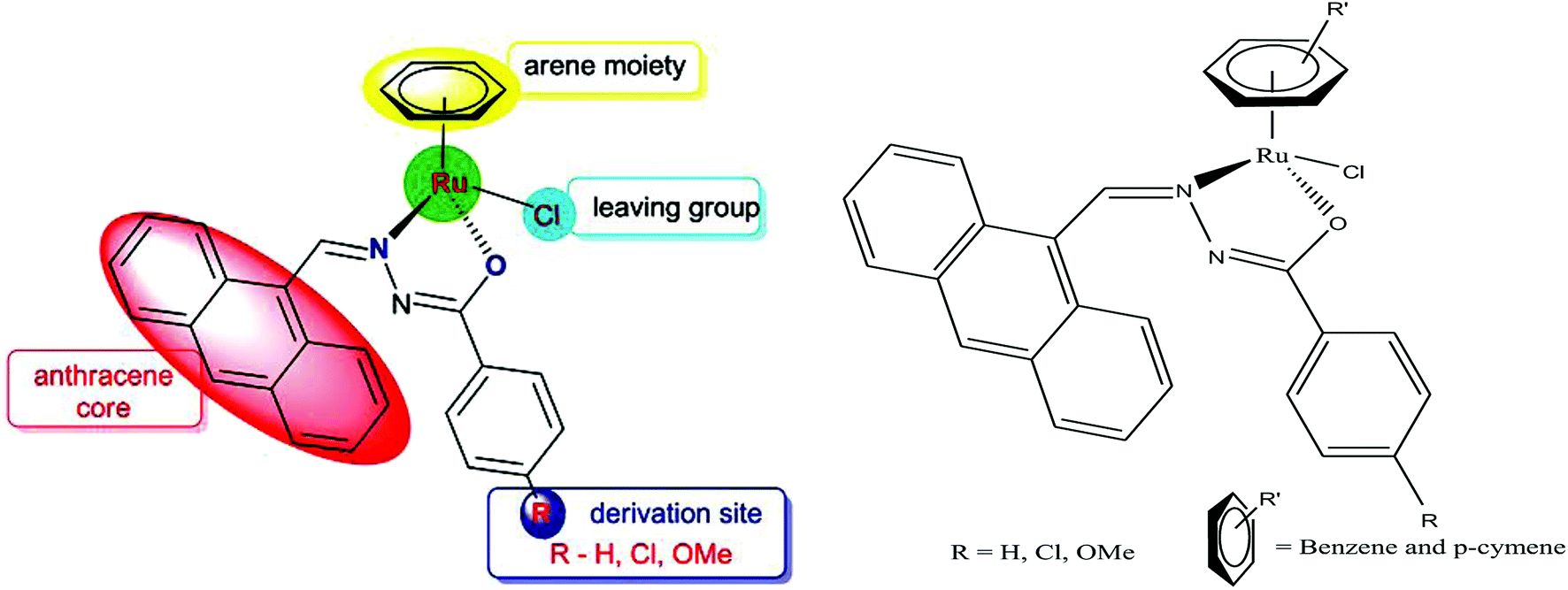 | ||
| Fig. 6 Design of ligand framework and chemical structure of organometallic ruthenium Ru(II) arene 9-anthraldehyde benzhydrazone complexes. This figure was reproduced from ref. 66 with permission from the Royal Society of Chemistry (Great Britain). | ||
Generally, the mechanism of action of these chemotherapeutic drugs involves apoptosis in cancer cells, but the effectiveness of chemotherapeutic treatments is frequently diminished because of multidrug-resistance (MDR) found in many types of cancer cells. Thus, to overcome the problem of multidrug resistance, we have to change our strategy by selecting non-apoptotic routes to induce cancer cell death. Non-apoptotic cell death can be induced by choosing organometallic facially-bound arene ligands since they can have drastic effects on the mechanism of action leading to cell death via non-apoptotic programmed cell death (PCD).67 In this regard, recently, W. H. Ang and C. Gaiddon et al.68 reported the combinatorial synthesis and evaluation of a new class of water-soluble/stable half-sandwich RuII arene Schiff-base (RAS) complexes, [Ru(η6-HMB)(MQMA)Cl]Cl and [Ru(η6-TBP)(MQMA)Cl]Cl {where, TPB = 1,3,5-triisopropybenzene, HMB = hexamethylbenzene, and MQMA = 4-methoxy-N-(2-quinolinyl-methylene)-aniline} (Fig. 7). The [Ru(η6-TBP)(MQMA)Cl]Cl complex contained triisopropylbenzene (TIPB) and iminoquinoline ligands, which are highly active against various cancer cell lines, distinct from classic alkylating agents, such as cisplatin and previously reported anticancer RuII complexes. It was observed that the [Ru(η6-TBP)(MQMA)Cl]Cl complex was stable against hydrolysis and did not interact directly with dGMP nucleotides. In addition, it did not induce upregulation in p53 expression, which is commonly associated with DNA damage. Since most of the classic drugs act through apoptosis, drugs capable of inducing alternative forms of programmed cell death (PCD) can potentially harnessed to bypass multidrug resistance (MDR). An alternative strategy is to bypass the mechanism of MDR entirely to induce cancer cell death via non-apoptotic programmed cell death (PCD). Therefore to unravel the mechanism of action of the [Ru(η6-HMB)(MQMA)Cl]Cl and [Ru(η6-TBP)(MQMA)Cl]Cl complexes, they were tested on AGS gastric cancer cells to determine their effect on cellular ROS levels. Both organoruthenium complexes induced non-apoptotic PCD through ER stress pathways, but their modes of action were quite different irrespective of their structural variations. The [Ru(η6-TBP)(MQMA)Cl]Cl complex acted through ROS-mediated ER stress; whereas, [Ru(η6-HMB)(MQMA)Cl]Cl exhibited ROS-independent activity (Fig. 8). This independent activation of both pathways led to non-apoptotic PCD in the treated cells, and thus, this strategy can be harnessed to overcome apoptosis resistance. These complexes were more active against apoptosis-resistant cells compared to certain clinical drugs, including oxaliplatin. This study, for the first time, provided a molecular basis for underpinning ER stress, demonstrating the role of structure-reactivity relationship to bypass apoptosis resistance.
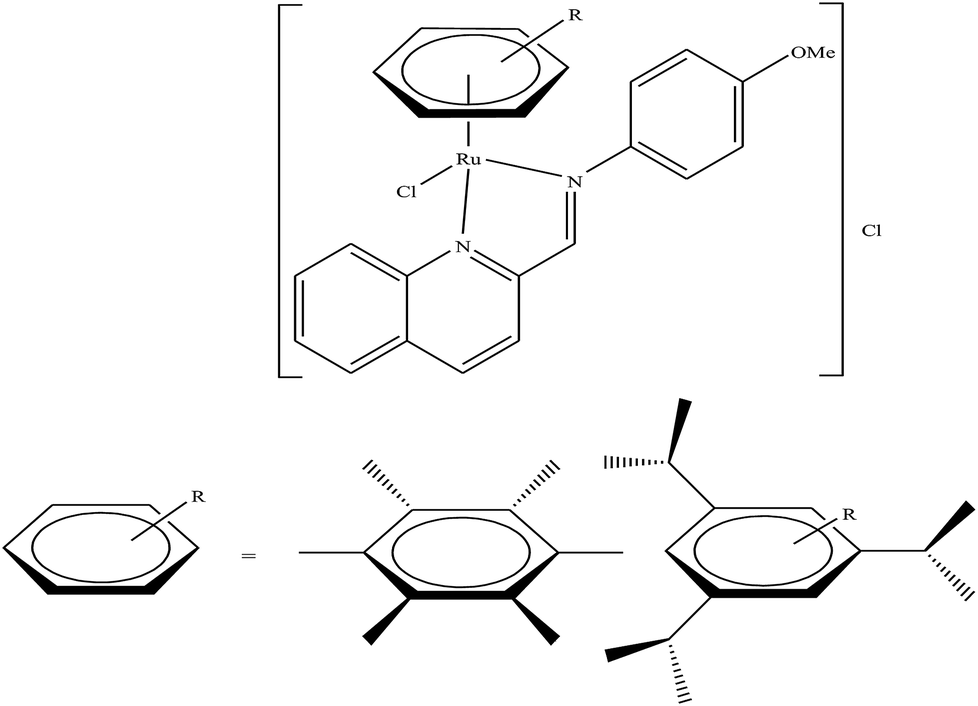 | ||
| Fig. 7 Chemical structure of ruthenium(II) Schiff-base (RAS) complexes [Ru(η6-HMB)(MQMA)Cl]Cl and [Ru(η6-TBP)(MQMA)Cl]Cl. | ||
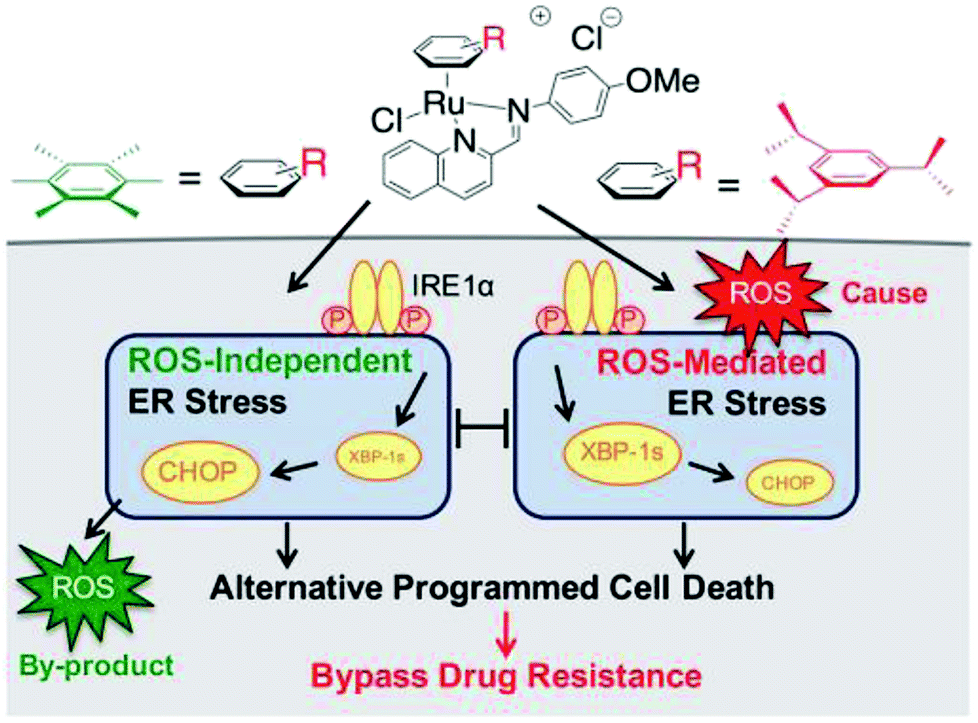 | ||
| Fig. 8 Differential ER stress pathway activation by the [Ru(η6-HMB)(MQMA)Cl]Cl and [Ru(η6-TBP)(MQMA)Cl]Cl complexes leads to an alternative (non-apoptotic) PCD, which bypasses drug resistance mechanisms. This figure was reproduced from ref. 68 [M. J. Chow, C. Licona, G. Pastorin, G. Mellitzer, W. H. Ang and C. Gaiddon, Chem. Sci., 2016, 7, 4117–4124.] with permission from The Royal Society of Chemistry. | ||
To improve the traditional paradigm of rational drug design, organometallic compounds have been considered as promising alternatives with improved efficacy and tolerability. In this regard, organometallic half-sandwich metal complexes show significantly enhanced potential in the development of antitumor agents. Further, it was also observed that hydrophobic arene ligands facilitate the diffusion of drug molecules through the cell membrane.69,70 Also, the selectivity of chemotherapeutic candidates can be improved by the implementation of different chelating ligands and functional groups. Therefore, the use of heterocyclic compounds as chelating ligands together with organometallic frameworks provides a new strategy for the development of promising organometallic drugs. Thus, J. Ruiz et al.71a designed (Fig. 9) and synthesized benzimidazole cyclometalated arene ruthenium complexes (Fig. 10), [Ru(η6-p-cymene)(Bnz-Bu)Cl] and [Ru(η6-p-cymene)(Bnz-Bz)Cl] (where, Bnz-Bu and Bnz-Bz are 2-phenyl-1H-benzimidazole-5-carboxylate derivatives with a butyl and benzyl group attached to the N atom, respectively), and evaluated their cytotoxicity against epithelial ovarian carcinoma A2780 and A2780cisR cells (acquired resistance to cisplatin), breast cancer cells (T47D) and colon cancer cells (HT29). It was observed that these complexes were much more effective on the HT29 and T47D cell lines than cisplatin with very low IC50 values of 2.18 μM and 5.48 μM, respectively. Furthermore, it was observed that the complex containing the butyl chain was more active compared to its benzyl analog on almost all cell lines. Further studies indicated that the [Ru(η6-p-cymene)(Bnz-Bu)Cl] complex specially induced apoptosis, good accumulation and S-phase cell cycle arrest together with strong binding to HSA sites I and II and weak binding in DNA minor grooves. Subsequently, J. Ruiz et al.71b also studied the effect of various substituents (H, Me, F, CF3, MeO, NO2 and Ph) at the R4 position of the phenyl ring of 2-phenylbenzimidazole on the therapeutic potential of the complexes (Fig. 10). It was observed that the rate of hydrolysis of the ruthenium–chlorido bond was very fast for the [Ru(η6-p-cymene)(Bnz-Bu Me)Cl], [Ru(η6-p-cymene)(Bnz-Bu F)Cl] and [Ru(η6-p-cymene)(Bnz-Bu·Ph)Cl] complexes. Furthermore, all the compounds were screened on a panel of human cancer cell lines (A2780, A427, 5637, LCLC, SISO and HT29) and results showed that the complex with phenyl and CF3 substitution was highly potent. The relative hydrophobicities according to RP-UPLC-QTOF-MS studies were in the order of [Ru(η6-p-cymene)(Bnz-Bu F)Cl] < [Ru(η6-p-cymene)(Bnz-Bu Me)Cl] < [Ru(η6-p-cymene)(Bnz-Bu·Ph)Cl]. Further, in vitro angiogenesis studies suggested that [Ru(η6-p-cymene)(Bnz-Bu·CF3)Cl] and [Ru(η6-p-cymene)(Bnz-Bu·Ph)Cl] were the most active angiogenic inhibitors in EA.hy926 cells at very low doses. Most of the new compounds were more active than CDDP in A427 and HT29 cells. Also, the complexes were more effective than cisplatin in A427 and HT29 cells and were able to kill A2780cisR cells with IC50 values in the range of 0.96–3.26 μM.
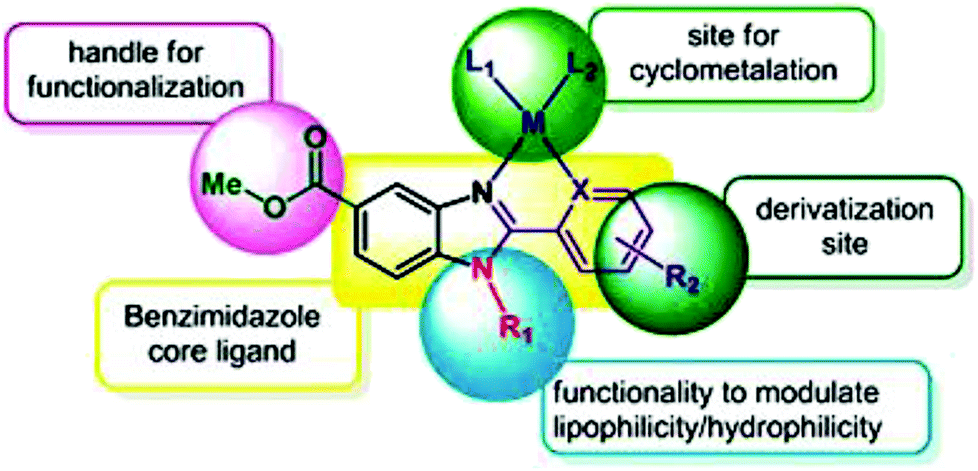 | ||
| Fig. 9 Design of a novel ligand for metallodrugs. This figure was reproduced from ref. 71a with permission from the Royal Society of Chemistry (Great Britain). | ||
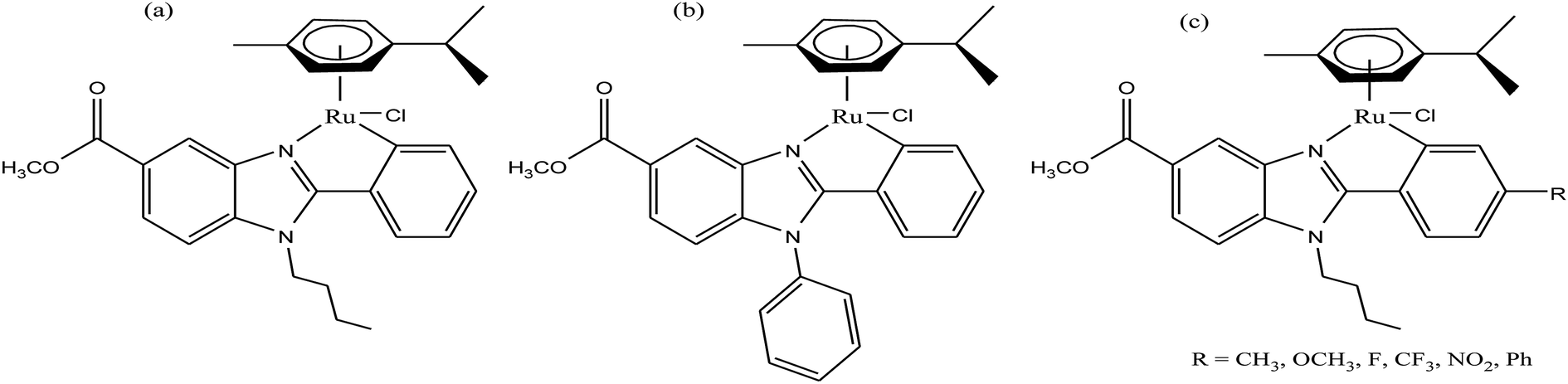 | ||
| Fig. 10 Synthesis of cyclometalated ruthenium complexes (a) [Ru(η6-p-cymene)(Bnz-Bu)Cl], (b) [Ru(η6-p-cymene)(Bnz-Bz)Cl] and (c) [Ru(η6-p-cymene)(Bnz-Bu·R)Cl]. | ||
Another approach to improve the therapeutic efficacy and preferential selectivity of ruthenium arene complexes towards cancer cells may be to tether them to a known compound of therapeutic value. Accordingly, R. Pettinari and P. J. Dyson et al.72 designed a series of novel water-soluble ruthenium(II) arene RAPTA-type complexes, [Ru(η6-p-cymene)(curc)-(PTA)]X and [Ru(hmb)(curc)(PTA)]X (where, curcH = curcumin, hmb = hexamethyl benzene, PTA = 1,3,5-triaza-7-phosphaadamantane, and X = SO3CF3−, and PF6−), containing curcumin-based ligands (Fig. 11). The in vitro antitumor activity of all the complexes was evaluated against human ovarian carcinoma cells (A2780 and A2780cisR), and non-tumorous human embryonic kidney (HEK293) cells. It was observed that all the complexes were 100-fold more cytotoxic compared to cisplatin on A2780 and A2780cisR cells with IC50 values typically ≤ 1 μM together with excellent selectivity towards cancer cells in comparison to non-tumorous human embryonic kidney cells, i.e. less cytotoxic on healthy cells. The presence or absence of peripheral methoxy groups in curcumin and the different arene rings did not strongly influence the cytotoxicity profiles of the complexes, despite leading to differences in hydrolysis rates. In contrast, the PTA ligand greatly enhanced the pharmacological properties and selectivity of the curcumin-based ruthenium(II)-arene complexes. To gain further insight into the mechanism of action and selectivity index of these complexes, further in vivo studies are required for modeling metal drugs for clinical therapeutic applications.
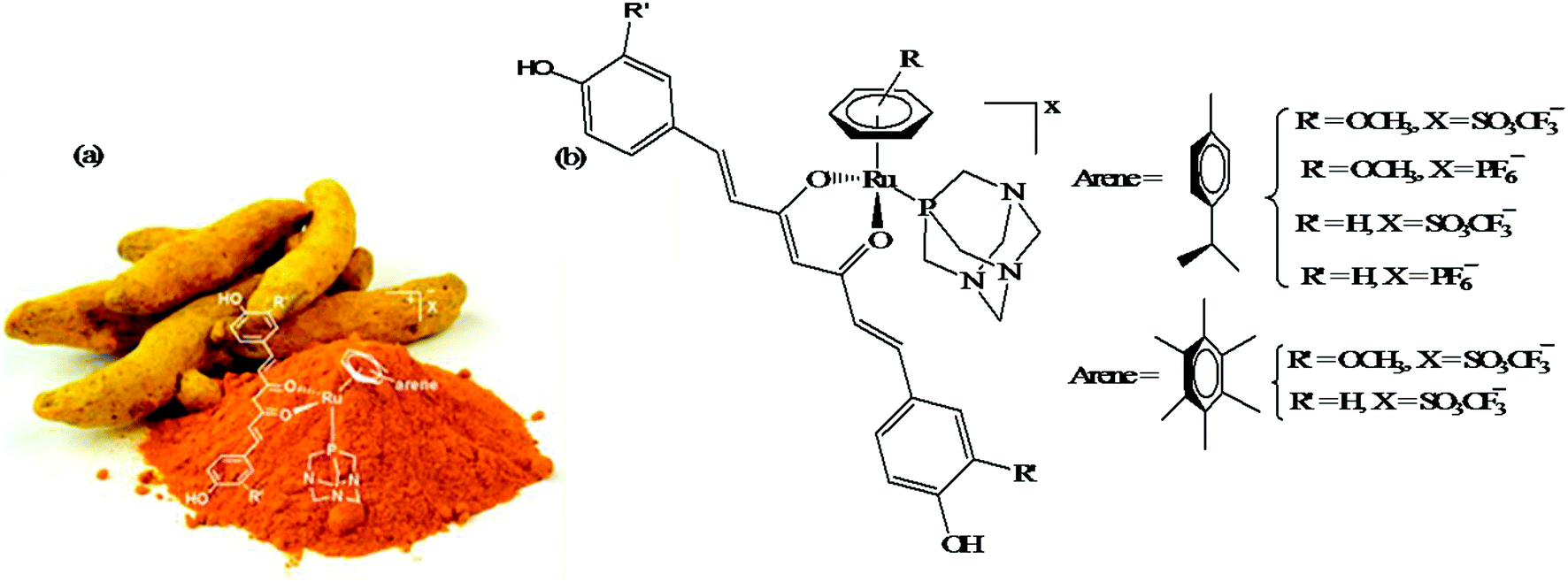 | ||
| Fig. 11 (a) Curcuma longa- (turmeric) and (b) ruthenium(II) arene RAPTA-type complexes [Ru(η6-p-cymene)(curc)-(PTA)]X and [Ru(hmb)(curc)(PTA)]X derived from curcumin. (a) was reproduced from its original paper after taking the copyright from ACS. This figure was taken from ref. 72, which is an open access article published under an ACS Author Choice License, which permits copying and redistribution of the article or any adaptations for non-commercial purposes. | ||
Similarly, to further improve the efficacy of half-sandwich complexes, W. Berger and I. Turel et al.73 reported the synthesis and antitumor properties of a series of ruthenium(II) chlorido complexes with fluorinated O, O3 ligands and phosphaadamantane (PTA) derivatives, [Ru(η6-p-cymene)(F3C-acac-Ar)Cl] and [Ru(η6-p-cymene)(F3C-acac-Ar)PTA]PF6 (where, F3C-acac-Ar = acetyl acetonate derivatives and PTA = 1,3,5-triaza-7-phosphaadamantane) (Fig. 12). They observed that all the complexes were efficacious against two cancer cell models (ovarian and osteosarcoma), but did not produce any toxic effects on nonmalignant keratinocytes. The PTA Ru(II) complexes showed lower cellular Ru accumulation, but higher efficacy, especially in the osteosarcoma cells compared to their chloride analogues. It was clearly observed that the chloride series [Ru(η6-p-cymene)(F3C-acac-Ar)Cl] exerted its antitumor activity via oxidative stress, DNA damage and apoptotic cell death in the G0/G1 phase. In contrast, for the PTA complexes, [Ru(η6-p-cymene)(F3C-acac-Ar)pta]PF6, there was no production of ROS and they blocked cell cycle progression in the G0/G1 phase. This revealed that there was a clear-cut shift from cytotoxic to cytostatic activity upon replacing the halido with the PTA ligand, confirming the structure-reactivity relationship with different modes of action. Also the reduction in glutathione levels by buthionine-sulfoximine (BSO) significantly enhanced the activity of all the compounds with the most pronounced effects observed for the PTA series, resulting in IC50 values in the nanomolar range. Finally, it can be concluded that proper tuning of the chemical structure of complexes by altering their ligand sphere allows better understanding of the selective modes of action of future organometallic chemotherapeutics.
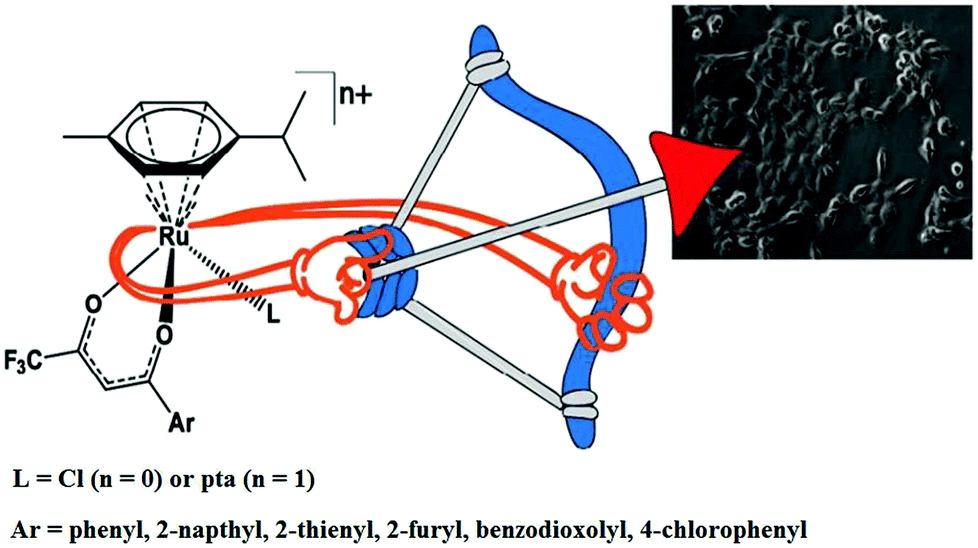 | ||
| Fig. 12 Design of RAPTA-type ruthenium(II) complexes [Ru(η6-p-cymene)(F3C-acac-Ar)Cl] and [Ru(η6-p-cymene)(F3C-acac-Ar)pta]PF6 containing the F3C-acac-Ar ligand. This figure was reproduced from ref. 73 with permission from the American Chemical Society. | ||
Organometallic titanium complexes with thioredoxin reductase (TrRx) as the target
To date, the search for antitumor chemotherapeutic agents that overcome the toxicity and intrinsic resistance of cisplatin has prompted the discovery of cytotoxic complexes based on various metals. The actual interest in organometallic therapeutics began after the discovery of titanocene dichloride (Cp2TiCl2) in 1979 (Fig. 13) by Kopf and Kopf-Maier.74,75 This complex exhibited much better activity than cisplatin and showed promising cytotoxic effects on various carcinoma lines, such as Ehrlich ascites tumor, B16 melanoma, colon 38 carcinoma and Lewis lung carcinoma. Additionally, titanocene dichloride was found to be highly effective on cisplatin-resistant cells with less toxic effects. | ||
| Fig. 13 Chemical structure of titanocene dichloride. | ||
Titanocene dichloride (Cp2TiCl2) displayed medium antiproliferative activity in vitro but promising results in vivo.76–78 Due to its high efficacy on tumors in vivo, this was the first organometallic complex to enter clinical trials in 1993 with a maximum tolerable dose of 315 mg m−2 per week.79,80 However, the efficacy of Cp2TiCl2 in Phase II clinical trials was too low to be pursued in patients with metastatic renal cell carcinoma and metastatic breast cancer81–84 due to its rapid hydrolysis to unidentified aggregates in the biological medium. This lack of hydrolytic stability at physiological pH restricted its further use as a chemotherapeutic drug for the treatment of cancer. In the literature, it is well established that uptake of albumin in cancer cells as a nutrient is very high to facilitate their rapid growth.85 Thus, anti-cancer drugs bound to albumin are of significant advantage since this can facilitate their uptake across the endothelial cell wall of blood vessels of tumors in an albumin receptor-mediated transport pathway. Besides, many organometallic titanocene complexes exert their antitumor effect by targeting the enzyme thioredoxin reductase (TrRx). Thioredoxin reductase (TrRx) is a selenoenzyme that maintains intracellular oxidative balance and is found in high concentration in many human tumor cells (in addition to cisplatin-resistant cells). Cancer cells are highly dependent on the concentration of this enzyme since it removes reactive oxygen species (ROS) and helps in cancer progression. Thus, if the function of TrRx is inhibited, then the concentration of ROS will increase in cancer cells to an extent that will lead to cell death or apoptosis. Due to its important role in cancer progression, TrRx is becoming an attractive target in the development of chemotherapeutic antitumor drugs.86–90
Thus, in the last two decades, there has been growing interest towards the development of new substituted titanocene dichloride derivatives by considering TrRx as a target. Interest in the development of other titanium organometallic complexes started after the synthesis of water-soluble and antitumor ring-substituted cationic titanocene dichloride derivatives with activity against ovarian cancers by P. C. McGowan et al.91–93 Another series of water soluble titanocene dichloride complexes with effective antitumor against lung and ovarian cancers was synthesized by M. C. Baird et al.94,99 Furthermore, new method for the development of substituted titanocene complexes starting from substituted fulvenes was recently reported by Tacke et al.95,96 Altogether, the discoveries in this field has led to the synthesis of substituted titanocenes that exhibit remarkable antitumor activity against the kidney carcinoma LLC-PK cell line, with IC50 values as low as 5.4 μM. Nevertheless, many researchers are on a quest for the development of effective antitumor agents using titanium organometallics with reduced side effects and non-cross resistance. In this review, we discuss the various substituted titanocene derivatives synthesized by these eminent scientists. Firstly, we focus on the discovery of P. C. McGowan et al.91–93 who synthesized a series of water-soluble and stable substituted titanocene salts via the direct reaction of neutral amino-substituted cyclopentadienes with TiCl4. This series included compounds of the type [Cp(CH2)2N(CH2)5]2TiBr2·2HBr and [Cp-R][Cp(CH2)2N(CH2)5]TiX2·HX, where, Cp = cyclopentadienyl moiety, R = H and (SiMe3)2 and X = Br and Cl, as depicted in Fig. 14. After evaluating the therapeutic potential of the entire series, it was well established that the most effective complex was [Cp-(SiMe3)2][Cp(CH2)2N(CH2)5]TiCl2·HCl, which contained two trimethylsilyl groups, resulting in a potent cytotoxic effect on a cisplatin-resistant ovarian tumor cell line. The prominent activity of this complex is mainly due to the formation of interstrand crosslinks with cellular DNA in the resistant and sensitive forms of the ovarian cancer cell line A2780. Further assessment of the antitumor activity of the other complexes against the A2780 and A2780 cisplatin-resistant cell lines led to the conclusion that the [Cp(CH2)2N(CH2)5]2TiBr2·2HBr, [Cp][Cp(CH2)2N(CH2)5]TiBr2·HBr and [Cp-(SiMe3)2][Cp(CH2)2N(CH2)5]TiCl2·HCl complexes showed promising activity. Generally, it can be estimated that ionic titanocenes exhibit much better activity and stability compared to non-functionalised titanocenes. It can be envisaged that these complexes follow structure-reactivity relationships and we can further increase the chemotherapeutic potential of ionic metallocenes by varying the: (i) transition metal, (ii) counterions, (iii) spacer linking the Cp and amino group, (iv) substituents attached to the Cp ring (which may affect the hydrophobicity and intercalating ability), and (v) pH of the metallocene dihalides depending on the substituent on the nitrogen atom and the counterion.
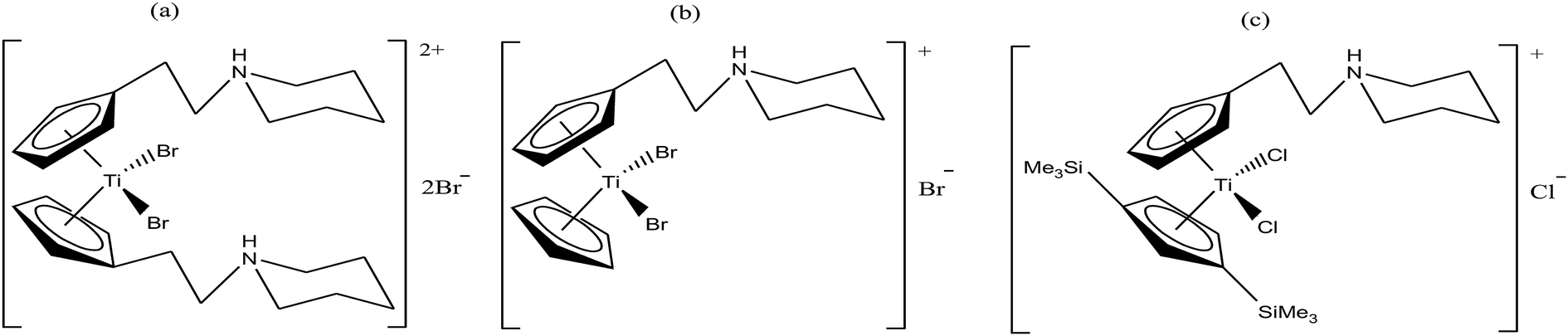 | ||
| Fig. 14 Chemical structure of substituted titanocene salts (a) [Cp(CH2)2N(CH2)5]2TiBr2·2HBr, (b) [Cp][Cp(CH2)2N(CH2)5]TiBr2·HBr and (c) [Cp-(SiMe3)2][Cp(CH2)2N(CH2)5]TiCl2·HCl. | ||
Another scientist, M. C. Baird et al.,97–99 synthesized a series of water-soluble titanocene dichlorides of the type [(η5-Cp-R1)(η5-Cp-R2)TiCl2] containing alkylammonium group pendants on one (monocationic complexes) or both (dicationic complexes) cyclopentadienyl rings (Fig. 15). All the complexes were screened for in vitro cytotoxicity against human lung cancer (H209, A549, and H209/CP) and ovarian cancer (A2780 and A2780/CP) cell lines, and the results were compared with both the classic drug cisplatin (cis-PtCl2(NH3)2) and a titanocene dichloride. Unfortunately, it was concluded that none of the complexes exhibited greater activity than cisplatin, but were superior to the clinical formulation of titanocene dichloride. After reviewing all the complexes, we can conclude that dicationic complexes are more active compared to their monocationic analogues, and derivatives containing protonated piperidinyl rings exhibit greater potency than compounds containing protonated 2-aminoethyl and 3-aminopropyl groups. Among them, the most active is the dicationic complex containing 3-picolylium groups, which showed the lowest IC50 value of 41 μM. Due to the structural differences in the side chains of the complexes, they showed different activities, which is mainly attributed to the different rates of hydrolysis of the Cp–Ti bonds, and hence uptake of the metal by transfer. However, the introduction of the electron withdrawing carbomethoxy group into the cyclopentadienyl rings increased the effectiveness of this class of drugs. The complex with two carbomethoxy groups [(η5-Cp-COOMe)(η5-Cp-COOMe)TiCl2] showed an IC50 value of 1 μg mL−1, which is comparable to that of cisplatin (IC50 = 2 μg mL−1) against SCLC NCI-H209.
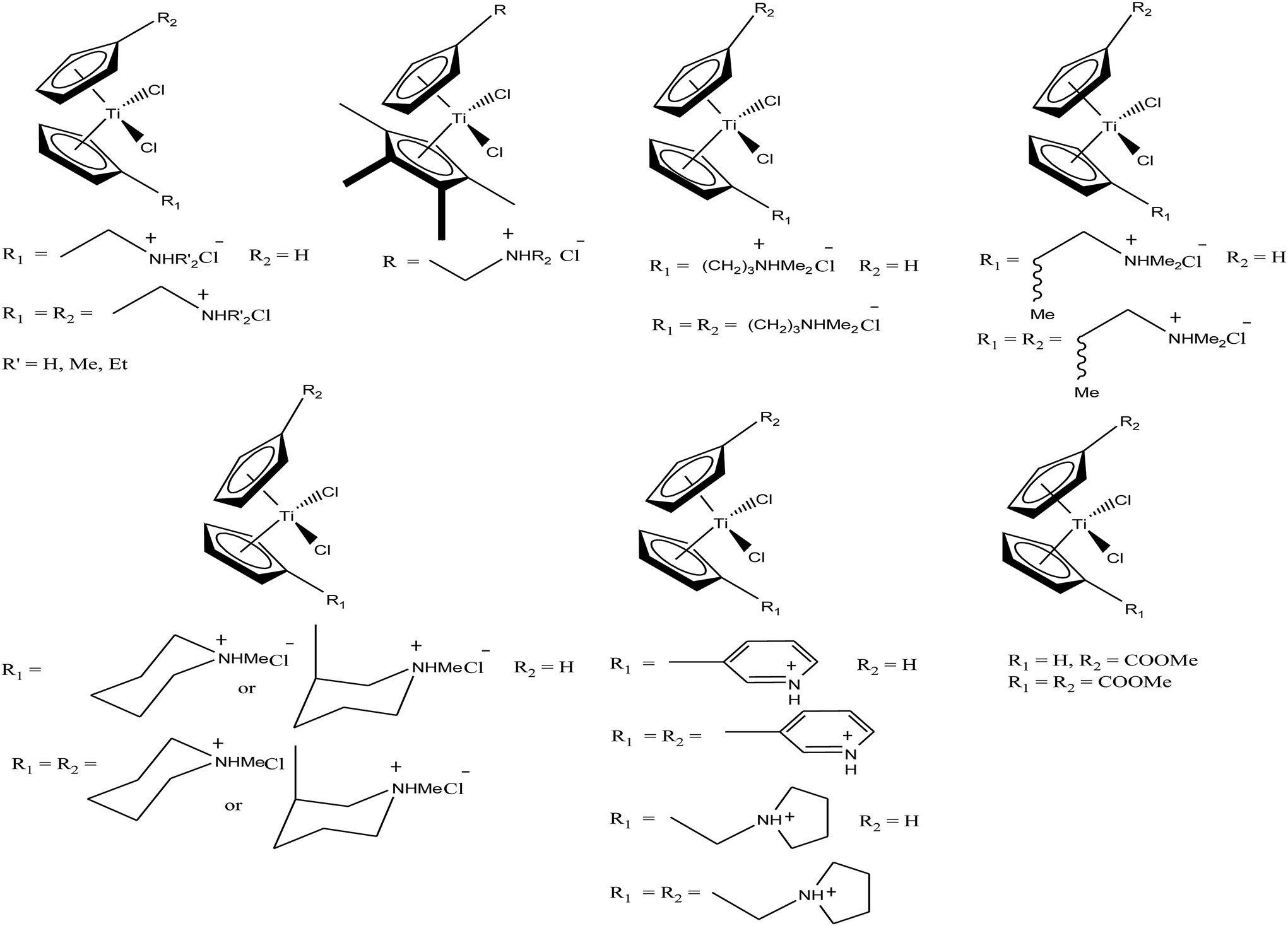 | ||
| Fig. 15 Chemical structure of a series of titanocene derivatives, [(η5-Cp-R1)(η5-Cp-R2)TiCl2], containing alkylammonium pendant groups on both or one ring. | ||
M. Tacke et al.100–102 established a novel process starting from titanium dichloride and fulvenes for direct access to highly substituted ansa-titanocenes and titanocenes containing a carbon–carbon bridge. They synthesized a series of ansa-derivatives using the above method with the general formula [1,2-(R)2C2H2{η5-Cp}2]TiCl2, where R = p-OMe-C6H4, 2′,4′,6′-(MeO)3-C6H2, and 3,3′-(MeO)2-C6H3; [1,2-(4-Me2N-C6H4)2C2H2{η5-Cp}2]TiX2 and [1,2-(4-Me5C6)2C2H2{η5-Cp}2]TiX2, where X = Cl and NCS; and [1,2-(2-C4H3X)2C2H2{η5-Cp}2]TiCl2, where X = O, S, and N-Me, as shown in Fig. 16. He also evaluated the cytotoxic activity of these complexes against pig kidney carcinoma (LLC-PK) cells with IC50 values in the range of 2.0 × 10−4 to 4.5 × 10−4 M. [1,2-(2-C4H3-NMe)2C2H2{η5-Cp}2]TiCl2 with an N-methyl-2-pyrrolyl ring was found to be the most cytotoxic in this series. M. Tacke et al.103 also synthesized novel benzyl substituted titanocene of the type [(η5-Cp-CH2-C6H4-R)]2TiCl2 (where, R = NMe2, OMe, and NHMe2Cl) via the reaction of Super-Hydride (LiB(Et)3H) with substituted fulvenes and TiCl4. When these complexes were screened for cytotoxicity against LLC-PK cells, it was found that they were more cytotoxic than the previously reported ansa-titanocenes. In this series, the lowest IC50 value of 2.1 × 10−5 M was obtained for the [(η5-Cp-CH2-C6H4-OMe)]2TiCl2 complex with two methoxy groups due to the loss of its stereocenter. Another method for the synthesis of analogous of unbridged titanocenes was established via the carbolithiation of 6-heteroarylfulvenes with aryl lithium species followed by transmetallation with TiCl4.104 Substituted titanocenes with the general formula {η5-Cp-CH-[C4H4-R]2}2TiCl2 (where, R = O, S, and NMe) synthesized using this method exhibited much better cytotoxicity on LLC-PK cells than the corresponding ansa-analogues and benzyl-substituted titanocenes with the promising IC50 value of 32 μM (cisplatin = 3.3 ± 0.5 μM) for the {η5-Cp-CH-[C4H4-NMe]2}2TiCl2 complex containing N-methyl amino pyrrolyl groups. Surprisingly, the change in the substitution pattern of titanocenes enhanced the cytotoxic activity of these substituted titanocenes probably due to their increased solubility and stabilization of the metal centre by different groups.
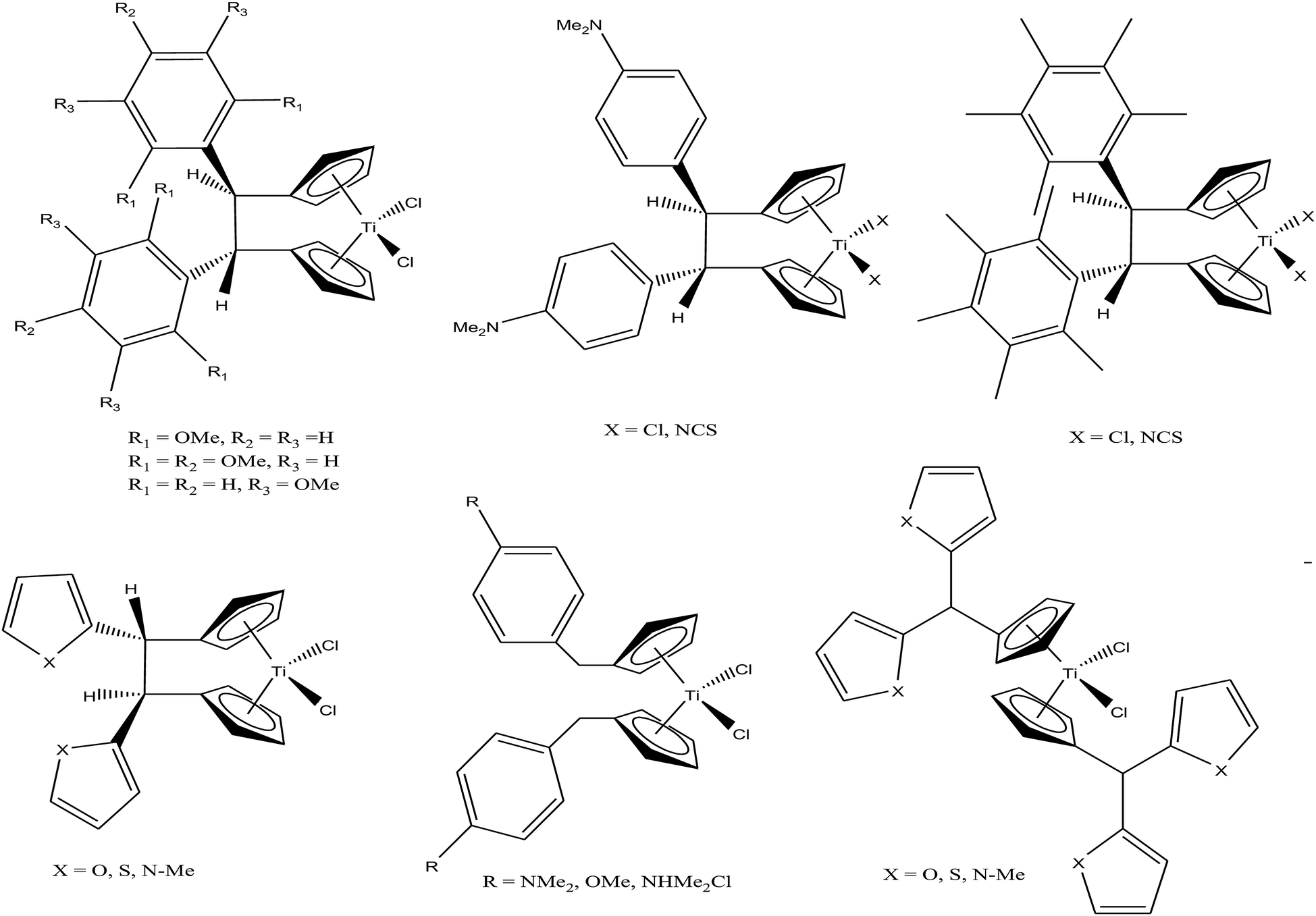 | ||
| Fig. 16 Chemical structure of a series of substituted ansa-titanocenes. | ||
Carbolithiation of 6 N,N-dimethyl amino fulvene with lithiated heteroaryl species followed by transmetallation is another method for the synthesis of new chiral heteroaryl-substituted and dimethylamino-functionalised metallocenes of the type {η5-Cp-CH[NMe2][C5H3Z]}2TiCl2, where, Z = S, O, and NMe.105 The complexes in this series showed the highest cytotoxicity against LLC-PK, indicating their high potential as anti-cancer drugs. The titanocene derivative {η5-Cp-CH[NMe2][C5H3NMe]}2TiCl2 was the most cytotoxic with an IC50 value of 5.5 μM, which is very close to that of cisplatin (3.3 μM) against LLC-PK and 400 times better than that of titanocene dichloride (Fig. 17). The increase in cytotoxicity is due to the methoxy and dimethylamino groups, which increase the solubility and potentially stabilize the metal centre via intramolecular coordination of the methoxy/dimethylamino substituents to the Ti metal centre upon displacement of the chloride ligands. A study on the cellular mechanism of the p-methoxy derivative in prostate cancer cells showed that this complex induced more apoptosis than cisplatin in a dose-dependent manner.
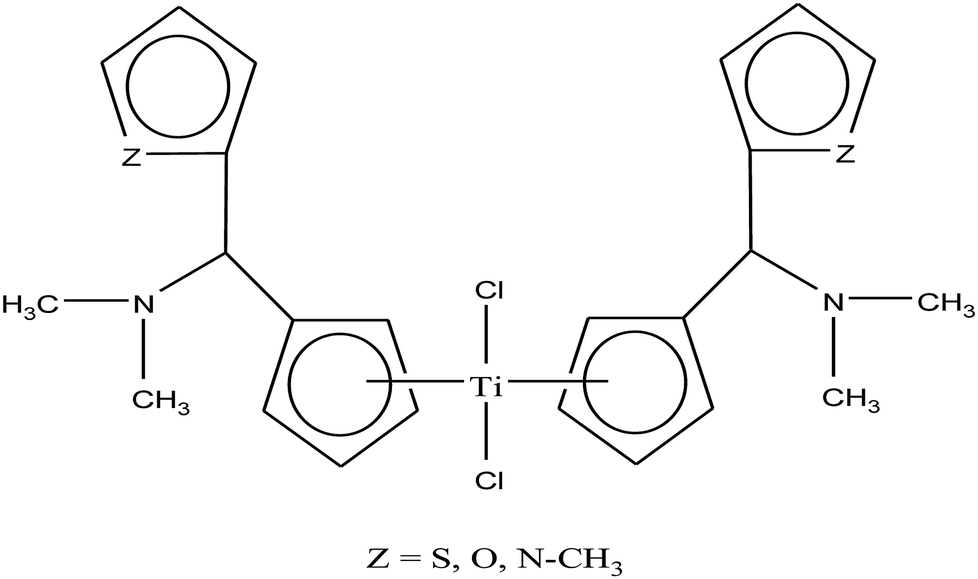 | ||
| Fig. 17 Chemical structure of dimethylamino-functionalised titanocene {η5-Cp-CH[NMe2][C5H3Z]}2TiCl2. | ||
Thus, it can be concluded that the activity of these ionic metallocenes can be increased by changing their counter ions, substituent linkers between the two cyclopentadienyl rings (cp) and the pH of the ionic metallocenes depending on the structure of the chemotherapeutic agent. Finally, we can conclude that there is a type of structure-reactivity relationship with thioredoxin reductase (TrRx) as target for the mode of action between these molecules, where slight changes in structure will drastically improve the chemotherapeutic efficiency of the compounds.
However, the problem of acquired resistance towards the chemotherapeutic drug still exists in cancerous cells, and thus, scientists are focusing on the development of heterometallic complexes to enhance the antitumor potential. It has been observed that the presence of two different metals in the same pharmacological framework improves its chemotherapeutic activity, probably due to their multiple targets. For example, titanocene dichloride is hydrolyzed at pH 7, liberating the Cp rings and binding to transferrin to be transported into tumor cells, and released by via a non-redox pathway.106–109 Thus, it can be expected that heterometallic compounds potentially break into monometallic species in physiological media or in vivo before reaching tumors. We hypothesized that incorporating a second metal to a ligand strongly bound to a titanium(IV) center will ensure that heterometallic Ti-M species remain after Ti-Cp hydrolysis occurs under physiological pH conditions.
Accordingly, M. Contel et al.110 synthesized stable trinuclear TiAu2 complexes of the type [Ti{η5-Cp(CH2)nPPh2(AuCl)}2]·2THF, where, n = 0, 2, and 3 based on a titanocene phosphine backbone (Fig. 18). The stability of these complexes in different deuterated solvents (including buffer solutions) was determined via 31P{1H} NMR spectroscopy. The cytotoxic studies revealed that these TiAu2 complexes were highly cytotoxic in vitro on HeLa human cervical carcinoma and DU-145 human prostate cancer cells. However, the most cytotoxic complex [Ti{η5-Cp(CH2)3PPh2(AuCl)}2]·2THF, which contained a propyl spacer between the P atom and Cp ring. This complex was more active on HeLa cells with an IC50 value of 1.12 μM than its parent titanocene dichloride and titanocene phosphine derivatives and second metal-related precursors. In addition, all the TiAu2 complexes interacted with DNA through the electrostatic binding mode. However, it was not confirmed that DNA is the ultimate biomolecular target for these complexes, and hence further studies on transport and mitochondrial proteins are warranted to determine the plausible mode of action of these types of heteronuclear complexes.
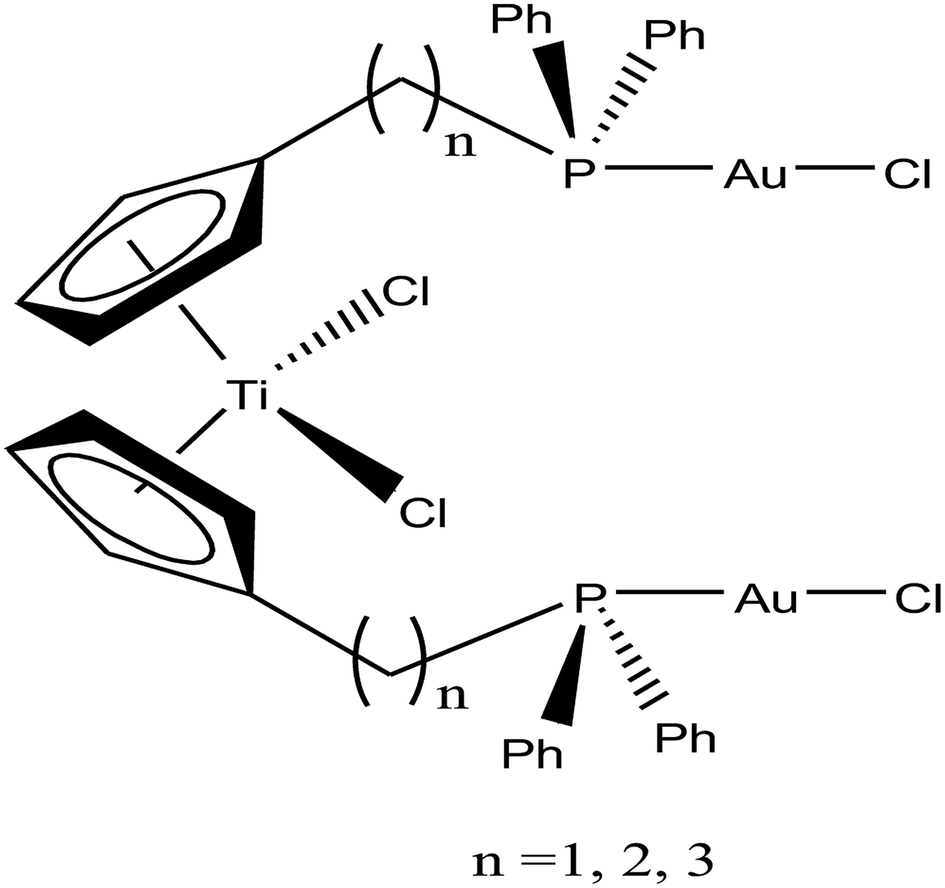 | ||
| Fig. 18 Chemical structure of titanocene–gold complexes [Ti{η5-Cp(CH2)nPPh2(AuCl)}2]·2THF. | ||
To further increase the stability of heterometallic Ti complexes, M. Contel et al.111 envisioned that carboxylate instead of the labile chlorine may be the ideal group to bind titanium(IV) centers (Fig. 19). Considering this, they synthesized new early-late transition metal TiAu2 heterometallic complexes [(η5-Cp)2Ti{OC(O)RPPh2AuCl}2 (where, R = CH2 and 4-C6H4) as potential anticancer agents in vitro against renal (A498, UO31, and Caki-1) and prostate cancer cell lines (PC3 and DU145). Both complexes were significantly more active than their monometallic titanocene dichloride (IC50 = >200 μM) and gold(I) [{HOC(O)RPPh2}AuCl] (R = –CH2 and –4-C6H4) derivatives with IC50 values in the range of 1.2 to 43 μM in renal cancer cell lines, revealing the synergistic effect of the heterometallic species in vitro. The cytotoxicity of the [(η5-Cp)2Ti{OC(O)-C6H4PPh2AuCl}2 complex on renal cancer cell lines (IC50 = 0.3 μM on UO31 cells) was extraordinarily higher than that of cisplatin and highly active titanocene Y, while being considerably less toxic to the non-tumorigenic human embryonic kidney cell line (HEK-293T). Furthermore, the cell death induced by the compounds was studied, which revealed apoptosis for [(η5-Cp)2Ti{OC(O)-C6H4PPh2AuCl}2. The lack of interaction of the compounds with plasmid (pBR322) DNA indicates that other bimolecular targets may be implicated in their cell death pathways. In addition, both complexes inhibited protein kinases of the AKT and MAPKAPK families with a higher selectivity toward MAPKAPK3 (IC50 for [(η5-Cp)2Ti{OC(O)CH2PPh2AuCl}2 = 91 nM and IC50 for [(η5-Cp)2Ti{OC(O)-C6H4PPh2AuCl}2 = 117 nM). The selectivity of complex [(η5-Cp)2Ti{OC(O)-C6H4PPh2AuCl}2 in vitro against renal cancer cell lines and its favorable preliminary toxicity profile on C57black6 mice proved that it is an excellent candidate for further development as a potential chemotherapeutic agent, and even higher target specificity can be achieved by rational modification of its ligand scaffolds.
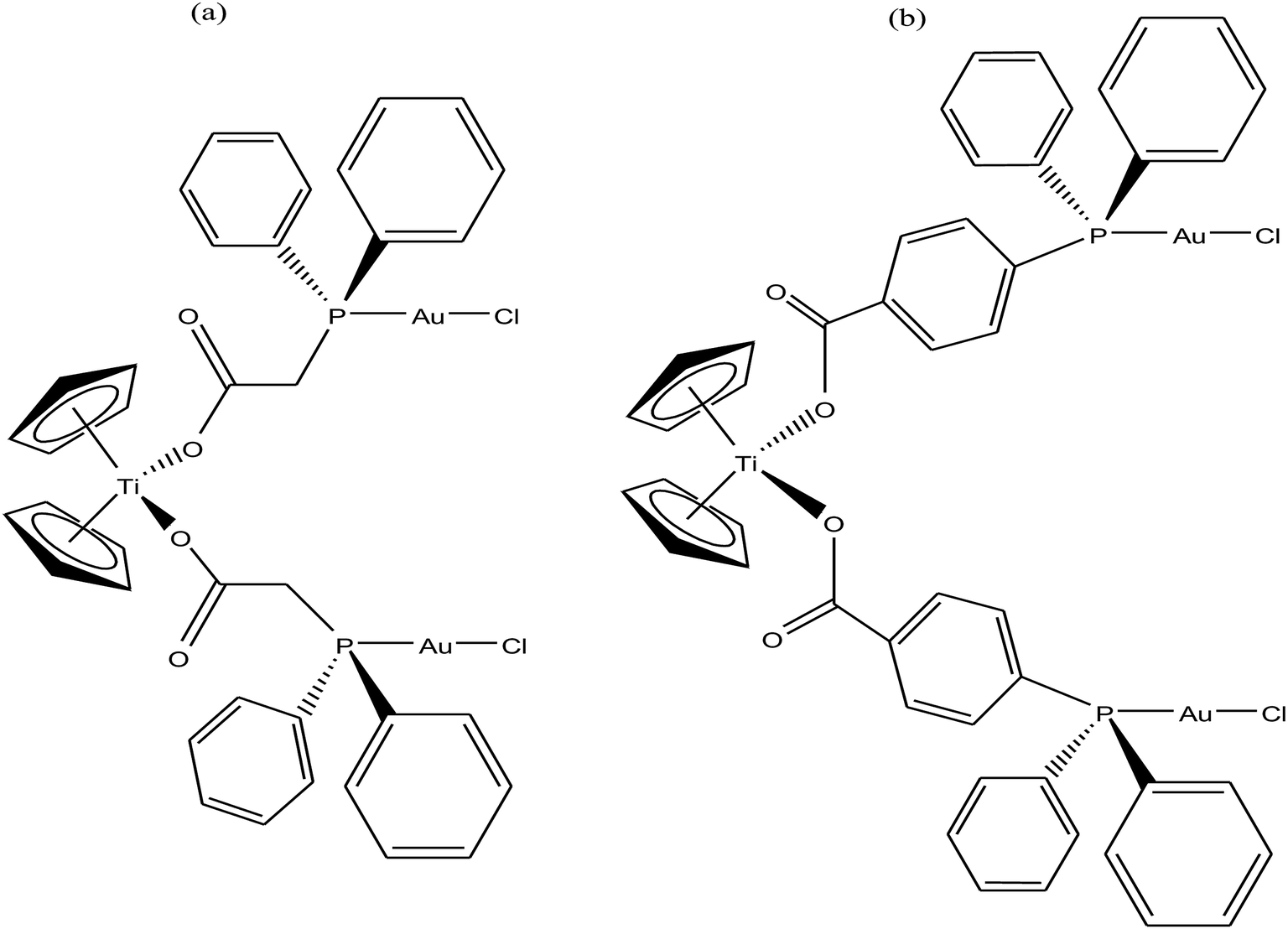 | ||
| Fig. 19 Chemical structure of titanocene–gold complexes (a) [(η5-Cp)2Ti{OC(O)CH2PPh2AuCl}2 and (b) [(η5-Cp)2Ti{OC(O)-C6H4PPh2AuCl}2. | ||
Following their work on heterometallic titanocene–gold complexes as potential chemotherapeutic agents for renal cancer, M. Contel et al.112 reported the synthesis of another new titanocene complex, [(η5-Cp)2TiMe(μ-mba)Au(PPh3)] (where, mba = S–C6H4–COO−), containing a methyl group and a carboxylate ligand bound to gold(I)-phosphane fragments (Fig. 20). This complex was found to be more stable and highly cytotoxic on human cancer renal cells (IC50 = 0.12 μM on Caki-1 cells) as compared to their previously reported complex [(η-Cp)2Ti{OC(O)-4-C6H4-P(Ph2)AuCl}2]. Mechanistic studies on [(η5-Cp)2TiMe(μ-mba)Au(PPh3)] revealed that it showed anti-invasive properties, i.e. it blocked the growth of renal cancer both in vitro and in vivo through the inhibition of thioredoxin reductase and decreased the expression of protein kinases known to drive cell migration (AKT, p90-RSK, and MAPKAPK3). In vivo studies on mice showed impressive tumor reduction (67%) with the heterometallic compound [(η5-Cp)2TiMe(μ-mba)Au(PPh3)] compared with their previously described complex [(η-Cp)2Ti{OC(O)-4-C6H4-P(Ph2)AuCl}2], which was non-inhibitory. These findings further indicated the structure-activity relationship via the inhibition of thioredoxin reductase, i.e. slight structural modifications in the ligand scaffold increased the in vivo efficacy of this class of compounds.
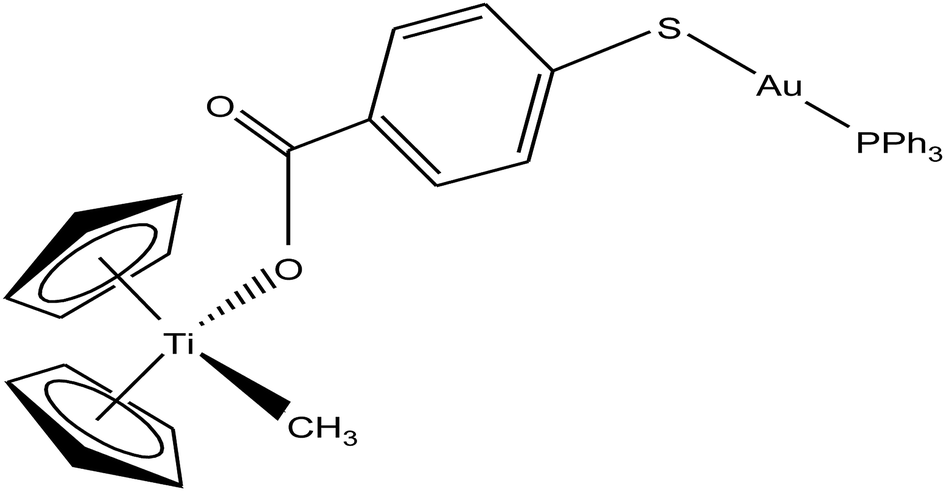 | ||
| Fig. 20 Chemical structure of titanocene–gold complex containing gold(I)-phosphane [(η5-Cp)2TiMe(μ-mba)Au(PPh3)]. | ||
Thus, to further improve the efficacy and stability of titanocene–gold derivatives as potential anticancer agents compared to their previously reported complexes, recently M. Contel et al.113 replaced the gold(I)-phosphane fragments with gold(I)-N-heterocyclic carbene moieties to obtain complexes of the type [(η5-Cp)2Ti(CH3){OC(O)-p-C6H4SAu(NHC)}] (where, NHC = IPr, IMes, and ICy), as shown in Fig. 21. The cytotoxic activity of all the heterometallic complexes was tested on the prostate PC3 and DU145, renal Caki-1, colon DLL1 and breast MDA-MB-231 cancer cell lines. Unfortunately, the IC50 values of these complexes were found to be higher than that of the previously described complexes of the same type. However, they exhibited higher selectivity with respect to non-cancerous cell lines in prostate and colon cancer cell lines. Similarly to other titanocene–gold complexes containing phosphanes, the new heterometallic carbene derivatives did not display significant interaction with plasmid (pBR322) DNA. The selective heterometallic complex [(η5-Cp)2Ti(CH3){OC(O)-p-C6H4SAu(IPr)}] was found to be highly apoptotic and inhibited thioredoxin reductase (TrRx) in the prostate PC3 cancer cell line. Finally, it can be concluded that substitution of PR3-gold(I) by NHC-gold(I) fragments in titanocene–gold complexes is not proven to be successful and further optimization of NHC ligands with more detailed mechanistic studies is necessary for the design of chemotherapeutic candidates with improved pharmacological properties.
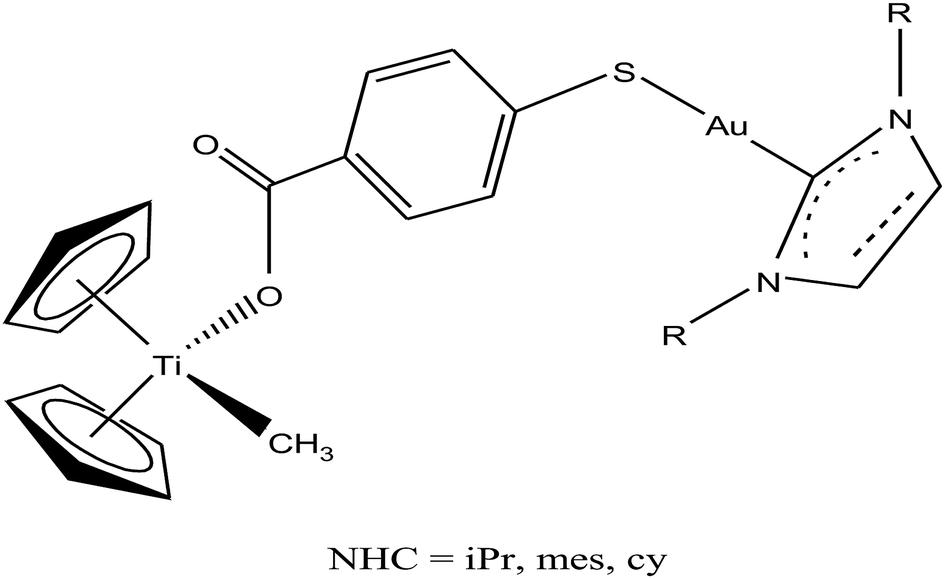 | ||
| Fig. 21 Chemical structure of heterobimetallic titanocene–gold complexes [(η5-Cp)2Ti(CH3){OC(O)-p-C6H4SAu(NHC)}]. | ||
Organometallic gold complexes with ER stress, mitochondria and TrRx as targets
For centuries gold has been utilized as a therapeutic agent for the treatment of various diseases. However, its chemotherapeutic potential as an antitumor agent was accelerated after the clinical entry of auranofin, a triethylphosphine gold(I) glucose-thiolate, which was used for the treatment of rheumatoid arthritis.114 The oral availability of this antiarthritic caused a major breakthrough in the field of cancer therapeutics. Subsequently, auranofin was found to possess potent in vitro antitumor properties and promising in vivo effects in a P388 leukemia mouse model.115 Many gold chemotherapeutics are also highly effective on cisplatin-resistant cell lines probably due to their different mechanisms of action compared to the classic drug cisplatin, which acts only on DNA.116,117 The literature revealed that antitumor gold chemotherapeutic agents exert their cytotoxic effect by interaction with different targets, such as endoplasmic reticulum (ER), mitochondria and thiol-containing proteins/enzymes, such as thioredoxin reductase (TrRx).118 The enzyme thioredoxin reductase (TrRx) is present in high quantities inside tumor cells because there is high production of ROS in tumor cells and this enzyme helps in maintaining the oxidative stress produced by ROS. Thus, if the function of the TrRx enzyme is inhibited in any way, due to the high accumulation of ROS, cancer cells will undergo apoptosis or cell death.87,90 Similarly, mitochondria are one of the main sources of intracellular ROS due to electron flow in the respiratory chain. Antitumor drugs targeting the mitochondria lead to the accumulation of drugs inside the mitochondria, which results in dysfunction in the various metabolic processes, such increases mitochondria permeability and the production of ROS. This ultimately leads to cell death or apoptosis without causing any adverse effects and toxicity issues.119–121 Besides, many drugs target the endoplasmic reticulum and cause ER-stress inside tumor cells, which activates the unfolded protein response (UPR). This response (UPR) either restores homeostasis and promotes survival of cells or activates cell death or apoptosis.122,123 Therefore, all these strategies are very important in the design and development of antitumor drugs. Recently, organometallic gold(I) N-heterocyclic carbene (NHC) complexes have been extensively investigated in the field of organometallic chemotherapeutics.124–127 This type of complex was found to inhibit the activity of thioredoxin reductase (TrRx) at low nanomolar levels, which is probably due to the delivery of the bioactive metal at the target site and stabilization of the metal ion by the NHC ligand under physiological conditions. Thus, organometallic gold complexes act as prospective drug candidates with potent antitumor activities on drug-resistant malignant tumors due to their different modes of action compared to classic drugs.128–133 Other scientists believed that mitochondria and the pathways of oxidative phosphorylation were the primary intracellular targets for organometallic gold chemotherapeutics.134,135 However, studies on many organometallic gold(I) and gold(III) complexes revealed that inhibition of thioredoxin reductase (TrRx) was their major mechanism of cytotoxic action, which ultimately led to apoptosis via the mitochondrial pathway and endoplasmic reticulum stress.136,137 Therefore, the mechanisms of action of anticancer gold complexes are different from that of cisplatin, where most cytotoxic gold complexes are also effective against cisplatin-resistant cancer cells, revealing the promising prospect in the development of gold chemotherapeutics to resolve the problem of cisplatin resistance. Some Au(I) and Au(III) complexes have also been demonstrated to display significant in vivo anticancer effects.Despite their promising therapeutic potential, there is a risk in developing chemotherapeutic Au(III) compounds due to their low stability in intracellular medium. To stabilize the Au(III) metal ion, researchers are focusing on the development of organometallic gold(III) complexes because presence of a direct carbon–gold bond greatly stabilizes the gold(III) oxidation state and guarantees more controlled chemical speciation in an aqueous environment. Generally, both organometallic gold(I) and gold(III) centers exhibit increased stability with respect to classical gold-based coordination complexes and are extremely suitable for the design of organometallic gold chemotherapeutic agents. In general, Au(III) complexes are redox active and can be easily reduced to Au(I)/Au(0). Thus, choosing the appropriate ligand to stabilize the Au(III) ion is of great interest. The bioactive organometallic gold compounds mainly include cyclometallated gold(III) complexes with C, N-donor ligands, gold(I) and gold(I/III) N-heterocyclic (NHC) carbene complexes, and gold(I) alkynyl complexes, which have promising anticancer activities. In normal gold coordination compounds, the Au(III) ion is reduced to metallic gold in biological media, which can be toxic to the human body. Thus, to prevent the reduction of Au(III), the organometallic complexes of gold are found to be promising due to the presence of M–C bonds, which provide high stability to the complex. Thus, in this review, we focus on the development of organometallic antitumor compounds due to their high stability. For the first time, R. V Parish et al.138,139 discovered the antitumor properties of gold(III) complexes of the type [Au(dmamp)X2] {where, X = Cl, and OAc and dmamp = 2-(dimethylaminomethy1)-phenyl}, as shown in Fig. 22. These complexes displayed similar cytotoxicity to that of cisplatin against several human tumor cell lines. Their acetato analogs, [Au(dmamp)(OAc)2], showed higher solubility in water compared to [Au(dmamp)Cl2]; thereby, exhibiting promising in vitro and moderate in vivo antitumor activity against ZR-75-1 human carcinoma xenografts.140,141 Thus, this suggests that changing the chloride ion to the acetate ion increases the hydrolysis of the complex similar to cisplatin and the these complexes present an example of structure-reactivity relationship by inhibiting proteases. Both complexes inhibited the cysteine proteases cathepsin B and K and were also very potent inhibitors of TrRx.142
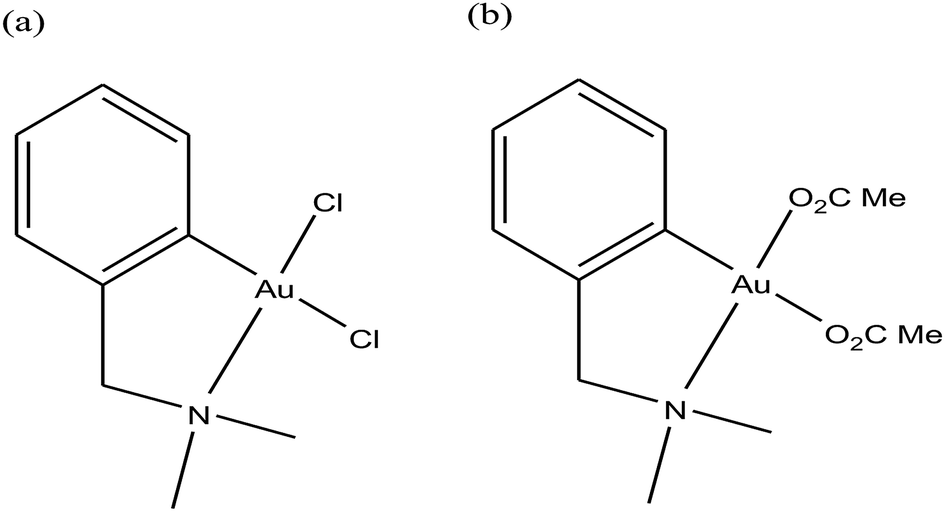 | ||
| Fig. 22 Chemical structure of Au(III) complexes of 2-(dimethylaminomethyl)phenyl (dmamp). (a) [Au(dmamp)Cl2] and (b) [Au(dmamp)(OAc)2]. | ||
It is well established that gold complexes containing the covalent Au–C bond are more difficult to decompose in physiological media. Further, these compounds are isoelectronic with the well-known classical drug cisplatin, and thereby show high activity due to their interaction with DNA. However, besides their interaction with DNA, organometallic gold complexes exert their antitumor activity by interacting with thiol-containing molecules such as TrRx activity and cancer cell killing capacity as potential antitumor agents. Therefore, Simon P. Fricker et al., Robert G. Buckley et al. and Yongbao Zhu et al.140,141,143,144 synthesized a series of square planar six-membered cycloaurated complexes of the type [Au(dmamp)(X–X)] {where, X–X = catecholate bis(amidate)}, [Au(R-C^N)(X–X)] {where, C^N = pyridinyl-phenyl unit, R = H and Me, and X–X = thiosalicylate and bis(amidate)} and [Au(R-N^E^C-R′)X2] {where, N^E^C = pyridinyl linker phenyl unit; E = CH2, CHPh, CO, NH, NMe, O, and S; X = Cl and OAc; X2 = CH2(COO)2 and thiosalicylate; R = H, Me, Et, Prn, CH2OMe, CH2Ph, and Ph; and R′ = H, Me, Et, Bun, Hexn, CH2Ph, Cl, and OR′′} (Fig. 23). All these complexes were found to inhibit the cathepsin cysteine proteases B and K in the same manner. However, only [Au(N^CH2^C)Cl2], [Au(N^NH^C)Cl2], [Au(N^O^C)Cl2], [Au(N^S^C)Cl2] and [Au(N^CH2^C)(THS)2], where, THS = thiosalicylate, were tested for in vitro activity against a panel of human tumor cell lines. The results revealed that the thiosalicylate compound [Au(N^CH2^C)(THS)2] was more active than the chloro compound [Au(N^CH2^C)Cl2], and their mechanism of cytotoxicity may be gold–protein interactions. Furthermore, it was also observed that an increase in the number of C–Au bonds and substitution at the 6 position of the pyridine ring caused the complexes to become more active and lipophilic, thereby facilitating their intracellular uptake. However, the ability of these complexes to interact with the thiols and selenol groups of proteins appeared to be reduced.
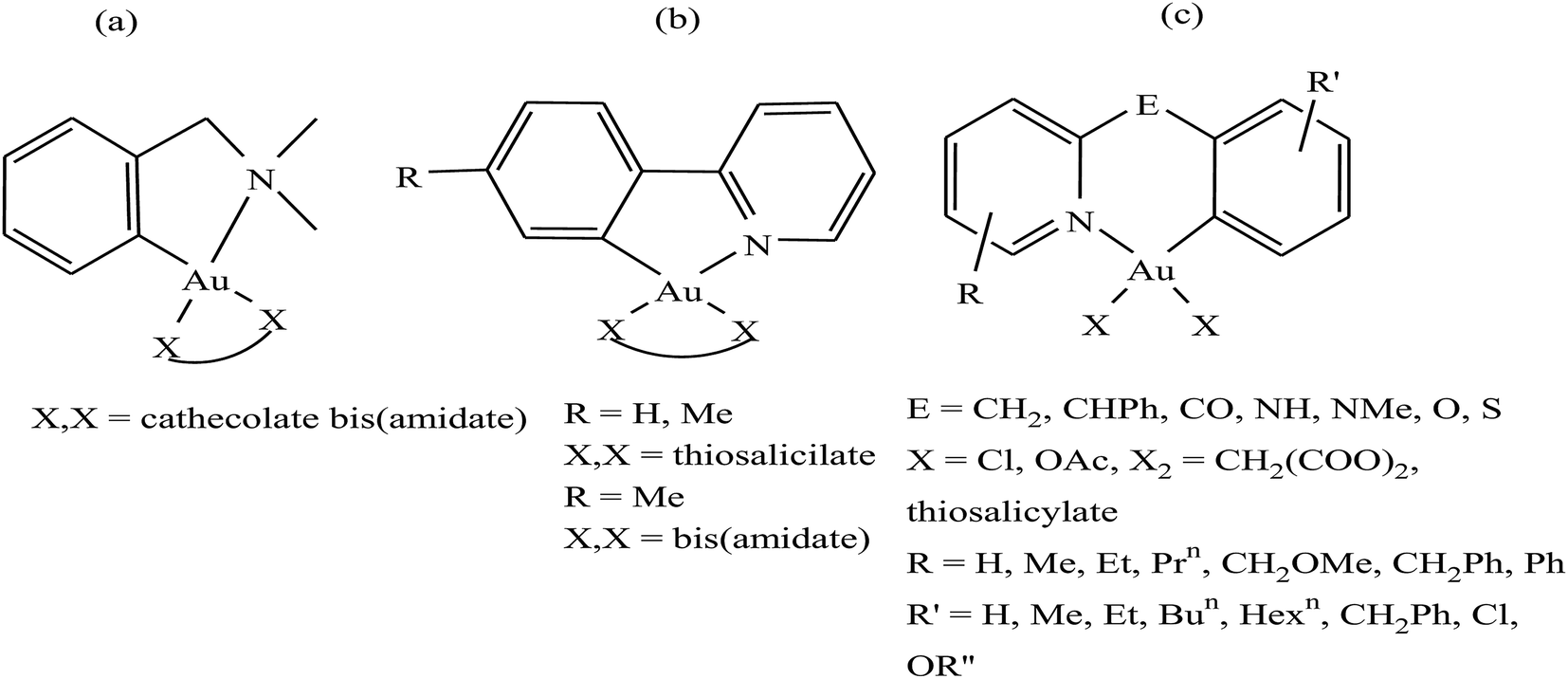 | ||
| Fig. 23 Chemical structure of a series of a series of cycloaurated complexes (a) [Au(dmamp)(X–X)], (b) [Au(R-C^N)(X–X)] and (c) [Au(R-N^E^C-R′)X2]. | ||
Moreover, there has been considerable interest in the development of organometallic lipophilic metal complex cations since they show promising cytotoxicity under in vitro and in vivo conditions in cancer cells. However, enhanced lipophilicity can lead to a decrease in aqueous solubility, resulting in lower bioavailability, which restricts their application. Thus, a balance between lipophilicity and aqueous solubility must be considered for the development of clinically useful organo–gold complexes. Accordingly, C. M. Che et al.145 reported gold(III) biguanide complexes with the general formula [AuIII(R-C^N)L]n+ (where, R = H and butyl, C^N = pyridinyl-phenyl unit, and L = biguanide (BG) and biuret (BU), with n = 0–1), which contained the lipophilic 2-(4-n-butylphenyl) pyridine moiety and hydrophilic chelating biguanide (Fig. 24). They modified the lipophilicity of the complexes by varying the R group and ensured their solubility by using a polar ligand in the synthesis of the organometallic gold(III) complexes. The [AuIII(nBu-C^N)(BG)]Cl and [AuIII(nBu-C^N)(BU)] complexes were more cytotoxic (IC50 = 1.5–17 μM) than cisplatin (IC50 = 12.0–66.7 μM) against HeLa cells due to their lipophilic n-butyl group(s), which enhances their cellular uptake. However, the [AuIII(nBu-C^N)(BG)]Cl complex exhibited high toxicity against HeLa cells (Fig. 25) and low toxicity towards normal lung fibroblast CCD-19Lu normal cells. Further studies confirmed that this complex interacted rapidly with GSH to form gold-GSH adduct(s), which induced ER stress, ER swelling and up-regulated ER-stress markers, such as CHOP and HSP70. These markers promote partial S-phase arrest in HeLa cells, and subsequently, apoptosis- and necrosis-independent cell death.
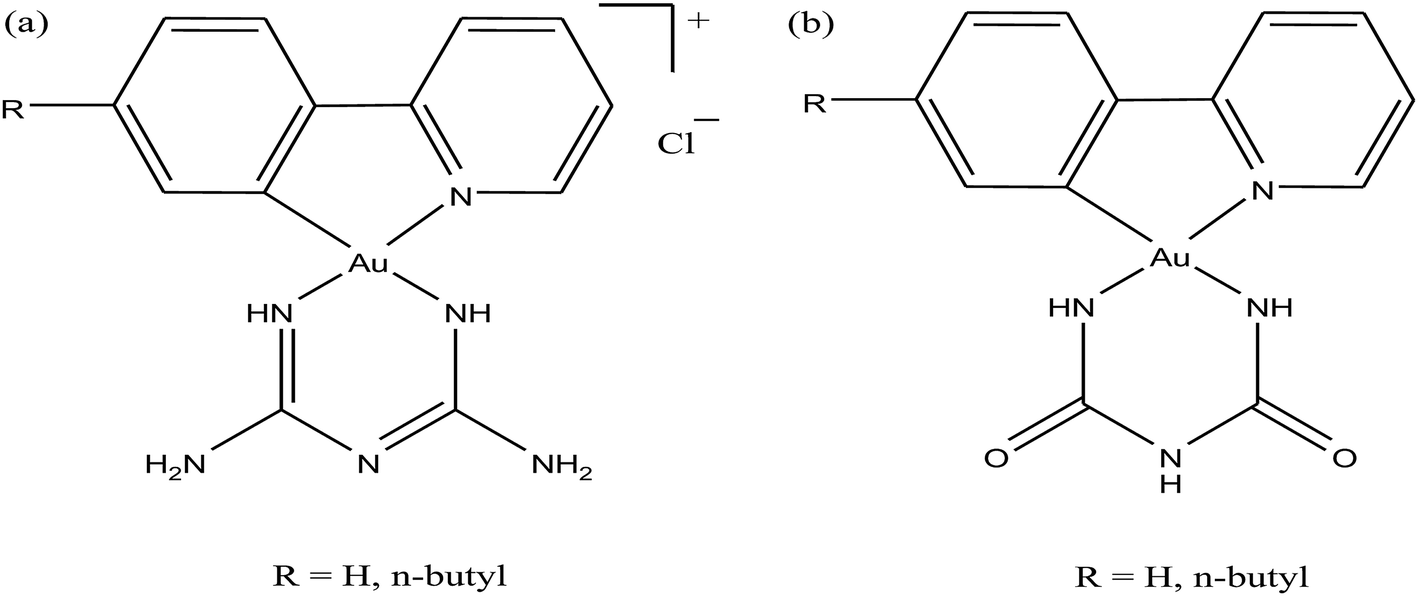 | ||
| Fig. 24 Chemical structure of gold(III) biguanide complexes (a) [AuIII(R-C^N)(BG)]Cl and (b) [AuIII(R-C^N)(BU)]. | ||
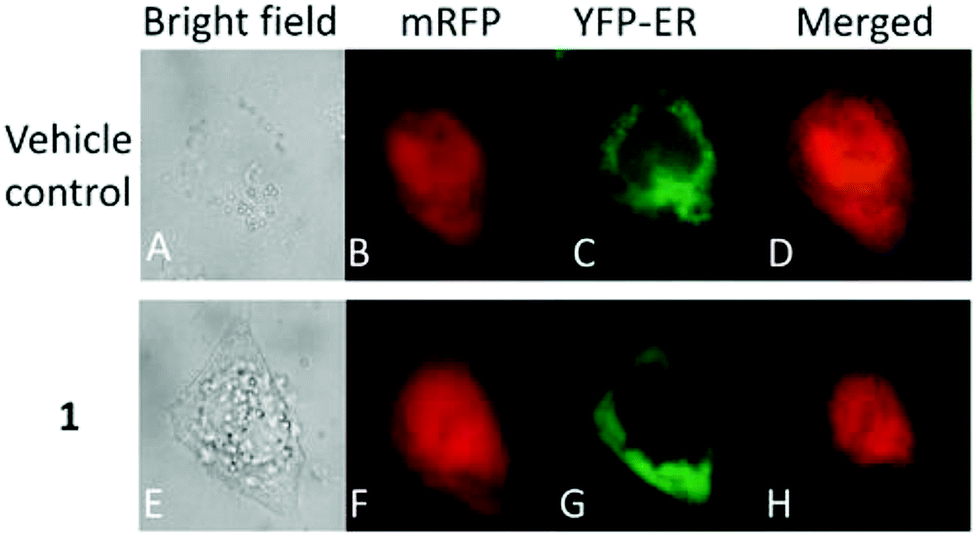 | ||
| Fig. 25 Fluorescence images of HeLa cells co-expressing mRFP and YFP-ER after treatment with or without [AuIII(nBu-C^N)(BG)]Cl (24 mM, 24 h). This figure was reproduced from ref. 145 with permission from the Royal Society of Chemistry (Great Britain). | ||
Further, attention has been directed towards the synthesis of organo-gold complexes because they mostly adopt a square planar geometry similar to that of cisplatin. However, cisplatin acts by covalent binding to DNA purine bases, while the mechanism of action of organo-gold(III) compounds is totally different. Organo-gold complexes containing a C, N-backbone are highly stable, and the lipophilicity/hydrophilicity of these complexes can be easily tuned by using different ligands and functional groups. Therefore, Brian K. Nicholson et al.,146 Isabel Marzo et al., and Maria Contel et al.147–149 synthesized organogold(III) complexes containing the “pincer” iminophosphorane Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NPh. Isabel Marzo et al. and Maria Contel et al. reported the biological activities of organogold(III) complexes of the type [Au{κ2-C,N-C6H4(PPh2N(C6H5)-2}Cl2], [Au{κ2-C,N-C6H4(PPh2N(C6H5)-2}(S2CN-R2)]PF6 (where, R = Me and CH2Ph), [Au{κ2-C,N-C6H4(PPh2N(C6H5)-2}(P{Cp(m-C6H4-SO3Na)2}3)Cl] and [Au{κ2-C,N-C6H4(PPh2N(C6H5)-2}(PR3)2Cl]PF6 (where, PR3 = PTA (1,3,5-triaza-7-phosphaadamantane) (Fig. 26). Isabel Marzo et al. observed that the [Au{κ2-C,N-C6H4(PPh2N(C6H5)-2}Cl2], C6H4(PPh2N(C6H5)-2}(S2CN-Me2)]PF6, and [Au{κ2-C,N-C6H4(PPh2N(C6H5)-2}(S2CN-(CH2Ph)2)]PF6 complexes induced mitochondrial depolarization preceding apoptotic or necrotic cell death. Also, these complexes were highly cytotoxic towards Jurkat T-cell acute lymphoblastic leukemia cells and B-CLL cells compared to normal T-lymphocytes. Furthermore, experiments with Bax/Bak-deficient Jurkat cells indicated that the [Au{κ2-C,N-C6H4(PPh2N(C6H5)-2}(S2CN-(CH2Ph)2)]PF6 complex induced apoptotic cell death; whereas the [Au{κ2-C,N-C6H4(PPh2N(C6H5)-2}Cl2] and C6H4(PPh2N(C6H5)-2}(S2CN-Me2)]PF6 complexes resulted in necrotic cell death. This difference in mechanism of cell death is probably due to the reduction of the Au(III) to Au(I) species [AuCl{Cp(m-C6H4-SO3Na)2}3)] in the [Au{κ2-C,N-C6H4(PPh2N(C6H5)-2}(S2CN-(CH2Ph)2)]PF6 complex. Similarly, Maria Contel et al. evaluated the activity of all their complexes against HeLa human cervical carcinoma and Jurkat-T acute lymphoblastic leukemia cells. All the complexes displayed prominent cytotoxic activity compared to cisplatin. However, the C6H4(PPh2N(C6H5)-2}(S2CN-Me2)]PF6 complex was the most cytotoxic, and apoptosis studies on HeLa cells revealed that it triggered cell death by activating not only apoptotic pathways (major) but also other death mechanisms such as necrosis. In addition these complexes did not show any interaction with DNA, indicating that their mechanism of action is different from DNA damage. Thus, determine their mode of action, the interaction of the C6H4(PPh2N(C6H5)-2}(S2CN-Me2)]PF6 complex with two model proteins (cytochrome c and thioredoxin reductase) was analyzed via spectroscopic methods (UV-vis and fluorescence). The C6H4(PPh2N(C6H5)-2}(S2CN-Me2)]PF6 complex displayed slow interaction with cytochrome c, but with protein thioredoxin reductase it induced irreversible denaturation. Generally, it can be concluded that these complexes do not interact with DNA and their mechanism of action may involve either direct mitochondrial damage or deregulation of the thioredoxin reductase/thioredoxin redox system.
NPh. Isabel Marzo et al. and Maria Contel et al. reported the biological activities of organogold(III) complexes of the type [Au{κ2-C,N-C6H4(PPh2N(C6H5)-2}Cl2], [Au{κ2-C,N-C6H4(PPh2N(C6H5)-2}(S2CN-R2)]PF6 (where, R = Me and CH2Ph), [Au{κ2-C,N-C6H4(PPh2N(C6H5)-2}(P{Cp(m-C6H4-SO3Na)2}3)Cl] and [Au{κ2-C,N-C6H4(PPh2N(C6H5)-2}(PR3)2Cl]PF6 (where, PR3 = PTA (1,3,5-triaza-7-phosphaadamantane) (Fig. 26). Isabel Marzo et al. observed that the [Au{κ2-C,N-C6H4(PPh2N(C6H5)-2}Cl2], C6H4(PPh2N(C6H5)-2}(S2CN-Me2)]PF6, and [Au{κ2-C,N-C6H4(PPh2N(C6H5)-2}(S2CN-(CH2Ph)2)]PF6 complexes induced mitochondrial depolarization preceding apoptotic or necrotic cell death. Also, these complexes were highly cytotoxic towards Jurkat T-cell acute lymphoblastic leukemia cells and B-CLL cells compared to normal T-lymphocytes. Furthermore, experiments with Bax/Bak-deficient Jurkat cells indicated that the [Au{κ2-C,N-C6H4(PPh2N(C6H5)-2}(S2CN-(CH2Ph)2)]PF6 complex induced apoptotic cell death; whereas the [Au{κ2-C,N-C6H4(PPh2N(C6H5)-2}Cl2] and C6H4(PPh2N(C6H5)-2}(S2CN-Me2)]PF6 complexes resulted in necrotic cell death. This difference in mechanism of cell death is probably due to the reduction of the Au(III) to Au(I) species [AuCl{Cp(m-C6H4-SO3Na)2}3)] in the [Au{κ2-C,N-C6H4(PPh2N(C6H5)-2}(S2CN-(CH2Ph)2)]PF6 complex. Similarly, Maria Contel et al. evaluated the activity of all their complexes against HeLa human cervical carcinoma and Jurkat-T acute lymphoblastic leukemia cells. All the complexes displayed prominent cytotoxic activity compared to cisplatin. However, the C6H4(PPh2N(C6H5)-2}(S2CN-Me2)]PF6 complex was the most cytotoxic, and apoptosis studies on HeLa cells revealed that it triggered cell death by activating not only apoptotic pathways (major) but also other death mechanisms such as necrosis. In addition these complexes did not show any interaction with DNA, indicating that their mechanism of action is different from DNA damage. Thus, determine their mode of action, the interaction of the C6H4(PPh2N(C6H5)-2}(S2CN-Me2)]PF6 complex with two model proteins (cytochrome c and thioredoxin reductase) was analyzed via spectroscopic methods (UV-vis and fluorescence). The C6H4(PPh2N(C6H5)-2}(S2CN-Me2)]PF6 complex displayed slow interaction with cytochrome c, but with protein thioredoxin reductase it induced irreversible denaturation. Generally, it can be concluded that these complexes do not interact with DNA and their mechanism of action may involve either direct mitochondrial damage or deregulation of the thioredoxin reductase/thioredoxin redox system.
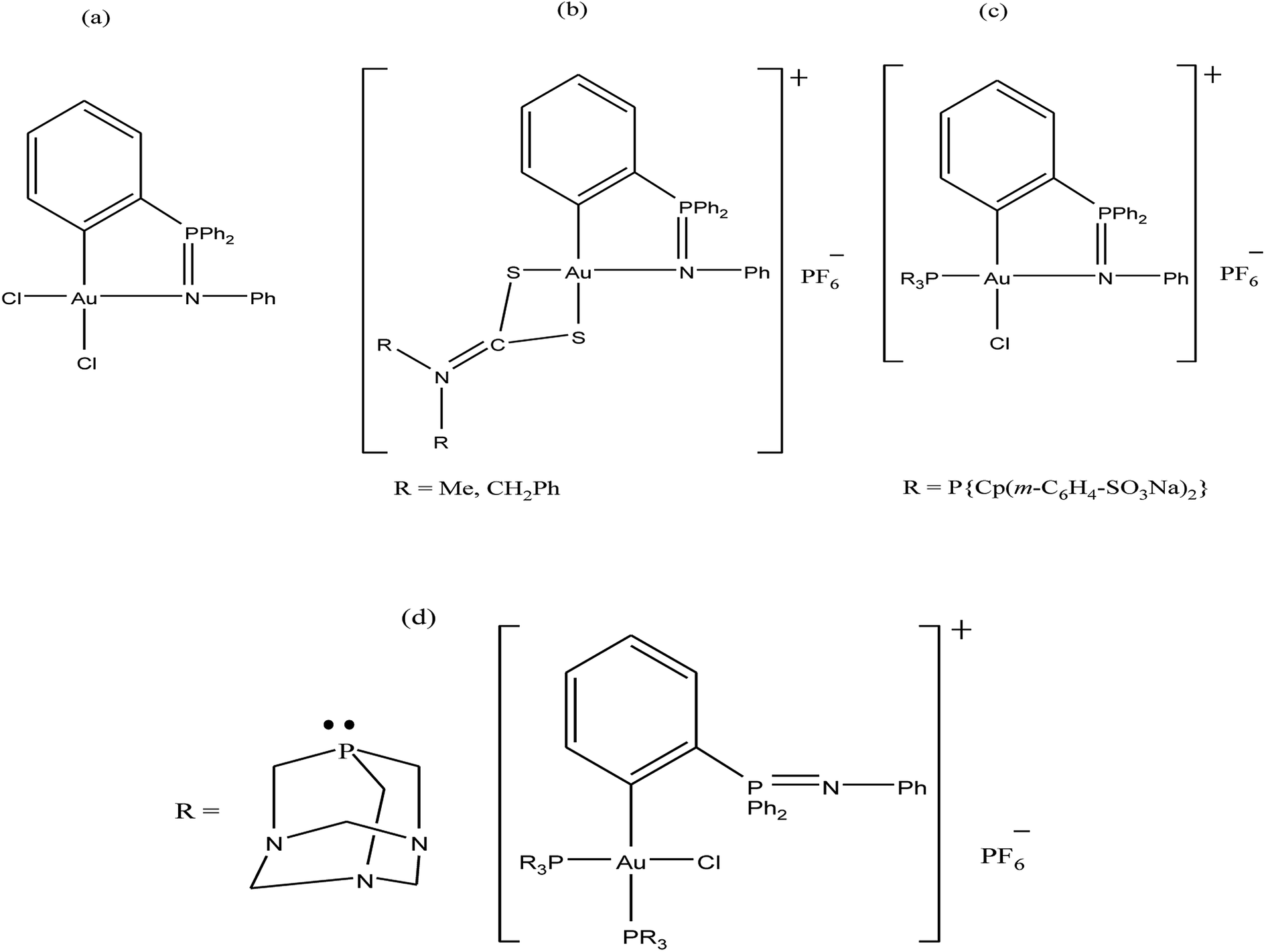 | ||
| Fig. 26 Chemical structure of organogold(III) complexes (a) [Au{κ2-C,N-C6H4(PPh2N(C6H5)-2}Cl2], (b) [Au{κ2-C,N-C6H4(PPh2N(C6H5)-2}(S2CN-R2)]PF6, (c) [Au{κ2-C,N-C6H4(PPh2N(C6H5)-2}(P{Cp(m-C6H4-SO3Na)2}3)Cl] and (d) [Au{κ2-C,N-C6H4(PPh2N(C6H5)-2}(PR3)2Cl]PF6. | ||
To enhance the stability and bioavailability of gold(III) complexes under physiological conditions, scientists have used multidentate and strong σ-donor C^N^C ligands to stabilize the gold(III) ion in organo-gold complexes. Accordingly, Chi-Ming Che et al.150 reported the stable gold(III)-phosphine complex [Au2(C^N^C)2(μ-dppp)](CF3SO3)2 (where, HC^N^CH = 2,6-diphenylpyridine and dppp = bis(diphenylphosphino)propane), as shown in Fig. 27. This complex displayed more potent in vitro cytotoxicity towards toward various human hepatocellular carcinoma (HCC) (Hep G2 and PLC) and human cervical epithelial carcinoma (HeLa) cell lines than its structural and iso-electronic platinum(II) analog [Pt2(C^N^N)2(μ-dppp)](CF3SO3)2 (where, HC^N^N = 6-phenyl-2,2′-bipyridine) and gold(III)-carbene complexes. Administration of the [Au2(C^N^C)2(μ-dppp)](CF3SO3)2 complex via intravenous (i.v.) injection suppressed the growth of tumor in nude mice models bearing human H22 hepatocellular carcinoma cells and human S180 sarcoma cells. In addition, its acute and sub-chronic toxicities were examined in mice and beagle dogs and the results revealed that there was no severe and irreversible side-effect in these animal models. Furthermore, transcriptomic and connectivity map analyses revealed that the transcriptional profile of [Au2(C^N^C)2(μ-dppp)](CF3SO3)2 is similar to that of inhibitors of thioredoxin reductase (TrRx) and inducers of endoplasmic reticulum (ER) stress such as CHOP.151 Endoplasmic reticulum (ER) stress activates apoptosis via multiple mechanisms including transcriptional down-regulation of the levels of the anti-apoptotic bcl-2 and up-regulation of DR5, which is a member of the death receptor protein family. DR5 is the receptor of the death ligand TRAIL152–154 and the binding of TRAIL to DR5 activates caspase, which ultimately leads to death-receptor-dependent apoptosis. Thus, we can conclude that [Au2(C^N^C)2(μ-dppp)](CF3SO3)2 is the only organo-gold(III) complex found in the literature to have promising in vivo anti-tumor activity in an orthotropic model (Fig. 28). Therefore, [Au2(C^N^C)2(μ-dppp)](CF3SO3)2 warrants further pre-clinical/clinical evaluations.
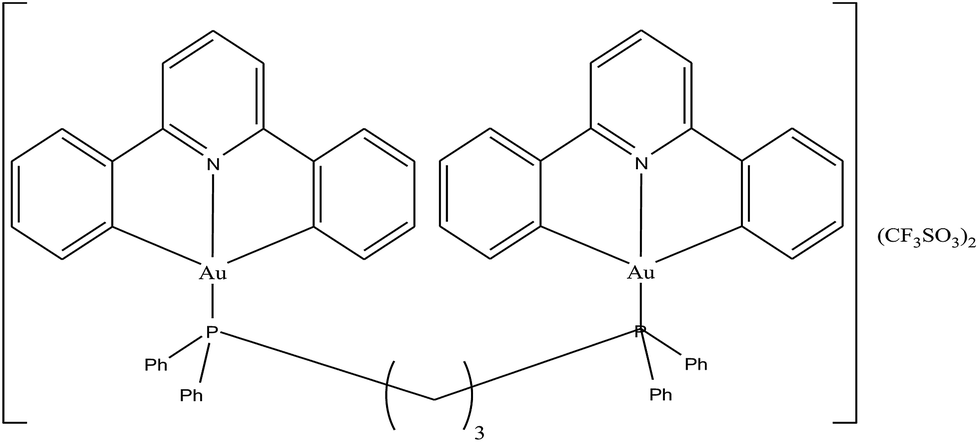 | ||
| Fig. 27 Chemical structure of gold(III)-phosphine complex [Au2(C^N^C)2(μ-dppp)](CF3SO3)2. | ||
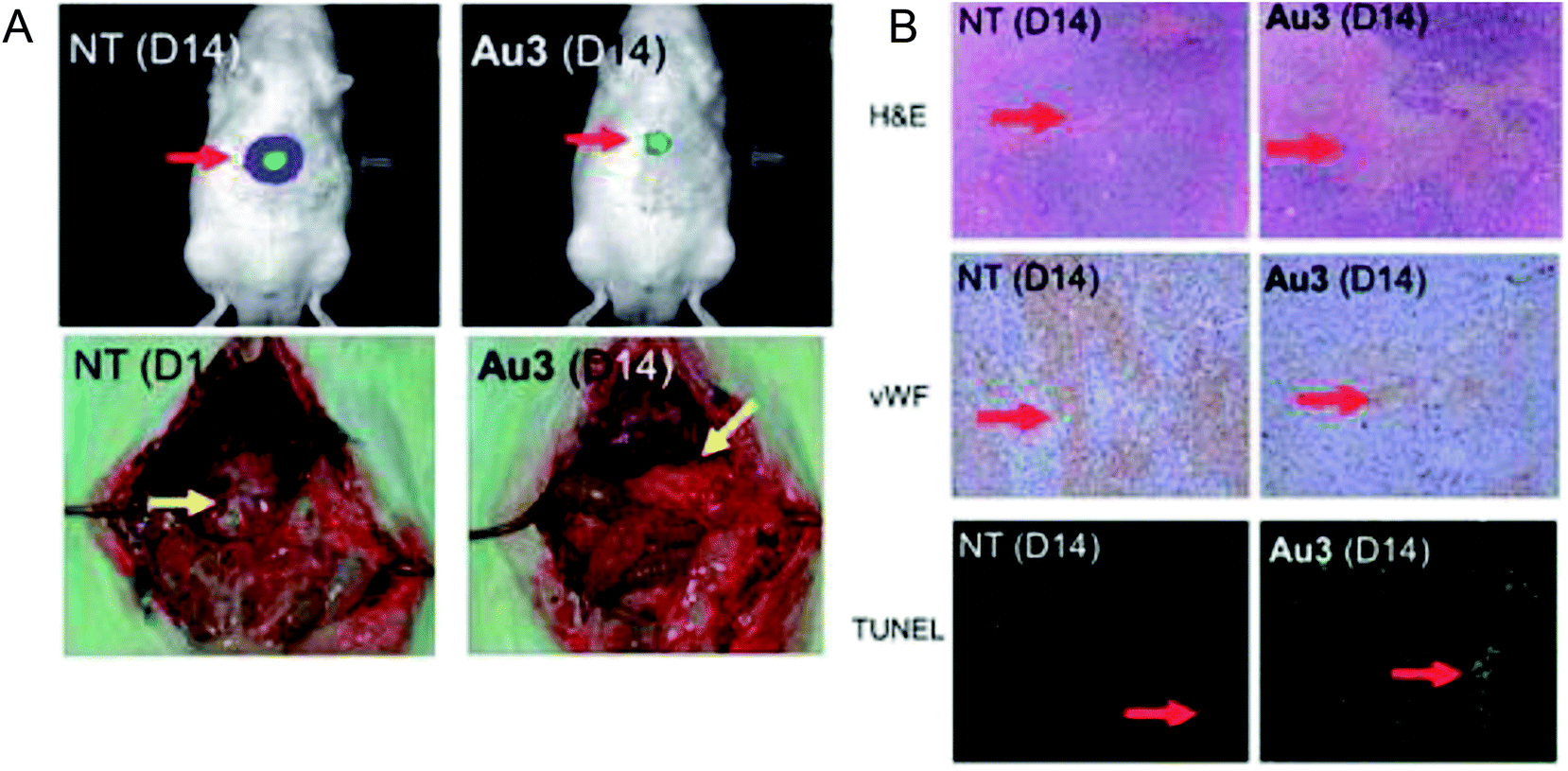 | ||
| Fig. 28 (A) The sizes of tumor nodules of the vehicle-control (NT, left) or [Au2(C^N^C)2(μ-dppp)](CF3SO3)2-treated (0.5 mg kg−1) rat examined by Xenogen Imaging System (upper) and dissect ion (lower). (B) H&E, vWF, and TUNEL staining of the tumor tissue of vehicle control (NT) and [Au2(C^N^C)2(μ-dppp)](CF3SO3)2-treated (0.5 mg kg–1) rats. This figure is reproduced from ref. 150 with permission from the Royal Society of Chemistry (Great Britain). | ||
To improve the biological properties of organometallic gold compounds, the proper choice of secondary ligands must be considered, such as PTA for increased water solubility and thio-sugar moieties to influence uptake and reduce exchange with biological nucleophiles. By considering this, recently M. A. Cinellu and A. Casini et al.155 reported the synthesis of a new series of (C^N) cyclometallated gold(III) complexes with general formula [Au(pyb-H)L1L2]n+ (where, pyb-H = C^N cyclometallated 2-benzylpyridine, L1 and L2 = chlorido 1,3,5-triaza-7-phosphaadamantane (PTA) and glucosethiolato (GST) ligands, respectively, and n = 0 or 10) (Fig. 29) bearing different ancillary ligands such as PTA for increased water-solubility and thio-sugar moieties to influence uptake and reduce exchange with biological nucleophiles. The X-ray structure of the [Au(pyb-H)(PTA)Cl]PF6 complex displayed the typical square-planar geometry of the gold(III) cation with the phosphane ligand trans to the nitrogen. All the complexes were screened in vitro in a panel of human cancer cell lines, including ovarian adenocarcinoma (A2780), mammary carcinoma (MCF-7), lung carcinoma (A549) and colon carcinoma overexpressing p53 (HCT116 p53+/+) or p53 knock-out (HCT116 p53−/−), as well as on healthy human embryonic kidney cells (HEK-293T). It was observed that the new cyclometallated complexes [Au(pyb-H)(PTA)Cl]PF6, [Au(pyb-H)(GST)Cl] and [Au(pyb-H)(GST)2] were more cytotoxic than their precursor [Au(pyb-H)Cl2] in all the cell lines except the A549 cell line. The most interesting toxicity profile was shown by the phosphane-containing complex [Au(pyb-H)(PTA)Cl]PF6, where this complex was twice as toxic as cisplatin against HCT116+/+ cells, which suggested its dependence on p53 pathways. Also the [Au(pyb-H)(PTA)Cl]PF6 complex was less toxic on healthy human embryonic kidney cells (HEK-293T). The complexes containing the tetra-acetylated β-D-glucose-1-thiolato ligand, [Au(pyb-H)(GST)Cl] and [Au(pyb-H)(GST)2], showed moderate antiproliferative activity. Furthermore, the [Au(pyb-H)Cl2] and [Au(pyb-H)(PTA)Cl]PF6 complexes were revealed to be potent inhibitors of the zinc finger protein PARP-1, which is involved in the mechanism of cisplatin resistance.
 | ||
| Fig. 29 Chemical structure of novel (C^N)gold(III) cyclometallated compounds (a) [Au(pyb-H)Cl2], (b) [Au(pyb-H)(PTA)Cl]PF6, (c) [Au(pyb-H)(GST)Cl] and (d) [Au(pyb-H)(GST)2]. | ||
Besides organometallic gold(III) complexes, the interest in neutral gold(I) complexes started when auranofin, a triethylphosphine gold(I) glucose-thiolate, was marketed in 1982 as an anti-rheumatic substance.156 The oral availability of this substance represented a major breakthrough in the field of gold organometallics. In past decades, it was well established that auranofin (Fig. 30) inhibited the growth of different cancer cells in vitro. However, in mice bearing the P388 leukemia model, auranofin was only effective when it was administered via the i.p. route and remained inactive when administered via the i.v. and s.c. routes.157 Detailed studies revealed that auranofin rapidly reacts with Cys-34 of serum albumin (free –SH concentration is ∼400 μM in the bloodstream).158 Despite the significant effort, auranofin was not very successful in tumor models in vivo. Thus, to further improve cytotoxicity in vivo, Kamei et al.159 reported the antitumor activity of another antiarthritic gold(I) drug, aurothiomalate (ATM). They observed that treatment of tumor-bearing mice with ATM at 30 mg per kg per day (s.c.) or by oral administration (p.o.) at 75 mg per kg per day significantly increased the survival time of the mice. Also, ATM was comparatively less toxic than cisplatin. Another group of scientists, Fields et al.,160 found that aurothioglucose (ATG) and ATM as potent inhibitors disrupt the interaction of protein kinase Cι (PKCι) (required for the transformed growth of human non-small-cell lung cancer (NSCLC)) with its downstream effector Par6. They also observed that ATG significantly reduced the tumor size in nude mice inoculated with human lung adenocarcinoma (A549).
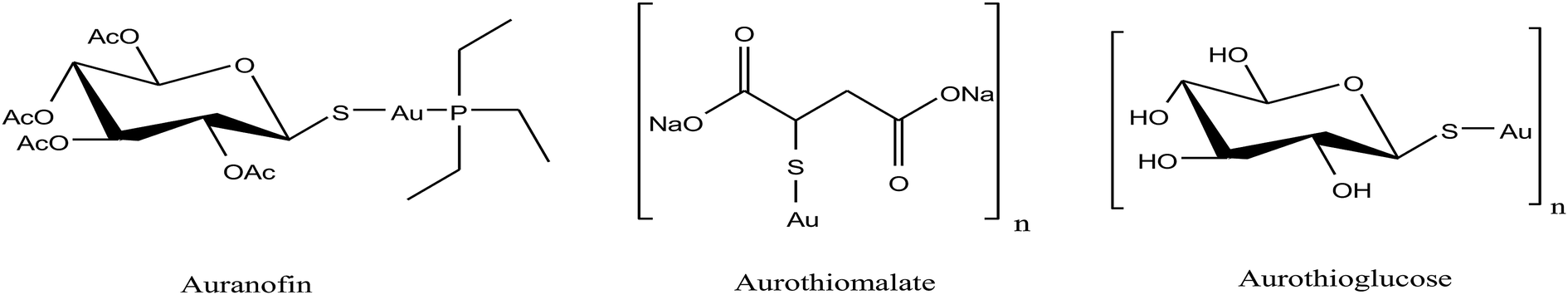 | ||
| Fig. 30 Chemical structures of the clinically used antirheumatic gold(I) complexes with promising anticancer potential. | ||
The traditional gold complexes such as auranofin and aurothioglucose are readily metabolized under physiological conditions by thiol-containing biomolecules and their ligands are easily lost before they reach the target enzyme. This means that these gold complexes are not stable in physiological conditions. Thus, to stabilize these complexes, scientists are focusing on the development of organometallic gold complexes because they are more stable and prevent the metabolization of complexes. In this regard, NHC ligands are more attractive due to their high stability and interesting biological properties, such as they induce apoptosis and inhibit thioredoxin reductase (TrRx) enzyme. Therefore, gold(I)-NHC complexes of the type [Au(NHC) L]n+ (where, NHC = N-heterocyclic carbene; L = –Cl, –NHC, or –PR3 (R = Me, Et, iPr, and Ph), and n = 0–1) (Fig. 31) have been extensively investigated in the past by B. Price et al. and I. Ott et al.161–164 as cytotoxic agents that interact with both DNA and the disulfide reductase enzyme thioredoxin reductase (TrRx). The majority of complexes binds to human serum albumin in the order [Au(NHC)Cl], [Au(NHC)2]I and [Au(NHC)PPh3]I. The chloro derivative [Au(NHC)Cl] was found to be the specific inhibitor of TrRx and showed intensive binding to albumin similar to auranofin. The cationic complex [Au(NHC)2]I with two NHC ligands exhibited the lowest inhibition of TrRx and was found to exhibit the lowest binding capacity to albumin. Finally, the triphenylphosphine derivative [Au(NHC)PPh3]I strongly inhibited TrRx and showed increased binding with protein. It was also observed that cationic complexes of [Au(NHC)2]I and [Au(NHC)PPh3]I are lipophilic, and thus called delocalized lipophilic cations (DLC). These DLCs can penetrate the hydrophobic barriers of cellular membranes and show selective accumulation in tumor cell mitochondria.165 Berners-Price et al. reported clearly that DLC effects increased the cellular uptake of gold(I) complexes in tumor cells.166 Therefore the cellular uptake of the [Au(NHC)2]I and [Au(NHC)PPh3]I complexes was enhanced compared to [Au(NHC)Cl]. The increased cellular and mitochondrial uptake of [Au(NHC)2]I and [Au(NHC)PPh3]I induced apoptosis and ROS formation in tumor cells. However, the cationic complex with the triphenylphosphine and NHC ligands, [Au(NHC)PPh3]I, was found to be the most active among them. Furthermore, the incorporation of the naphthalimide moiety increased the intercalation of the [Au(NHC)(NAP)Cl] complex (where, NAP = naphthalimide moiety) with DNA together with the TrRx inhibition. Hence, the conjugation of naphthalimides with gold(I) NHC moieties provides a useful strategy for the design of chemotherapeutic agents with multiple (nonrelated) modes of action.
 | ||
| Fig. 31 Chemical structures of (a) [Au(NHC)Cl], (b) [Au(NHC)2]I (c) [Au(NHC)PR3]I and (d) [Au(NHC)(NAP)Cl] complexes. | ||
In continuation of this, C.-M. Che et al.167 recently reported a dinuclear gold(I) complex containing a bridging bis(N-heterocyclic carbene) ligand as the first gold(I) complex reported to inhibit cancer stem cell activity and inhibit in vivo angiogenesis in a tumor model (Fig. 32). The [Au(B-NHC)(B-DP)](PF6)2 complex (where, B-NHC = bis-(NHC) bridging ligands and B-DP = bridging diphosphine ligand) was quite stable, as envisaged from its low reactivity towards blood thiols, and potently inhibited TrRx activity in the presence of GSH in a time- and concentration-dependent manner. Therefore, the activity of [Au(B-NHC)(B-DP)](PF6)2 can be attributed to its high thiol reactivity and inhibition of TrRx. Subsequently, using a model TrRx C-terminal GCUG (glycine-cysteine-selenocysteine-glycine) peptide, high-resolution ESI-MS together with 1H NMR experiments revealed that the dinuclear gold(I) complex formed a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adduct with the GCUG motif. Altogether, these studies confirmed the simultaneous coordination of the dinuclear [Au2(dcpm)]+ unit to the corresponding S(Cys) and Se(Sec) of GCUG, accompanied by the release of bis(NHC), but not the dcpm ligand. Further studies showed that the complex inhibited the tumor growth of HeLa xenografts and highly aggressive mouse B16-F10 melanoma with no mouse death or bodyweight loss. It also inhibited angiogenesis in the tumor models (Fig. 33) and in vitro sphere formation in cancer stem cells. The toxicology results revealed that [Au(B-NHC)(B-DP)](PF6)2 did not show systemic anaphylaxis on guinea pigs and localized irritation on rabbits. Thus, it can be inferred that gold(I)-NHC complexes are very promising candidates for the development of chemotherapeutic agents and warrant clinical investigations.
:1 adduct with the GCUG motif. Altogether, these studies confirmed the simultaneous coordination of the dinuclear [Au2(dcpm)]+ unit to the corresponding S(Cys) and Se(Sec) of GCUG, accompanied by the release of bis(NHC), but not the dcpm ligand. Further studies showed that the complex inhibited the tumor growth of HeLa xenografts and highly aggressive mouse B16-F10 melanoma with no mouse death or bodyweight loss. It also inhibited angiogenesis in the tumor models (Fig. 33) and in vitro sphere formation in cancer stem cells. The toxicology results revealed that [Au(B-NHC)(B-DP)](PF6)2 did not show systemic anaphylaxis on guinea pigs and localized irritation on rabbits. Thus, it can be inferred that gold(I)-NHC complexes are very promising candidates for the development of chemotherapeutic agents and warrant clinical investigations.
 | ||
| Fig. 32 Chemical structure of the dinuclear gold(I) complex [Au(B-NHC)(B-DP)](PF6)2. | ||
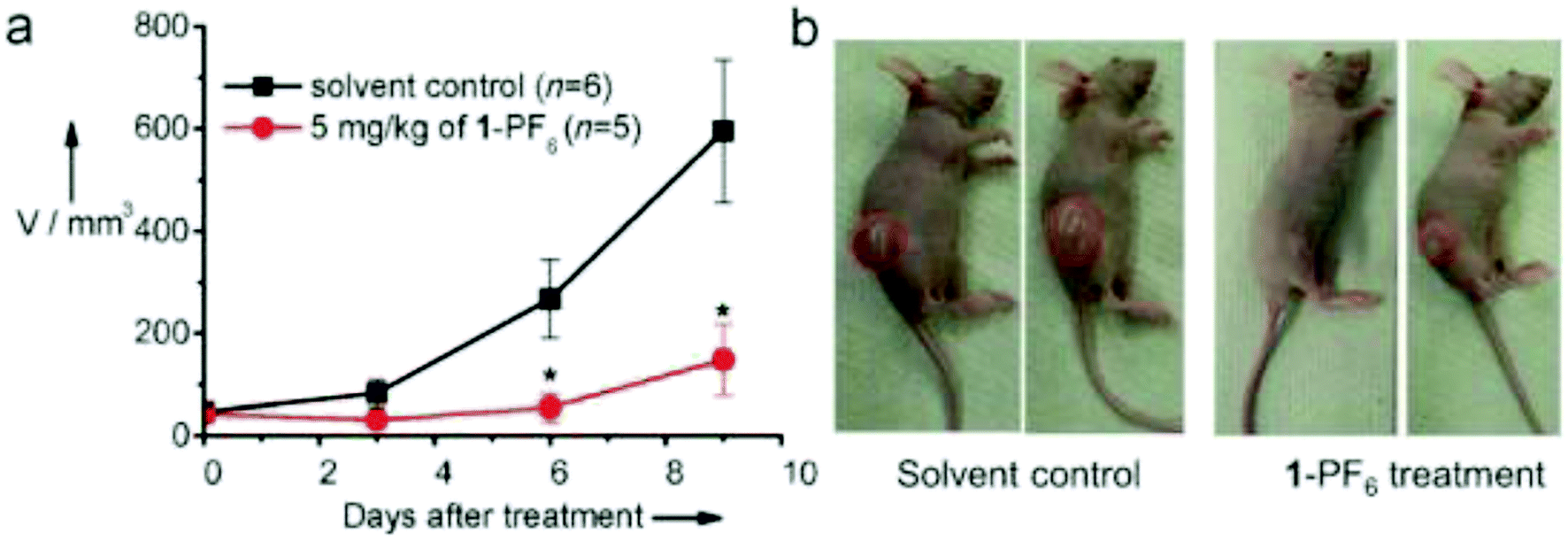 | ||
| Fig. 33 Antitumor effect of [Au(B-NHC)(B-DP)](PF6)2 on mice bearing HeLa xenografts. (a) Changes in tumor volume (V) after treatment with [Au(B-NHC)(B-DP)](PF6)2,*p < 0.05. (b) Representative mouse photos after 9 days of treatment. This figure was reproduced from ref. 167 with permission from John Wiley and Sons. | ||
Similar to organometallic gold(I)-NHC complexes, gold(I) complexes are also stabilized via the use of alkyne ligands due to the presence of the Au(I)-carbon bond. These organometallic gold(I) complexes show more pronounced antiproliferative activity compared to the classical drug cisplatin. Recently, organometallic gold(I) derivatives with an alkyne ligand displayed promising in vitro and in vivo anticancer activity. M. Laguna et al.168 synthesized an alkyne gold(I) complex, [Au(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCH2S pyridine)(PTA)] (where, CCCH2Spyridine = alkyl S-pyridine and PTA = 1,3,5-triaza-7-phosphaadamantane), as shown in Fig. 34, and investigated its anticancer activity against the colon cancer cell line Caco-2 (PD7 and TC7 clones). This complex showed strong antiproliferative effects, which were even more pronounced than cisplatin but similar to auranofin. The interaction of this complex with bovine serum albumin (BSA) was also studied via fluorescence spectroscopy, and moderate binding constant values were obtained. Thermodynamically, the [Au(CCCH2Spyridine)(PTA)] complex interacts with protein through van der Waals interactions or hydrogen bonding. Furthermore, [Au(CCCH2Spyridine)(PTA)] showed inhibition in colon cancer proliferation of the HTC-116-luc2 cell line via the apoptotic pathway and S-phase arrest of the cell cycle. Intraperitoneal injection of [Au(CCCH2Spyridine)(PTA)] in athymic nude mice inoculated with HTC-116-luc2 cells prolonged their survival and resulted in moderate inhibition of the tumor growth with no subsequent organ (kidney and liver) damage (Fig. 35).
CCH2S pyridine)(PTA)] (where, CCCH2Spyridine = alkyl S-pyridine and PTA = 1,3,5-triaza-7-phosphaadamantane), as shown in Fig. 34, and investigated its anticancer activity against the colon cancer cell line Caco-2 (PD7 and TC7 clones). This complex showed strong antiproliferative effects, which were even more pronounced than cisplatin but similar to auranofin. The interaction of this complex with bovine serum albumin (BSA) was also studied via fluorescence spectroscopy, and moderate binding constant values were obtained. Thermodynamically, the [Au(CCCH2Spyridine)(PTA)] complex interacts with protein through van der Waals interactions or hydrogen bonding. Furthermore, [Au(CCCH2Spyridine)(PTA)] showed inhibition in colon cancer proliferation of the HTC-116-luc2 cell line via the apoptotic pathway and S-phase arrest of the cell cycle. Intraperitoneal injection of [Au(CCCH2Spyridine)(PTA)] in athymic nude mice inoculated with HTC-116-luc2 cells prolonged their survival and resulted in moderate inhibition of the tumor growth with no subsequent organ (kidney and liver) damage (Fig. 35).
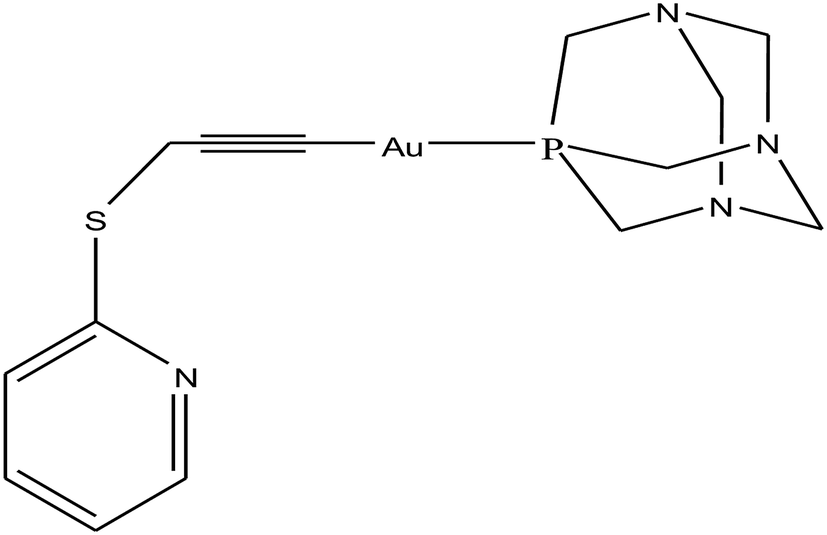 | ||
| Fig. 34 Chemical structure of the alkyne gold(I) complex [Au(CCCH2Spyridine)(PTA)]. | ||
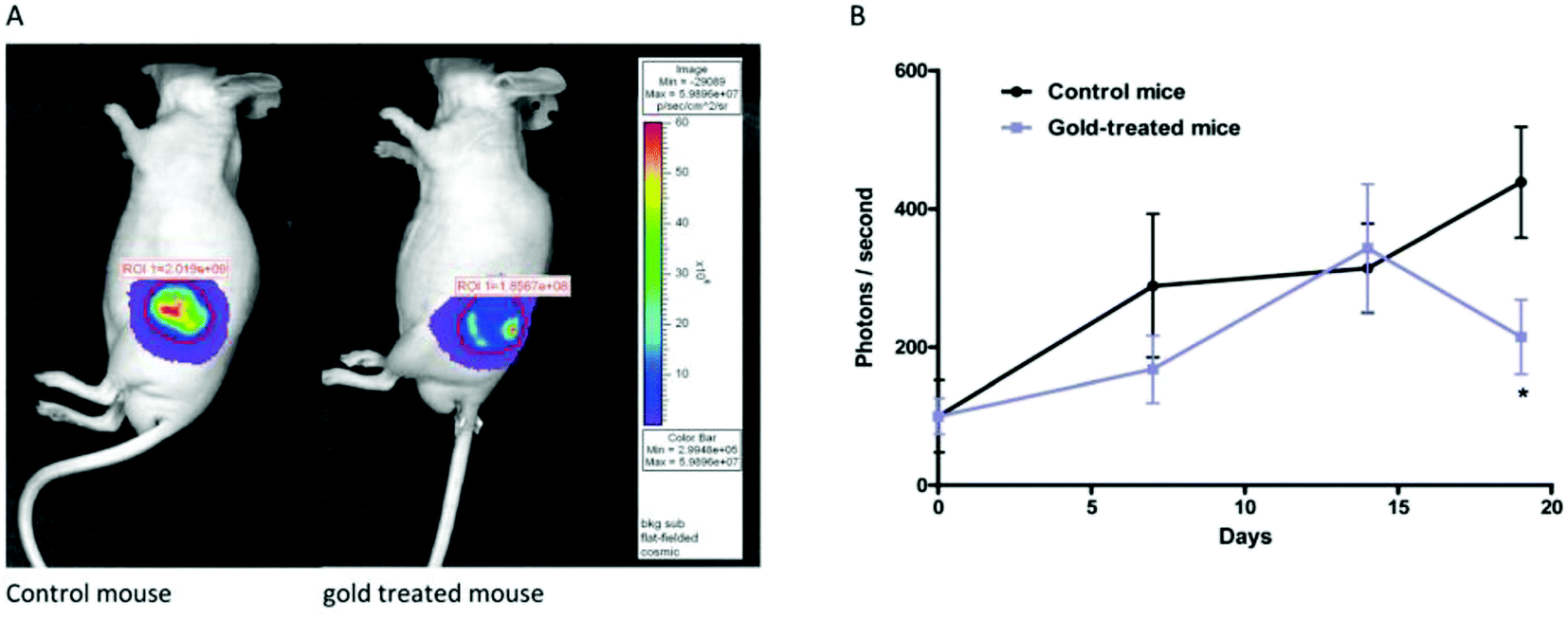 | ||
| Fig. 35 (A) Bioluminescence images after injection of luciferin to mice infected with HCT-116-luc2. (B) Results showing the changes in the HCT-116-luc2 tumor volume against number of days in mice treated with [Au(CCCH2Spyridine)(PTA)] in comparison with the control mice. *P < 0.05 vs. control. This figure was reproduced from ref. 168 with permission from the Royal Society of Chemistry (Great Britain). | ||
Since the organometallic alkynyl gold(I) complexes displayed much better activity than cisplatin both in vitro and in vivo, no evidence of necrosis was observed in the vital organs of the mice. Therefore, P. J. Dyson et al.169 also reported alkynyl phosphane gold(I) derivatives, [Au(CCPh)(DAPTA)], [Au(CC-3-SC4H3)(DAPTA)] and N[Au(CCCH2)(DAPTA)]3, containing the water-soluble 1,3,5-triaza-7-phosphaadamantane (PTA) and 3,7-diacetyl-1,3,7-triaza-5-phosphabicyclo[3.3.1]-nonane (DAPTA) phosphane ligands (Fig. 36). These complexes displayed cytotoxicity on A2780 ovarian cancer cells and on the cisplatin-resistant cell line A2780cisR similar to that of auranofin. Furthermore, due to their highly luminescent nature, their cellular uptake was observed via fluorescence microscopy and the images shows that the complexes were efficiently and rapidly taken up by the cancer cells.169 However, these complexes did not interact with DNA like auranofin, which suggests they have a different target of action. Although the exact mechanism of Au(I) phosphane-induced cytotoxicity is unknown, some studies suggest involvement of mitochondria.170 Also the high affinity of Au(I) for S- and Se-donor ligands indicates that proteins, including enzymes and transport proteins, may be possible targets.
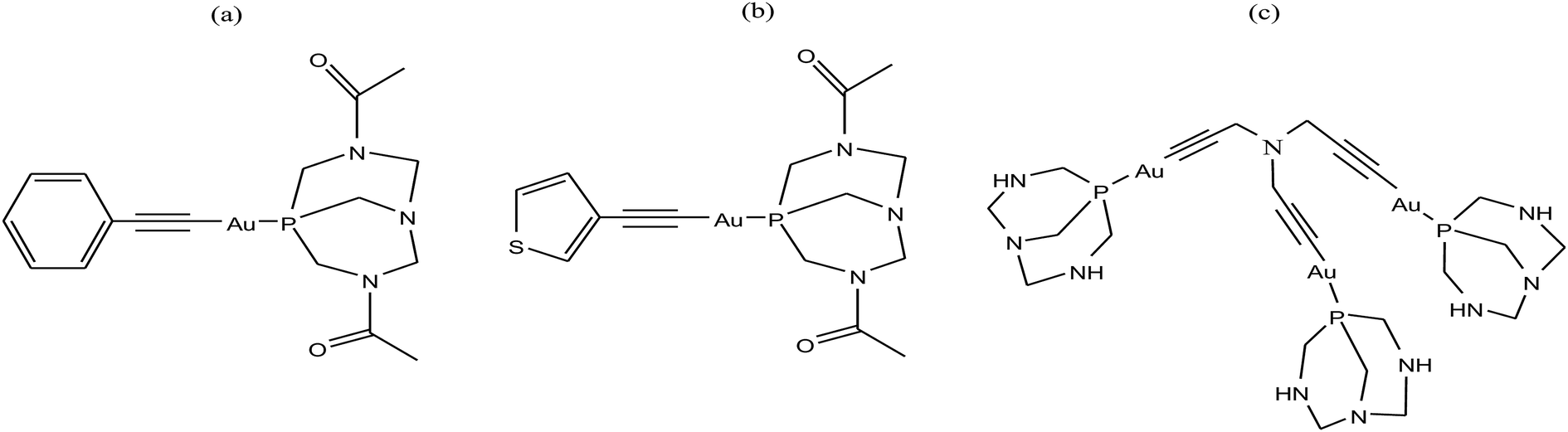 | ||
| Fig. 36 Chemical structure of (a) [Au(CCPh)(DAPTA)], (b) [Au(CC-3-SC4H3)(DAPTA)] and (c) N[Au(CCCH2)(DAPTA)]3. | ||
Ingo Ott et al.171 also reported the synthesis of mononuclear alkynyl gold(I) complexes, [Au(CC-BnZO)PPh3] and [Au(CC-MeOPh)PPh3] (where, BnZO = benzyloxy group and MeOPh = methoxyphenyl group) by reacting the respective alkynes with chloro(triphenylphosphine)gold(I), as depicted in Fig. 37. These complexes, particularly [Au(CC-BnZO)PPh3] and [Au(CC-MeOPh)PPh3], exhibited good antiproliferative effects and were generally strong inhibitors of thioredoxin reductase (TrRx) with high selectivity for the related enzyme glutathione reductase, where TrRx is an enzyme with various physiological and pathophysiological functions.172,173 These complexes also showed effects against tumor cell metabolism and mitochondrial respiration, thus preventing tumor formation. Furthermore, they showed prominent anti-angiogenic properties in zebrafish embryos, i.e. inhibited the formation of blood vessels in growing tumors, resulting in cell death, compared to the ligand, which is inactive. This approach led to the development of a human cancer model in in vivo assays using developing zebra fish embryos (Fig. 38).174 These models provide fast and reliable results and have practical applicability for drug screening procedures and are already being utilized in academia and industry. Thus, attention has been further stimulated by the amenability of zebra fish, which allows the visualization of cancer progression and angiogenesis in live animals.
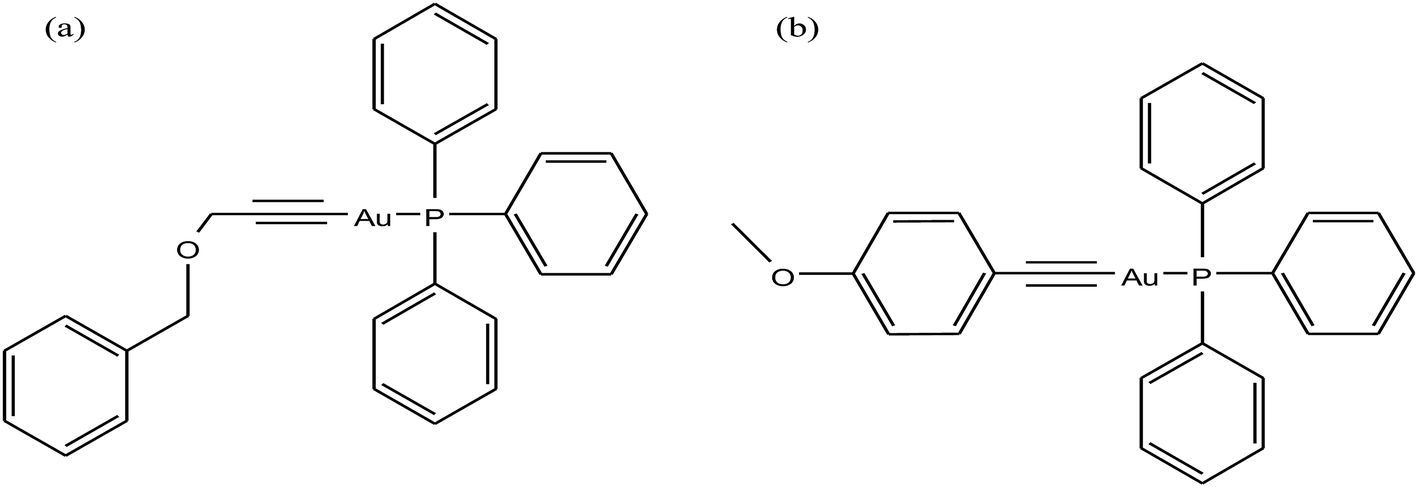 | ||
| Fig. 37 Chemical structure of alkynyl gold(I) complexes (a) [Au(CC-BnZO)PPh3] and (b) [Au(CC-MeOPh)PPh3]. | ||
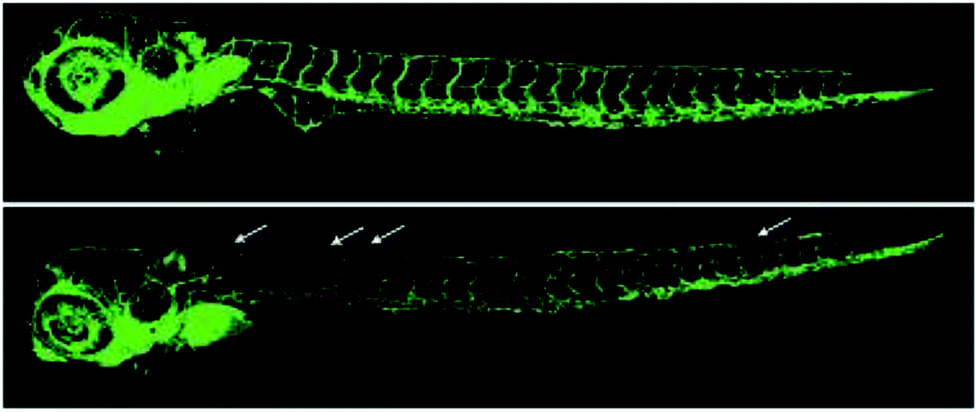 | ||
| Fig. 38 Blood vessel formation in developing zebrafish embryos (transgenic zebrafish line, Tg:fli1/eGFP) was monitored three days after fertilization; top: ligand HCC-BnZO (0.1 μm) and bottom: [Au(CC-MeOPh)PPh3] (0.1 μm). Examples of effects on vessel formation are marked with arrows. This figure was reproduced from ref. 171 with permission from John Wiley and Sons. | ||
Organometallic gold(I)-alkynyl complexes have been considered as motifs for the design of antitumor drugs due to their potential stability, luminescence and apoptosis-inducing properties. Subsequently, Simon J. A. Pope et al.175 synthesized a series of highly luminescent mono- and dimetallic Au(I) triphenylphosphine complexes, [Au(1-DOQ)PPh3], [Au(1,4-DOQ)PPh3] and [Au(1,8-DOQ)PPh3] (where DOQ = 1,1,4- and 1,8-dialkynyloxyanthraquinone), as shown in Fig. 39. The ligands generally showed lower cytotoxicity compare to the complexes, which were considerably more toxic to MCF-7 cells. The MCF-7 cell line has higher mitochondrial mass, which indicates that mitochondria may be the intracellular target of gold complexes. Also the observed cytotoxicity of these complexes is associated with their gold alkynyl unit rather than the biological properties of the anthraquinones. Cellular imaging studies showed that these complexes accumulated in a significant concentration across the entire cytoplasm, including all organelles, confirming that the mode of gold cytotoxicity involves mitochondrial inhibition (Fig. 40).
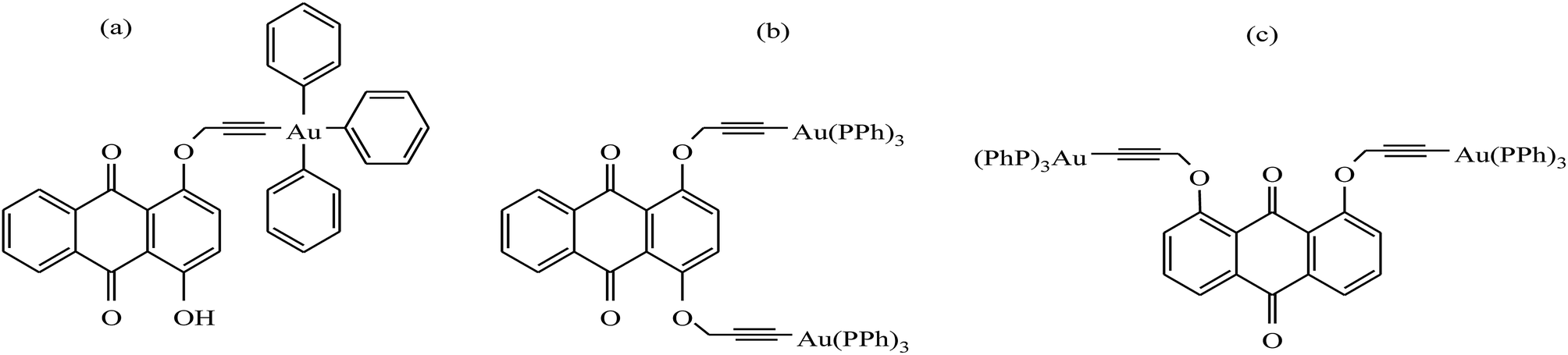 | ||
| Fig. 39 Chemical structure of Au(I) triphenylphosphine complexes (a) [Au(1-DOQ)PPh3], (b) [Au(1,4-DOQ)PPh3] and (c) [Au(1,8-DOQ)PPh3]. | ||
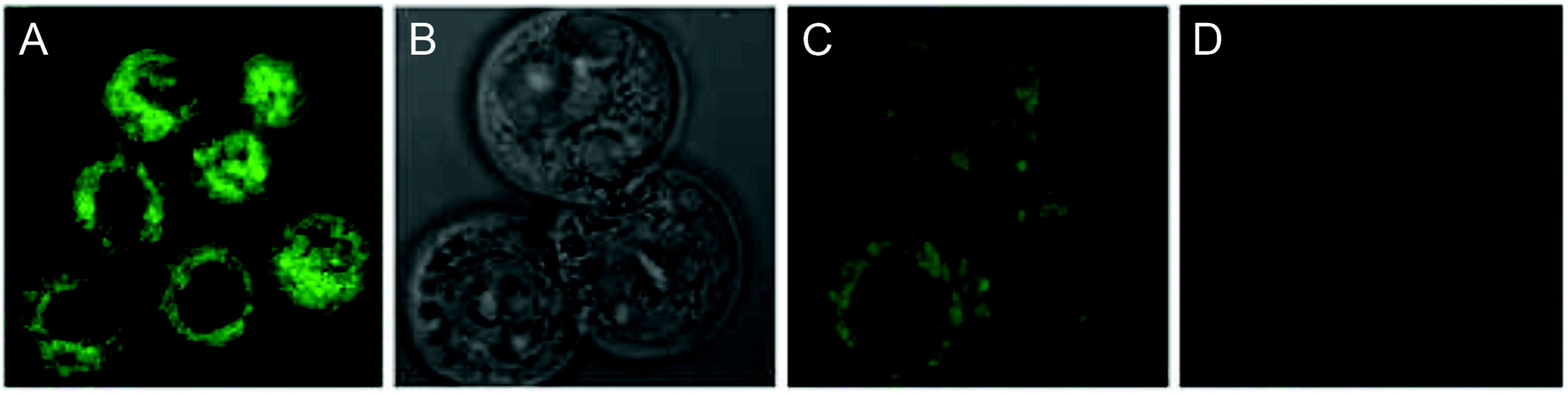 | ||
| Fig. 40 Images of MCF-7 cells incubated with [Au(1,4-DOQ)PPh3] (100 μg mL−1, 4 °C, 30 min), excited at 405 nm, acquired at 530–580 nm showing: (A) cytoplasmic distribution (overlaid luminescence and transmitted light), (B) appearance of vacuoles upon irradiation (transmitted light only) and (C and D) photobleaching (luminescence only). This figure was reproduced from ref. 171 with permission from the American Chemical Society. | ||
Organometallic iron complexes with thioredoxin reductases (TrRxs) as targets
Generally, iron is mostly found in heme complexes, which are basically the oxygen binding cytochrome proteins, such as hemoglobin, myoglobin and leghemoglobin. Therefore iron, is required by cancer cells for their proliferation.176,177 One of the well-known iron-containing glycopeptide antitumor drugs is Bleomycin, which acts on cancer cells by causing the oxidative cleavage of DNA, leading to cell death.178–180 The high activity of Bleomycin has led to the search for other iron-containing complexes as chemotherapeutic agents by many scientists. In this regard, an organometallic iron metallocene named ferrocene has attracted attention due to its small size, relative lipophilicity, ease of chemical modification, and redox potential.181–183 Many ferrocenyl derivatives showed promising activities, such as antineoplastic, antimalarial, antifungal, antibacterial and DNA-cleaving activities.184–188 Very early in 1984, oxidized ferrocene, denoted as ferrocenium salt, exhibited in vivo and in vitro antiproliferative activity.189 The mechanism of the antitumor activity of the ferrocenium derivatives was mainly due to the oxidative cleavage of DNA by reactive oxygens species through the Fenton mechanism.190,191 It was also found that the ferrocenyl group induced lymphocyte activation and antitumor activity mediated by redox-sensitive signaling.192 Currently, the incorporation of the ferrocenyl moiety into existing drug molecules has become a pioneering strategy for the enhancement of therapeutic effectiveness in tumor cells. Some examples are the antiestrogen tamoxifen,193,194 polyphenols,195–197 steroidal androgens,198 and the commercial antiandrogen nilutamide and benzo[b]thiophene derivative raloxifene.199 Moreover, ferrocene containing alcohols with longer side chains and low formal reduction potential was found to be cytotoxic on HeLa cancer cell lines.200 Furthermore, other iron organometallics demonstrated potent antitumor activities and some of them can potentially overcome the intrinsic resistance caused by classical drugs.201–203 Therefore, it was thought that iron organometallics with different ligand frameworks and functional groups may act as ideal chemotherapeutic agents with different mechanisms of action. Currently, many organometallic iron complexes are being developed worldwide in the quest to fight cancer cells effectively without causing adverse effects on normal cells.The discovery of iron organometallics as antitumor agents was first revealed by Kopf and Kopf-Mayer in 1984. They reported that ferrocenium picrate and ferricenium trichloroacetate salts (Fig. 41) exhibited antineoplastic properties with optimal cure rates due to oxidative DNA damage.185,189,204 However, the exploration of the anticancer efficacies of ferrocene derivatives can be traced back to the early studies by S. Brynes et al., who reported the synthesis and anticancer activity of ferrocenyl complexes with amine or amide groups against lymphocytic leukemia P-388 cells.205 Specifically, the amide ferrocenyl complexes displayed significant antitumor activity on P-388 cells, which is probably due to their binding with the tumor cell surface nucleic acid. This was enough to provide a platform for the development of several classes of ferrocenyl complexes with interesting anticancer activities.206–209 Later, this field gained attention when ferrocene was incorporated in tamoxifen derivatives, which were called ferrocifens. The ferrocifens exhibited highly promising antiproliferative activity towards breast cancer due to the redox behavior of iron.210–212 Thus, A. C. de Oliveira et al.213 examined the anticancer potential of the [Fe(η5-cp)2(TAM)] complex (where, cp = cyclopentadienyl and TAM = tamoxifen derivative) on a panel of cell lines such as HL-60 acute promyelocytic leukemia, HCT-8 human colon cancer, SF-295 human glioblastoma, OVCAR-8 ovarian carcinoma and GBM glomerular basement membrane non-cancerous cells. This complex (Fig. 42) was not cytotoxic toward GBM monkey cells, but was able to decrease, in a concentration-dependent manner, the number of viable cells in the HL-60 line. Usually, in the presence of DNA damage, cells can trigger a complex sequence of events, including checkpoint activation, leading to cell-cycle arrest, thus allowing DNA repair. Moreover, it induces cell-cycle arrest at the G0/G1 phase despite the low ER expression, followed by an increase in the number of cells with DNA fragmentation. Indeed, cells treated with [Fe(η5-cp)2(TAM)] underwent characteristic apoptotic cell death, as demonstrated by the morphology of the treated cells, increase in number of cells with DNA fragmentation, PS externalization and caspase 3 activation, all phenotypic and biochemical features related to apoptosis. Thus, tethering ferrocene to tamoxifen derivatives for targeting receptors and enzymes open a new panoramic view for modeling chemotherapeutic drugs with diverse and simultaneous functions.
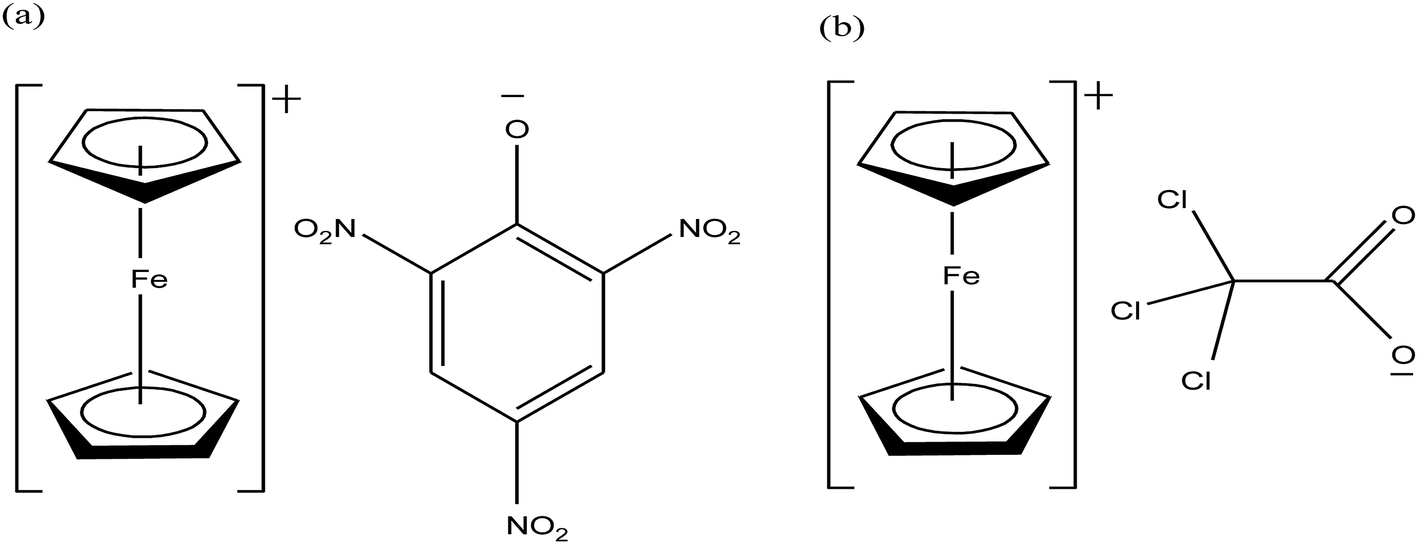 | ||
| Fig. 41 Chemical structures of (a) ferrocenium picrate and (b) ferrocenium trichloroacetate salts. | ||
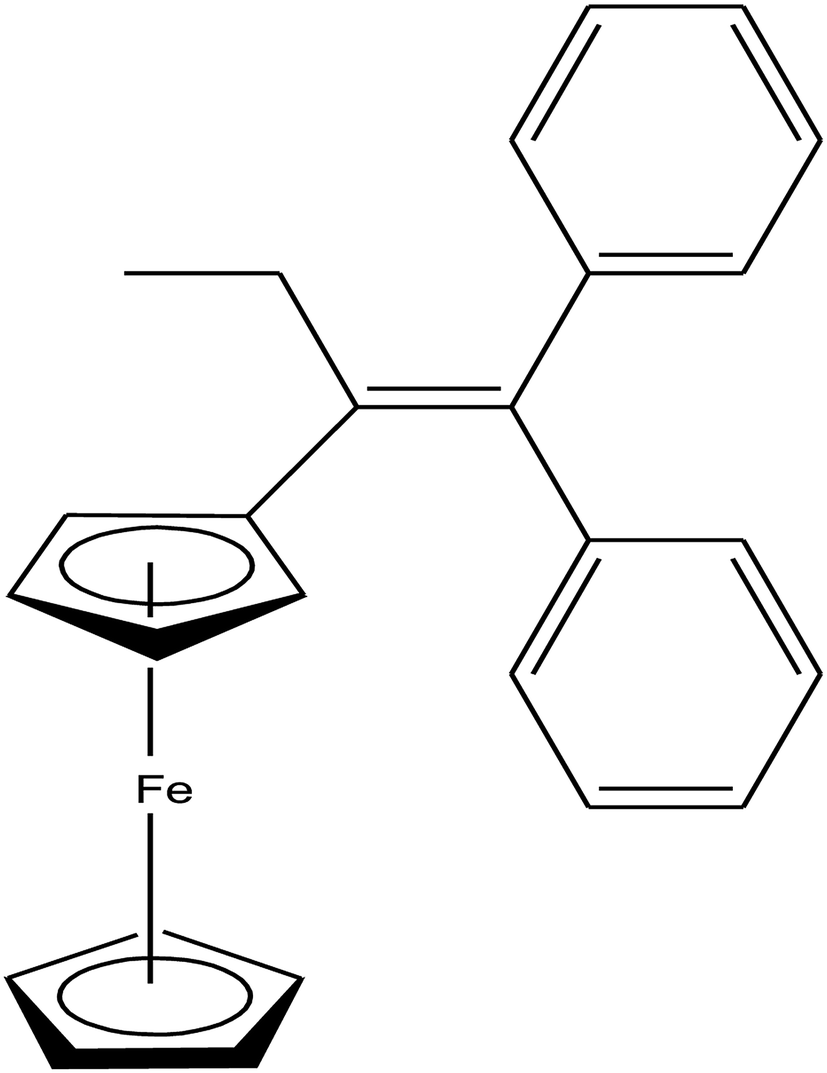 | ||
| Fig. 42 Chemical structure of 2-ferrocenyl-1,1-diphenylbut-1-ene complex [Fe(η5-cp)2(TAM)]. | ||
The effectiveness of the available chemotherapeutic drugs is still limited due to the intrinsic resistance inside cancer cells. This intrinsic resistance can be overcome via the use of organometallic redox active complexes such as ferrocene, which are highly stable and redox active. Furthermore, biological activity can be improved by tethering them with pharmacologically active molecules. It was observed that ferrocene analogs are strong inhibitors of thioredoxin reductases (TrRxs), which are selenoenzymes; thus, they have been suggested as molecular targets. Accordingly, M. P. Rigobello et al.214 reported two TrRx-targeting hydroxyferrocifens, [Fe(η5-cp)2(TAM-OH)] and [Fe(η5-cp)2(TAM-OH R)] and their corresponding quinone methides [Fe(η5-cp)2(QM)] and [Fe(η5-cp)2(QM R)] (where, TAM-OH = the hydroxyl derivative of tamoxifen, QM = quinone methides, and R = O(CH2)3N(CH3)2)) (Fig. 43). It was observed that the quinone methides, [Fe(η5-cp)2(QM)] and [Fe(η5-cp)2(QMR)] (IC50 ≈ 2.5 μM), were significantly more cytotoxic to TrRx in vitro than the hydroxyferrocifens, [Fe(η5-cp)2(TAM-OH)] and [Fe(η5-cp)2(TAM-OH R)] (IC50 ≈ 15 μM). The [Fe(η5-cp)2(TAM-OHR)] and [Fe(η5-cp)2(QM R)] complexes inhibited TrRxs almost to the same extent; thereby, causing the accumulation of oxidized forms of cytosolic and mitochondrial thioredoxin in Jurkat cancer cells. However, [Fe(η5-cp)2(TAM-OH)] and [Fe(η5-cp)2(QM)] were almost inactive on Jurkat cancer cells. This differential behaviour of ferrocenyl derivatives was mainly attributed to the competitive transformation of [Fe(η5-cp)2(QM)] into an inactive indene in protic medium. This study for the first time substantiated the unique role of ferrocenyl quinone methides in the field of anticancer activity.
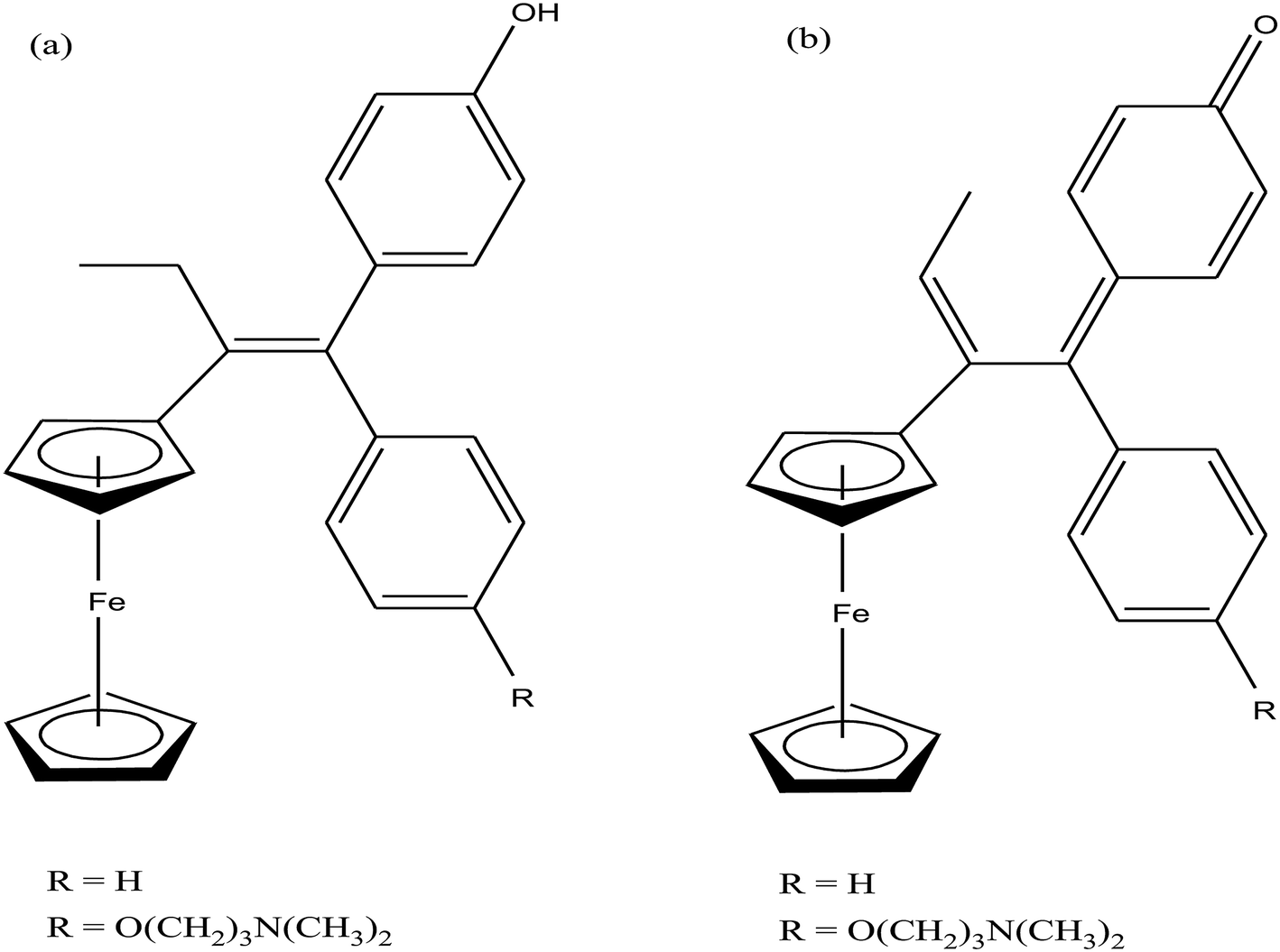 | ||
| Fig. 43 Chemical structure of (a) hydroxyferrocifens [Fe(η5-cp)2(TAM-OH)], [Fe(η5-cp)2(TAM-OH·R)] and their (b) ferrocenyl quinone methides [Fe(η5-cp)2(QM)] and [Fe(η5-cp)2(QM·R)]. | ||
The organic drug tamoxifen was found to be highly effective against breast cancer cells due to its anti-proliferative action on estrogen receptor positive (ER+) cancers. However, the use of tamoxifen is limited since it does not show any effect on tumors that do not express the estrogen receptor (ER−).215,216 Also, the long-term use of tamoxifen increases the risk of uterine and endometrial cancer because tamoxifen and its analogs are further metabolized to catechol 3,4-di hydroxytamoxifen.217 Further, the combination of the ferrocenyl group with catechol via a conjugated system increased its in vitro anti-proliferative activity on hormone-independent (MDA-MB-231) breast cancer cells, as reported by G. Jaouen et al.218 They synthesized a series of complexes with the general formula [Fe(η5-cp)2(CTAM)] and [Fe(η5-cp)2-μ-C3H5(CTAM)] (Fig. 44) by tethering organometallic biovectors (where, CTAM = catechol 3,4-di hydroxytamoxifen). The catechol complexes discussed here exhibited significant anti-proliferative potency (IC50 = 0.48–1.21 μM) in comparison to their corresponding phenolic analogues (0.57–12.7 μM). However, among the catechol complexes, [Fe(η5-cp)2-μ-C3H5(CTAM)], with the [3]ferrocenophane motif, displayed the highest anti-proliferative activity. Finally, these results supported the hypothesis that the ferrocenyl group also plays an important role in the oxidation of catechol derivatives. This molecule can be transformed into a quinone methide or orthoquinone, which is responsible for the anti-proliferative activity of these complexes.
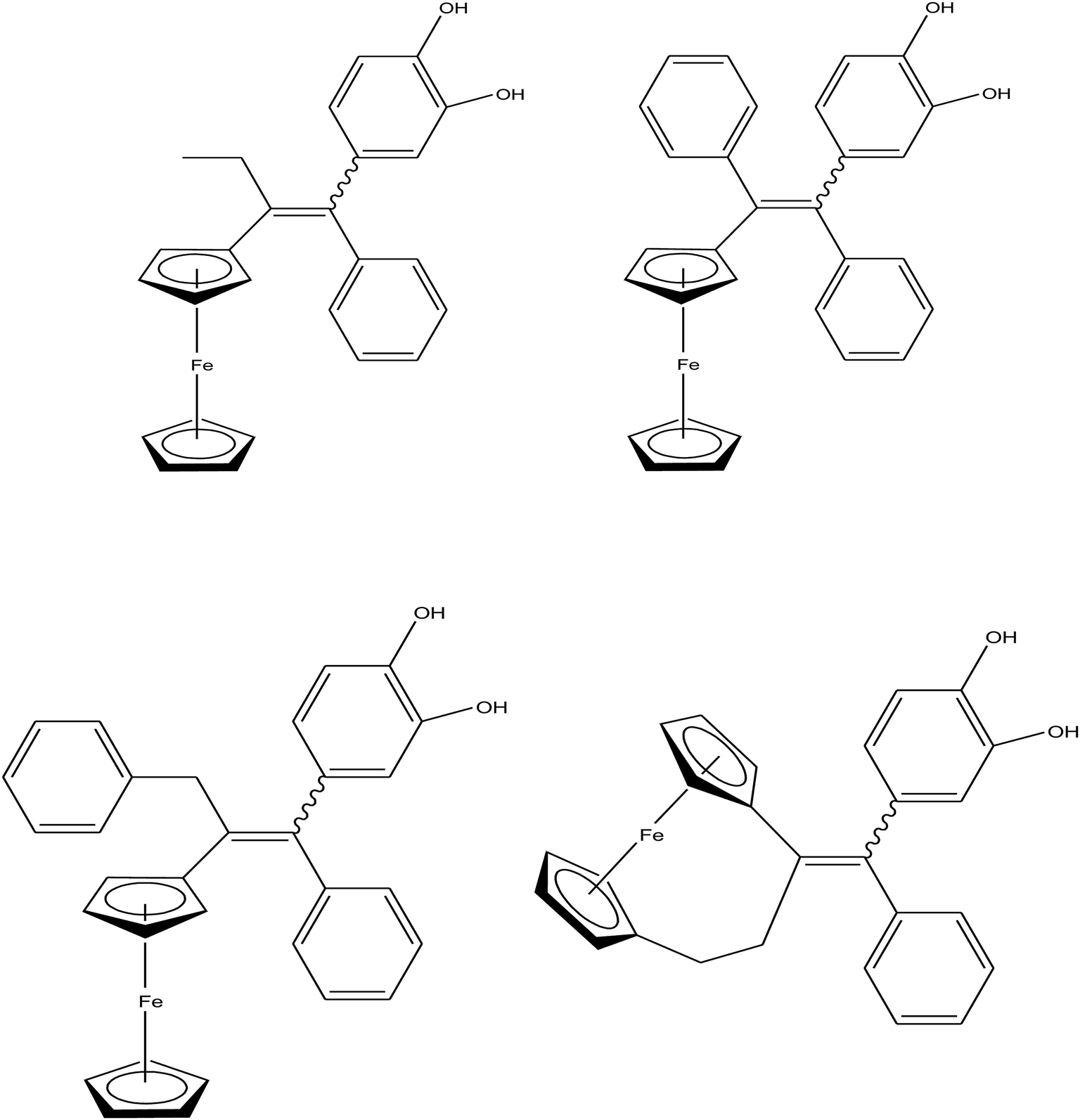 | ||
| Fig. 44 Chemical structure of ferrocenyl tamoxifen derivatives [Fe(η5-cp)2(CTAM)] and [Fe(η5-cp)2-μ-C3H5(CTAM)]. | ||
The attachment of a covalently-grafted organometallic unit to a normal metal complex prevents ligand substitution, but it potentiates the activity of biomolecules either by modification of their pharmacokinetic profile or by acting as structural mimics.219 Therefore, G. Jaouen et al.220 synthesized and compared the antitumor activity of a series of ferrocene, [Fe(η5-cp)2(DPME)], and [3]ferrocenophane, [Fe(η5-cp)2-μ-C3H5(DPME)] {where, DPME = 1-(diphenylmethylidenyl)ethyl substituted derivatives}, tetrasubstituted olefin derivatives (Fig. 45). All the complexes were screened for their activity against the MDA-MB-231 human breast cancer cell line, which is a hormone-independent line. The results revealed that the [3]ferrocenophane complexes, [Fe(η5-cp)2-μ-C3H5(DPME), were comparatively more active against the MDA-MB-231 cell line than the corresponding ferrocenyl complexes. This is ascribed to the smaller HOMO–LUMO gap present in the oxidized form of the [3]ferrocenophane compounds. Moreover, the complexes that could form quinone or imine methides were the most active among the [3]ferrocenophane and ferrocene series. Furthermore, the complexes from the [3]ferrocenophanyl and ferrocenyl series possessing two phenol groups were screened on the NCI/DTP 60-cell line panel. The results showed that the [Fe(η5-cp)2-μ-C3H5(DPME-OH)] complex with the [3]ferrocenophanyl motif exhibited greater cell line selectivity compared to the ferrocenyl analog on leukemia, CNS cancer, and renal cancer cells. The low systemic toxicity and lack of interaction with DNA suggested their different modes of action.
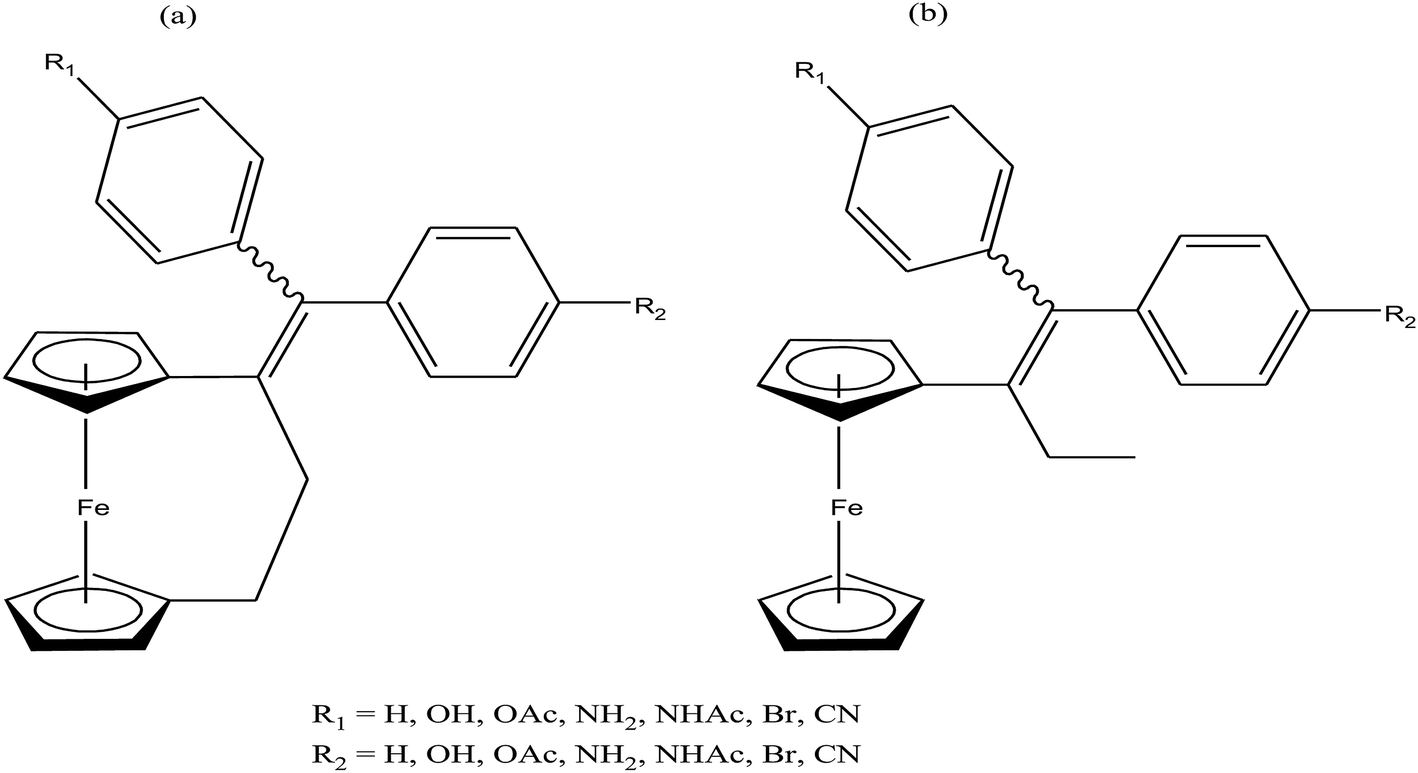 | ||
| Fig. 45 Chemical structure of ferrocene (a) [Fe(η5-cp)2(DPME)] and (b) [3]ferrocenophane [Fe(η5-cp)2-μ-C3H5(DPME)] and tetrasubstituted olefin derivatives. | ||
Furthermore, C. Passirani and G. Jaouen et al.221 tested the [3]ferrocenophanyl complex with two phenol groups, [Fe(η5-cp)2-μ-C3H5(DPME-OH)] (Fig. 46), for brain tumor therapy by encapsulating it in 40 nm-sized stealth lipid nanocapsules (LNCs). This complex displayed a potent effect on 9L glioma cells (IC50 = 0.1 μM), which is related with oxidative stress and dose-dependent alteration of the cell cycle. Also inhibition in tumor growth was observed in rats bearing ectopic glioma upon repeated intravenous administration of the stealth [Fe(η5-cp)2-μ-C3H5(DPME-OH)] LNCs. In addition, there was no liver damage in the treated rats, indicating that the complex-loaded stealth [Fe(η5-cp)2-μ-C3H5(DPME-OH)] LNCs may represent as an alternative approach to treat cancer and may comply with the current needs in oncology.
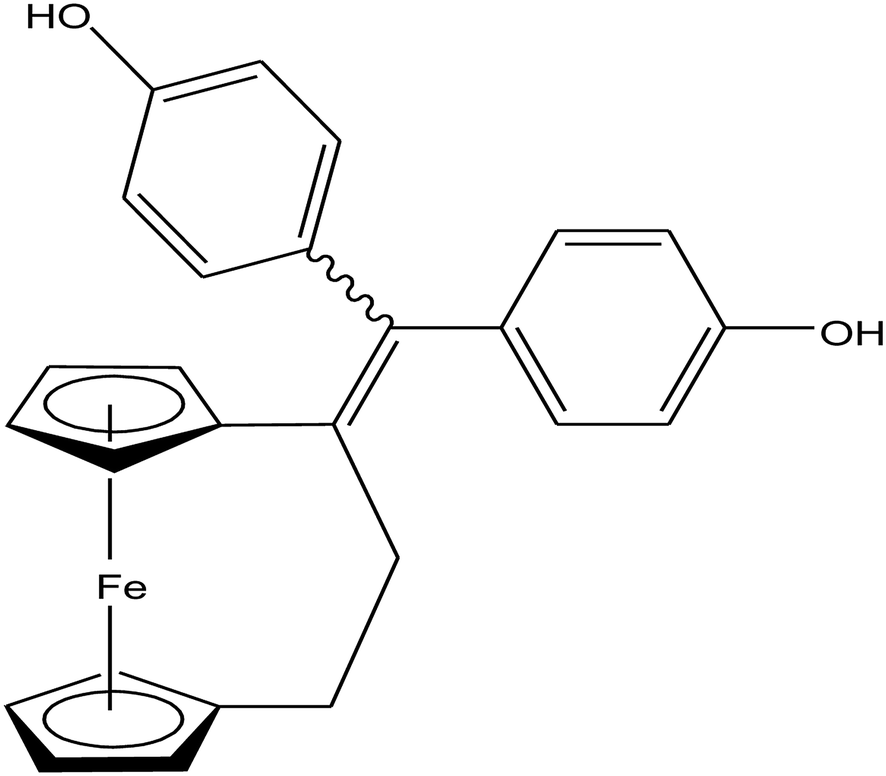 | ||
| Fig. 46 Chemical structure of ansa-FcdiOH molecule [Fe(η5-cp)2-μ-C3H5(DPME-OH)]. | ||
However, the incorporation of the ferrocenyl moiety does not always increase the cytotoxic activity of chemotherapeutic drug molecules. To further improve the cytotoxic activity of organometallic biovectors, cytotoxic moieties need to be introduced, such the nonsteroidal antiandrogen nilutamide bearing an organometallic ferrocenyl substituent. Thus, G. Jaouen et al.206,222 reported the synthesis and antitumor activity of [Fe(η5-cp)2(NL)] and [Fe(η5-cp)2(TAM-OH·R)] (where, NL = substituted nilutamide TAM-OH = hydroxyl derivative of tamoxifen and R = O(CH2)3N(CH3)2)) complexes (Fig. 47). The C31H22F6FeN4O2 complex displayed promising activity on prostate cancer (PC3) cells with a low IC50 value of 5.4 μM. This complex mainly acted through the recognition of cannabinoid receptors since a higher level of these receptors is found in prostate cancer cells compared to healthy organs. Another complex, C33H41FeNO2, exhibited strong cell growth inhibition in three melanoma cells, WM9, WM35 and WM793, compared to normal melanocytes. Mechanistic studies suggested that there was no production of ROS species as found for ferrocenyl derivatives in breast cancer cells. Therefore, the results clearly indicated that the complex was actively metabolized into quinone methides by rat liver microsomes or recombinant human cytochromes. Chemotherapy-induced alopecia (hair loss) is a common side effect attributable to the high proliferative rate of follicular matrix cells, which makes hair follicles highly sensitive. Thus, the potential toxicity of the complex C33H41FeNO2 was explored on hair follicle growth, and the results predicted hair loss as a potential side effect of treatment.
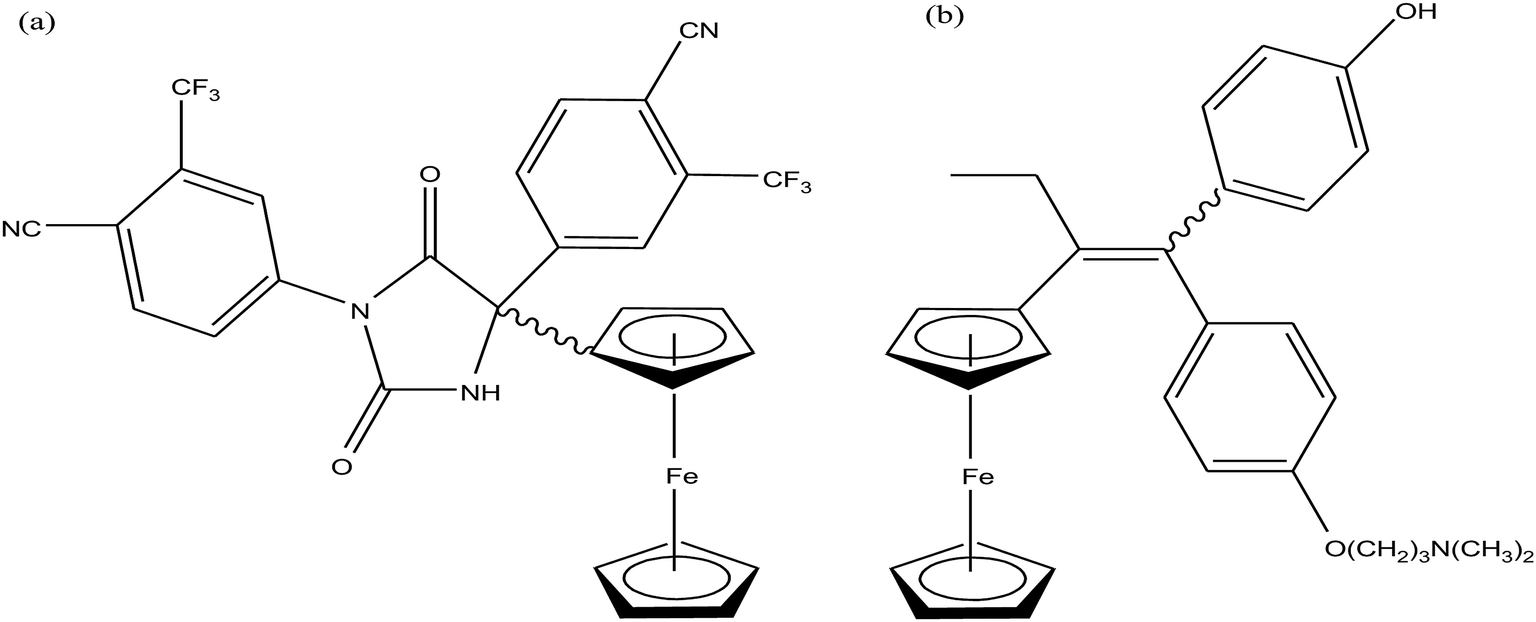 | ||
| Fig. 47 Chemical structure of (a) [Fe(η5-cp)2(NL)] and (b) [Fe(η5-cp)2(TAM-OH·R)]. | ||
Another approach to investigate the effect of the organometallic ferrocene unit in biological systems is to incorporate it as building blocks into peptide chains. Thus, H. G. Schmalz et al.223,224 reported the synthesis of HUNI 068 as a class of ferrocene-derived compounds with clinically promising activity (Fig. 48). This complex, [Fe(η5-cp)2(Fmoc-AA)] (HUNI 068) (where, Fmoc-AA = Fmoc-protected amino acid building blocks), induced strong apoptosis, DNA fragmentation and phosphatidylserine exposure without significantly affecting membrane integrity. It also overcame multiple drug resistance (MDR), which is a major problem in the treatment of relapsed malignant diseases. Generally, tumor cells develop resistance via the overexpression of p-glycoprotein and Bcl-2, which are common causes of MDR. Therefore, to improve the current therapies, more selective drugs with the ability to overcome mechanisms of MDR are urgently required. Besides, this complex was active against Burkitt-like lymphoma (BJAB) and human B cell precursor leukemia (Nalm-6 cells) cells with IC50 values in the range of 30–40 μM. It induced apoptosis via the intrinsic mitochondrial pathway probably by acting independently of the Bcl-2 protein. Experiments on primary lymphoblasts also showed that the apoptotic activity of HUNI 068 was higher than that of other drugs, which was tested in patients with childhood leukemia. Thus, it can be concluded that HUNI 068 is quite useful in the therapy of childhood leukemia when conventional compounds fail.
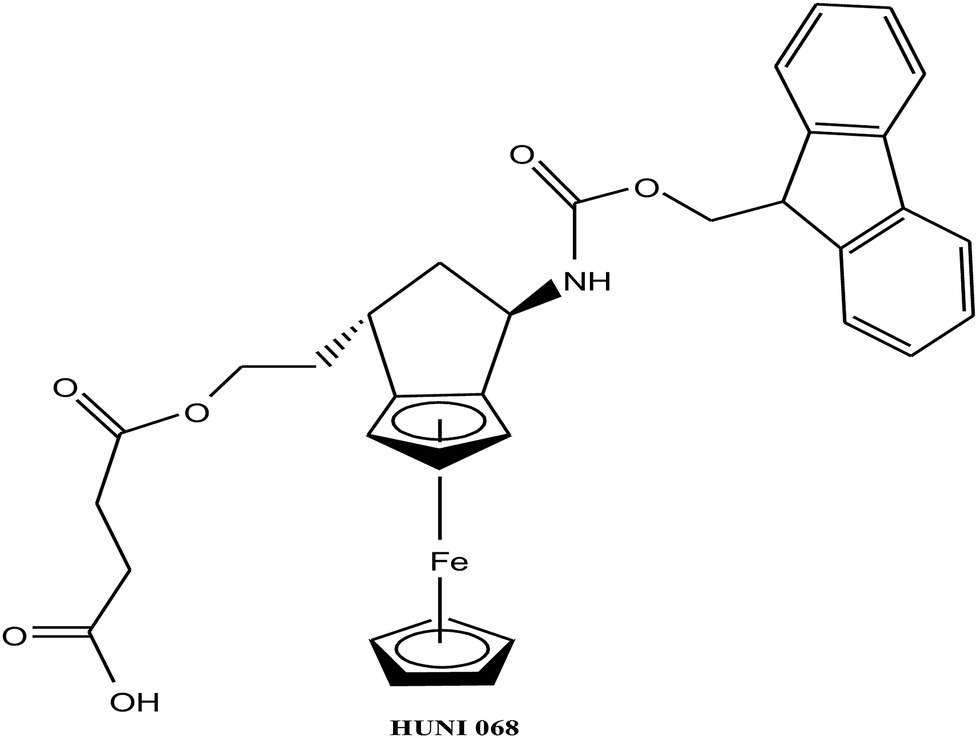 | ||
| Fig. 48 Chemical structure of the ferrocene-derived HUNI 068 [Fe(η5-cp)2(Fmoc-AA)]. | ||
To further explore the biological potential of metal-containing nucleosides, the stable organometallic ferrocene moiety was incorporated into bioactive nucleoside analogs. In this endeavor, H.-G. Schmalz et al.225 reported the synthesis of a series of novel carbocyclic nucleoside analogues with a ferrocenocyclopentene backbone (Fig. 49). The cytotoxic activity of these new ferrocenyl nucleoside complexes, [Fe(η5-cp)2(Nuc-tBu)] and [Fe(η5-cp)2(Nuc-DTS)] (where, Nuc-tBu = carbocyclic nucleoside derivative with tertiary butyl group and Nuc-DTS = carbocyclic nucleoside derivative with dimethylthexylsilyl group), were investigated in vitro and ex vivo using BJAB tumor cells (Burkitt-like lymphoma cells) and primary lymphoblasts from children with acute lymphoblastic leukemia (ALL). Both complexes exhibited apoptosis-inducing activity at low concentrations (LD50 = 10–20 μM). Moreover, the cells treated with both complexes displayed characteristic apoptosis behavior, such as changes in cell shape, cytoplasm condensation, nuclear envelope changes, and blebbing. Surprisingly, the [Fe(η5-cp)2(Nuc-tBu)] complex was able to overcome resistance in primary lymphoblasts isolated from children ex vivo with a relapse. Therefore, it can be concluded that ferrocenyl nucleoside complexes are suitable for the development of organometallic antitumor agents with pronounced apoptosis-inducing activity, which is related to their structure-activity dependency. To determine the mechanism of action of these complexes and also to explore the specific role of the metal fragment for biological activity further studies are required.
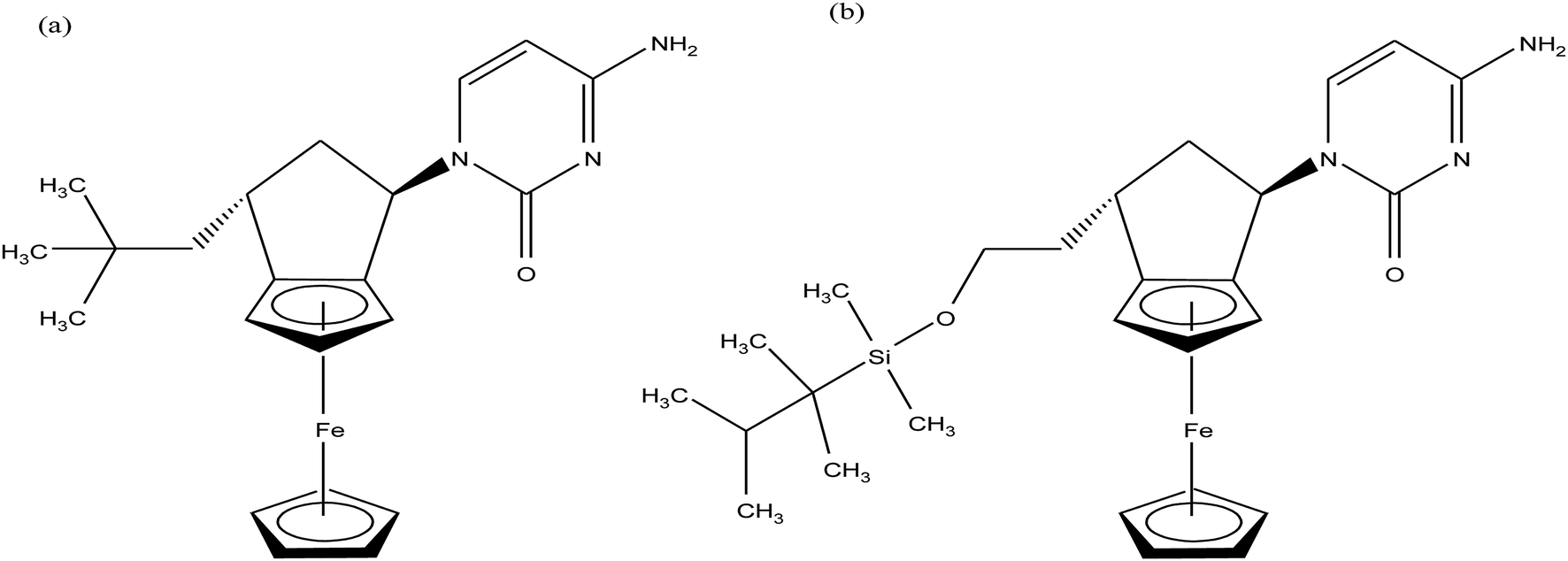 | ||
| Fig. 49 Chemical structure of the carbocyclic nucleoside analogues (a) [Fe(η5-cp)2(Nuc-tBu)] and (b) [Fe(η5-cp)2(Nuc-DTS)]. | ||
After obtaining encouraging results with organometallic half-sandwich ruthenium complexes and their ability to overcome the limitations of cisplatin, enormous effort has been directed towards the development of half-sandwich iron complexes since they displayed enhanced antitumor activities. Accordingly, M. H. Garcia et al.226 synthesized four new half-sandwich iron(II) complexes of the type [FeCp(dppe)L][CF3SO3] (Fig. 50) containing derivatives of imidazoles substituted ligands, and ethylenebis(diphenylphosphane) (dppe) for the first time. All four complexes, [Fe(η5-cp)(dppe)(1-BuIm)]CF3SO3, [Fe(η5-cp)(dppe)(1-BI)]CF3SO3, [Fe(η5-cp)(dppe)(ImH)]CF3SO3 and [Fe(η5-cp)(dppe)(1-HmIm)]CF3SO3 (where, 1-BuIm = 1-butylimidazole, 1-BI = benzyl imidazole, ImH = imidazole and 1-HmIm = N-hydroxymethylimidazole), displayed promising cytotoxic activity against the ovarian A2780 (cisplatin sensitive), breast MCF7 and cervical HeLa human carcinoma (ATCC) cell lines with much lower IC50 values than that for cisplatin, particularly for MCF7 cells. Furthermore, the highest cytotoxicity for A2780 cells was found for the [Fe(η5-cp)(dppe)(1-BuIm)]CF3SO3 complex; whereas, for MCF7 and HeLa cells, the [Fe(η5-cp)(dppe)(1-BuIm)]CF3SO3 and [Fe(η5-cp)(dppe)(1-BI)]CF3SO3 complexes showed equal values of cytotoxicity. Thus, it was concluded that the [Fe(η5-cp)(dppe)(1-BuIm)]CF3SO3 and [Fe(η5-cp)(dppe)(1-BI)]CF3SO3 complexes were 3.5 to 7 times more cytotoxic than the well-known drug cisplatin. Therefore, these complexes are as promising anticancer drugs and further warrant in vivo studies.
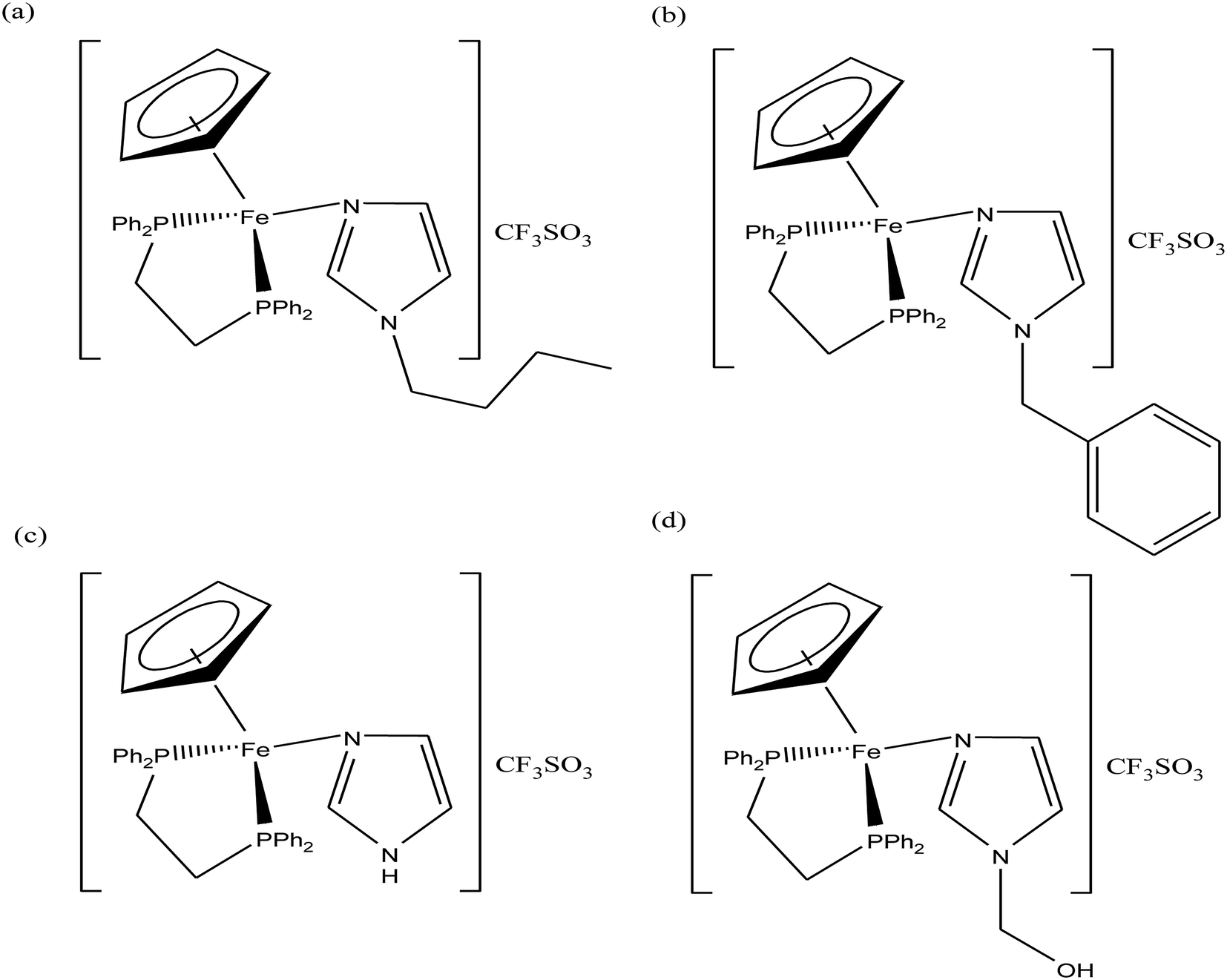 | ||
| Fig. 50 Chemical structure of the (a) [Fe(η5-cp)(dppe)(1-BuIm)]CF3SO3, (b) [Fe(η5-cp)(dppe)(1-BI)]CF3SO3, (c) [Fe(η5-cp)(dppe)(ImH)]CF3SO3 and (d) [Fe(η5-cp)(dppe)(1-HmIm)]CF3SO3 complexes. | ||
Since tumors are known to develop vasculature and interact with the stroma, in vivo activity investigations allow the evaluation of actual toxicity and pharmacokinetic activity of drugs. The development of drugs for the treatment of cancer requires studies on preclinical models such as mice, wherein important parameters of effectiveness such as an increase in lifetime and tumor growth delay in tumor-bearing mice are monitored according to standard protocols. Unfortunately, in vivo studies of only a few iron complexes have been carried out in detail. A. A Simenel et al.227 synthesized a ferrocenylmethyl thymine complex [Fe(η5-cp)2(THY)] (where, THY = thymine) (Fig. 51) and demonstrated its in vivo antitumor activity on solid tumor models, such as carcinoma 755 (Ca755), melanoma B16 (B16) and Lewis lung carcinoma (LLC). This complex showed a strong antitumor effect at small doses against carcinoma 755 cells with the maximum level of tumor growth inhibition of 70% compared to the control. Moreover, the [Fe(η5-cp)2(THY)] complex displayed a synergistic therapeutic effect with the well-known antitumor drug cyclophosphamide against carcinoma 755 cells, which suggests ferrocene compounds should be investigated as potential organometallic drugs for antitumor polychemotherapy.
 | ||
| Fig. 51 Chemical structure of N-(ferrocenylmethyl)thymine [Fe(η5-cp)2(THY)]. | ||
The organometallic ferrocene moiety has been incorporated in many drug molecules due to its small size, lipophilicity, ease of chemical modification, redox potential and promising bioactivities. Using this strategy, H. Yan and X. Wang et al.228 reported the synthesis and in vitro antineoplastic activities of ferrocene-substituted dithio-o-carborane conjugates [Fe(η5-cp)2(SB1)], [Fe(η5-cp)2(SB2)] and [Fe(η5-cp)2(SBCO)] where, SB1, SB2 and SBCO are dithio-o-carborane units (Fig. 52). The results of electrochemical and MTT experiments revealed that the highly hydrophobic ferrocenyl and carboranyl groups allow the conjugates to rapidly enter human hepatocellular carcinoma (SMMC-7721) and human hepatocellular carcinoma (Hep G2) cancer cells and exert acute cytotoxicity after 4 h incubation in serum-free media. All the complexes exhibited high inhibition activity on the proliferation of SMMC-7721 and Hep G2 cancer cells via the G0/G1 arrest mechanism. Specifically, [Fe(η5-cp)2(SB1)] and [Fe(η5-cp)2(SB2)] displayed a potent inhibition effect on the proliferation of SMMC-7721 and Hep G2 cancer cells, but showed almost no effect on the normal cell line, the human embryonic lung fibroblast (HELF) cells. The observed order of anticancer activity was found to be [Fe(η5-cp)2(SB2)] > [Fe(η5-cp)2(SB1)] > [Fe(η5-cp)2(SBCO)], which was consistent with the order of redox potential in the ferrocenyl moiety of the conjugates. All these studies demonstrated the anticancer structure-reactivity relationship of carborane-ferrocene conjugates, which may become prospective candidates for the development of novel anticancer drugs.
 | ||
| Fig. 52 Chemical structures of the ferrocene-carborane conjugates (a) [Fe(η5-cp)2(SB1)], (b) [Fe(η5-cp)2(SB2)] and (c) [Fe(η5-cp)2(SBCO)]. | ||
Organometallic osmium complexes with DNA and mitochondria as the target
A number of potential organometallic osmium compounds have been discovered over the past few years as chemotherapeutic agents. They provide a new platform for the development of antitumor agents together with specific recognition towards the molecular target DNA. For decades, DNA has been a well-known target because inside cancer cells there is uncontrolled division of DNA, and if the further replication of DNA is stopped, then we stop the further growth of tumor cells. Similarly, mitochondria are considered as recent targets for antitumor drugs due to their high production of ROS, where if the proper functioning of mitochondria is altered, then due to very high production of ROS, cancer cells will undergo mitochondria-induced apoptosis or cell death.229Osmium-arene complexes were first developed by Paul J. Dyson, Peter J. Sadler and Bernhard K. Keppler as analogs of Ru(II)/Ru(III) drugs, which included Os-RAPTA-C, Os-RM175 and Os-NAMI-A (Fig. 53).230–233 It has been found that in organometallic ruthenium-PTA complexes, the loss of aromatic arene takes place in physiological conditions; whereas, in organometallic osmium complexes, the loss of arene is not observed. Os-RAPTA-C binds to the oligonucleotide molecule with the loss of a chloride ligand, thereby providing more stability to the osmium–arene bond. However, unfortunately, Os-RM175 complexes do not show promising cytotoxicity towards A2780 ovarian cancer cells due to the formation of inactive hydroxo-adduct at physiological pH. Osmium–arene complexes are relatively inert, but the kinetics of ligand substitution reactions of Os-arene complexes in aqueous solution can be controlled by varying the chelating ligand. On the other hand, amazingly Os-NAMI-A showed reasonable in vitro antiproliferative activity against the HT-29 (colon carcinoma) and SK-BR-3 (mammary carcinoma) cell lines. It was observed that the Os-NAMI-A complexes were markedly more inert than their related ruthenium compounds towards ligand substitution reactions, such as hydrolysis and interaction with DNA bases. Due to their inert behavior, they partially show higher antiproliferative activity than their related ruthenium analogs.
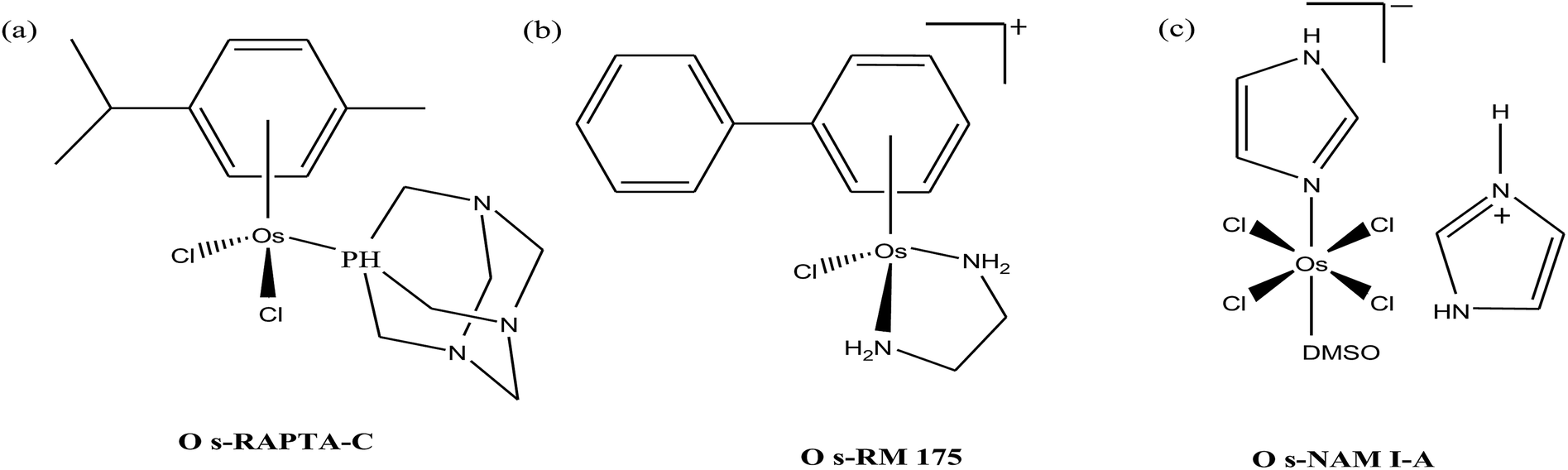 | ||
| Fig. 53 Chemical structures of the osmium complexes (a) Os-RAPTA-C, (b) Os-RM175 and (c) Os-NAMI-A. | ||
This unpredictable activity and inertness of osmium-arene complexes provide a new platform for the design of novel antitumor agents using the osmium-arene framework and changing the other ligands. Therefore, Paul J. Dyson and B. K. Keppler et al.234 further explored the antitumor properties of organometallic osmium complexes of the type [Os(η6-p-cymene)(BIG-R)Cl2] (where, BIG = 3,5,6-bicyclophosphite-1,2-o-isopropylidene-α-D-glucofuranoside and R = C(CH3)2 and C6H10) by choosing carbohydrate-derived phosphorus-containing ligands as secondary ligands (Fig. 54). It was observed that the [Os(η6-p-cymene)(BIG-C6H10)Cl2] complex exhibited slightly higher IC50 values than its Ru analogs on the human ovarian cancer (CH1) cell line. This complex also lacked reactivity towards 5′-GMP and exhibited a slow hydrolysis rate, which may be one of the reasons for its low cytotoxicity. Thus, this suggests that the further design of organometallic osmium complexes is warranted to improve the chemotherapeutic potential.
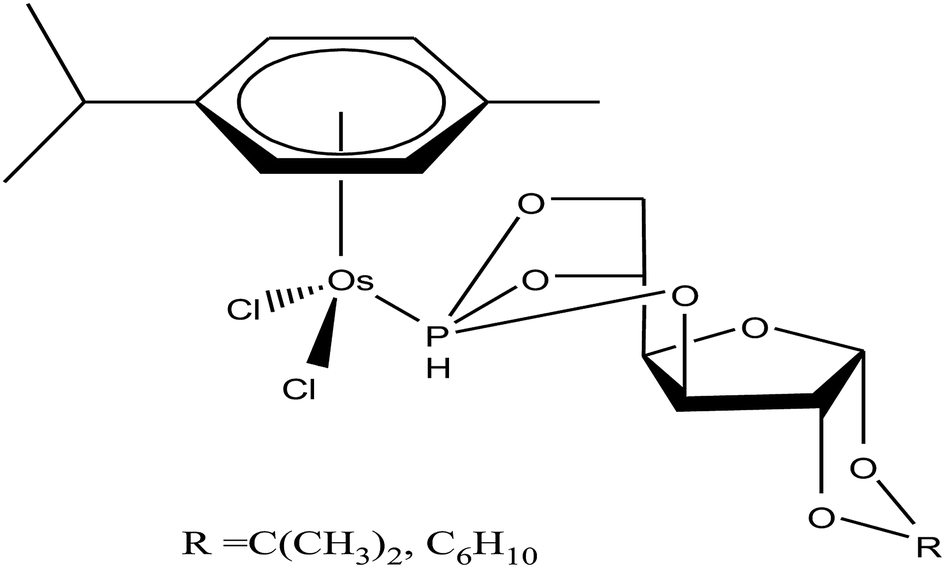 | ||
| Fig. 54 Chemical structures of the osmium-arene complexes [Os(η6-p-cymene)(BIG-R)Cl2] containing P-derived sugar ligands. | ||
Organometallic complexes offer many advantages over normal coordination complexes; however, to date, they have not been successful in the development of clinically approved antitumor drugs. This is mainly due to their relative inertness, and the lack of understanding of the aqueous chemistry of organometallic complexes is found to be the major obstacle in their further development. Accordingly, P. C. McGowan and P. J. Sadler et al.235,236 controlled the aqueous chemistry by designing osmium-arene N, O- and N,N-coordinated [Os(η6-bip)(pico)Cl] and [Os(η6-p-cym)(NPP)Cl] complexes (where, pico = picolinic acid and NPP = N-4-nitro-Ph-picolinamide), as depicted in Fig. 55. They observed that the N, O–coordinated complex, [Os(η6-bip)(pico)Cl], hydrolyzed much more slowly than the N,N-coordinated [Os(η6-p-cym)(NPP)Cl] complex. Also, the [Os(η6-bip)(pico)Cl] complex displayed no reactivity towards guanine or adenine nucleobases, but showed prominent activity towards the A2780 cancer cell line (IC50 = 4.2 μM), which suggests its target of action may not be DNA. However, the [Os(η6-p-cym)(NPP)Cl] complex exhibited a better rate of hydrolysis and strong DNA binding affinity with selectivity towards guanine base. As a result, [Os(η6-p-cym)(NPP)Cl] displayed very promising activity in the HCT116(colon) and A2780cis (ovarian cisplatin resistant) cell lines.
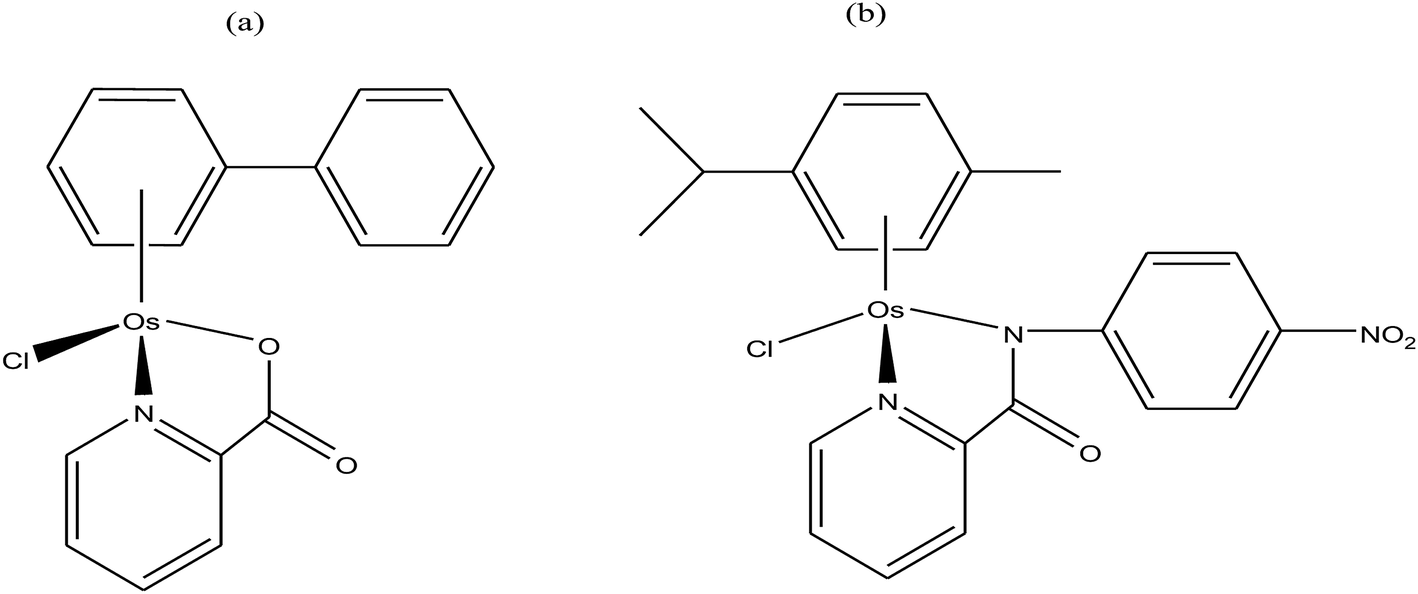 | ||
| Fig. 55 Chemical structures of the osmium-arene complexes (a) [Os(η6-bip)(pico)Cl] and (b) [Os(η6-p-cym)(NPP)Cl]. | ||
Although organometallic osmium complexes are kinetically more inert than their analogous ruthenium complexes, the aqueous chemistry of osmium-arene complexes can be controlled/increased by changing the chelating ligands. To further improve the hydrolysis rate, Peter J. Sadler et al.237 changed the N, O- and N,N-chelating ligands to an anionic O,O-chelating ligand in the [M(η6-p-cym)(mal)Cl] complex (where, mal = maltolato), as shown in Fig. 56. In can be seen that the reactivity of the Os–Cl bond drastically increased compared to the complexes with N,N-chelating ligands. They hydrolysed and bound to guanine nucleobases rapidly compared to their ruthenium analogs. However, they were still deactivated to a significant extent at particular concentrations due to the formation of inert hydroxo-bridged dimers. Similarly, B. K. Keppler and C. G. Hartinger et al.238 used another O,O-chelating ligand in the same osmium-arene framework to synthesize a new complex, [Os(η6-p-cymene)(NEHP)Cl] (where, NEHP = N-[(ethoxycarbonyl)methyl]-3-hydroxy-2-(1H)-pyridone), as shown in Fig. 56. The hydrolysis rate of this complex was also found to be high together with the quick binding with the proteins biquitin and cytochrome c, forming adducts of type [protein + Os(η6-p-cymene)]. However, due to their binding with the proteins, the pyridone ligands were cleaved from the [Os(η6-p-cymene)(NEHP)Cl] complex, but the cleavage of these ligands was found to be less for the osmium complex compared to its ruthenium analogs. Further, the [Os(η6-p-cymene)(NEHP)Cl] complex showed good in vitro activity due to its strong binding with the N7 atom of the guanine nucleobase. These findings give insights into the aqueous solution chemistry of metal-arene complexes under biologically relevant conditions and further aid in the rational design of organometallic chemotherapeutic agents.
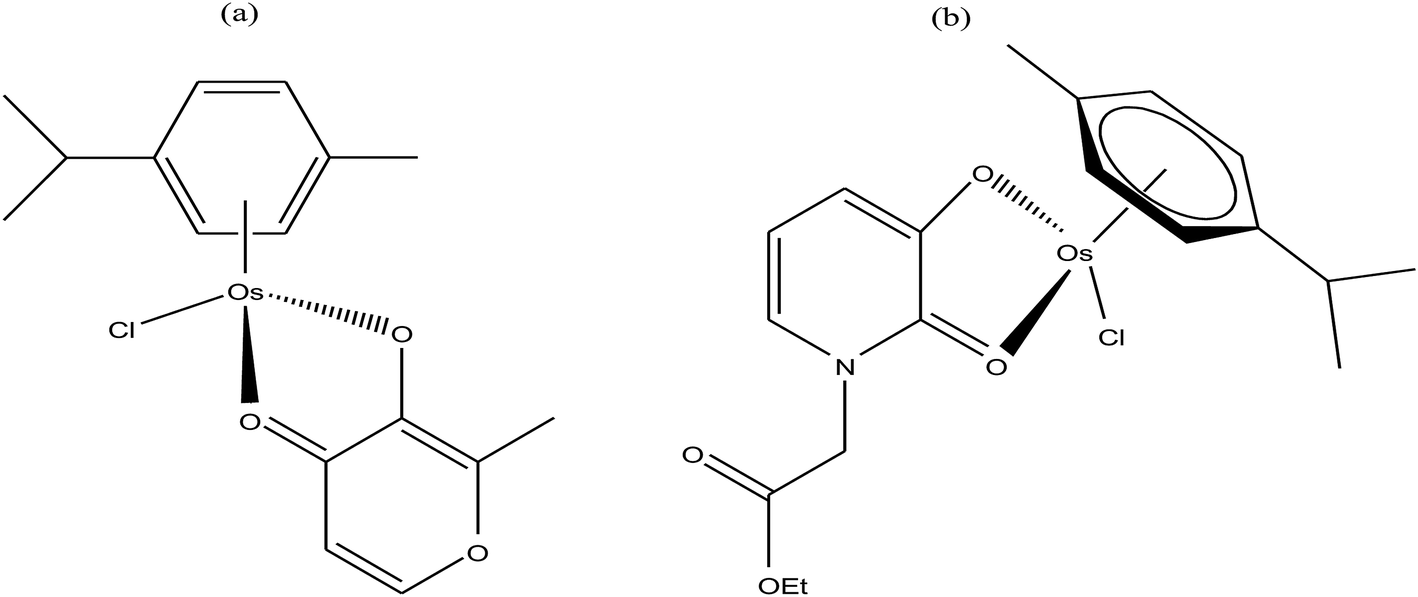 | ||
| Fig. 56 Chemical structure of the osmium-arene complexes (a) [M(η6-p-cym)(mal)Cl] and (b) [Os(η6-p-cymene)(NEHP)Cl] containing an anionic O,O-chelating ligand. | ||
Further, to enhance the antitumor profile and structure-reactivity relationship of osmium arene complexes, W. Kandioller and B. K. Keppler et al.239 synthesized cyclometalated osmium(II) complexes of the type [Ru(η6-p-cym)(κ2-C^N-R)Cl] (where, R = p-OMe Bn and (CH2)2CH3), as indicated in Fig. 57. They observed that the osmium(II) cyclometalates with C, N-coordination exhibited higher antiproliferative activities than their ruthenium(II) counterparts in three human cancer cell lines (A549, SW480, and CH1/PA-1). However, the antiproliferative activity of the complex with the methoxy butyl R group [Ru(η6-p-cym)(κ2-C^N-p-OMe Bn)Cl] was found to be extraordinarily higher compared to that with propyl R group, [Ru(η6-p-cym)(κ2-C^N-(CH2)2CH3))Cl], which further shed light on their structure-reactivity relationship. Specifically, the cytotoxicity increases with an increase in the lipophilicity of these complexes, leading to their higher accumulation inside cancer cells. Investigations on their mode of action revealed that the prepared organometallics were unable to inhibit topoisomerase Iiα, which further warrants investigation.
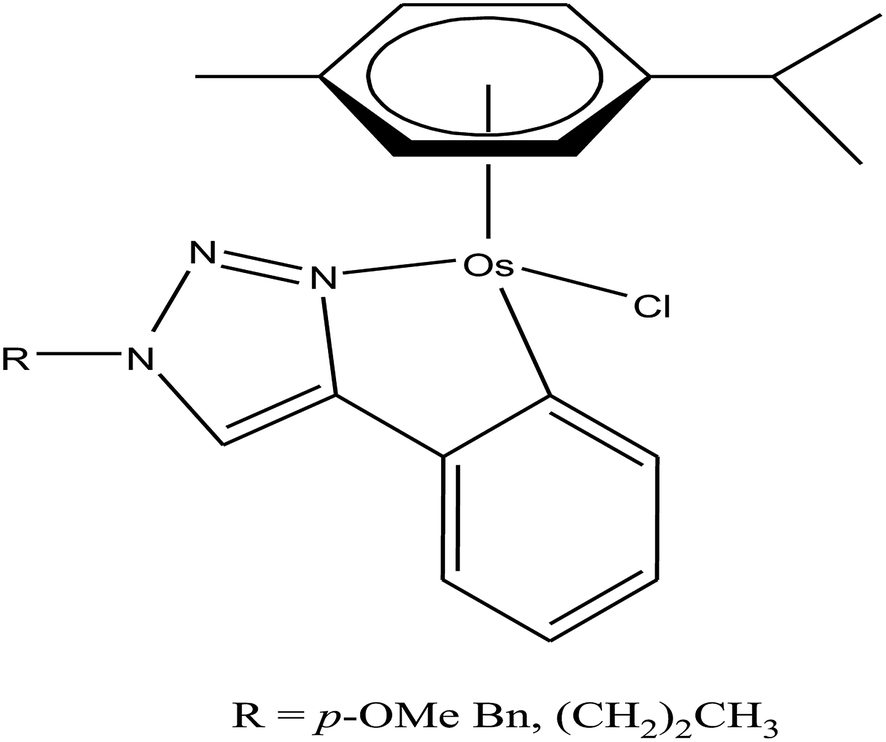 | ||
| Fig. 57 Chemical structure of the cyclometalated osmium-arene complexes [Ru(η6-p-cym)(κ2-C^N-R)Cl]. | ||
Further, the coordination was changed to S,N- in osmium-arene complexes by B. K. Keppler and C. G. Hartinger et al.240 They synthesized a series of osmium-arene complexes of the type [Os(η6-p-cym)(PCA)Cl]Cl (where, PCA = N-substituted 2-pyridinecarbothioamides) (Fig. 58). To explore the possible reasons for the structure-reactivity relationships with the mode of action as protein histidine, the lipophilicity of these compounds was determined. They observed that the highly lipophilic compound with a fluorine R group showed the most potent cytotoxic activities in human ovarian cancer (CH1), colon carcinoma (SW480) and non-small cell lung cancer (A549) cell lines. The other complexes with morpholinyl and benzoyl R groups were expected to show higher lipophilicity; however, they found to be sterically demanding and a exhibited lower antiproliferative effect. Thus, it can be concluded that the most lipophilic and smallest congeners are the most effective with IC50 values in the low micromolar range. Further these complexes form adducts with histidine protein located on the nucleosome surface, suggesting interference with chromatin activity as the possible mode of action of these compounds. The osmium–arene complex with a fluorine R group displayed slow ligand exchange kinetics, high in vitro cytotoxicity and high stability in HCl, which make it an excellent candidate as the first orally active osmium organometallic.
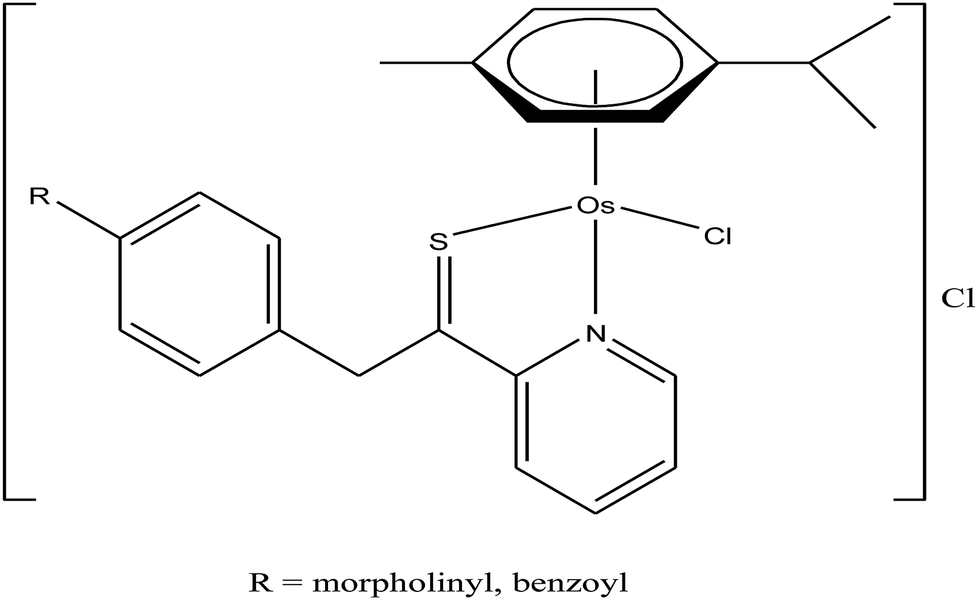 | ||
| Fig. 58 Chemical structure of the S, N-coordinated osmium-arene complexes of the type [Os(η6-p-cym)(PCA)Cl]Cl. | ||
The coordination was further changed to N,N- by Peter J. Sadler et al.241 to obtain the complexes [Os(η6-p-cym)(Impy-NMe2)Cl]PF6 and [Os(η6-p-cym)(Impy-OH)Cl]PF6 (where, Impy = N,N′-chelated phenyliminopyridine) (Fig. 59). Both complexes showed outstanding antitumor activity against A2780 human ovarian and A549 human lung cancer cell lines. However, the [Os(η6-p-cym)(Impy-NMe2)Cl]PF6 complex (IC50 = 6.7 μM) exhibited higher activity than the classical drug cisplatin (IC50 = 8.7 μM) on A549 human lung cancer cells. Further, [Os(η6-p-cym)(Impy-NMe2)Cl]PF6 strongly interacted with guanine base of DNA and induced the formation of ROS in A549 lung cancer cells, which account for its higher antitumor activity.
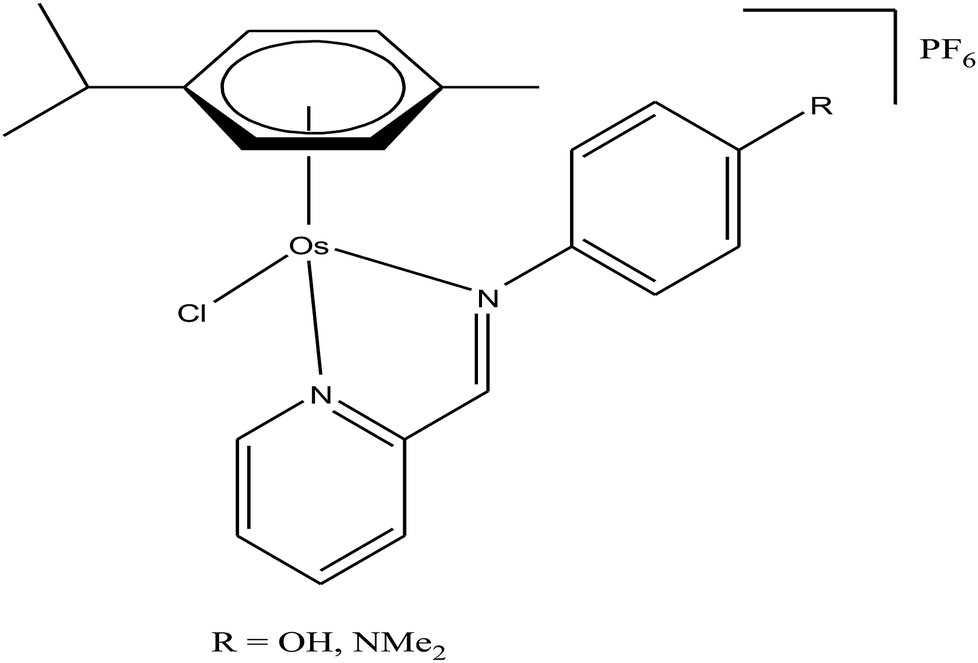 | ||
| Fig. 59 Chemical structure of the N,N-coordinated osmium-arene complexes of the type [Os(η6-p-cym)(Impy-R)Cl]PF6. | ||
In continuation of the work by P. J. Sadler et al., B. K. Keppler et al.242 also designed new N,N-coordinated osmium-arene complexes bearing 3-(1H-benzimidazol-2-yl)-1H-quinoxalin-2-one with the general formula [Os(η6-p-cym)(BOQ)Cl]Cl and [Os(η6-p-cym)(BTQ)Cl]Cl (where, BOQ = 3-(benzoxazol-2-yl)-1H-quinoxalin-2-one and BTQ = 3-(benzothiazole-2-yl)-1H-quinoxalin-2-one) (Fig. 60). All the complexes exhibited promising cytotoxicity against A549, CH1, SW480 cancer cell lines, but the complex [Os(η6-p-cym)(BTQ)Cl]Cl showed much higher activity than [Os(η6-p-cym)(BOQ)Cl]Cl. The lower cytotoxicity of [Os(η6-p-cym)(BOQ)Cl]Cl containing benzoxazole derivatives was probably due to its lower thermodynamic and kinetic stability. However, it was observed that both complexes induced apoptosis in the low micromolar concentration range, suggesting that other effects not affecting the cell cycle contribute to their activity in vitro. All these findings should be considered for the future design of new osmium-arene complexes, which certainly open new possibilities for the development of cancer chemotherapeutics.
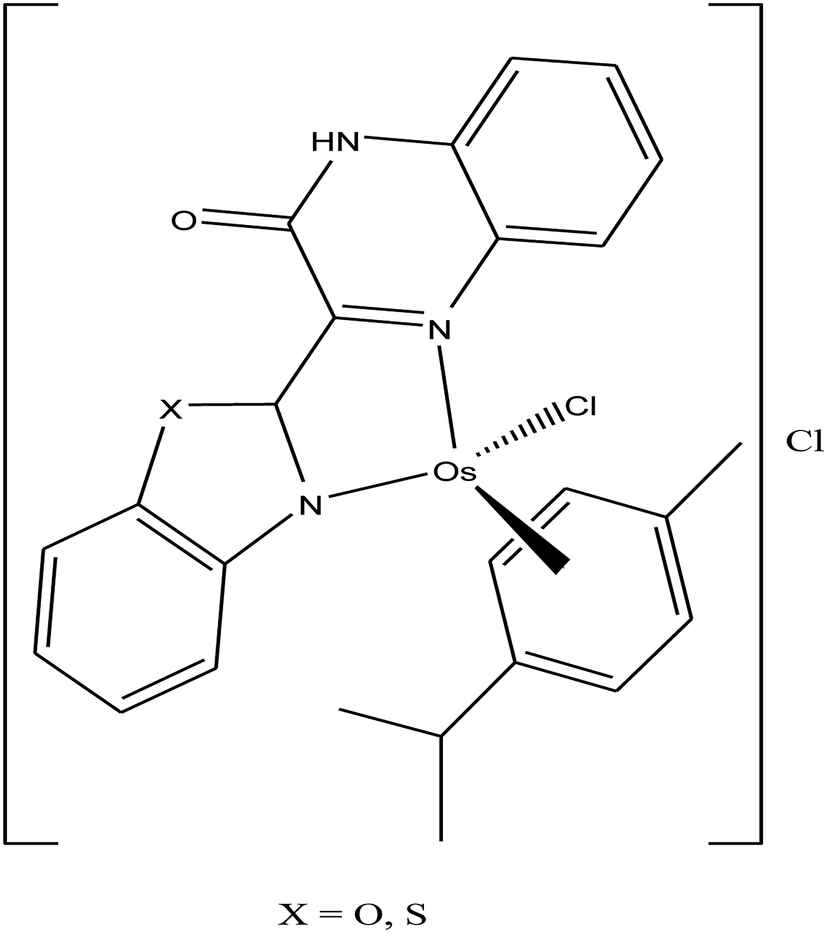 | ||
| Fig. 60 Chemical structure of the N,N-coordinated osmium-arene complexes bearing 3-(1H-benzimidazol-2-yl)-1H-quinoxalin-2-one with the general formula [Os(η6-p-cym)(BOQ)Cl]Cl and [Os(η6-p-cym)(BTQ)Cl]Cl. | ||
It has been observed that tethering an organometallic framework to pharmacologically active ligands increases the affinity of these complexes towards the target together with an increase in cellular uptake. This strategy for the design of chemotherapeutic drugs also helps in overcoming the acquired resistance caused by other classical metal-based drugs. Accordingly, V. B. Arion and B. K. Keppler et al.243,244 designed two osmium-arene complexes by using the bioactive paullone-based organic ligand of the general formula [Os(η6-p-cymene)L1Cl]Cl and [Os(η6-p-cymene)L2Cl]Cl (where, L1 and L2 are the paullone-based organic ligands), as indicated in Fig. 61. The antiproliferative activity of both complexes was tested against A549, CH1, and SW480 cancer cells. Both complexes exhibited promising antiproliferative activity, but the most potent activity reported to date was observed for [Os(η6-p-cymene)LCl]Cl against CH1 cancer cells with an IC50 value of 0.75 μM. However, these complexes did not show strong binding with 5′-GMP, but high antiproliferative activity, which suggests that they exert their effect either by binding to DNA non-covalently, i.e. intercalation, or by binding to proteins.
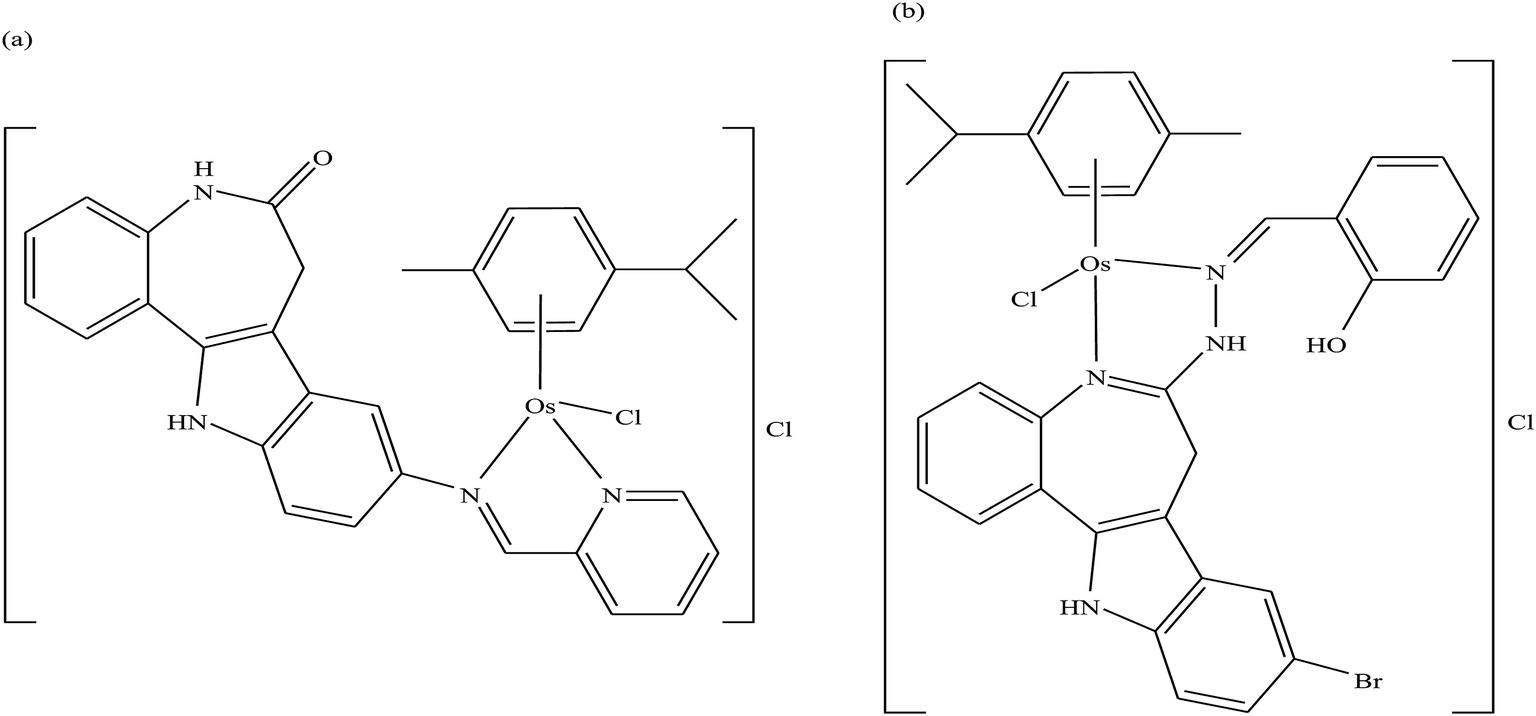 | ||
| Fig. 61 Chemical structure of the N,N-coordinated osmium-arene complexes containing a bioactive paullone-based ligand of the general formula (a) [Os(η6-p-cymene)L1Cl]Cl and (b) [Os(η6-p-cymene)L2Cl]Cl. | ||
Conclusion
The field of antitumor metal-based drugs attracted significant interest after the discovery of cisplatin and its analogues, which proved the therapeutic potential of metal complexes for the design of efficient chemotherapeutic agents. The classical antitumor dugs show serious side effects such as systematic toxicity and intrinsic resistance. Therefore, scientists are focusing towards the development of new chemotherapeutic agents by employing new strategies and different metal ions. However, there are many obstacles in the design of chemotherapeutic agents, and to achieve the perfect antitumor drugs we have to focus not only cytotoxic activity but also on the mechanism of action on tumor cells. Generally, classical drugs target DNA, but since DNA is present in healthy cells also, we have to design drugs that target only tumor cells, leaving healthy cells unaffected. Besides, proper strategies are required for the use of metal-based drugs, selection of the target and interaction of the drug with biomolecules. Therefore, it is important to understand the behavior of metal ions and the reactivity of ligands towards the recognition of the target site, also called the structure-reactivity relationship. Presently, with advanced techniques and computational molecular orbital calculations, it is much easier to understand the structure-reactivity relationship of metal complexes towards tumor cells. However, unfortunately, to date, we have not achieved 100% success in this field and it requires further thrust. In the search for new therapeutic strategies, scientists are focusing on organometallic complexes of transition metals, which may provide a breakthrough. Organometallic complexes are highly stable and have been used as catalysts for centuries. However, the advent of the organometallic framework in the field of chemotherapeutic drugs provides rational drug design with high potential towards a selective target and less toxicity. Currently, many organometallic drugs are in clinical trials, providing new paradigms for the design of novel chemotherapeutic drugs for cancer. The main target for organometallic drugs is not only DNA, and they act via other targets and mechanisms. Some of them act by causing endoplasmic reticulum stress and mitochondrial stress, while some target proteins and various enzymes such as thioredoxin reductases (TrRxs) leading to cell death. The focus of scientists working in this field is the synthesis of effective antitumor agents that will target only tumor cells, which can be accomplished by changing or modifying the ligand framework and utilizing new metal ions. In this review, we summarized the novel antitumor organometallics such as ruthenium, titanium, gold, iron and osmium complexes with excellent properties, which will provide a new platform for the development of further organometallic chemotherapeutics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Mehvash Zaki is gratefully acknowledged the financial support received from the King Abdulaziz University, Jeddah, Saudi Arabia. Also I am highly thankful to Dr Mohd Afzal, Department of Chemistry, Jadavpur University, Kolkata for their generous attention and support to finish a complete draft. Prof. Elham S. Aazam also expresses her gratitude towards King Abdulaziz University, Jeddah, Saudi Arabia. Dr Suboot Hairat is highly thankful for Wachemo University, Ethiopia for the support. Fig. 7 and 8 are reproduced from ref. 68 with permission from the Royal Society of Chemistry.References
- G. Jaouen, Bioorganometallics, Wiley-VCH, Weinheim (Germany), 2006 Search PubMed.
- G. Jaouen and P. Dyson, in Comprehensive Organometallic Chemistry III, ed. R. H. Crabtree and D. M. P. Mingos, Elsevier Ltd., Oxford, 2007, vol. 12, p. 445 Search PubMed.
- C. G. Hartinger, N. Metzler-Nolte and P. J. Dyson, Organometallics, 2012, 31, 5677–5685 CrossRef CAS.
- C. S. Allardyce, A. Dorcier, C. Scolaro and P. J. Dyson, Appl. Organomet. Chem., 2005, 19, 1–10 CrossRef CAS.
- (a) P. Kopf-Maier, J. Struct. Biol., 1990, 105, 35–45 CrossRef CAS PubMed; (b) P. Kopf-Maier and H. Kopf, in Metal compounds in cancer therapy, ed. S. P. Fricker, Chapman and Hall, London, 1994, p. 109 Search PubMed.
- P. Kopf-Maier, Antican. Res., 1999, 19, 493–504 CAS.
- C. V. Christodoulou, A. G. Eliopoulos, L. S. Young, L. Hodgkins, D. R. Ferry and D. J. Kerr, Br. J. Cancer, 1998, 77, 2088–2097 CrossRef CAS PubMed.
- M. Cini, T. D. Bradshaw and S. Woodward, Chem. Soc. Rev., 2017, 46, 1040–1051 RSC.
- I. Romero-Canelon and P. J. Sadler, Inorg. Chem., 2013, 52, 12276–12291 CrossRef CAS PubMed.
- Z. Liu and P. J. Sadler, Acc. Chem. Res., 2014, 47, 1174–1185 CrossRef CAS PubMed.
- D. N. Akbayeva, L. Gonsalvi, W. Oberhauser, M. Peruzzini, F. Vizza, P. Brüggeller, A. Romerosa, G. Sava and A. Bergamo, Chem. Commun., 2003, 264–265 RSC.
- S. Top, A. Vessieres, C. Cabestaing, I. Laios, G. Leclercq, C. Provot and G. Jaouen, J. Organomet. Chem., 2001, 637–639, 500–506 CrossRef CAS.
- G. Jaouen, S. Top, A. Vessieres, G. Leclercq, J. Quivy, L. Jin and A. Croisy, C. R. Acad. Sci., Ser. IIc: Chim., 2000, 3, 89–93 CrossRef CAS.
- U. Ndagi, N. Mhlongo and M. E. Soliman, Drug Des., Dev. Ther., 2017, 11, 599–616 CrossRef CAS PubMed.
- K. Strohfeldt and M. Tacke, Chem. Soc. Rev., 2008, 37, 1174–1187 RSC.
- Y. K. Yan, M. Melchart, A. Habtematiam and P. J. Sadler, Chem. Commun., 2005, 4764–4776 RSC.
- S. Schfer, I. Ott, R. Gust and W. S. Sheldrick, Eur. J. Inorg. Chem., 2007, 2007, 3034–3046 CrossRef.
- M. Melchart and P. J. Sadler, in Bioorganometallics, ed. G. Jaouen, WileyVCH, 2005, p. 39 Search PubMed.
- N. Metzler-Nolte, Angew. Chem., Int. Ed., 2001, 40, 1040–1043 CrossRef CAS PubMed.
- R. Alberto and R. Motterlini, Dalton Trans., 2007, 1651–1660 RSC.
- R. Motterlini and L. E. Otterbein, Nat. Rev. Drug Discovery, 2010, 9, 728–743 CrossRef CAS PubMed.
- S. H. van Rijt and P. J. Sadler, Drug Discovery Today, 2009, 14, 1089–1097 CrossRef CAS PubMed.
- Y. Yamamoto, K. Yamashita and M. Nakamura, Organometallics, 2010, 29, 1472–1478 CrossRef CAS.
- A. Alama, B. Tasso, F. Novelli and F. Sparatore, Drug Discovery Today, 2009, 14, 500–508 CrossRef CAS PubMed.
- H. Struthers, A. Hagenbach, U. Abram and R. Schibli, Inorg. Chem., 2009, 48, 5154–5163 CrossRef CAS PubMed.
- L. Wei, J. Babich, W. C. Eckelman and J. Zubieta, Inorg. Chem., 2005, 44, 2198–2209 CrossRef CAS PubMed.
- (a) D. Schlawe, A. Majdalani, J. Velcicky, E. Heßler, T. Wieder, A. Prokop and H.-G. Schmalz, Angew. Chem., 2004, 116, 1763–1766 CrossRef; (b) D. Schlawe, A. Majdalani, J. Velcicky, E. Hessler, T. Wieder, A. Prokop and H. G. Schmalz, Angew. Chem., Int. Ed., 2004, 43, 1731–1734 CrossRef CAS PubMed.
- J. C. Franke, M. Plçtz, A. Prokop, C. C. Geilen, H.-G. Schmalz and J. Eberle, Biochem. Pharmacol., 2010, 79, 575–586 CrossRef CAS PubMed.
- J. Ruiz, J. Lorenzo, L. Sanglas, N. Cutillas, C. Vicente, M. D. Villa, F. X. Aviles, G. Lopez, V. Moreno, J. Perez and D. Bautista, Inorg. Chem., 2006, 45, 6347–6360 CrossRef CAS PubMed.
- G. Jaouen and N. Metzler-Nolte, Medicinal Organometallic Chemistry, in Topics in Organometallic Chemistry, Springer-Verlag, 2010, vol 32 Search PubMed.
- W. Kandioller, E. Balsano, S. M. Meier, U. Jungwirth, S. Goschl, A. Roller, M. A. Jakupec, W. Berger, B. K. Keppler and C. G. Hartinger, Chem. Commun., 2013, 49, 3348–3350 RSC.
- T. Gianferrara, I. Bratsos and E. Alessio, Dalton Trans., 2009, 7588–7598 RSC.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed.
- E. A. Hillard and G. Jaouen, Organometallics, 2011, 30, 20–27 CrossRef CAS.
- K. B. Garbutcheon-Singh, M. P. Grant, B. W. Harper, A. M. Krause-Heuer, M. Manohar, N. Orkey and J. R. Aldrich-Wright, Curr. Top. Med. Chem., 2011, 11, 521–542 CrossRef CAS PubMed.
- R. H. Fish, Aust. J. Chem., 2010, 63, 1505–1513 CrossRef CAS.
- A. Casini, C. G. Hartinger, A. A. Nazarov and P. J. Dyson, Top. Organomet. Chem., 2010, 32, 57–80 CrossRef CAS.
- D. Dive and C. Biot, ChemMedChem, 2008, 3, 383–391 CrossRef CAS PubMed.
- T. W. Hambley, Dalton Trans., 2007, 4929–4937 RSC.
- S. van Zutphen and J. Reedijk, Coord. Chem. Rev., 2005, 249, 2845–2853 CrossRef CAS.
- C. Lottner, R. Knuechel, G. Bernhardt and H. Brunner, Cancer Lett., 2004, 203, 171–180 CrossRef CAS.
- N. P. E. Barry and P. J. Sadler, Chem. Commun., 2013, 49, 5106–5131 RSC.
- G. Suss-Fink, Dalton Trans., 2010, 39, 1673–1688 RSC.
- W. H. Ang, A. Casini, G. Sava and P. J. Dyson, J. Organomet. Chem., 2011, 696, 989–998 CrossRef CAS.
- A. L. Noffke, A. Habtemariam, A. M. Pizarro and P. J. Sadler, Chem. Commun., 2012, 48, 5219–5246 RSC.
- R. E. Aird, J. Cummings, A. A. Ritchie, M. Muir, R. E. Morris, H. Chen, P. J. Sadler and D. I. Jodrell, Br. J. Cancer, 2002, 86, 1652–1657 CrossRef CAS PubMed.
- A. Weiss, R. H. Berndsen, M. Dubois, C. Muller, R. Schibli, A. W. Griffioen, P. J. Dyson and P. Nowak-Sliwinska, Chem. Sci., 2014, 5, 4742–4748 RSC.
- C. Scolaro, A. Bergamo, L. Brescacin, R. Delfino, M. Cocchietto, G. Laurenczy, T. J. Geldbach, G. Sava and P. J. Dyson, J. Med. Chem., 2005, 48, 4161–4171 CrossRef CAS PubMed.
- E. Alessio, G. Mestroni, A. Bergamo and G. Sava, Curr. Top. Med. Chem., 2004, 4, 1525–1535 CrossRef CAS PubMed.
- G. Palermo, A. Magistrato, T. Riedel, T. von Erlach, C. A. Davey, P. J. Dyson and U. Rothlisberger, ChemMedChem, 2016, 11, 1199–1210 CrossRef CAS PubMed.
- A. Levina, A. Mitra and P. A. Lay, Metallomics, 2009, 1, 458–470 RSC.
- W. H. Ang, E. Daldini, C. Scolaro, R. Scopelliti, L. Juillerat-Jeannerat and P. J. Dyson, Inorg. Chem., 2006, 45, 9006–9013 CrossRef CAS PubMed.
- M. A. Jakupec, M. Galanski, V. B. Arion, C. G. Hartinger and B. K. Keppler, Dalton Trans., 2008, 183–194 RSC.
- G. Sava, A. Bergamo and P. J. Dyson, Dalton Trans., 2011, 40, 9069–9075 RSC.
- F. Y. Wang, A. Habtemariam, E. P. L. van der Geer, R. Fernandez, M. Melchart, R. J. Deeth, R. Aird, S. Guichard, F. P. A. Fabbiani, P. Lozano-Casal, I. D. H. Oswald, D. I. Jodrell, S. Parsons and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18269–18274 CrossRef CAS PubMed.
- P. J. Dyson and G. Sava, Dalton Trans., 2006, 16, 1929–1933 RSC.
- Z. Adhireksan, G. E. Davey, P. Campomanes, M. Groessl, C. M. Clavel, H. Yu, A. A. Nazarov, H. F. Yeo, W. H. Ang, P. Droge, U. Rothlisberger, P. J. Dyson and C. A. Davey, Nat. Commun., 2014, 5, 3462 CrossRef PubMed.
- A. Casini, C. Gabbiani, F. Sorrentino, M. P. Rigobello, A. Bindoli, T. J. Geldbach, A. Marrone, N. Re, C. G. Hartinger, P. J. Dyson and L. Messori, J. Med. Chem., 2008, 51, 6773–6781 CrossRef CAS PubMed.
- C. X. Zhang and S. J. Lippard, Curr. Opin. Chem. Biol., 2003, 7, 481–489 CrossRef CAS PubMed.
- G. S. Smitha and B. Therrien, Dalton Trans., 2011, 40, 10793–10800 RSC.
- I. F. Tannock and D. Rotin, Cancer Res., 1989, 49, 4373–4384 CAS.
- C. M. Hackl, B. Schoenhacker-Alte, M. H. M. Klose, H. Henke, M. S. Legina, M. A. Jakupec, W. Berger, B. K. Keppler, O. Bruggemann, I. Teasdale, P. Heffeter and W. Kandioller, Dalton Trans., 2017, 46, 12114–12124 RSC.
- P. Chellan, K. M. Land, A. Shokar, A. Au, S. H. An, D. Taylor, P. J. Smith, T. Riedel, P. J. Dyson, K. Chibale and G. S. Smith, Dalton Trans., 2014, 43, 513–526 RSC.
- H. Chen, J. A. Parkinson, R. E. Morris and P. J. Sadler, J. Am. Chem. Soc., 2003, 125, 173–186 CrossRef CAS PubMed.
- M. K. M. Subarkhan, R. Ramesh and Y. Liu, New J. Chem., 2016, 40, 9813–9823 RSC.
- M. K. M. Subarkhan and R. Ramesh, Inorg. Chem. Front., 2016, 3, 1245–1255 RSC.
- S. Fulda and K. M. Debatin, Oncogene, 2006, 25, 4798–4811 CrossRef CAS PubMed.
- M. J. Chow, C. Licona, G. Pastorin, G. Mellitzer, W. H. Ang and C. Gaiddon, Chem. Sci., 2016, 7, 4117–4124 RSC.
- C. G. Hartinger and P. J. Dyson, Chem. Soc. Rev., 2009, 38, 391–401 RSC.
- W. Kandioller, C. G. Hartinger, A. A. Nazarov, C. Bartel, M. Skocic, M. A. Jakupec, V. B. Arion and B. K. Keppler, Chem.–Eur. J., 2009, 15, 12283–12291 CrossRef CAS PubMed.
- (a) G. S. Yellol, A. Donaire, J. G. Yellol, V. Vasylyeva, C. Janiak and J. Ruiz, Chem. Commun., 2013, 49, 11533–11535 RSC; (b) J. Yellol, S. A. Perez, A. Buceta, G. Yellol, A. Donaire, P. Szumlas, P. J. Bednarski, G. Makhloufi, C. Janiak, A. Espinosa and J. Ruiz, J. Med. Chem., 2015, 58, 7310–7327 CrossRef CAS PubMed.
- R. Pettinari, F. Marchetti, F. Condello, C. Pettinari, G. Lupidi, R. Scopelliti, S. Mukhopadhyay, T. Riedel and P. J. Dyson, Organometallics, 2014, 33, 3709–3715 CrossRef CAS.
- S. Sersen, J. Kljun, K. Kryeziu, R. Panchuk, B. Alte, W. Korner, P. Heffeter, W. Berger and I. Turel, J. Med. Chem., 2015, 58, 3984–3996 CrossRef CAS PubMed.
- P. Kopf-Maier, W. Wagner and H. Kopf, Cancer Chemother. Pharmacol., 1981, 5, 237–241 CrossRef CAS PubMed.
- P. Kopf-Maier, Eur. J. Clin. Pharmacol., 1994, 47, 1–16 CrossRef CAS PubMed.
- E. Melendez, Crit. Rev. Oncol.-Hematol., 2002, 42, 309–315 CrossRef PubMed.
- H. Kopf and P. Kopf-Maier, Angew. Chem., Int. Ed., 1979, 18, 477–478 CrossRef CAS PubMed.
- O. Oberschmidt, A.-R. Hanauske, F.-J. K. Rehmann, K. Strohfeldt, N. Sweeney and M. Tacke, Anti-Cancer Drugs, 2005, 16, 1071–1073 CrossRef CAS PubMed.
- C. V. Christodoulou, D. R. Ferry, D. W. Fyfe, A. Young, J. Doran, T. M. Sheehan, A. Eliopoulos, K. Hale, J. Baumgart, G. Sass and D. J. Kerr, J. Clin. Oncol., 1998, 16, 2761–2769 CrossRef CAS PubMed.
- A. Korfel, M. E. Scheulen, H. J. Schmoll, O. Grundel, A. Harstrick, M. Knoche, M. Fels, M. Skorzec, F. Bach, J. Baumgart, G. Sass, S. Seeber, E. Thiel and W. E. Berdel, Clin. Cancer Res., 1998, 4, 2701–2708 CAS.
- M. Gielen and E. R. T. Tiekink, Metallotherapeutic Drugs and Metal-Based Diagnostic Agents: The Use of Metals in Medicine, John Wiley & Sons, Ltd, 2005, pp. 125–142 Search PubMed.
- F. Caruso and M. Rossi, Met. Ions Biol. Syst., 2004, 42, 353–384 CAS.
- G. Lummen, H. Sperling, H. Luboldt, T. Otto and H. Rubben, Cancer Chemother. Pharmacol., 1998, 42, 415–417 CrossRef CAS PubMed.
- N. Kroger, U. R. Kleeberg, K. Mross, L. Edler and D. K. Hossfeld, Oncologie, 2000, 23, 60–62 Search PubMed.
- A. Vessieres, M.-A. Plamont, C. Cabestaing, J. Claffey, S. Dieckmann, M. Hogan, H. Muller-Bunz, K. Strohfeldt and M. Tacke, J. Organomet. Chem., 2009, 694, 874–879 CrossRef CAS.
- L. He, S. Ji, H. Lai and T. Chen, J. Mater. Chem. B, 2015, 3, 8383–8393 RSC.
- D. Mustacich and G. Powis, Biochem. J., 2000, 346, 1–8 CrossRef CAS PubMed.
- T. F. Whayne Jr, N. Parinandi and N. Maulik, Can. J. Physiol. Pharmacol., 2015, 93, 903–911 CrossRef PubMed.
- Z. Luo, L. Yu, F. Yang, Z. Zhao, B. Yu, H. Lai, K.-H. Wong, S.-M. Ngai, W. Zheng and T. Chen, Metallomics, 2014, 6, 1480–1490 RSC.
- C. H. Williams Jr, L. D. Arscott, S. Muller, B. W. Lennon, M. L. Ludwig, P.-F. Wang, D. M. Veine, K. Becker and R. H. Schirmer, Eur. J. Biochem., 2000, 267, 6110–6117 CrossRef.
- O. R. Allen, A. L. Gott, J. A. Hartley, J. M. Hartley, R. J. Knox and P. C. McGowan, Dalton Trans., 2007, 5082–5090 RSC.
- O. R. Allen, L. Croll, A. L. Gott, R. J. Knox and P. C. McGowan, Organometallics, 2004, 23, 288–292 CrossRef CAS.
- M. McGowan and P. C. McGowan, Inorg. Chem. Commun., 2000, 3, 337–340 CrossRef CAS.
- G. D. Potter, M. C. Baird and S. P. C. Cole, J. Organomet. Chem., 2007, 692, 3508–3518 CrossRef CAS.
- M. Tacke, J. P. Dunne, S. Fox, G. Linti and R. Teuber, J. Mol. Struct., 2001, 570, 197–202 CrossRef CAS.
- S. Fox, J. P. Dunne, M. Tacke and J. F. Gallagher, Inorg. Chim. Acta, 2004, 357, 225–234 CrossRef CAS.
- P. W. Causey and M. C. Baird, Organometallics, 2004, 23, 4486–4494 CrossRef CAS.
- G. D. Potter, M. C. Baird, M. Chan and S. P. C. Cole, Inorg. Chem. Commun., 2006, 9, 1114–1116 CrossRef CAS.
- J. R. Boyles, M. C. Baird, B. G. Campling and N. Jain, J. Inorg. Biochem., 2001, 84, 159–162 CrossRef CAS PubMed.
- M. Tacke, L. P. Cuffe, W. M. Gallagher, Y. Lou, O. Mendoza, H. Muller-Bunz, F.-J. K. Rehmann and N. Sweeney, J. Inorg. Biochem., 2004, 98, 1987–1994 CrossRef CAS PubMed.
- M. Tacke, L. T. Allen, L. Cuffe, W. M. Gallagher, Y. Lou, O. Mendoza, H. Muller-Bunz, F.-J. K. Rehmann and N. Sweeney, J. Organomet. Chem., 2004, 689, 2242–2249 CrossRef CAS.
- F.-J. K. Rehmann, L. P. Cuffe, O. Mendoza, D. K. Rai, N. Sweeney, K. Strohfeldt, W. M. Gallagher and M. Tacke, Appl. Organomet. Chem., 2005, 19, 293–300 CrossRef CAS.
- N. J. Sweeney, O. Mendoza, H. Muller-Bunz, C. Pampillon, F.-J. K. Rehmann, K. Strohfeldt and M. Tacke, J. Organomet. Chem., 2005, 690, 4537–4544 CrossRef CAS.
- C. Pampillon, N. J. Sweeney, K. Strohfeldt and M. Tacke, Inorg. Chim. Acta, 2006, 359, 3969–3975 CrossRef CAS.
- C. Pampillon, N. J. Sweeney, K. Strohfeldt and M. Tacke, J. Organomet. Chem., 2007, 692, 2153–2159 CrossRef CAS.
- X. Chen and L. Zhou, J. Mol. Struct., 2010, 940, 45–49 CrossRef CAS.
- U. Olszewski and G. Hamilton, Anti-Cancer Agents Med. Chem., 2010, 10, 302–311 CrossRef CAS PubMed.
- K. M. Buettner, R. C. Snoeberger III, V. S. Batista and A. M. Valentine, Dalton Trans., 2011, 40, 9580–9588 RSC.
- C. J. P. P. Siburt, E. M. Lin, S. J. Brandt, A. D. Tinoco, A. M. Valentine and A. L. Crumbliss, J. Inorg. Biochem., 2010, 104, 1006–1009 CrossRef PubMed.
- J. F. Gonzalez-Pantoja, M. Stern, A. A. Jarzecki, E. Royo, E. Robles-Escajeda, A. Varela-Ramirez, R. J. Aguilera and M. Contel, Inorg. Chem., 2011, 50, 11099–11110 CrossRef CAS PubMed.
- J. Fernandez-Gallardo, B. T. Elie, F. J. Sulzmaier, M. Sanau, J. W. Ramos and M. Contel, Organometallics, 2014, 33, 6669–6681 CrossRef CAS PubMed.
- J. Fernandez-Gallardo, B. T. Elie, T. Sadhukha, S. Prabha, M. Sanau, S. A. Rotenberg, J. W. Ramos and M. Contel, Chem. Sci., 2015, 6, 5269–5283 RSC.
- Y. F. Mui, J. Fernandez-Gallardo, B. T. Elie, A. Gubran, I. Maluenda, M. Sanau, O. Navarro and M. Contel, Organometallics, 2016, 35, 1218–1227 CrossRef CAS PubMed.
- S. R. Block, J. Rheumatol., 1986, 13, 663–664 CAS.
- T. M. Simon, D. H. Kunishima, G. J. Vibert and A. Lorber, Cancer Res., 1981, 41, 94–97 CAS.
- K. Palanichamy, N. Sreejayan and A. C. Ontko, J. Inorg. Biochem., 2012, 106, 32–42 CrossRef CAS PubMed.
- S. J. Berners-Price, Angew. Chem., Int. Ed., 2011, 50, 804–805 CrossRef CAS PubMed.
- T. Zou, C. T. Lum, C.-N. Lok, J.-J. Zhang and C.-M. Che, Chem. Soc. Rev., 2015, 44, 8786–8801 RSC.
- R. W. Y. Sun and C. M. Che, Coord. Chem. Rev., 2009, 253, 1682–1691 CrossRef CAS and references therein.
- Y. Wang, Q. Y. He, R. W. Y. Sun, C. M. Che and J. F. Chiu, Cancer Res., 2005, 65, 11553–11564 CrossRef CAS PubMed.
- K. H. M. Chow, R. W. Y. Sun, J. B. B. Lam, C. K. L. Li, A. M. Xu, D. L. Ma, R. Abagyan, Y. Wang and C. M. Che, Cancer Res., 2010, 70, 329–337 CrossRef CAS PubMed.
- C. Giampietri, S. Petrungaro, S. Conti, A. Facchiano, A. Filippini and E. Ziparo, Mediators Inflammation, 2015, 2015, 1–11 CrossRef PubMed.
- F. Martinon, Acta Oncol., 2012, 51, 822–830 CrossRef CAS PubMed.
- I. Ott, Coord. Chem. Rev., 2009, 253, 1670–1681 CrossRef CAS.
- W. Liu and R. Gust, Chem. Soc. Rev., 2013, 42, 755–773 RSC.
- T. Zou, C.-N. Lok, Y. M. E. Fung and C.-M. Che, Chem. Commun., 2013, 49, 5423–5425 RSC.
- S. B. Aher, P. N. Muskawar, T. Thenmozhi and P. R. Bhagat, Eur. J. Med. Chem., 2014, 81, 408–419 CrossRef CAS PubMed.
- K. M. Hindi, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Youngs, Chem. Rev., 2009, 109, 3859–3884 CrossRef CAS PubMed.
- A. John and P. Ghosh, Dalton Trans., 2010, 39, 7183–7206 RSC.
- J. J. Yan, A. L.-F. Chow, C.-H. Leung, R. W.-Y. Sun, D.-L. Ma and C.-M. Che, Chem. Commun., 2010, 46, 3893–3895 RSC.
- R. W.-Y. Sun, A. L.-F. Chow, X.-H. Li, J. J. Yan, S. S.-Y. Chui and C.-M. Che, Chem. Sci., 2011, 2, 728–736 RSC.
- L. Oehninger, R. Rubbiani and I. Ott, Dalton Trans., 2013, 42, 3269–3284 RSC.
- (a) F. Cisnetti and A. Gautier, Angew. Chem., 2013, 125, 12194–12196 CrossRef; (b) F. Cisnetti and A. Gautier, Angew. Chem., Int. Ed., 2013, 52, 11976–11978 CrossRef CAS PubMed.
- S. J. Berners-Price and A. Filipovska, Metallomics, 2011, 3, 863–873 RSC.
- O. Rackham, A. M. Shearwood, R. Thyer, E. McNamara, S. M. Davies, B. A. Callus, A. Miranda-Vizuete, S. J. Berners-Price, Q. Cheng, E. S. Arner and A. Filipovska, Free Radicals Biol. Med., 2011, 50, 689–699 CrossRef CAS PubMed.
- A. Bindoli, M. P. Rigobello, G. Scutari, C. Gabbiani, A. Casini and L. Messori, Coord. Chem. Rev., 2009, 253, 1692–1707 CrossRef CAS.
- A. G. Cox, K. K. Brown, E. S. J. Arner and M. B. Hampton, Biochem. Pharmacol., 2008, 76, 1097–1109 CrossRef CAS PubMed.
- R. V. Parish, B. P. Howe, J. P. Wright, J. Mack, R. G. Pritchard, R. G. Buckley, A. M. Elsome and S. P. Fricker, Inorg. Chem., 1996, 35, 1659–1666 CrossRef CAS PubMed.
- R. V. Parish, J. Mack, L. Hargreaves, J. P. Wright, R. G. Buckley, A. M. Elsome, S. P. Fricker and B. R. C. Theobald, Dalton Trans., 1996, 69–74 RSC.
- R. G. Buckley, A. M. Elsome, S. P. Fricker, G. R. Henderson, B. R. C. Theobald, R. V. Parish, B. P. Howe and L. R. Kelland, J. Med. Chem., 1996, 39, 5208–5214 CrossRef CAS PubMed.
- S. P. Fricker, R. M. Mosi, B. R. Cameron, I. Baird, Y. Zhu, V. Anastassov, J. Cox, P. S. Doyle, E. Hansell, G. Lau, J. Langille, M. Olsen, L. Qin, R. Skerlj, R. S. Y. Wong, Z. Santucci and J. H. McKerrow, J. Inorg. Biochem., 2008, 102, 1839–1845 CrossRef CAS PubMed.
- L. Engman, M. McNaughton, M. Gajewska, S. Kumar, A. Birmingham and G. Powis, Anticancer Drugs, 2006, 17, 539–544 CrossRef CAS PubMed.
- Y. Zhu, B. R. Cameron, R. Mosi, V. Anastassov, J. Cox, L. Qin, Z. Santucci, M. Metz, R. T. Skerlj and S. P. Fricker, J. Inorg. Biochem., 2011, 105, 754–762 CrossRef CAS PubMed.
- Y. Zhu, B. R. Cameron and R. T. Skerlj, J. Organomet. Chem., 2003, 677, 57–72 CrossRef CAS.
- J. J. Zhang, R. W. Y. Sun and C. M. Che, Chem. Commun., 2012, 48, 3388–3390 RSC.
- S. D. Brown, W. Henderson, K. J. Kilpin and B. K. Nicholson, Inorg. Chim. Acta, 2007, 360, 1310–1315 CrossRef CAS.
- L. Vela, M. A. Contel, L. Palomera, G. Azaceta and I. Marzo, J. Inorg. Biochem., 2011, 105, 1306–1313 CrossRef CAS PubMed.
- N. Shaik, A. Martinez, I. Augustin, H. Giovinazzo, A. Varela-Ramirez, M. Sanau, R. J. Aguilera and M. Contel, Inorg. Chem., 2009, 48, 1577–1587 CrossRef CAS PubMed.
- D. Aguilar, M. Contel, R. Navarro and E. P. Urriolabeitia, Organometallics, 2007, 26, 4604–4611 CrossRef CAS.
- R. W.-Y. Sun, C.-N. Lok, T. T.-H. Fong, C. K.-L. Li, Z. F. Yang, T. Zou, A. F.-M. Siu and C.-M. Che, Chem. Sci., 2013, 4, 1979–1988 RSC.
- J. Boelens, S. Lust, F. Offner, M. E. Bracke and B. W. Vanhoecke, In Vivo, 2007, 21, 215–226 CAS.
- G. Pan, J. Ni, Y.-F. Wei, G.-l. Yu, R. Gentz and V. M. Dixit, Science, 1997, 277, 815–818 CrossRef CAS PubMed.
- H. Walczak, M. A. Degli-Esposti, R. S. Johnson, P. J. Smolak, J. Y. Waugh, N. Boiani, M. S. Timour, M. J. Gerhart, K. A. Schooley, C. A. Smith, R. G. Goodwin and C. T. Rauch, EMBO J., 1997, 16, 5386–5397 CrossRef CAS PubMed.
- J. P. Sheridan, S. A. Marsters, R. M. Pitti, A. Gurney, M. Skubatch, D. Baldwin, L. Ramakrishnan, C. L. Gray, K. Baker, W. I. Wood, A. D. Goddard, P. Godowski and A. Ashkenazi, Science, 1997, 277, 818–821 CrossRef CAS PubMed.
- B. Bertrand, S. Spreckelmeyer, E. Bodio, F. Cocco, M. Picquet, P. Richard, P. Le Gendre, C. Orvig, M. A. Cinellu and A. Casini, Dalton Trans., 2015, 44, 11911–11918 RSC.
- P. Davis, Can. Fam. Physician, 1988, 34, 445–452 Search PubMed.
- C. K. Mirabelli, R. K. Johnson, C. M. Sung, L. Faucette, K. Muirhead and S. T. Crooke, Cancer Res., 1985, 45, 32–39 CAS.
- P. J. Sadler and R. E. Sue, Met.-Based Drugs, 1994, 1, 107–144 CrossRef CAS PubMed.
- H. Kamei, T. Koide, T. Kojima, Y. H ashimoto and M. Hasegawa, Cancer Biother.Radiopharm., 1998, 13, 403–406 CrossRef CAS PubMed.
- M. Stallings-Mann, L. Jamieson, R. P. Regala, C. Weems, N. R. Murray and A. P. Fields, Cancer Res., 2006, 66, 1767–1774 CrossRef CAS PubMed.
- (a) R. Rubbiani, I. Kitanovic, H. Alborzinia, S. Can, A. Kitanovic, L. A. Onambele, M. Stefanopoulou, Y. Geldmacher, W. S. Sheldrick, G. Wolber, A. Prokop, S. Wolfl and I. Ott, J. Med. Chem., 2010, 53, 8608–8618 CrossRef CAS PubMed; (b) P. J. Barnard and S. J. Berners-Price, Coord. Chem. Rev., 2007, 251, 1889–1902 CrossRef CAS.
- (a) R. Rubbiani, S. Can, I. Kitanovic, H. Alborzinia, M. Stefanopoulou, M. Kokoschka, S. Monchgesang, W. S. Sheldrick, S. Wolfl and I. Ott, J. Med. Chem., 2011, 54, 8646–8657 CrossRef CAS PubMed; (b) J. L. Hickey, R. A. Ruhayel, P. J. Barnard, M. V. Baker, S. J. Berners-Price and A. Filipovska, J. Am. Chem. Soc., 2008, 130, 12570–12571 CrossRef CAS PubMed.
- (a) R. Rubbiani, L. Salassa, A. de Almeida, A. Casini and I. Ott, ChemMedChem, 2014, 9, 1205–1210 CrossRef CAS PubMed; (b) P. J. Barnard, M. V. Baker, S. J. Berners-Price and D. A. Day, J. Inorg. Biochem., 2004, 98, 1642–1647 CrossRef CAS PubMed.
- (a) A. Meyer, L. Oehninger, Y. Geldmacher, H. Alborzinia, S. Wolfl, W. S. Sheldrick and I. Ott, ChemMedChem, 2014, 9, 1794–1800 CAS; (b) M. V. Baker, P. J. Barnard, S. J. Berners-Price, S. K. Brayshaw, J. L. Hickey, B. W. Skelton and A. H. White, Dalton Trans., 2006, 3708–3715 RSC.
- J. S. Modica-Napolitano and J. R. Aprille, Adv. Drug Delivery Rev., 2001, 49, 63–70 CrossRef CAS PubMed.
- M. M. Jellicoe, S. J. Nichols, B. A. Callus, M. V. Baker, P. J. Barnard, S. J. Berners-Price, J. Whelan, G. C. Yeoh and A. Filipovska, Carcinogenesis, 2008, 29, 1124–1133 CrossRef CAS PubMed.
- T. Zou, C. T. Lum, C.-N. Lok, W.-P. To, K.-H. Low and C.-M. Che, Angew. Chem., Int. Ed., 2014, 53, 5810–5814 CrossRef CAS PubMed.
- E. Garcia-Moreno, A. Tomas, E. Atrian-Blasco, S. Gascon, E. Romanos, M. Jesus Rodriguez-Yoldi, E. Cerrada and M. Laguna, Dalton Trans., 2016, 45, 2462–2475 RSC.
- E. Vergara, E. Cerrada, A. Casini, O. Zava, M. Laguna and P. J. Dyson, Organometallics, 2010, 29, 2596–2603 CrossRef CAS.
- L. M. McKeage and S. J. Berners-Price, Coord. Chem. Rev., 2002, 232, 127–135 CrossRef.
- A. Meyer, C. P. Bagowski, M. Kokoschka, M. Stefanopoulou, H. Alborzinia, S. Can, D. H. Vlecken, W. S. Sheldrick, S. Wölfl and I. Ott, Angew. Chem., Int. Ed., 2012, 51, 8895–8899 CrossRef CAS PubMed.
- A. Holmgren and J. Lu, Biochem. Biophys. Res. Commun., 2010, 396, 120–124 CrossRef CAS PubMed.
- E. S. J. Arner, Biochim. Biophys. Acta, Gen. Subj., 2009, 1790, 495–526 CrossRef CAS PubMed.
- K. Stoletov and R. Klemke, Oncogene, 2008, 27, 4509–4520 CrossRef CAS PubMed.
- R. G. Balasingham, C. F. Williams, H. J. Mottram, M. P. Coogan and S. J. A. Pope, Organometallics, 2012, 31, 5835–5843 CrossRef CAS.
- C. P. Siegers, D. Bumann, H. D. Trepkau, B. Schadwinkel and G. Baretton, Prog. Clin. Biol. Res., 1991, 369, 439–444 CAS.
- A. Kicic, A. C. G. Chua and E. Baker, Anti Cancer Drug Des., 2001, 16, 195–207 CAS.
- L. M. Mir, O. Tounekti and S. Orlowski, Gen. Pharmacol., 1996, 27, 745–748 CrossRef CAS PubMed.
- J. Stubbe and J. W. Kozarich, Chem. Rev., 1987, 87, 1107–1136 CrossRef CAS.
- S. T. Hoehn, H. D. Junker, R. C. Bunt, C. J. Turner and J. Stubbe, Biochemistry, 2001, 40, 5894–5905 CrossRef CAS PubMed.
- D. R. van Staveren and N. Metzler-Nolte, Chem. Rev., 2004, 104, 5931–5986 CrossRef CAS PubMed.
- M. F. R. Fouda, M. M. Abd-Elzaher, R. A. Abdelsamaia and A. A. Labib, Appl. Organomet. Chem., 2007, 21, 613–625 CrossRef CAS.
- R. H. Fish and G. Jaouen, Organometallics, 2003, 22, 2166–2177 CrossRef CAS.
- G. Caldwell, M. G. Meirim, E. W. Neuse and C. E. J. van Rensburg, Appl. Organomet. Chem., 1998, 12, 793–799 CrossRef CAS.
- C. Biot, G. Glorian, L. A. Maciejewski, J. S. Brocard, O. Domarle, G. Blampain, P. Millet, A. J. Georges, H. Abessolo, D. Dive and J. Lebibi, J. Med. Chem., 1997, 40, 3715–3718 CrossRef CAS PubMed.
- C. Biot, N. Francois, L. Maciejewski, J. Brocard and D. Poulain, Bioorg. Med. Chem. Lett., 2000, 10, 839–841 CrossRef CAS PubMed.
- J. T. Chantson, M. V. V. Falzacappa, S. Crovella and N. Metzler-Nolte, ChemMedChem, 2006, 1, 1268–1274 CrossRef CAS PubMed.
- Y. N. V. Gopal, D. Jayaraju and A. K. Kondapi, Arch. Biochem. Biophys., 2000, 376, 229–235 CrossRef PubMed.
- P. Kopf-Maier, H. Kopf and E. W. Neuse, J. Cancer Res. Clin. Oncol., 1984, 108, 336–340 CrossRef CAS PubMed.
- G. Tabbi, C. Cassino, G. Cavigiolio, D. Colangelo, A. Ghiglia, I. Viano and D. Osella, J. Med. Chem., 2002, 45, 5786–5796 CrossRef CAS PubMed.
- D. Osella, M. Ferrali, P. Zanello, F. Laschi, M. Fontani, C. Nervi and G. Cavigiolio, Inorg. Chim. Acta, 2000, 306, 42–48 CrossRef CAS.
- R. Kovjazin, T. Eldar, M. Patya, A. Vanichkin, H. M. Lander and A. Novogrodsky, FASEB J., 2003, 17, 467–469 CrossRef CAS PubMed.
- G. Jaouen, S. Top, A. Vessieres, G. Leclercq and M. J. McGlinchey, Curr. Med. Chem., 2004, 11, 2505–2517 CrossRef CAS PubMed.
- E. Hillard, A. Vessieres, L. Thouin, G. Jaouen and C. Amatore, Angew. Chem., Int. Ed., 2006, 45, 285–290 CrossRef CAS PubMed.
- A. Vessieres, S. Top, P. Pigeon, E. Hillard, L. Boubeker, D. Spera and G. Jaouen, J. Med. Chem., 2005, 48, 3937–3940 CrossRef CAS PubMed.
- E. Hillard, A. Vessieres, F. Le Bideau, D. Plazuk, D. Spera, M. Huche and G. Jaouen, ChemMedChem, 2006, 1, 551–559 CrossRef CAS PubMed.
- E. A. Hillard, P. Pigeon, A. Vessieres, C. Amatore and G. Jaouen, Dalton Trans., 2007, 5073–5081 RSC.
- S. Top, C. Thibaudeau, A. Vessieres, E. Brule, F. Le Bideau, J.-M. Joerger, M.-A. Plamont, S. Samreth, A. Edgar, J. Marrot, P. Herson and G. Jaouen, Organometallics, 2009, 28, 1414–1424 CrossRef CAS.
- A. P. Ferreira, J. L. Ferreira da Silva, M. T. Duarte, M. F. Minas da Piedade, M. P. Robalo, S. G. Harjivan, C. Marzano, V. Gandin and M. M. Marques, Organometallics, 2009, 28, 5412–5423 CrossRef CAS.
- R. F. Shago, J. C. Swarts, E. Kreft and C. E. J. Van Rensburg, Anticancer Res., 2007, 27, 3431–3434 CAS.
- P. V. Bernhardt, L. M. Caldwell, T. B. Chaston, P. Chin and D. R. Richardson, J. Biol. Inorg Chem., 2003, 8, 866–880 CrossRef CAS PubMed.
- D. S. Kalinowski, Y. Yu, P. C. Sharpe, M. Islam, Y.-T. Liao, D. B. Lovejoy, N. Kumar, P. V. Bernhardt and D. R. Richardson, J. Med. Chem., 2007, 50, 3716–3729 CrossRef CAS PubMed.
- J. L. Buss, B. T. Greene, J. Turner, F. M. Torti and S. V. Torti, Curr. Top. Med. Chem., 2004, 4, 1623–1635 CrossRef CAS PubMed.
- L. Delhaes, C. Biot, L. Berry, P. Delcourt, A. Maciejewski Lucien, D. Camus, S. Brocard Jacques and D. Dive, ChemBioChem, 2002, 3, 418–423 CrossRef CAS PubMed.
- V. J. Fiorina, R. J. Dubois and S. Brynes, J. Med. Chem., 1978, 21, 393–395 CrossRef CAS PubMed.
- Q. Michard, G. Jaouen, A. Vessieres and B. A. Bernard, J. Inorg. Biochem., 2008, 102, 1980–1985 CrossRef CAS PubMed.
- J. Spencer, A. P. Mendham, A. K. Kotha, S. C. Richardson, E. A. Hillard, G. Jaouen, L. Malec and M. B. Hursthouse, Dalton Trans., 2009, 918–921 RSC.
- D. Hamels, P. M. Dansette, E. A. Hillard, S. Top, A. Vessieres, P. Herson, G. Jaouen and D. Mansuy, Angew. Chem., 2009, 121, 9288–9290 CrossRef.
- A. Arezki, G. Chabot, L. Quentin, D. Scherman, G. Jaouen and E. Brule, MedChemComm, 2011, 2, 190–195 RSC.
- S. Top, K. E. l Bachir, A. Vessieres, G. Leclercq, I. Laios, M. Ourevitch, C. Deuschel, M. J. McGlinchey and G. Jaouen, ChemBioChem, 2003, 4, 754–761 CrossRef CAS PubMed.
- A. Vessieres, S. Top, W. Beck, E. Hillard and G. Jaouen, Dalton Trans., 2006, 529–541 RSC.
- S. Top, J. Tang, A. Vessieres, D. Carrez, C. Provot and G. Jaouen, Chem. Commun., 1996, 955–956 RSC.
- A. C. de Oliveira, E. G. da Silva, D. D. Rocha, E. A. Hillard, P. Pigeon, G. Jaouen, F. A. R. Rodrigues, F. C. de Abreu, F. da Rocha Ferreira, M. O. F. Goulart and L. V. Costa-Lotufo, ChemMedChem, 2014, 9, 2580–2586 CrossRef CAS PubMed.
- A. Citta, A. Folda, A. Bindoli, P. Pigeon, S. Top, A. Vessieres, M. Salmain, G. Jaouen and M. P. Rigobello, J. Med. Chem., 2014, 57, 8849–8859 CrossRef CAS PubMed.
- L. L. Smith and I. N. White, Oncology, 1998, 12, 14–22 CAS.
- M. A. Killackey, T. B. Hakes and V. K. Price, Cancer Treat. Rep., 1985, 69, 237–238 CAS.
- G. K. Poon, B. Walter, P. E. Lonning, M. N. Holton and R. McCague, Drug Metab. Dispos., 1995, 23, 377–382 CAS.
- Y. L. K. Tan, P. Pigeon, S. Top, E. Labbe, O. Buriez, E. A. Hillard, A. Vessieres, C. Amatore, W. K. Leonge and G. Jaouen, Dalton Trans., 2012, 41, 7537–7549 RSC.
- E. Meggers, Chem. Commun., 2009, 1001–1010 RSC.
- M. Gormen, P. Pigeon, S. Top, E. A. Hillard, M. Huche, C. G. Hartinger, F. de Montigny, M. A. Plamont, A. Vessieres and G. Jaouen, ChemMedChem, 2010, 5, 2039–2050 CrossRef PubMed.
- A. L. Laine, A. Clavreul, A. Rousseau, C. Tetaud, A. Vessieres, E. Garcion, G. Jaouen, L. Aubert, M. Guilbert, J. P. Benoit, R. A. Toillon and C. Passirani, Nanomedicine, 2014, 10, 1667–1677 CrossRef CAS PubMed.
- O. Payen, S. Top, A. Vessieres, E. Brule, M. A. Plamont, M. J. McGlinchey, H. Muller-Bunz and G. Jaouen, J. Med. Chem., 2008, 51, 1791–1799 CrossRef CAS PubMed.
- B. Kater, A. Hunold, H. G. Schmalz, L. Kater, B. Bonitzki, P. Jesse and A. Prokop, J. Cancer Res. Clin. Oncol., 2011, 137, 639–649 CrossRef CAS PubMed.
- A. Hunold, I. Neundorf, P. James, J. Neudorfl and H. G. Schmalz, Eur. J. Org. Chem., 2009, 2009, 4429–4440 CrossRef.
- P. James, J. Neudorfl, M. Eissmann, P. Jesse, A. Prokop and H.-G. Schmalz, Org. Lett., 2006, 8, 2763–2766 CrossRef CAS PubMed.
- A. C. Goncalves, T. S. Morais, M. P. Robalo, F. Marques, F. Avecilla, C. P. Matos, I. Santos, A. I. Tomaza and M. H. Garcia, J. Inorg. Biochem., 2013, 129, 1–8 CrossRef CAS PubMed.
- A. A. Simenel, E. A. Morozova, L. V. Snegur, S. I. Zykova, V. V. Kachal, L. A. Ostrovskaya, N. V. Bluchterova and M. M. Fomina, Appl. Organomet. Chem., 2009, 23, 219–224 CrossRef CAS.
- C. Wu, H. Ye, W. Bai, Q. Li, D. Guo, G. Lv, H. Yan and X. Wang, Bioconjugate Chem., 2011, 22, 16–25 CrossRef CAS PubMed.
- P. Zhang and H. Huang, Dalton Trans., 2018, 47, 14841–14854 RSC.
- A. Dorcier, P. J. Dyson, C. Gossens, U. Rothlisberger, R. Scopelliti and I. Tavernelli, Organometallics, 2005, 24, 2114–2123 CrossRef CAS.
- A. Dorcier, W. H. Ang, S. Bolano, L. Gonsalvi, L. Juillerat-Jeannerat, G. Laurenczy, M. Peruzzini, A. D. Phillips, F. Zanobini and P. J. Dyson, Organometallics, 2006, 25, 4090–4096 CrossRef CAS.
- A. F. A. Peacock, A. Habtemariam, R. Fernandez, V. Walland, F. P. A. Fabbiani, S. Parsons, R. E. Aird, D. I. Jodrell and P. J. Sadler, J. Am. Chem. Soc., 2006, 128, 1739–1748 CrossRef CAS PubMed.
- B. Cebrian-Losantos, A. A. Krokhin, I. N. Stepanenko, R. Eichinger, M. A. Jakupec, V. B. Arion and B. K. Keppler, Inorg. Chem., 2007, 46, 5023–5033 CrossRef CAS PubMed.
- M. Hanif, A. A. Nazarov, C. G. Hartinger, W. Kandioller, M. A. Jakupec, V. B. Arion, P. J. Dyson and B. K. Keppler, Dalton Trans., 2010, 39, 7345–7352 RSC.
- A. F. A. Peacock, S. Parsons and P. J. Sadler, J. Am. Chem. Soc., 2007, 129, 3348–3357 CrossRef CAS PubMed.
- S. H. van Rijt, A. J. Hebden, T. Amaresekera, R. J. Deeth, G. J. Clarkson, S. Parsons, P. C. McGowan and P. J. Sadler, J. Med. Chem., 2009, 52, 7753–7764 CrossRef CAS PubMed.
- A. F. A. Peacock, M. Melchart, R. J. Deeth, A. Habtemariam, S. Parsons and P. J. Sadler, Chem.–Eur. J., 2007, 13, 2601–2613 CrossRef CAS PubMed.
- M. Hanif, H. Henke, S. M. Meier, S. Martic, M. Labib, W. Kandioller, M. A. Jakupec, V. B. Arion, H.-B. Kraatz, B. K. Keppler and C. G. Hartinger, Inorg. Chem., 2010, 49, 7953–7963 CrossRef CAS PubMed.
- C. A. Riedl, L. S. Flocke, M. Hejl, A. Roller, M. H. M. Klose, M. A. Jakupec, W. Kandioller and B. K. Keppler, Inorg. Chem., 2017, 56(1), 528–541 CrossRef CAS PubMed.
- S. M. Meier, M. Hanif, Z. Adhireksan, V. Pichler, M. Novak, E. Jirkovsky, M. A. Jakupec, V. B. Arion, C. A. Davey, B. K. Keppler and C. G. Hartinger, Chem. Sci., 2013, 4, 1837–1846 RSC.
- Y. Fu, M. J. Romero, A. Habtemariam, M. E. Snowden, L. Song, G. J. Clarkson, B. Qamar, A. M. Pizarro, P. R. Unwin and P. J. Sadler, Chem. Sci., 2012, 3, 2485–2494 RSC.
- W. Ginzinger, G. Muhlgassner, V. B. Arion, M. A. Jakupec, A. Roller, M. Galanski, M. Reithofer, W. Berger and B. K. Keppler, J. Med. Chem., 2012, 55, 3398–3413 CrossRef CAS PubMed.
- W. F. Schmid, R. O. John, G. Muhlgassner, P. Heffeter, M. A. Jakupec, M. Galanski, W. Berger, V. B. Arion and B. K. Keppler, J. Med. Chem., 2007, 50, 6343–6355 CrossRef CAS PubMed.
- W. F. Schmid, R. O. John, V. B. Arion, M. A. Jakupec and B. K. Keppler, Organometallics, 2007, 26, 6643–6652 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |