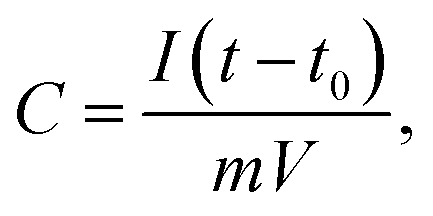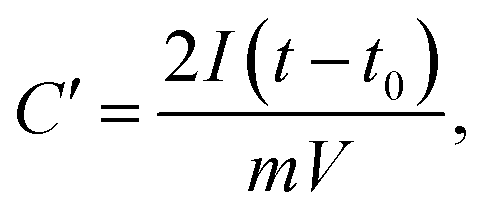DOI:
10.1039/C8RA07587E
(Paper)
RSC Adv., 2019,
9, 6419-6428
Preparation of cellulose acetate derived carbon nanofibers by ZnCl2 activation as a supercapacitor electrode†
Received
12th September 2018
, Accepted 15th February 2019
First published on 22nd February 2019
Abstract
Porous carbon nanofibers are fabricated by one-step carbonization and activation of electrospun cellulose acetate (CA) nanofibres. Electrospun CA nanofibers were obtained by the electrospinning of a CA/DMAC/acetone solution, followed by deacetylation in NaOH/ethanol solution. One-step carbonization and activation was achieved by dipping the as-spun fibers in ZnCl2 solution, followed by one-step high temperature treatment. The effects of the concentration of the dipping solution on the microstructure of the CA-based carbon nanofibers (CACNFs), including the morphology, crystal structure, porous structure, specific surface area and surface chemical properties, have been investigated. The coating of ZnCl2 effectively improves the thermal stability of electrospun CA nanofibers and obviously enhances the oxygen-containing surface groups of the CACNFs. The CACNFs have a norrow pore size distribution (0.6–1.2 nm) and a high specific surface area (∼1188 m2 g−1). Electrochemical performances of the CACNFs were evaluated as supercapacitor electrodes in 6 M KOH solution. The CACNFs demonstrate high specific capacitance (202 F g−1 at 0.1 A g−1) and excellent rate capability (61% of the retention from 0.1 to 20 A g−1). After 5000 cycles of the electrode, the capacitance is maintained at 92%, and the coulombic efficiency is close to 100%, showing high electrochemical stability and reversibility. The renewable features and excellent performance make CACNFs quite a promising alternative to efficient supercapacitor electrodes for energy storage applications.
1. Introduction
With the excessive consumption of fossil fuels, new energy technology and secondary energy (electric energy) storage and conversion devices are urgently required to ease environmental pollution and the energy crisis.1,2 Supercapacitors, as a new-type electricity storage device, possess much higher power densities, much faster charge–discharge rate and a longer cycle life than batteries, showing great potential for portable electronics, electrical vehicles and solar cells.3,4 Electrodes play a key role in deciding the electrochemical performance of supercapacitors.5,6 Porous carbon materials are considered ideal electrode materials for supercapacitors because of their large specific surface area, relatively high conductivity and favorable chemical/thermal stabilities.7,8 In the past few years, porous carbons in various patterns, for example, activated carbons, carbon nanofibers, graphene and carbon aerogels, have been studied extensively.9–12 Among them, porous carbon nanofibers prepared by electrospinning have attracted interest as electrode materials for supercapacitors because of their ultrafine diameter, three-dimensional network structure, high surface area and diversified pore structure.13 Up to now, most of electrospun porous carbon nanofibers (ECNFs) are fabricated using polymers as carbon precursors that rely on unsustainable resources (fossil fuels), including polyacrylonitrile (PAN), phenolic resin, polyvinylpyrrolidone, polyvinyl alcohol, pitch, etc.14–16 From the cost and sustainability point of view, renewable resources should be considered as future alternative to these synthetic polymers and pitch. Cellulose as a renewable resource can be obtained from the plant and the product of fermentation by microorganisms. Therefore, cellulose could be an inexhaustible source for the synthesis of carbon nanofibers.17,18 However, cellulose has poor solubility in the most common solvents, making it hard to fabricate fibers by electrospinning.19,20 Cellulose acetate (CA), a kind of cellulose derivatives, by contrast, is much easier to dissolve and electrospin. It is more promising and has been widely used as precursor for carbon nanofibers by electrospinning.21,22 To achieve porous carbon fibers, polymer blending and activation methods have been widely used. For example, Cai et al.23 have prepared inter-bonded carbon nanofibers as supercapacitor electrodes using neat CA as precursor by CO2 activation. Time-consuming and complicated post-activation was involved, and a relatively limited specific surface area was obtained. Considering researches on the CA based porous carbon nanofibers as supercapacitors is few, exploiting a convenient and feasible approach to CA-based porous carbon nanofibers (CACNFs) with high specific surface area is still of significance for development of supercapacitors.
In the present work, porous carbon nanofibers were fabricated by one-step carbonization and activation of electrospun CA nanofibers with ZnCl2 as activating agent, which was coated on the surface of deacetylated electrospun CA fibers by dipping in ZnCl2 solution. The effects of concentration of ZnCl2 solution on the microstructure of resultant carbon nanofibers, including the morphology, porous structure and surface chemical properties, were investigated. Results showed that coating of ZnCl2 improved the thermal stability of the deacetylated electrospun CA fibers. The ZnCl2 activation of rendered carbon nanofibers a norrow pore size distribution (0.6–1.2 nm) and a high specific surface area (∼1188 m2 g−1). The electrochemical performances of the resulting porous carbon nanofibers as supercapacitor electrode were investigated using three- and two-electrode in 6 M KOH electrolyte. Results showed that CACNFs had a high specific capacitance (202 F g−1 at 0.1 A g−1) and excellent rate capability (61% of the retention from 0.1 to 20 A g−1). The relationship between electrochemical performance and structure of CACNFs was discussed.
2. Experimental
2.1. Materials
Dimethylacetamide (DMAC, 99.5%), CA (Mw = 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 39.5 wt% aceyl groups) and acetone were from Shanghai Macklin Biochemical Co. Ltd. Shanghai, China. ZnCl2 was from Tianjin Kemiou Chemical Reagent Co. Ltd. Tianjin, China. All the chemical reagents in this work were of analytical grade and used without further purification.
000, 39.5 wt% aceyl groups) and acetone were from Shanghai Macklin Biochemical Co. Ltd. Shanghai, China. ZnCl2 was from Tianjin Kemiou Chemical Reagent Co. Ltd. Tianjin, China. All the chemical reagents in this work were of analytical grade and used without further purification.
2.2. Preparation of porous carbon nanofibers
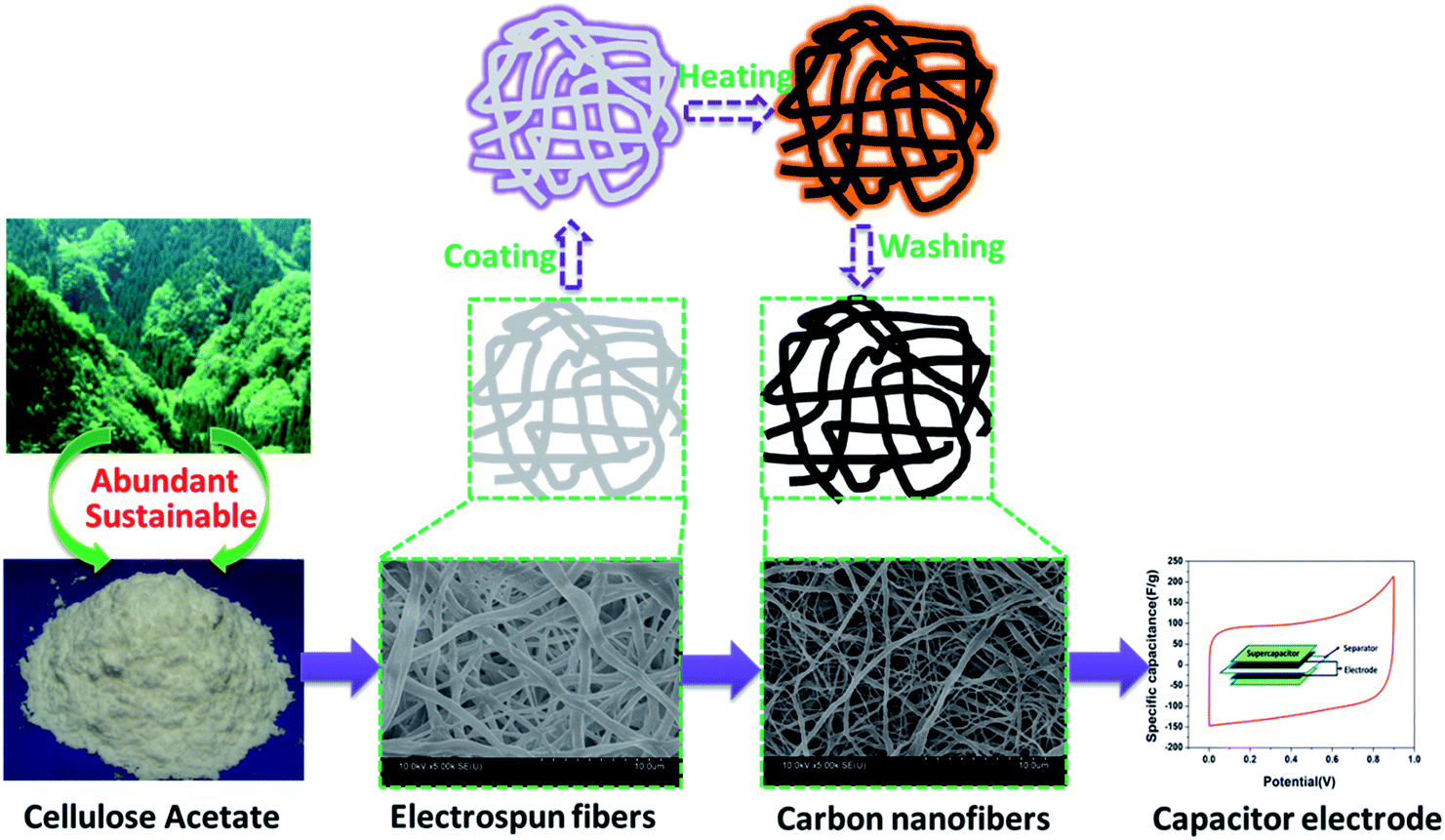
The pure CA electrospinning solution (CA = 8 wt%) was prepared by dissolving 2 g CA in 8 g DMAC/acetone-mixed solvent (the volume ratio of DMAC/acetone = 2:1) and stirring at 55 °C for 2 h. The as-prepared solution was electrospun using a purpose-made a single-nozzle electrospinner setup. During electrospinning, a positive voltage of 20 kV was applied to the needle, the feeding rate of spin dope was 2 mL h−1, the distance between the needle and fiber collection plate was 20 cm and the relative humidity of the electrospinning environment was 40–60%. Electrospun CA fibrous mats were then placed in a 0.05 M NaOH/ethanol solution at room temperature for 12 h to convert CA into cellulose. After being rinsed with deionized water, the fiber mats were immersed in a 2–20 wt% aqueous ZnCl2 solution for 2 hours, then the samples were dried in an oven at 80 °C for 12 hours. The pretreated fiber mat were stabilized under air flow using the following heating procedure: first, temperature was increased from room temperature to 200 °C at a heat rate of 3 °C min−1 and kept at 200 °C for 1 h; second, temperature was increased from 200 to 220 °C at a heat rate of 2 °C min−1 and kept at 220 °C for 1 h; finally, sample was cooled down to room temperature. The stabilized samples were carbonized at 700 °C for 1 h at a heat rate of 3 °C min−1 in flowing nitrogen. The as-carbonized fibers were treated with excess diluted hydrochloric acid and repeatedly washed by deionized water until the pH of the test filtrate reached about 7. The resultant porous carbon nanofibers were named CACNF-ZnCl2-X, where X corresponds to concentration of ZnCl2 solution, namely 2%, 5%, 10% and 20%. The sample without ZnCl2 activation was labeled as CACNF-0. The synthesis route to CACNFs is depicted in Fig. 1.
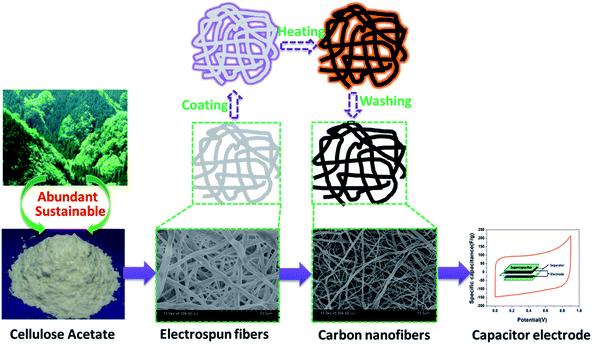 |
| | Fig. 1 A schematic illustration depicting the synthesis route to CA derived carbon nanofibers. | |
2.3. Characterization methods
The morphology of carbon fibers was characterized by scanning electron microscopy (SEM, Hitachi S4800, Japan). The crystallographic structure of carbon nanofiber was examined by X-ray diffraction (XRD, Rigaku, Japan) equipped with Cu-Kα radiation (λ = 0.15456 nm). The surface chemical species of carbon nanofibers were measured using a high-performance Al-Kα radiation (1486.6 eV, 15 kV) XPS (ESCALAB 250, ThermoFisher Scientific, USA). Nitrogen adsorption/desorption isotherms at 77 K were obtained by an automatic gas-adsorption system (JW-BK200C, JWGB SCI&TECH, China) to investigate specific surface area and the porous texture of the carbon nanofibers. The samples were degassed at 300 °C under vacuum for 6 h before the measurements. The specific surface area of the carbon nanofibers was calculated using the Brunauer–Emmett–Teller (BET) equation, and the pore size distribution was calculated using the BJH and HK method. The total pore volume (Vtotal) was estimated from single-point adsorption at a relative pressure P/P0 = 0.995. The micropore volume (Vmicro) and micropore surface area (Smicro) were calculated using the t-plot method.
2.4. Electrochemical measurements
All the electrochemical tests, including three-electrode electrochemical tests and two-electrode electrochemical tests, were performed using a CHI 660C (Shanghai Chenhua Apparatus Co. Ltd.) electrochemical workstation in 6 M KOH aqueous solution at room temperature. The resulting carbon fibrous-mats were cut directly into 10 × 10 mm square (about 1.5 mg) and then pressed on the foamed nickel at a pressure of 8 MPa for 10 s. A Pt plate and Hg/HgO electrode were used as counter electrode and reference electrode, respectively. Galvanostatic charge/discharge (GCD) was performed at a current density ranging from 0.1 to 10 A g−1, and cyclic voltammetry (CV) carried out at scan rate from 5 to 100 mV s−1. The potential window was 0.9 to 0 V versus Hg/HgO for all GCD and CV tests.
3. Results and discussion
3.1. Structural characterizations
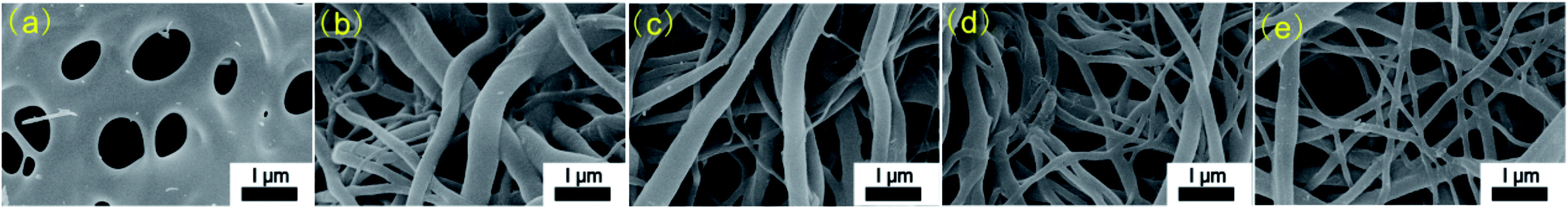
Fig. 2 shows the SEM images of CACNF-0, CACNF-ZnCl2-2%, CACNF-ZnCl2-5%, CACNF-ZnCl2-10% and CACNF-ZnCl2-20%, respectively. It can be seen that CACNFs without coating of ZnCl2 (Fig. 2a) still show melting phenomenon in spite of undergoing deacetylation and air-stabilization. It is speculated that the partial melting of CACNF is due to tincomplete deacetylation, which might be associated to a short soaking time in NaOH/ethanol solution. It is well known that a direct carbonized CA film undergoes melting, and the shape of the fiber cannot be maintained. Thus, the electrospun CA fibers are typically regenerated into stable cellulose by deacetylation with a NaOH/ethanol solution.24–27 The base-catalyzed deacetylation process primarily involves a two-step hydrolysis mechanism of the carboxylic acid ester (see ESI Fig. S1†). The commonly accepted explanation is that in the first step, OH− attacks the carbonyl groups in the acyl group to form a transition state until the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond is opened, which is a reversible process and the reaction process is slow. The second step is to remove the acetyl group, which is an instantaneous process.28,29 However, CACNF-ZnCl2-Xs exhibited an obviously-improved fibrous morphology. It indicates that ZnCl2 contributes to the conversion of the carbonyl group in the acyl group to a transition state. In addition, ZnCl2 can also play an active role in improving thermal stability and structural stability by ion crosslinking of macromolecules and promoting dehydrogenation. The carbonized fiber films (CACNF-ZnCl2-20%) has a certain extent flexibility and mechanical strength (see ESI Fig. S2†). The diameter of CACNF-ZnCl2-Xs was obtained by randomly measuring 100 carbon fibers and the average diameter was calculated. The average fiber diameter is 338 nm for CACNF-ZnCl2-2%, 293 nm for CACNF-ZnCl2-5%, 260 nm for CACNF-ZnCl2-10% and 213 nm for CACNF-ZnCl2-20%, respectively. As the concentration of ZnCl2 solution increases, the carbon fiber becomes a little smaller. It may be because the ZnCl2 coating restrains softening by surface solidification. ZnCl2 can enhance structure stability by promoting oxidative cross-linking of as-spun fibers during the pre-oxidation stage.30 Fibers are entangled with each other to form a large number of contact points and open porous network, which can provide a short transfer path for the electrolyte ions.
O double bond is opened, which is a reversible process and the reaction process is slow. The second step is to remove the acetyl group, which is an instantaneous process.28,29 However, CACNF-ZnCl2-Xs exhibited an obviously-improved fibrous morphology. It indicates that ZnCl2 contributes to the conversion of the carbonyl group in the acyl group to a transition state. In addition, ZnCl2 can also play an active role in improving thermal stability and structural stability by ion crosslinking of macromolecules and promoting dehydrogenation. The carbonized fiber films (CACNF-ZnCl2-20%) has a certain extent flexibility and mechanical strength (see ESI Fig. S2†). The diameter of CACNF-ZnCl2-Xs was obtained by randomly measuring 100 carbon fibers and the average diameter was calculated. The average fiber diameter is 338 nm for CACNF-ZnCl2-2%, 293 nm for CACNF-ZnCl2-5%, 260 nm for CACNF-ZnCl2-10% and 213 nm for CACNF-ZnCl2-20%, respectively. As the concentration of ZnCl2 solution increases, the carbon fiber becomes a little smaller. It may be because the ZnCl2 coating restrains softening by surface solidification. ZnCl2 can enhance structure stability by promoting oxidative cross-linking of as-spun fibers during the pre-oxidation stage.30 Fibers are entangled with each other to form a large number of contact points and open porous network, which can provide a short transfer path for the electrolyte ions.
 |
| | Fig. 2 SEM images of carbon fibers. (a) CACNF-0, (b) CACNF-ZnCl2-2%, (c) CACNF-ZnCl2-5%, (d) CACNF-ZnCl2-10%, (e) CACNF-ZnCl2-20%. | |
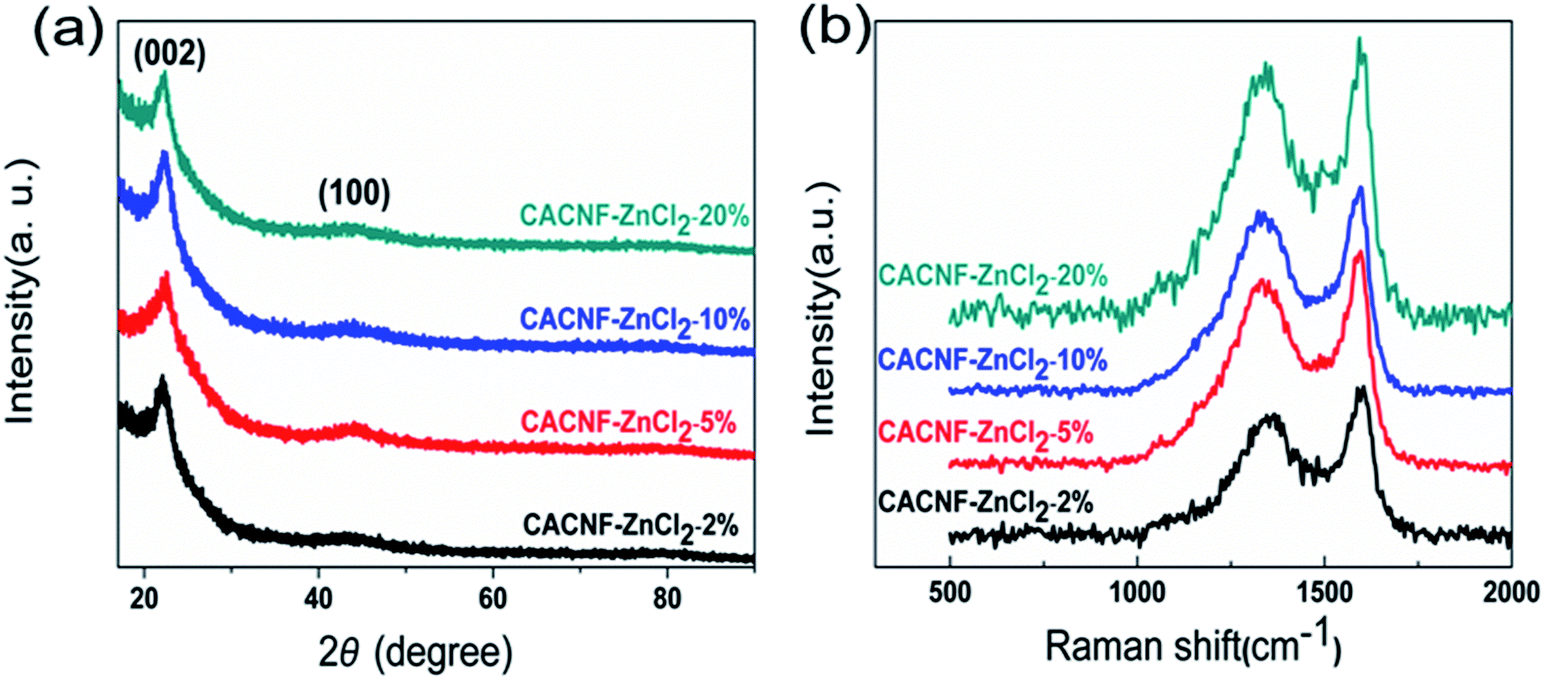
To reveal the influence of activation process on the crystal structure, XRD measurement was performed. Fig. 3a shows XRD profiles of all samples. All samples present an obviously diffraction peak at around 23° and an inconspicuous peak at around 44°, which are assigned to the disordered graphitic (002) plane and (100) plane.31–34 The peak at 20–25° range is broad. The peak at 44° is weak. Both are typical diffraction peaks of the amorphous carbons. With the increase of ZnCl2 solution concentration, the intensity and angle of both diffraction peaks do not change significantly. This phenomenon shows that ZnCl2 activation does not cause significant influence on the order degree of carbon. Raman spectroscopy is employed to characterize the degree of graphitization of CACNFs, as shown in Fig. 3b. It shows two characteristic bands, including a D band near 1340 cm−1 and a G band at 1580 cm−1.35–38 Among them, D-bands is related to disorders, including defects, curved sheets, and dangling bonds in carbon structures, and G-bands is associated with the in-plane vibration of the sp2 hybridized carbon atoms in a graphitic layer.39–42 In addition, the relative intensity of the D band and G band (ID/IG) could reflect the degree of disorder in the graphitic structure.43 The smaller the ID/IG ratio is, the higher the degree of graphitization is. The values of ID/IG for CACNF-ZnCl2-2%, CACNF-ZnCl2-5%, CACNF-ZnCl2-10% and CACNF-ZnCl2-20% are 0.84, 0.93, 0.92, and 0.99, respectively. These results indicate that ZnCl2 activation brings about more defects and turbostratic structure.
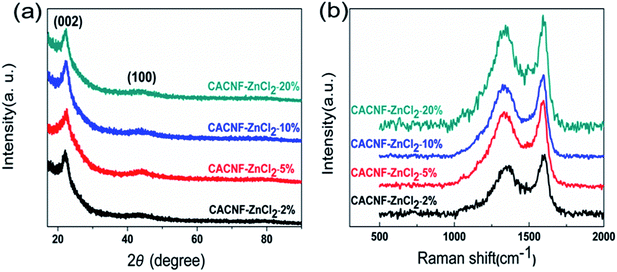 |
| | Fig. 3 (a) XRD and (b) Raman profiles of all samples. | |
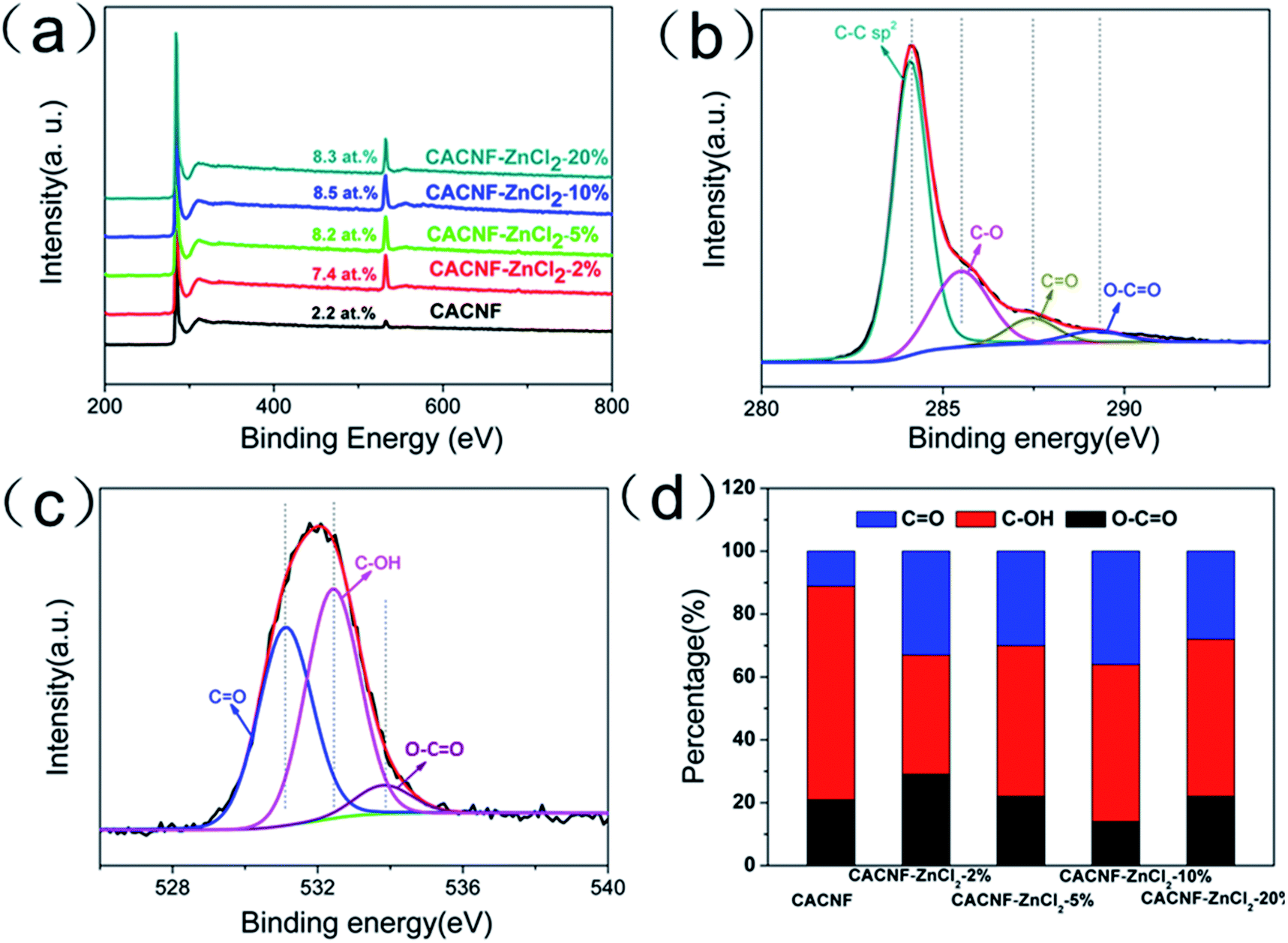
A large number of studies have shown that the functional groups on the surface of the material are closely related to its electrochemical properties. The surface chemical compositions of all samples were analyzed by XPS. XPS survey spectra are displayed in Fig. 4a. All the samples have two obvious peaks at around 283.8 and 532.4 eV that correspond to characteristic spectra of C 1s and O 1s orbital, respectively. No Zn peak is observed, demonstrating the Zn species has been removed completely. The O content is 2.2 at% for CACNF-0, 7.4 at% for CACNF-ZnCl2-2%, 8.2 at% for CACNF-ZnCl2-5%, 8.5 at% for CACNF-ZnCl2-10% and 8.3 at% for CACNF-ZnCl2-20%, respectively. The activated CACNFs have rich oxygen-containing functionalities on the surface, much higher than that of non-activated CACNFs. It may be attributed to that the ZnCl2 costing prevents oxygen from escaping. The further increase of ZnCl2-activator concentration did not bring about more oxygen-containing groups. It is because ZnCl2 has a low melting point (365 °C), which enables a limited amount of ZnCl2 to achieve complete coating at low temperature.30,44–46 Fig. 4b shows the fitting results of the C 1s peaks of CACNFs. The results show the presence of CC (284.5 eV, graphite carbon), C–O (286.5 eV), CO (287.6 eV) and OC–O (289.2 eV) bond, suggesting the partial graphitization and the existence of O functional groups, which also coincide with the XRD results. The O 1s spectrum can be fitted to three peaks as shown in Fig. 4c. There are peaks at the binding energies of 531.3 ± 0.2, 532.4 ± 0.3, 533.9 ± 0.9, which are assigned to CO quinone groups, C–OH phenol groups and/or C–O–C ether groups, and COOH carboxylic groups, respectively.47–49 Fig. 4d shows the change in the percentage of oxygen-containing O functional groups for all samples. It can be found that as the ZnCl2 increases, the percentage of CO groups are substantially unchanged, while the C–OH group presented a slight increasing trend. It has been widely revealed that the oxygen-containing functional groups facilitate the electrolyte ion diffusion and the CO groups contribute extra pseudocapacitance by providing hydrophilic polar sites and the electrochemical redox activity.1,44,46
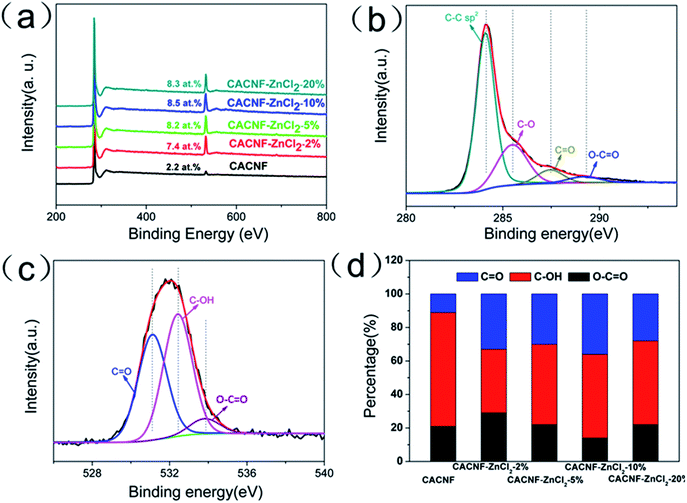 |
| | Fig. 4 (a) XPS survey spectra of all samples. (b) High-resolution XPS C 1s spectra. (c) High-resolution XPS O 1s spectra. (d) Percentage of O-containing species. | |
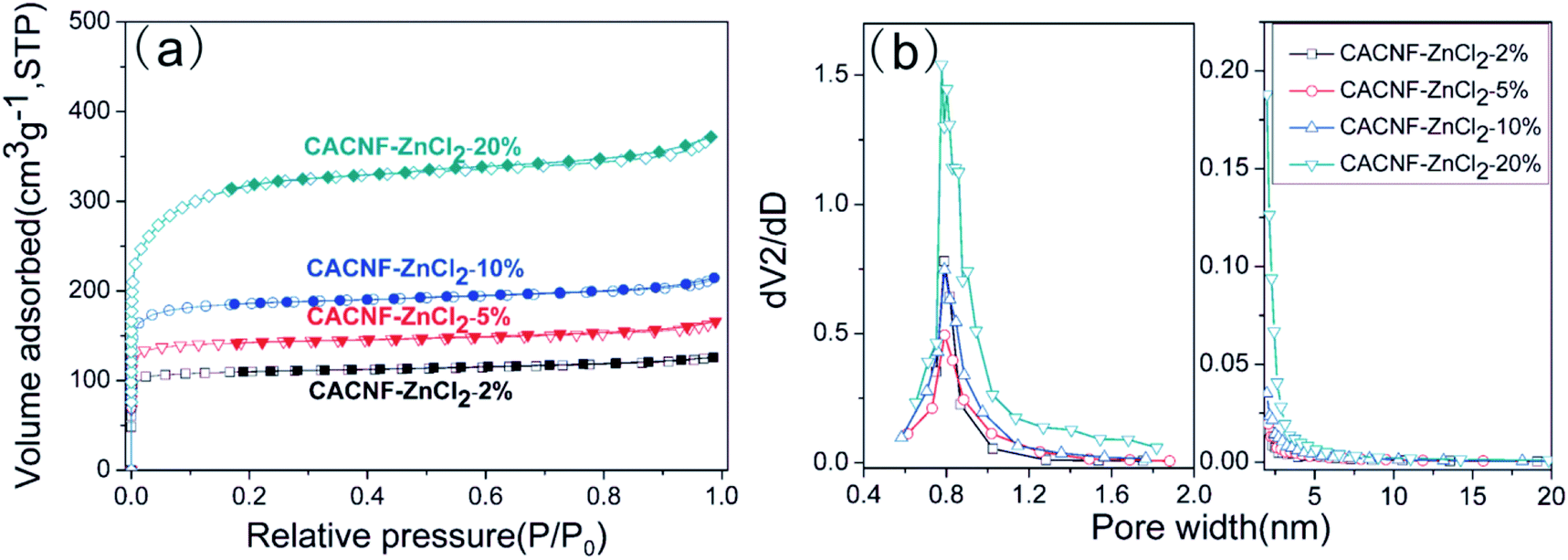
The textural properties and porosities of CACNF-ZnCl2 are verified by N2 adsorption–desorption isotherms and the corresponding pore size distribution curves. Fig. 5a shows the N2 adsorption/desorption isotherms of CACNF-ZnCl2-Xs. All the samples exhibit type-I adsorption isotherms according to the International Union of Pure and Applied Chemistry (IUPAC) nomenclature, which are typical pore characteristics for microporous materials.50–52 The mesopore/micropore size distribution was calculated using the BJH and HK method and showed in Fig. 5b. It can be seen that the micropores are mainly distributed between 0.6 and 1.2 nm. With the increase of the concentration of ZnCl2 solution, micropores turn more and wider. CACNF-ZnCl2-20% displays obvious small mesopores. It suggests that ZnCl2-activation not only creates more new micropores, but also widens the as-formed pores. Some reports have revealed that 0.6–1.2 nm micropores favor for charge accumulation in aqueous electrolytes.53 The textural parameters of all samples are listed in Table 1. As the concentration of ZnCl2 solution increases, the SBET of the as-prepared carbon nanofibers increases from 420 to 551, 716 and 1188 m2 g−1, and the Vt grows from 0.195 to 0.257, 0.332 and 0.575 cm3 g−1. The specific surface area and pore volume are dependent on ZnCl2 dosage. It is consistent with that of the ZnCl2 activated granular carbons.45,54–56 Interestingly, the ratio of relative surface area and pore volume produced by the micropores is not increased monotonously with ZnCl2 dosage, the microporosity of CACNF-ZnCl2-10% and CACNF-ZnCl2-20% are lower than CACNF-ZnCl2-5%, which may be attributed to collapse of micropore caused by ZnCl2 activation.
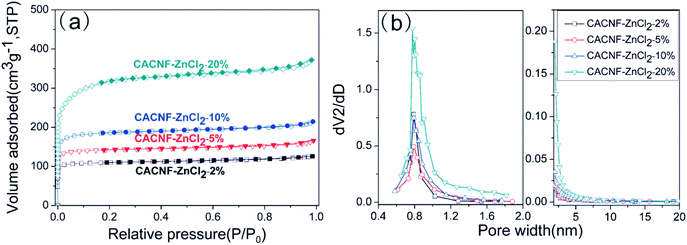 |
| | Fig. 5 (a) N2 adsorption/desorption isotherms and (b) pore size distributions for all samples. | |
Table 1 Sample pore structure parametersa
| Sample |
SBET (m2 g−1) |
Sm (m2 g−1) |
Sμ (%) |
Vt (cm3 g−1) |
Vm (cm3 g−1) |
Vμ (%) |
Dp (nm) |
| SBET: BET specific surface area; Sm: micropore surface area; Sμ (%): (Sm/SBET) × 100; Vt: total pore volume, Vm: micropore volume; Vμ (%): (Vm/Vt) × 100, Dp: mean pore diameter. |
| CACNF-ZnCl2-2% |
420 |
390 |
92.8 |
0.195 |
0.157 |
68.1 |
1.85 |
| CACNF-ZnCl2-5% |
552 |
513 |
92.9 |
0.257 |
0.203 |
79.0 |
1.86 |
| CACNF-ZnCl2-10% |
717 |
656 |
91.6 |
0.332 |
0.261 |
75.6 |
1.85 |
| CACNF-ZnCl2-20% |
1188 |
999 |
84.0 |
0.575 |
0.406 |
70.6 |
1.94 |
3.2. Electrochemical performance
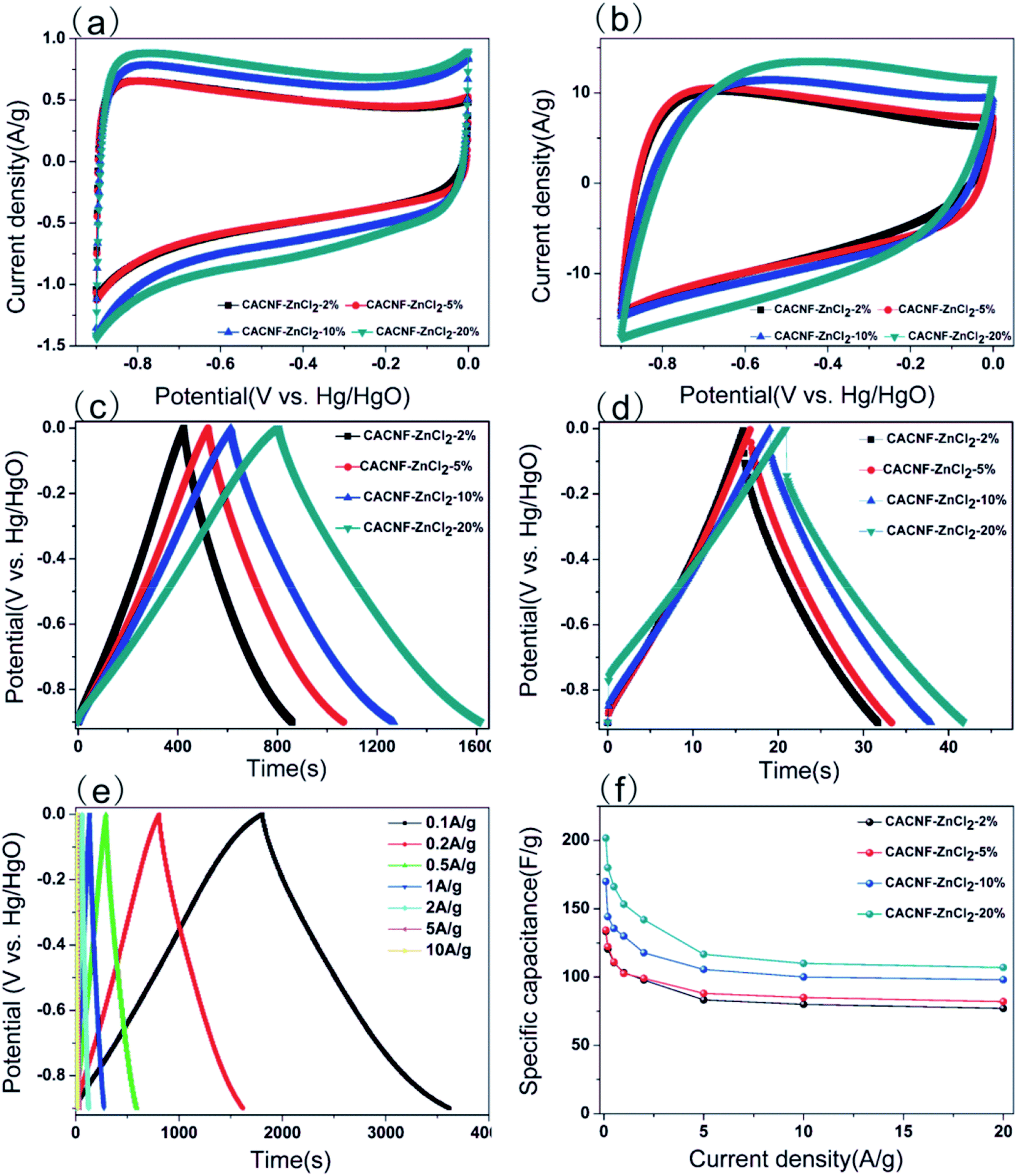
To assess the capacitive performance of CACNFs, the cyclic voltammograms (CV) tests and galvanostatic charge/discharge (GCD) were measured using a three-electrode system in 6 M KOH solution. Because CACNFs still maintain certain flexibility after ZnCl2 activation, it can be used as a free-standing electrode for supercapacitor without the addition of binders. The cyclic voltammograms (CV) of the carbon nanofiber electrodes were recorded in the potential window from −0.9 to 0 V at scan rates of 5 and 100 mV s−1, as shown in Fig. 6a and b, respectively. It is worth noting that the CV curves of all CACNFs samples display nearly rectangular shapes, which show a typical characteristic of energy storage in the double layer mechanism. In addition, the induced current and the loop areas of CACNF-ZnCl2-20% are significantly larger than other samples, indicating that the capacitance is substantially improved by ZnCl2 activation. However, the CV curve of all samples showed a slightly deformed rectangular shape at a large scan rate, unlike a standard electric double layer capacitor, indicating that the presence of the faradaic reaction is related to the surface oxygen-containing functional groups.1,10,57–59 The excellent electrochemical performance of CACNFs is attributed to the large surface area and narrow pore size distribution. The specific capacitance of electrode can be estimated from galvanostatic charging/discharging curves. Fig. 6c and d show the GCD curves of carbon nanofiber electrodes at current densities of 0.2 A g−1 and 5 A g−1, respectively. It can be observed that the charge–discharge curves of the carbon nanofiber electrodes activated by different dosage of ZnCl2 are approximate symmetrical triangular shape with linear voltage vs. time profile.60 The CACNF-ZnCl2-20% electrode shows maximum discharge times, either at high current densities or at low current densities, indicating the best electrochemical performance. Fig. 6e shows the galvanostatic charge/discharge curves for CACNF-ZnCl2-20% electrode carried out in the potential window of −0.9 to 0 V at different current densities of 0.1 to 10 A g−1. All the charge and discharge curves are a nearly symmetric triangle shape, indicating that the CACNF-ZnCl2-20% electrode has high reversibility at high current loading. In addition, only slight IR drop is observed at high current loading, indicating the high-rate capability and little internal resistance.19,61 According to the GCD measurements, the gravimetric capacitance C (F g−1) was calculated via following formula:
where I is the current (A), t0 is starting time of discharge, t is ending time of discharge, m is the mass of square electrode and V is applied potential difference. Fig. 6f shows the specific capacitance change of all electrodes at the current density from 0.1 to 20 A g−1. The specific capacitance value is the average value of three tests. At current density of 0.1 A g−1, the specific capacitance was calculated to be 133 F g−1, 134 F g−1, 170 F g−1 and 202 F g−1 for CACNF-ZnCl2-2%, CACNF-ZnCl2-5%, CACNF-ZnCl2-10% and CACNF-ZnCl2-20%, respectively. The results clearly show that the concentration of ZnCl2 solution has a significant effect on the specific capacitance of CACNFs. It is due to that ZnCl2 activation increases the specific surface area of the carbon nanofibers, and thus provides more active sites. The specific capacitance of all electrodes decreases as the current density of all electrodes increases. It is common for carbon-based electrodes and is mainly because that the small micropores restricts the entrance of ions and reduce surface availability of the pores. It has to be mentioned that the specific capacitance becomes stable for all samples when the current density is higher than 2 A g−1. When the current density increased from 0.1 to 20 A g−1, the capacitance decreases to 77 F g−1 (58%), 82 F g−1 (61%), 98 F g−1 (58%) and 107 F g−1 (53%) for CACNF-ZnCl2-2%, CACNF-ZnCl2-5%, CACNF-ZnCl2-10%, and CACNF-ZnCl2-20% electrode, respectively, indicating an excellent rate capability.62–64 It should be due to the nanometer-scaled fiber diameter and abundant oxygen-containing groups, which shorten the ion transport distance and accelerate ion transfer/diffusion. CACNF-ZnCl2-5% and CACNF-ZnCl2-10% have high capacity retention, maybe because of the more mesoporoses, which reduce the transfer/diffusion resistance of electrolyte ions.
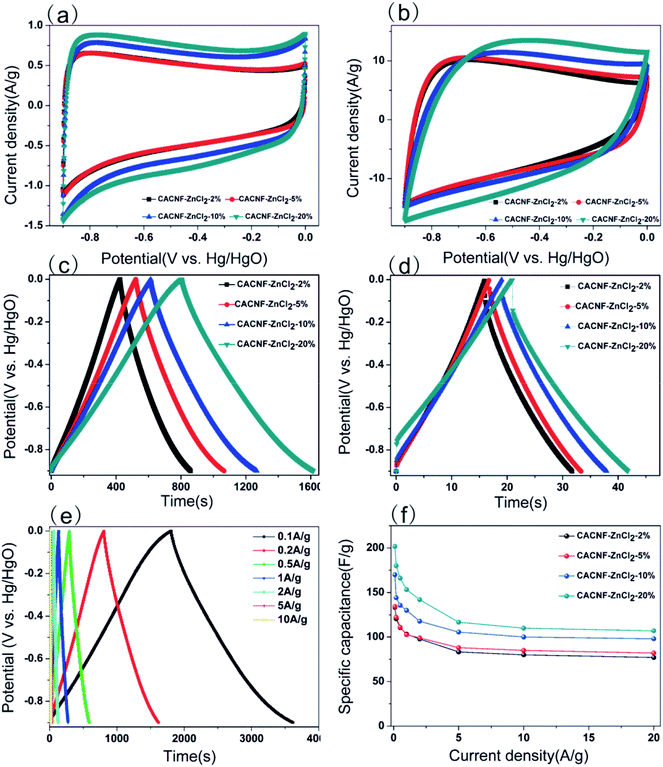 |
| | Fig. 6 Electrochemical performance from three-electrode test in 6 M KOH. (a) CV curves for all samples at scan rate of 5 mV s−1; (b) CV curves for all samples at scan rate of 100 mV s−1; (c) charge and discharge curves of all samples at 0.2 A g−1. (d) Charge and discharge curves of all samples at 5 A g−1; (e) charge and discharge curves of CACNF-ZnCl2-20% at various current densities; (f) variation of the specific capacitance of all samples as a function of current density. | |
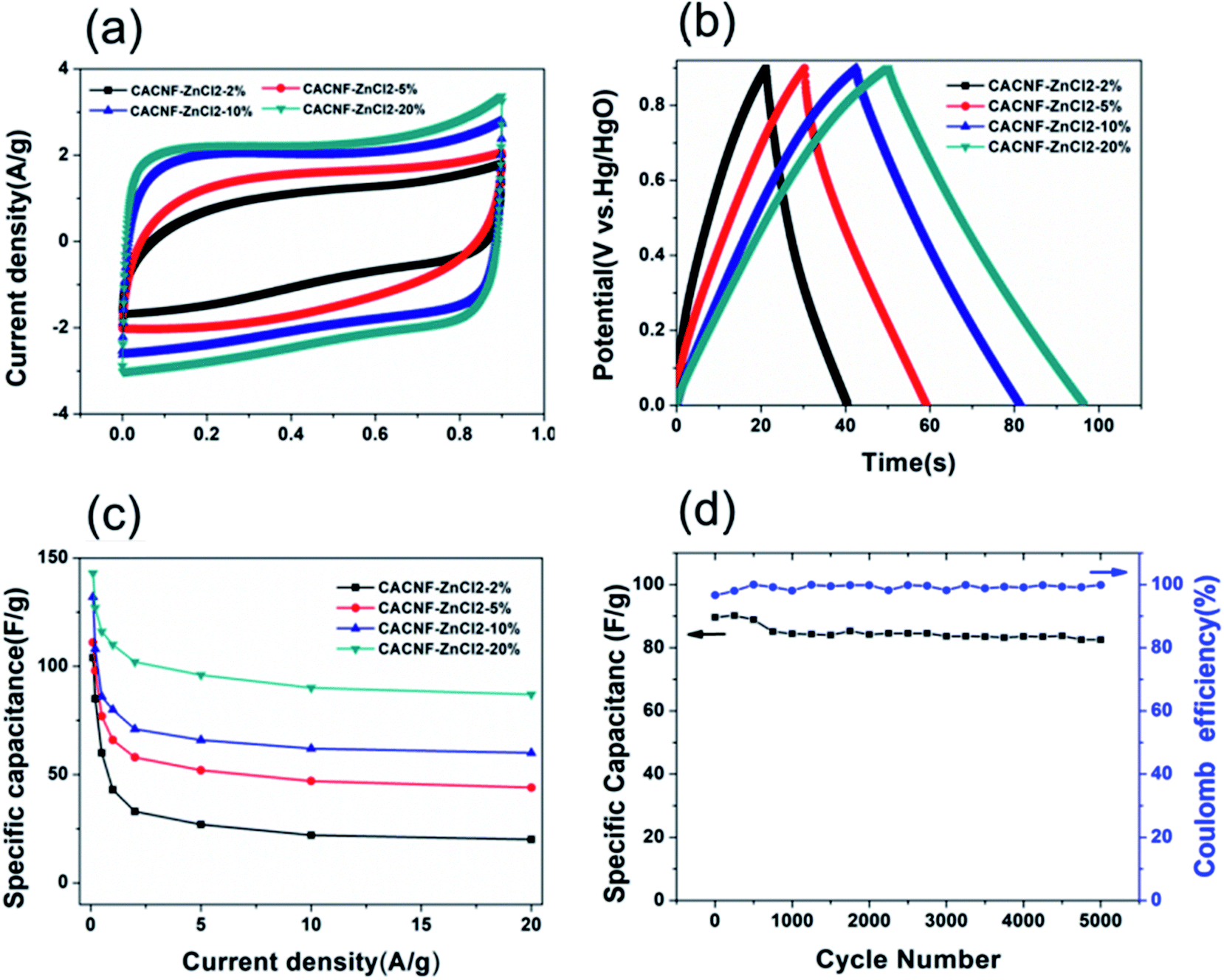
In order to obtain results closer to the real supercapacitor, the electrochemical test was performed using a two-electrode system, and the results are shown in Fig. 7. The cyclic voltammograms (CV) tests and galvanostatic charge/discharge (GCD) were measured in 6 M KOH solution. The CV curves of the all samples at a scan rate of 50 mV s−1 are shown in Fig. 7a. It is obvious that the shapes of all CV curves were similar with the rectangle, indicating that the sample exhibits typical carbon electrochemical behavior. The CACNF-ZnCl2-20% sample displays the largest plateau currents and the loop areas, which suggests that the significantly improved capacitance by ZnCl2 activation. Fig. 7b shows the GCD curves at 1 A g−1. All the charge/discharge curves are almost symmetrical, indicating an ideal capacitive characteristics and well-reversible capacitive characteristics. The specific capacitance was calculated via following equation:
where
I is the current (A),
t0 is starting time of discharge,
t is ending time of discharge,
m is the mass of square electrode and
V (0–0.9V) is applied potential difference.
Fig. 7c shows the variation of specific capacitance with increasing current density for all electrodes. It can be seen that the specific capacitance increases with the increase of the activator, and the CACNF-ZnCl
2-20% have a higher specific capacitance at different current densities, which is consistent with three-electrode system test results. The specific capacitance decreases with the increase of the current density, which is a common feature of porous carbons. At current density of 0.1 A g
−1, the specific capacitance was calculated to be 104 F g
−1 (CACNF-ZnCl
2-2%), 111 F g
−1 (CACNF-ZnCl
2-5%), 132 F g
−1 (CACNF-ZnCl
2-10%) and 143 F g
−1 (CACNF-ZnCl
2-20%), respectively. The highest specific capacitance of MRCNFs should benefit from the higher specific surface area and the 1D structure of carbon nanofibers. Cycle stability is a crucial indicator of supercapacitors in practical applications.
Fig. 7d shows a charge–discharge curve of CACNF-ZnCl
2-20% repeated 5000 cycles at a current density of 5 A g
−1. After 5000 continuous cycles, the capacitance maintains 92%, indicating good cycle stability. In addition, the coulombic efficiencies of all electrodes are stable and close to 100%, demonstrating the high electrochemical stability and reversibility.
Table 2 shows the specific capacitance and electrochemical stability of cellulose-derived carbon materials reported in the literature. The specific capacitance of the post-treated electrode material is mostly between 50 F g
−1 and 200 F g
−1. The CACNFs prepared in this work have relatively superior specific capacitance and cycle stability as the electrode material of the supercapacitor, showing greater potential for the development of electrode materials in the future.
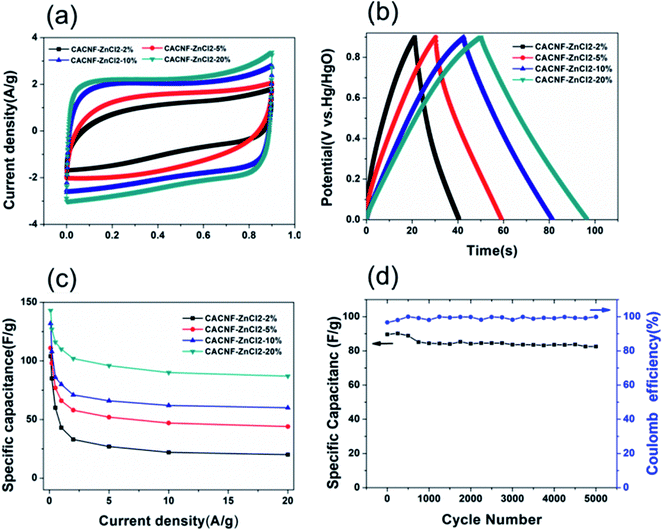 |
| | Fig. 7 Electrochemical performance from two-electrode test in 6 M KOH. (a) CV curves at 5 mV s−1; (b) galvanostatic charge/discharge curves at 1 A g−1; (c) specific capacitance at different current densities; (d) cycling stability of CACNF-ZnCl2-20% at 5 A g−1. | |
Table 2 Specific capacitance and cycle stability of cellulose carbon materials are reported in the literaturea
| Electrode materials |
Electrolyte |
Cs |
Current density |
Stability |
Cycles |
Ref. |
| Cs: specific capacitance; CNFs: carbon nanofibers; NFs: nanofibers; CA: cellulose acetate; NCFs: nanocomposite fibers; CNTs: carbon Nanotubes; PPy: polypyrrole; PAN: polyacrylonitrile; MMC: Micro- and meso-porous carbon; 3D: three-dimensional. |
| CNFs from partially hydrolyzed CA NFs |
6 M KOH |
241 F g−1 |
1 A g−1 |
99% (200 mV s−1) |
10000 |
23 |
| NCFs from cellulose and CNTs |
6 M KOH |
145 F g−1 |
10 A g−1 |
94% (2 A g−1) |
1000 |
24 |
| CNFs from PPy-coated cellulose NFs |
6 M KOH |
171 F g−1 |
1 A g−1 |
98% (100 mV s−1) |
5000 |
25 |
| CNFs from PAN/CA |
6 M KOH |
280 F g−1 |
0.1 A g−1 |
96.8% (1 A g−1) |
2000 |
63 |
| 3D porous carbon from activated cellulose |
6 M KOH |
162 F g−1 |
0.5 A g−1 |
99.8% (10 A g−1) |
5000 |
65 |
| Carbon material from natural cellulose |
5 M KCl |
107 F g−1 |
1 A g−1 |
90% (1 A g−1) |
2000 |
66 |
| Cellulose-derived composite electrodes |
6 M KOH |
101 F g−1 |
5 mV s−1 |
96.6% (1.5 A g−1) |
2000 |
67 |
| MMC via carbonizing cellulose aerogel |
1 M H2SO4 |
160 F g−1 |
0.2 A g−1 |
90% (1 A g−1) |
10000 |
68 |
| 3D interwoven structural film prepared from graphene and cellulose |
1 M H2SO4 |
120 F g−1 |
1 mV s−1 |
99% (50 mV s−1) |
5000 |
69 |
| CACNFs from activated CA nanofiber |
6 M KOH |
143 F g−1 |
0.1 A g−1 |
92% (5 A g−1) |
5000 |
This work |
4. Conclusions
In this work, we have successfully prepared microporous carbon nanofibers by one-step carbonization and activation of electrospun CA nanofibers. The ZnCl2 coating not only plays a positive role in maintaining the fibrous morphology but also significantly increases the surface oxygen-containing groups. With the concentration of ZnCl2 increasing, the specific surface area and pore volume increase gradually. Dramatic activation results in collapse of micropores and then reduce micropore amount and widen pore size. The CA-based carbon nanofibers (CACNFs) possesses the highest specific surface area of 1188 m2 g−1 and the largest pore volume of 0.575 cm3 g−1. CACNFs can be directly cut into electrodes for supercapacitors. CACNFs have high specific capacitance (202 F g−1 at 0.1 A g−1) and excellent rate performance (61% of the retention from 0.1 to 20 A g−1). Two-electrode tests showed that CACNFs-ZnCl2-20% had the largest specific capacitance (143 F g−1 at a current density of 0.1 A g−1). After 5000 cycles of the electrode, the capacitance retention rate was 92%, and the coulombic efficiency was close to 100%, indicating that the electrode has high electrochemical stability and reversibility. Considering the renewability of cellulose and excellent electrochemical performance, CACNFs are likely to have good application prospect as electrode materials of supercapacitors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors acknowledge financial support from the University of Science and Technology Development Fund Planning Project of Tianjin (2017KJ072), Natural Science Foundation of Tianjin Province (Grant No. 16JCQNJC06300), Joint Funds of the National Natural Science Foundation of China (Grant No. U1510134), National Nature Science Foundation of China (Grant No. 51502201), National Key R&D Program of China (Grant No. 2017YFB0308205).
References
- C. Wang, D. Wu, H. Wang, Z. Gao, F. Xu and K. Jiang, J. Colloid Interface Sci., 2018, 523, S0021979718302510 Search PubMed.
- S. Li, N. Zhang, H. Zhou, J. Li, N. Gao, Z. Huang, L. Jiang and Y. Kuang, Appl. Surf. Sci., 2018, 453, 63–72 CrossRef CAS.
- J. Sheng, C. Ma, Y. Ma, H. Zhang, R. Wang, Z. Xie and J. Shi, Int. J. Hydrogen Energy, 2016, 41, 9383–9393 CrossRef CAS.
- X. Bing, Y. Wei, W. Mei, X. Sheng, D. Long, J. Wang, W. Qiao and L. Ling, J. Colloid Interface Sci., 2017, 488, 207–217 CrossRef CAS PubMed.
- D. Nan, Z. H. Huang, R. Lv, L. Yang, J. G. Wang, W. Shen, Y. Lin, X. Yu, L. Ye and H. Sun, J. Mater. Chem. A, 2014, 2, 19678–19684 RSC.
- M. Xie, Z. Xu, S. Duan, Z. Tian, Y. Zhang, K. Xiang, M. Lin, X. Guo and W. Ding, Nano Res., 2018, 11, 216–224 CrossRef CAS.
- X. J. Dang, G. L. Zhang and G. B. Lv, Carbon Techniques, 2014 Search PubMed.
- T. T. Chen, W. L. Song and L. Z. Fan, Electrochim. Acta, 2015, 165, 92–97 CrossRef CAS.
- Y. Xu, L. Wang, P. Cao, C. Cai, Y. Fu and X. Ma, J. Power Sources, 2016, 306, 742–752 CrossRef CAS.
- C. Ma, Y. Song, J. Shi, D. Zhang, M. Zhong, Q. Guo and L. Liu, Mater. Lett., 2012, 76, 211–214 CrossRef CAS.
- Y. J. Li, X. Y. Ni, J. Shen, D. Liu and N. P. Liu, Appl. Mech. Mater., 2013, 302, 158–164 Search PubMed.
- J. Gamby, P. L. Taberna, P. Simon, J. F. Fauvarque and M. Chesneau, J. Power Sources, 2001, 101, 109–116 CrossRef CAS.
- L. Zhang, Y. Jiang, L. Wang, C. Zhang and S. Liu, Electrochim. Acta, 2016, 196, 189–196 CrossRef CAS.
- D. G. Yu, X. F. Zhang, X. X. Shen, C. Brandford-White and L. M. Zhu, Polym. Int., 2010, 58, 1010–1013 CrossRef.
- A. Laforgue, J. Power Sources, 2011, 196, 559–564 CrossRef CAS.
- N. N. Bui, B. H. Kim, K. S. Yang, M. E. D. Cruz and J. P. Ferraris, Carbon, 2009, 47, 2538–2539 CrossRef CAS.
- G. Zhe, Z. Hongli, G. Eleanor, H. Xiaogang, G. W. Rubloff, H. Liangbing and L. S. Bok, ACS Nano, 2013, 7, 6037 CrossRef PubMed.
- J. Cai, W. Xu, Y. Liu, Z. Zhu, G. Liu, W. Ding, G. Wang, H. Wang and Y. Luo, Eng. Sci., 2019, 5, 21–29 Search PubMed.
- Z. Li, J. Liu, K. Jiang and T. Thundat, Nano Energy, 2016, 25, 161–169 CrossRef CAS.
- F. Hermanutz, F. Gähr, E. Uerdingen, F. Meister and B. Kosan, Macromol. Symp., 2010, 262, 23–27 CrossRef.
- P. Lu and Y. L. Hsieh, ACS Appl. Mater. Interfaces, 2010, 2, 2413–2420 CrossRef CAS PubMed.
- S. Gaan, L. Mauclaire, P. Rupper, V. Salimova, T. T. Tran and M. Heuberger, J. Anal. Appl. Pyrolysis, 2011, 90, 33–41 CrossRef CAS.
- C. Jie, H. Niu, H. Wang, S. Hao, F. Jian, J. He, H. Xiong, C. Ma and L. Tong, J. Power Sources, 2016, 324, 302–308 CrossRef.
- D. Libo, R. J. Young, I. A. Kinloch, A. M. Abdelkader, S. M. Holmes, H. D. Rio and S. J. Eichhorn, ACS Appl. Mater. Interfaces, 2013, 5, 9983–9990 CrossRef PubMed.
- J. Cai, H. Niu, Z. Li, Y. Du, P. Cizek, Z. Xie, H. Xiong and T. Lin, ACS Appl. Mater. Interfaces, 2015, 7, 14946–14953 CrossRef CAS PubMed.
- L. Deng, R. J. Young, I. A. Kinloch, Y. Zhu and S. J. Eichhorn, Carbon, 2013, 58, 66–75 CrossRef CAS.
- W. K. Son, J. H. Youk, T. S. Lee and W. H. Park, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 5–11 CrossRef CAS.
- K. S. Kang, K. Y. Cho, H. K. Lim and J. Kim, J. Phys. D: Appl. Phys., 2008, 41, 3080–3090 Search PubMed.
- J. F. Mata-Segreda, J. Am. Chem. Soc., 2002, 124, 2259–2262 CrossRef CAS PubMed.
- A. Kumar and H. M. Jena, Appl. Surf. Sci., 2015, 356, 753–761 CrossRef CAS.
- W. Yang, F. Ding, G. Shao, L. Sang, W. Yang and Z. Ma, Carbon, 2017, 111, 419–427 CrossRef CAS.
- B. H. Kim, K. S. Yang and D. J. Yang, Electrochim. Acta, 2013, 109, 859–865 CrossRef CAS.
- V. A. Davydov, A. V. Rakhmanina, V. Agafonov, B. Narymbetov, J. P. Boudou and H. Szwarc, Carbon, 2004, 42, 261–269 CrossRef CAS.
- M. Xie, J. Yang, J. Liang, X. Guo and W. Ding, Carbon, 2014, 77, 215–225 CrossRef CAS.
- N. M. Rodriguez, J. Mater. Res., 1993, 8, 3233–3250 CrossRef CAS.
- C. Kim and K. S. Yang, Appl. Phys. Lett., 2003, 83, 1216–1218 CrossRef CAS.
- Y. Gao, L. Li, Y. Jin, Y. Wang, C. Yuan, Y. Wei, G. Chen, J. Ge and H. Lu, Appl. Energy, 2015, 153, 41–47 CrossRef CAS.
- Z. F. Tian, M. J. Xie, Y. Shen, Y. Z. Wang and X. F. Guo, Chin. Chem. Lett., 2017, 04, 203–207 Search PubMed.
- C. Young, J. Lin, J. Wang, B. Ding, X. Zhang, S. M. Alshehri, T. Ahamad, R. R. Salunkhe, S. A. Hossain and J. H. Khan, Chemistry, 2018, 24, 6127–6132 CrossRef CAS PubMed.
- H. M. Luo, H. Chen, Y. Z. Chen, P. Li, J. Q. Zhang and X. Zhao, J. Appl. Electrochem., 2016, 46, 703–712 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095–14107 CrossRef CAS.
- A. Duszová, J. Morgiel, Z. Bastl, J. Mihály and J. Dusza, Arch. Metall. Mater., 2013, 58, 459–463 Search PubMed.
- H. J. Liu, W. J. Cui, L. H. Jin, C. X. Wang and Y. Y. Xia, J. Mater. Chem., 2009, 19, 3661–3667 RSC.
- M. Kunowsky, J. P. Marco-Lozar, A. Oya and A. Linares-Solano, Carbon, 2011, 50, 1407–1416 CrossRef.
- J. Liu, S. Wang, J. Yang, J. Liao, M. Lu, H. Pan and L. An, Desalination, 2014, 344, 446–453 CrossRef CAS.
- X. Yang, D. Wu, X. Chen and R. Fu, J. Phys. Chem. C, 2010, 114, 8581–8586 CrossRef CAS.
- D. Hulicova-Jurcakova, M. Seredych, G. Q. Lu and T. J. Bandosz, Adv. Funct. Mater., 2009, 19, 438–447 CrossRef CAS.
- L. Zhang, Z. Su, F. Jiang, L. Yang, J. Qian, Y. Zhou, W. Li and M. Hong, Nanoscale, 2014, 6, 6590–6602 RSC.
- Z. Yu, T. Qu, K. Xiang, S. Yu, S. Chen, M. Xie and X. Guo, J. Mater. Chem. A, 2018, 6, 2353–2359 RSC.
- A. Kamari, S. N. M. Yusoff, F. Abdullah and W. P. Putra, J. Environ. Chem. Eng., 2014, 2, 1912–1919 CrossRef CAS.
- K. S. W. Sing, Pure Appl. Chem., 1985, 57, 603–619 CAS.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. RodriguezReinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 38, 25 Search PubMed.
- G. P. Hao, A. H. Lu, W. Dong, Z. Y. Jin, X. Q. Zhang, J. T. Zhang and W. C. Li, Adv. Energy Mater., 2013, 3, 1421–1427 CrossRef CAS.
- A. M. Noor and M. A. B. M. Nawi, J. Phys. Sci., 2008, 19, 93–104 Search PubMed.
- J. S. Im, S. J. Park, T. J. Kim, Y. H. Kim and Y. S. Lee, J. Colloid Interface Sci., 2008, 318, 42–49 CrossRef CAS PubMed.
- T. V. Tran, Q. T. P. Bui, T. D. Nguyen, N. T. H. Le and G. B. Long, Adsorpt. Sci. Technol., 2016, 35, 650–661 Search PubMed.
- M. Liu, L. Gan, W. Xiong, F. Zhao, X. Fan, D. Zhu, Z. Xu, Z. Hao and L. Chen, Energy Fuels, 2013, 27, 1168–1173 CrossRef CAS.
- A. Rosa, H. Michael, W. Sabine, B. Raoul, L. Martin, M. James, P. Edward, Z. J. Axel, G. Lynn, K. Axel, S. Robert and D. Su, J. Am. Chem. Soc., 2010, 132, 9616–9630 CrossRef PubMed.
- M. Xie, S. Duan, Y. Shen, K. Fang, Y. Wang, M. Lin and X. Guo, ACS Energy Lett., 2016, 1, 814–819 CrossRef CAS.
- X. Liu, L. Zhou, Y. Zhao, L. Bian, X. Feng and Q. Pu, ACS Appl. Mater. Interfaces, 2013, 5, 10280–10287 CrossRef CAS PubMed.
- Q. Zhao, X. Wang, C. Wu, J. Liu, H. Wang, J. Gao, Y. Zhang and H. Shu, J. Power Sources, 2014, 254, 10–17 CrossRef CAS.
- K. S. Kesavan, K. Surya and M. S. Michael, Solid State Ionics, 2018, 321, 15–22 CrossRef.
- C. Ma, J. Chen, Q. Fan, J. Guo, W. Liu, E. Cao, J. Shi and Y. Song, J. Mater. Sci., 2018, 53, 4527–4539 CrossRef CAS.
- L. Wang, M. Inagaki and M. Toyoda, Carbon, 2010, 48, 1323 CrossRef CAS.
- D. I. Abouelamaiem, G. He, I. P. Parkin, T. Neville, A. B. Jorge, J. Shan, R. Wang, M. Titirici, P. Shearing and B. Dan, Sustainable Energy Fuels, 2018, 2, 772–785 RSC.
- L. Jiang, G. W. Nelson, S. O. Han, H. Kim, I. N. Sim and J. S. Foord, Electrochim. Acta, 2016, 192, 251–258 CrossRef CAS.
- V. Kuzmenko, A. M. Saleem, H. Staaf, M. Haque, A. Bhaskar, M. Flygare, K. Svensson, V. Desmaris and P. Enoksson, J. Micromech. Microeng., 2016, 26, 124001 CrossRef.
- T. Xun, Z. Shan, J. Peng, Y. Zuo, W. Gang, X. Guo, N. Zhao, Y. Ma and M. Lei, Electrochim. Acta, 2017, 241, 170–178 CrossRef.
- Z. Weng, Y. Su, D.-W. Wang, F. Li, J. Du and H.-M. Cheng, Adv. Energy Mater., 2011, 1, 917–922 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra07587e |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Chang Ma
a,
Chang Ma