DOI:
10.1039/C8RA06647G
(Paper)
RSC Adv., 2019,
9, 753-760
Experimental and theoretical investigation of intramolecular cooperativity in cyclic benzene trimer motif†
Received
7th August 2018
, Accepted 18th December 2018
First published on 4th January 2019
Abstract
A series of new symmetrical tripodal molecules 1a–4b with a central benzene scaffold substituted with methyl/ethyl groups and three benzimidazolyl units having a bithiophene/biphenyl/5-alkylthiophene motif at the 2-position via a –CH2– unit were synthesized and characterized by elemental analysis, HR-MS, and NMR spectroscopy. NMR spectral data reveal that all molecules adopt a cyclic benzene trimer (CBT) using three benzimidazolyl units. Intramolecular cooperative edge-to-face C–H⋯π interactions stabilize the CBT motif in solution and are strong in ethyl substituted molecules (1b–4b) compared to methyl substituted (1a–4a) ones. However, the strength of the CBT unit in the tripodal molecule is independent of the length of the substituent at the 2-position of the benzimidazolyl unit. The relative 1H NMR chemical shift calculated at the MPW1PW91/6-311+G(d,p) level of theory corroborates the experimental values, and the calculations predict the distribution of the structures into syn isomers. The relative change in the NMR chemical shift is justified by the relative change in the magnitude of the (3,+3) critical point (CP) in the molecular electrostatic potential (MESP) topography. Also, a linear correlation of the intramolecular C–H⋯π interactions evaluated at M062X/6-311+G(d,p) with the relative NMR chemical shift suggest the latter as a measure of intramolecular cooperativity.
Introduction
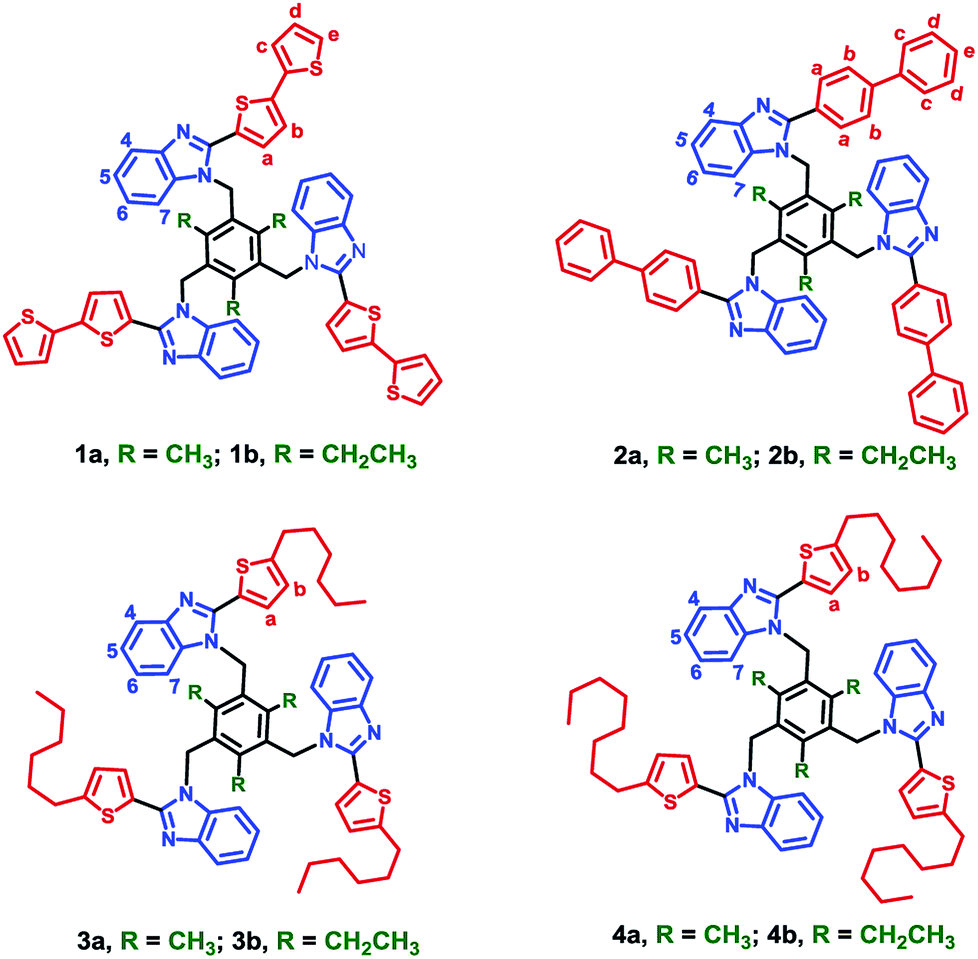
Cooperative noncovalent interactions are vital for determining the structure and properties of molecules and materials.1,2 Among the several interactions, O–H⋯O, O–H⋯N, N–H⋯N, N–H⋯O, C–H⋯O, C–H⋯π hydrogen bonding interactions and π⋯π aromatic stacking interactions, cooperative edge-to-face C–H⋯π interactions between the aromatic units provide a significant contribution in stabilizing molecular structure.3 The cyclic benzene trimer (CBT) motif, i.e., three benzene units arranged in a cyclic manner, is the smallest cyclic cluster stabilized through cooperative C–H⋯π interactions in various aromatic clusters and is the basic building unit for higher order aromatic clusters.1,4 Due to its importance, the cooperative edge-to-face C–H⋯π interactions in the CBT motif are studied theoretically.5 The non-additive behavior of the C–H⋯π interactions within CBT motifs adds to the intramolecular cooperativity.6 Several complexes possessing CBT motif in the solid state are not noticed or reported. However, reports on molecules possessing the CBT motif both in solution and the solid state has started to appear recently.7 We found a simple strategy to engineer CBT motif in a tripodal molecule which consists of central spacer and aryl/heteroaryl ring substituted benzimidazolyl cores.8 Our earlier studies8 reveal that both alkyl group on the central benzene spacer and the aromatic group at 2-position of benzimidazolyl core are necessary to arrange the tripodal molecules with benzimidazolyl based CBT motif. Up to now, furan, thiophene, phenyl, pyridyl, methoxy groups substituted phenyl are used as a steric group at 2-position of benzimidazolyl unit, to force the tripodal molecule to adopt CBT motif. To understand the stability of the CBT motif, we engineered the tripodal molecule by increasing the length of heteroaryl/aryl and alkylthiophene at 2-position of benzimidazolyl unit (Fig. 1).
 |
| | Fig. 1 Tripodal molecules 1a–4b. | |
Herein, a series of 2-substituted bithiophene, biphenyl and alkylthiophene benzimidazoles (L1–L4) and tripodal molecules 1a–4b were prepared and characterized by elemental analysis, high-resolution mass spectrometry (HR-MS) and NMR spectroscopic methods. Though there are experimental and theoretical works on cooperative interactions, quantitative intramolecular cooperativity measurements are scarce in the literature.9–17 Therefore, theoretically, it is challenging and interesting to come up with a scheme to partition the CBT motifs to calculate the intramolecular interactions and to correlate with experimental relative NMR chemical shifts.
Results and discussion
Preparations
2-(2,2′-Bithiophen-5-yl)benzimidazole (L1), 2-(1,1′-biphenyl-4-yl)benzimidazole (L2), 2-(5-hexylthiophen-2-yl)benzimidazole (L3) and 2-(5-octylthiophen-2-yl)benzimidazole (L4) were synthesized by the reaction of o-phenylenediamine with corresponding aldehydes.8c Subsequent treatment of Ln with 1,3,5-tris(bromomethyl)mesitylene or 1,3,5-tris(bromomethyl)-2,4,6-triethylbenzene in THF by using NaH as base yield tripodal molecules 1a–4b. All the benzimidazoles and the tripodal molecules (1a–4b) are air and moisture stable and soluble in organic solvents. Elemental analysis, HR-MS and NMR spectroscopic methods were used for characterizing the molecules. All the ligands and the tripodal molecules display molecular ion peaks (m/z for [M + H]+) those match the theoretical values (vide infra experimental section).
NMR studies
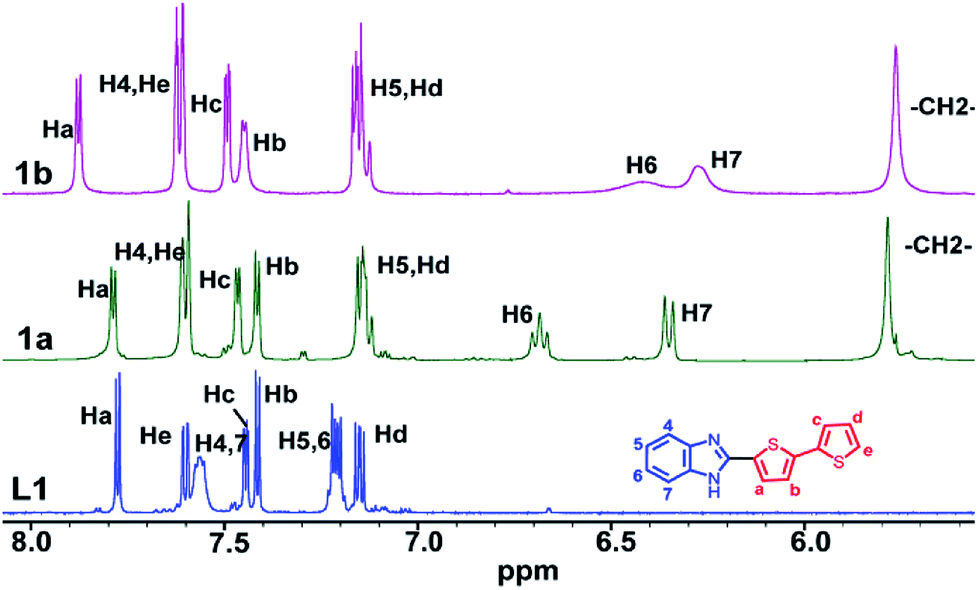
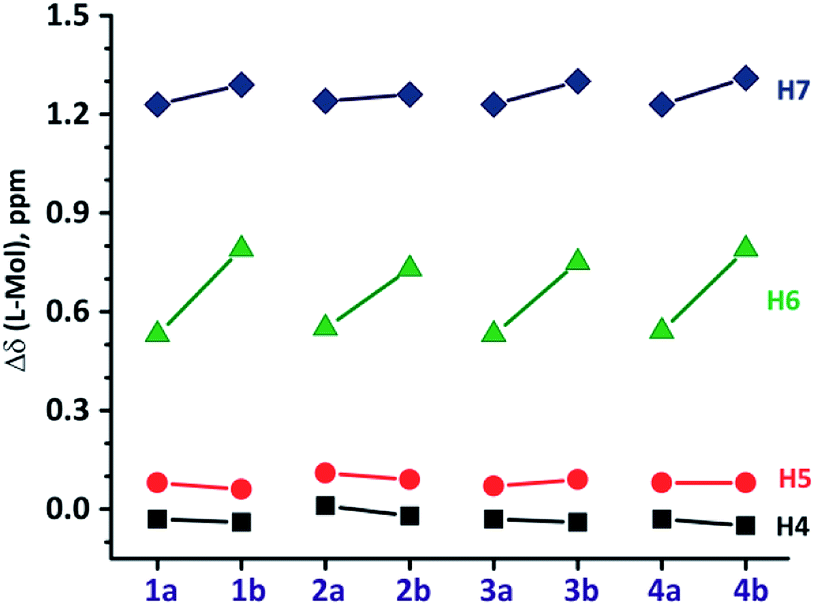
NMR spectra of L1–L4 and 1a–4b were recorded in d6-DMSO solvent (Fig. S7–S32 in ESI†). Molecule 1a displays a single set of chemical resonances for all protons. A new peak at δ 5.6 ppm and 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :27 methylene:aromatic protons ratio confirms the formation of 1a (Fig. 2). The chemical resonances for H6 and H7 protons of benzimidazolyl were significantly upfield shifted compare to those in L1 (Fig. 2 and 3). H4 and H5 protons of benzimidazolyl and bithiophene protons (Ha–He) either remain in the same position or downfield shifted relative to L1. These data reveal that 1a adopts a syn-conformer with strong CBT motif with three benzimidazolyl units, and all bithiophene units are pointing away from each other. The NMR spectrum of ethyl substituted molecule 1b shows a similar pattern like that of 1a, except for H6 and H7 protons, which are broad and upfield shifted compared to 1a (Fig. 2). This data indicates that 1b has a stronger CBT motif compared to 1a.8
:27 methylene:aromatic protons ratio confirms the formation of 1a (Fig. 2). The chemical resonances for H6 and H7 protons of benzimidazolyl were significantly upfield shifted compare to those in L1 (Fig. 2 and 3). H4 and H5 protons of benzimidazolyl and bithiophene protons (Ha–He) either remain in the same position or downfield shifted relative to L1. These data reveal that 1a adopts a syn-conformer with strong CBT motif with three benzimidazolyl units, and all bithiophene units are pointing away from each other. The NMR spectrum of ethyl substituted molecule 1b shows a similar pattern like that of 1a, except for H6 and H7 protons, which are broad and upfield shifted compared to 1a (Fig. 2). This data indicates that 1b has a stronger CBT motif compared to 1a.8
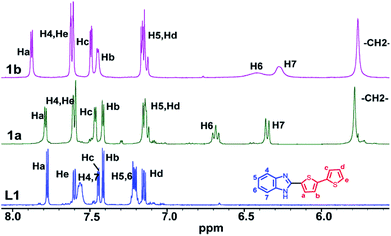 |
| | Fig. 2 Partial 1H NMR spectra of L1, 1a and 1b in d6-DMSO solvent. | |
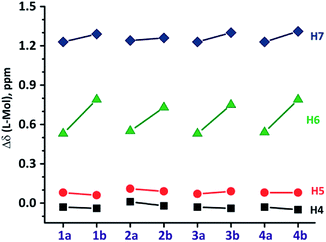 |
| | Fig. 3 The relative 1H-NMR chemical shift (in ppm) of H4–H7 protons (Mol = 1a–4b and L = L1–L4). | |
The 1H NMR spectrum of molecule 2a showed no significant shift for H4 and H5 protons of benzimidazolyl, whereas, H6 and H7 displayed an upfield shift compared to those in L2 (Fig. S1 in ESI†). The biphenyl protons (Hb–He) of 2a displayed no significant shift compared to L2. Thus, 2a adopt a strong CBT motif with syn-conformation similar to 1a. The 1H NMR spectrum of molecule 2b displays similar pattern as in 2a, except H6 and H7 protons of benzimidazolyl unit. Latter showed an upfield shift of δ 0.73 ppm and δ 1.25 ppm relative to L2, respectively (Fig. 3). This data indicates that 2b also adopt a syn-conformation, but a stronger CBT motif than 2a. Molecules 3 and 4, possessing alkylthiophene units display 1H NMR pattern similar to those of 1–2 (Fig. S3 and S4 in ESI†).
1H NMR data indicate that modulating five-membered heteroaryl units to six-membered aromatic or alkyl substituted five-membered heteroaryl at 2-position of benzimidazolyl unit does not alter the strength of edge-to-face interactions (CBT motif) in the tripodal molecules (Fig. S4 and S5 in ESI†).8 This data supports that increasing the length of alkyl chain on thiophene at 2-position of benzimidazolyl unit does not influence the nature of edge-to-face C–H⋯π interactions in the CBT motif as observed in previously studied molecules.8c However, alkyl substituted molecules (3–4) possess stronger CBT motif than aryl substituted molecules (1–2).
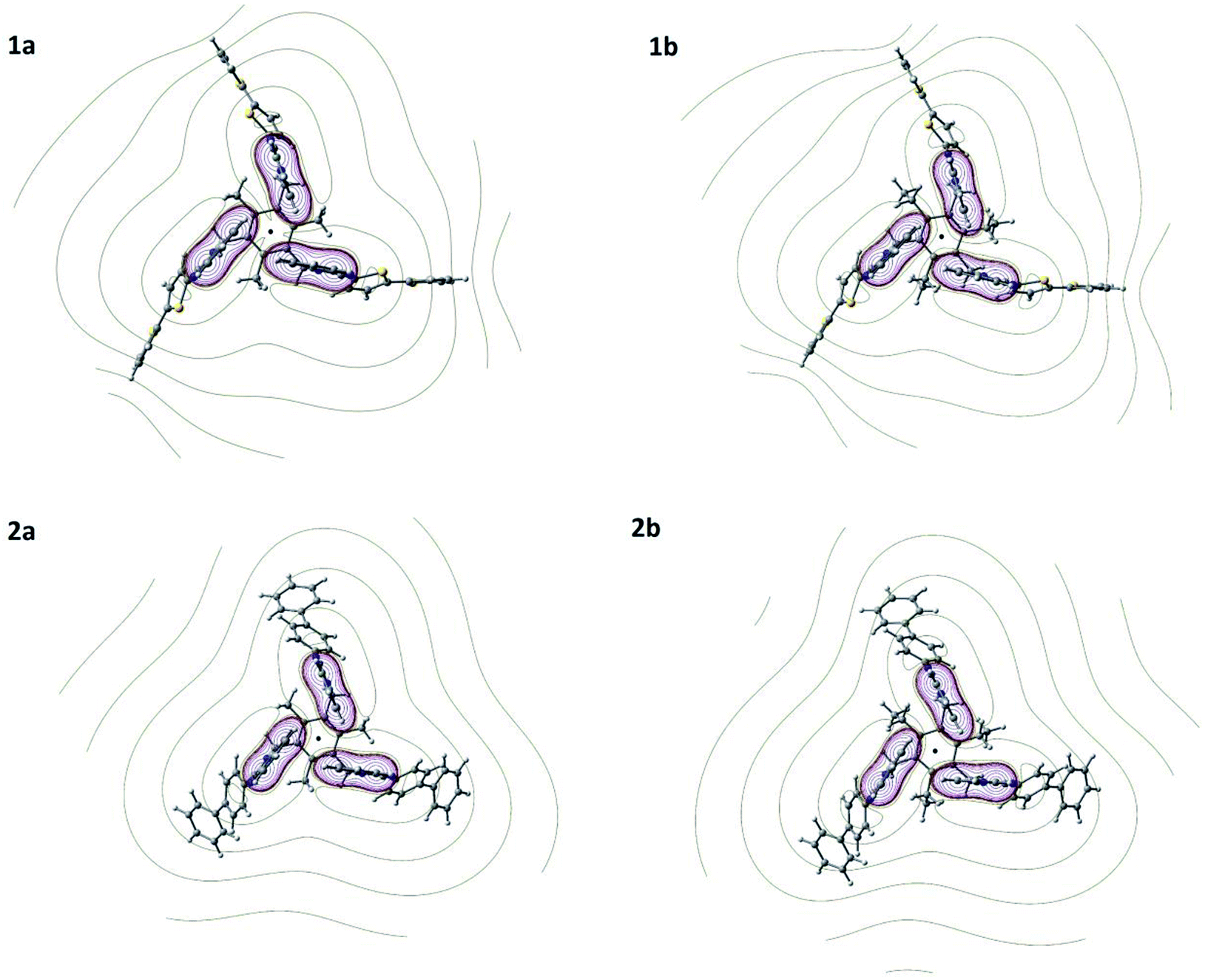
Molecular modelling
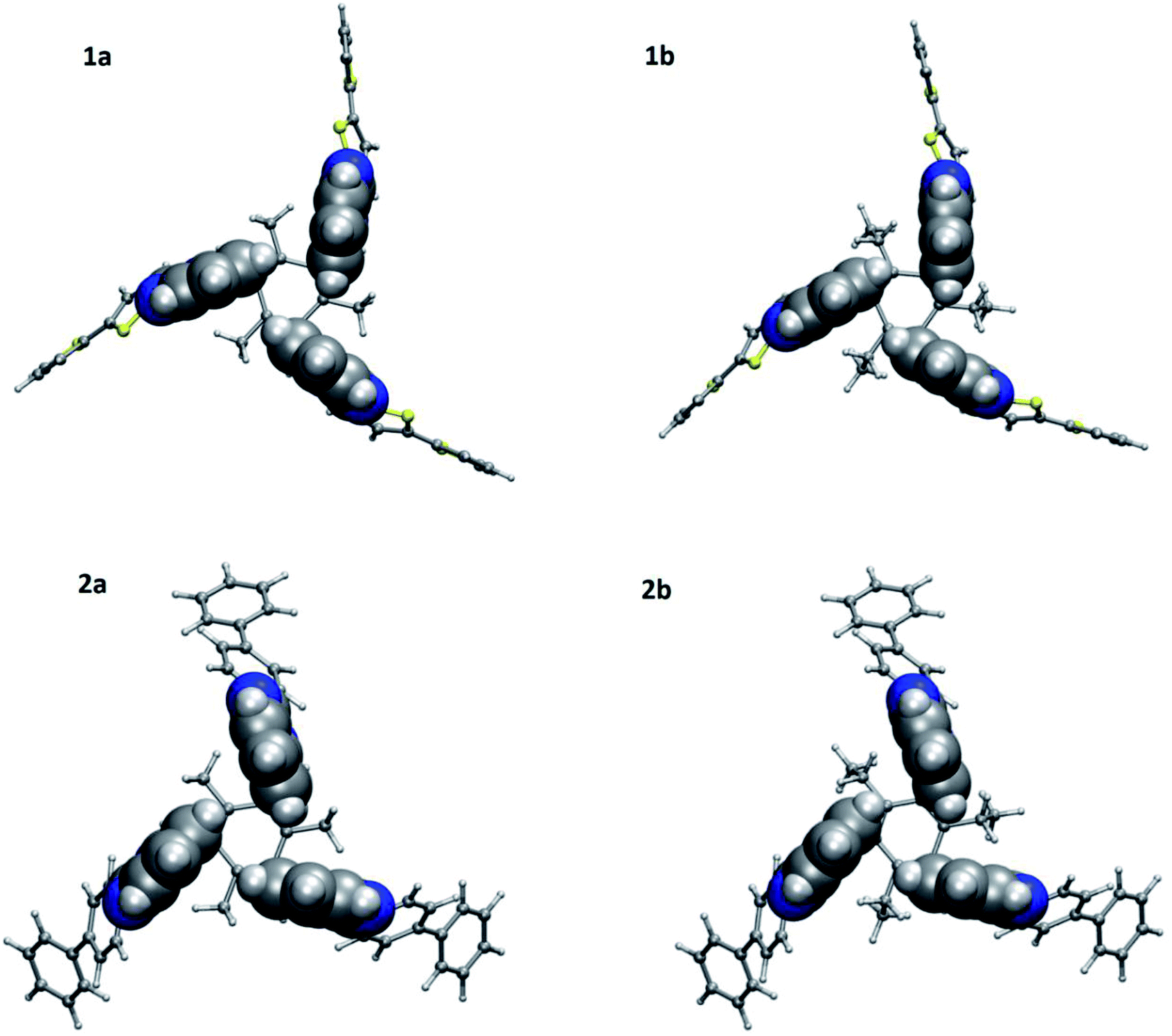
To further understand the geometry as well as intramolecular interactions for 1a–1b and 2a–2b, DFT calculations were performed with Gaussian09 suite of packages18 at MPW1PW91/6-311G(d,p) level of theory and basis function. All four molecules adopt an edge-to-face syn-conformation, i.e., CBT motif with three benzene rings of benzimidazolyl units (Fig. 4). The geometrical distance between H7 to the center of benzene ring in benzimidazolyl unit is shorter in ethyl substituted molecules (3.55 Å for 1b and 3.53 Å for 2b) compared to methyl substituted (3.66 Å for 1a and 3.60 Å for 2a) molecules.
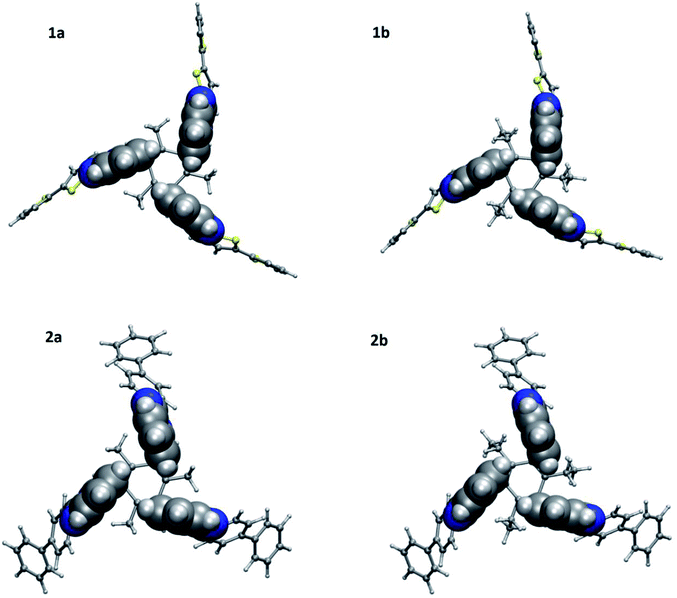 |
| | Fig. 4 Molecular structures of 1a, 1b, 2a and 2b evaluated at MPW1PW91/6-311+G(d,p) level of theory and basis function showing the cyclic benzene trimer (CBT) between three benzene rings of benzimidazolyl units. | |
We have followed the scheme depicted in Fig. S6 in ESI† for calculating the intramolecular C–H⋯π interaction energies,19 performed with the optimized geometries at MPW1PW91/6-311G(d,p). The calculated intramolecular energies at M062X/6-311+G(d,p) [MPW1PW91/6-311+G(d,p)] levels of theory and basis function are reported in Table 1. The calculated intramolecular C–H⋯π interaction energy between benzene C–H with neighboring benzene π-electron cloud in the CBT core indicates that the intramolecular interaction energy is higher for ethyl substituted molecules (1b and 2b) than methyl substituted ones (1a and 2a).
Table 1 Fragment energies, total energy (Hartrees) and intramolecular energies (kcal mol−1) at M062X [MPW1PW91]/6-311+G(d,p) levels of theory and basis function calculated for 1a, 1b, 2a and 2b, as per the scheme depicted in Fig. S6 in ESI
| Mol |
F1 |
F2 |
F3 |
ETotal |
E3F1−3F2+F3 |
EIntra-hydrogen |
| 1a |
−3392.63315 |
−1871.33900 |
−350.04673 |
−4913.92920 |
−4913.92731 |
−1.18[−0.10] |
| 1b |
−3510.52082 |
−1989.22580 |
−467.93282 |
−5031.81789 |
−5031.81585 |
−1.28[−0.12] |
| 2a |
−2109.61118 |
−1229.82792 |
−350.046850 |
−2989.39661 |
−2989.39443 |
−1.37[−0.04] |
| 2b |
−2227.49869 |
−1347.71468 |
−467.93286 |
−3107.28491 |
−3107.28271 |
−1.38[−0.05] |
The relative magnitude of the NMR chemical shift of 1a, 1b, 2a and 2b in DMSO solvent were calculated relative to the ligands (L1 and L2) using SMD solvent model20 (Table 2). The H6 and H7 protons of benzimidazolyl unit shows a higher upfield shift, while the remaining H4–H5 protons and Ha–He protons of bithiophene/biphenyl unit are unaffected on the formation of the cyclic-trimer motifs (Table 2). The theoretical results corroborated well with the experiment. To justify the relative NMR chemical shift and to quantify the cooperative C–H⋯π interaction, the molecular electrostatic potential (MESP) topography is calculated21 and is defined at a point r, as the amount of energy required to bring a unit positive charge from infinity to that point.22–24 MESP contours in the plane passing through the center of the benzene rings are depicted in Fig. 5. The (3,+3) MESP critical point at the center of the cyclic trimer ring indicates the formation of CBT motif in 1a, 1b, 2a, and 2b. The MESP minimum is more negative for ethyl substituted molecules (−0.024 a.u for 1b and −0.030 a.u. for 2b) than methyl substituted ones (−0.020 a.u for 1a and −0.027 a.u. for 2a). An increase in the negative value of the MESP value within the benzene trimer motifs shows an increase in the electronic contribution in this region. Also, the magnitude of the MESP (3,+3) CP indicates the formation of a stable intramolecular CBT motif.
Table 2 Experimental and theoretical [MPW1PW91/6-311+G(d,p)] relative 1H chemical shift (in ppm) of 1a, 1b, 2a and 2b with the corresponding ligand (L1–L2) in d6-DMSO solvent
| Mol |
H4 |
H5 |
H6 |
H7 |
Ha |
Hb |
Hc |
Hd |
He |
| 1a |
−0.03 [−0.02] |
0.08 [0.01] |
0.53 [0.79] |
1.22 [1.25] |
0.00 [0.05] |
0.00 [−0.11] |
−0.02 [−0.05] |
0.08 [−0.02] |
0.00 [ 0.02] |
| 1b |
−0.04 [−0.01] |
0.06 [0.06] |
0.79 [1.04] |
1.29 [1.42] |
−0.09 [0.00] |
−0.03 [−0.13] |
−0.05 [−0.05] |
0.07 [−0.02] |
−0.01 [ 0.02] |
| 2a |
0.01 [0.00] |
0.11 [0.39] |
0.55 [0.94] |
1.24 [1.47] |
0.43 [−0.02] |
0.09 [−0.01] |
0.04 [ 0.06] |
0.01 [0.03] |
0.02 [−0.04] |
| 2b |
−0.02 [0.00] |
0.09 [0.07] |
0.73 [1.16] |
1.26 [1.53] |
0.38 [0.20] |
0.06 [−0.09] |
0.02 [−0.06] |
0.00 [0.05] |
−0.01 [−0.04] |
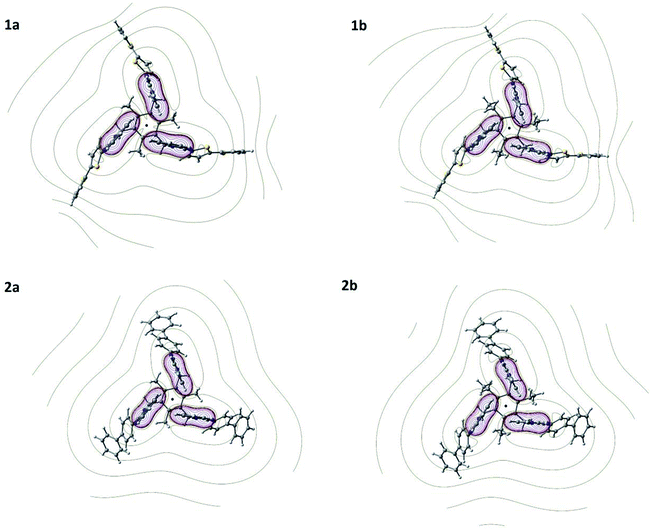 |
| | Fig. 5 MESP topography distribution of 1a, 1b, 2a and 2b evaluated at MPW1PW91/6-311+G(d,p) level of theory and basis set. The consecutive contours are separated by 0.02 a.u. | |
Conclusions
Bithiophene, biphenyl, alkylthiophene based benzimidazoles (L1–L4), new tripodal molecules (1–4) possessing these benzimidazolyl units and benzene spacer with methyl/ethyl group were prepared and characterized. These molecules adopt a strong CBT motif in solution. A stronger edge-to-face intramolecular C–H⋯π interactions is observed in ethyl substituted molecules (1b–4b) than the corresponding methyl ones (1a–4a) in solution. A similar formation of strong CBT motif stabilized by cooperative edge-to-face interactions in molecules (1–2) in DMSO solution is observed by using DFT calculations. Also, the formation of CBT is explained by intramolecular cooperative energy, relative 1H NMR chemical shift and (3,+3) critical point in MESP topology in molecules (1–2). The results indicate that increasing the length of aromatic units or alkyl chain on thiophene and benzene group at 2-position of benzimidazolyl unit is not enough to break the CBT motif, stabilized by the cooperative edge-to-face C–H⋯π interactions, in solution. Therefore, this quantitative study may facilitate crystal engineering of molecules with CBT motifs for desired properties.
Experimental section
General experimental methods
Biphenyl-4-carboxaldehyde, NaHSO3, o-phenylenediamine, NaH, 1,3,5-tris(bromomethyl)mesitylene and 1,3,5-tris(bromomethyl)-2,4,6-triethyl- benzene were purchased from commercial sources and used as received. 2,2′-Bithiophene-5-carboxaldehyde, 5-hexylthiophene-2-carboxaldehyde, and 5-octylthiophene-2-carboxaldehyde were prepared from previously reported methods.25 All solvents were purchased from commercial sources and distilled prior to use. All reactions were carried out in an inert atmosphere. NMR spectra were recorded on Bruker Avance III 400 and 500 MHz instruments. The chemical shifts (δ) were reported in parts per million (ppm) relative to the residual solvent signal. HR-MS were recorded on a Bruker maXis mass spectrometer. Elemental analysis was performed on a Flash EA series 1112 CHNS analyzer.
General procedure for the synthesis of ligands (L1–L4). A stirred solution of o-phenylenediamine (1 equiv.), aldehyde (1 equiv.) and NaHSO3 (3 equiv.) in DMF were heated at 80 °C for 7–8 h. The progress of the reaction was monitored by TLC. After completion of reaction, the crude mixture was poured into ice-cold water. The precipitated solid was filtered, washed with water and dried under vacuum to get crude product.
2-(2,2′-Bithiophen-5-yl)benzimidazole (L1). Orange solid product (1.8 g, 93%) was prepared from 2,2′-bithiophene-5-carboxaldehyde (1.37 g, 7.05 mmol), o-phenylenediamine (762 mg, 7.05 mmol) and NaHSO3 (2.21 g, 21.15 mmol). 1H NMR (500 MHz, d6-DMSO, ppm): δ 13.04 (1H, br, NH), 7.78 (1H, d, JHH = 3.9 Hz, Ha), 7.60 (1H, dd, JHH = 5.0, 1.1 Hz, He), 7.57 (2H, q, JHH = 3.0 Hz, H4,7), 7.44 (1H, dd, JHH = 3.5, 1.1 Hz, Hc), 7.41 (1H, d, JHH = 3.8 Hz, Hb), 7.21 (2H, q, JHH = 3.0 Hz, H5,6) and 7.15 (1H, dd, JHH = 3.6, 1.4 Hz, Hd). 13C NMR (125 MHz, d6-DMSO, ppm): δ 146.93, 139.14, 136.34, 132.44, 129.03, 128.15, 126.82, 125.44, 125.31 and 122.86 (aromatic and bithiophene). HR-MS (m/z): calc. for C15H11N2S2 (M + H)+: 283.0364; found, 283.0363. Anal. calc. for C15H10N2S2: C, 63.80; H, 3.57; N, 9.92; S, 22.71 found: C, 63.71; H, 3.51; N, 10.07; S, 22.71.
2-(1,1′-Biphenyl-4-yl)benzimidazole (L2). White solid product (2.3 g, 92%) was prepared from biphenyl-4-carboxaldehyde (1.69 g, 9.25 mmol), o-phenylenediamine (1.0 g, 9.25 mmol) and NaHSO3 (2.89 g, 27.75 mmol). 1H NMR (500 MHz, d6-DMSO, ppm): δ 8.28 (2H, dt, JHH = 8.5, 1.8 Hz, Ha), 7.88 (2H, dt, JHH = 8.5, 1.8 Hz, Hb), 7.78 (2H, dd, JHH = 7.2, 1.2 Hz, Hc), 7.63 (2H, q, JHH = 3.0 Hz, H4,7), 7.51 (2H, t, JHH = 7.4 Hz, Hd), 7.42 (1H, t, JHH = 7.3 Hz, He), 7.23 (2H, q, JHH = 3.0 Hz, H5,6). 13C NMR (100 MHz, d6-DMSO, ppm): δ 151.36, 141.83, 139.70, 129.52, 129.43, 128.46, 128.40, 127.64, 127.51, 127.17 and 122.72 (aromatic and biphenyl). HR-MS (m/z): calc. for C19H15N2 (M + H)+: 271.1235; found 271.1239. Anal. calc. for C19H14N2: C, 84.42; H, 5.22; N, 10.36 found: C, 84.32; H, 5.15, N, 10.36.
2-(5-Hexylthiophen-2-yl)benzimidazole (L3). Yellow solid product (2.6 g, 98%) was prepared from 5-hexylthiophene-2-carboxaldehyde (1.81 g, 9.23 mmol), o-phenylenediamine (1.0 g, 9.25 mmol) and NaHSO3 (2.88 g, 27.67 mmol). 1H NMR (500 MHz, d6-DMSO, ppm): δ 12.82 (1H, br s, NH), 7.63 (1H, d, JHH = 3.6 Hz, Ha), 7.52 (2H, br s, H4,7), 7.17 (2H, q, JHH = 3.0 Hz, H5,6), 6.94 (1H, d, JHH = 3.6 Hz, Hb), 2.83 (2H, t, JHH = 7.5 Hz, thio–CH2–), 1.68–1.62 (2H, quint, JHH = 7.4 Hz, thio–C–CH2–), 1.36–1.25 (6H, m, –(CH2)3–), 0.87 (3H, t, JHH = 7.05 Hz, –C–CH3). 13C NMR (100 MHz, CDCl3, ppm): δ 149.65, 148.07, 138.94, 130.25, 127.18, 125.28, 122.73, 114.84 (aromatic and thiophene), 31.50, 31.40, 30.19, 28.67, 22.54 and 14.06 (hexyl). HR-MS (m/z): calc. for C17H21N2S (M + H)+: 285.1425, found, 285.1429. Anal. calc. for C17H20N2S: C, 71.79; H, 7.09; N, 9.85; S, 11.27 found: C, 71.68; H, 7.15; N, 9.76; S, 11.41.
2-(5-Octylthiophen-2-yl)benzimidazole (L4). Yellow solid product (2.1 g, 85%) was prepared from 5-octylthiophene-2-carboxaldehyde (1.80 g, 8.0 mmol), o-phenylenediamine (865 mg, 8.0 mmol) and NaHSO3 (2.5 g, 24.0 mmol). 1H NMR (500 MHz, d6-DMSO, ppm): δ 12.83 (1H, br, NH), 7.64 (1H, d, JHH = 3.7 Hz, Ha), 7.52 (2H, br s, H4,7), 7.17 (2H, q, JHH = 3.0 Hz, H5,6), 6.94 (1H, d, JHH = 3.7 Hz, Hb), 2.83 (2H, t, JHH = 7.5 Hz, thio–CH2–), 1.65 (2H, quint, JHH = 7.4 Hz, thio–C–CH2–), 1.36–1.25 (10H, m, –(CH2)5–), 0.85 (3H, t, JHH = 7.0 Hz, –CH3). 13C NMR (100 MHz, CDCl3, ppm): δ 149.71, 147.71, 130.21, 126.67, 125.21, 122.81 (aromatic and thiophene), 31.84, 31.48, 30.22, 29.29, 29.20, 29.04, 22.65, 14.10 (octyl). HR-MS (m/z): calc. for C19H25N2S (M + H)+: 313.1728; found, 313.1736. Anal. calc. for C19H24N2S: C, 73.03; H, 7.74; N, 8.97; S, 10.26 found: C, 73.15; H, 7.68; N, 8.89; S, 10.28.
General procedure for the synthesis of tripodal molecules (1–4). To a stirred solution of ligand (L1–L4) in THF at 0 °C, NaH (1.2 equiv.) was added and allowed to stir at room temperature for 1 h. To this solution, solid tribromo compound(s) were added and stirring continued. The progress of the reaction was monitored by TLC. After completion of reaction, the solvent was removed under reduced pressure and ice water was added to the resulting mixture. The precipitated solid was collected by filtration under vacuum, washed with hexane and air dried.
1,3,5-Tris(2-(2,2′-bithiophen-5-yl)benzimidazol-1-ylmethyl)-2,4,6-trimethylbenzene (1a). Yellow solid product (436 mg, 87%) was prepared from L1 (424 mg, 1.5 mmol), NaH (45 mg, 2.0 mmol) and 1,3,5-tris(bromomethyl)mesitylene (200 mg, 0.5 mmol). 1H NMR (400 MHz, d6-DMSO, ppm): δ 7.78 (3H, d, JHH = 3.9 Hz, Ha), 7.60 (6H, d, JHH = 6.3 Hz, H4 & He), 7.46 (3H, d, JHH = 3.5 Hz, Hc), 7.41 (3H, d, JHH = 3.8 Hz, Hb), 7.15–7.11 (6H, m, H5 & Hd), 6.68 (3H, t, JHH = 7.7 Hz, H6), 6.35 (3H, d, JHH = 8.3 Hz, H7) 5.78 (6H, s, –CH2–) and 2.05 (9H, s, –CH3). 13C NMR (100 MHz, d6-DMSO, ppm): δ 147.72, 143.17, 139.96, 138.95, 136.06, 136.52, 131.58, 131.24, 130.70, 129.04, 126.98, 125.65, 125.20, 123.15, 122.40, 119.58, 111.68 (aromatic and bithiophene), 46.56 (–CH2–) and 16.84 (–CH3). HR-MS (m/z): calc. for C57H43N6S6 (M + H)+: 1003.1873; found 1003.1873. Anal. calc. for C57H42N6S6: C, 68.23; H, 4.22; N, 8.38; S, 19.17 found: C, 68.36; H, 4.26, N, 8.27, S, 19.05.
1,3,5-Tris(2-(2,2′-bithiophen-5-yl)benzimidazol-1-ylmethyl)-2,4,6-triethylbenzene (1b). Yellow solid product (492 mg, 83%) was prepared from L1 (480 mg, 1.7 mmol), NaH (52 mg, 2.1 mmol) and 1,3,5-tris(bromomethyl)-2,4,6-triethylbenzene (250 mg, 0.56 mmol). 1H NMR (400 MHz, d6-DMSO, ppm): δ 7.87 (3H, d, JHH = 3.9 Hz, Ha), 7.61 (6H, dd, JHH = 5.2, 1.3 Hz, H4 & He), 7.49 (3H, dd, JHH = 3.6, 1.2 Hz, Hc), 7.44 (3H, d, JHH = 3.4 Hz, Hb), 7.17–7.12 (6H, m, H5 & Hd), 6.42 (3H, br, H6), 6.28 (3H, br s, H7), 5.76 (6H, s, –CH2–), 2.58 (6H, d, JHH = 6.8 Hz, –CH2–CH3) and 0.72 (9H, s, –CH2–CH3). 13C NMR (125 MHz, d6-DMSO, ppm): δ 147.64, 145.75, 143.31, 140.07, 136.06, 135.58, 131.06, 130.67, 129.06, 126.99, 125.69, 125.16, 122.98, 122.29, 119.67, 112.28 (aromatic and bithiophene), 45.28 (–CH2–), 23.55 (–CH2–CH3) and 14.75 (–CH2–CH3). HR-MS (m/z): calc. for C60H49N6S6 (M + H)+: 1045.2343; found 1045.2342. Anal. calc. for C60H48N6S6: C, 68.93; H, 4.63; N, 8.04; S, 18.40 found: C, 68.83; H, 4.71; N, 8.12; S, 18.34.
1,3,5-Tris(2-(1,1′-biphenyl-4-yl)benzimidazol-1-ylmethyl)-2,4,6-trimethylbenzene (2a). White solid product (214 mg, 89%) was prepared from L2 (270 mg, 0.75 mmol), NaH (22 mg, 0.9 mmol) and 1,3,5-tris(bromomethyl)mesitylene (100 mg, 0.25 mmol). 1H NMR (500 MHz, d6-DMSO, ppm): δ 7.85 (6H, d, JHH = 8.2 Hz, Ha), 7.79 (6H, d, JHH = 8.2 Hz, Hb), 7.74 (6H, d, JHH = 7.3 Hz, Hc), 7.62 (3H, d, JHH = 8.0 Hz, H4), 7.50 (6H, t, JHH = 7.6 Hz, Hd), 7.40 (3H, t, JHH = 7.3 Hz, He), 7.12 (3H, t, JHH = 7.8 Hz, H5), 6.68 (3H, t, JHH = 7.7 Hz, H6), 6.39 (3H, d, JHH = 8.2 Hz, H7), 5.56 (6H, s, –CH2–) and 1.89 (9H, s, –CH3). 13C NMR (100 MHz, d6-DMSO, ppm): δ 154.04, 143.22, 141.63, 139.64, 138.40, 135.13, 131.56, 130.68, 129.94, 128.42, 127.24, 127.17, 122.85, 121.97, 119.71, 111.66 (aromatic and biphenyl), 46.25 (–CH2–) and 16.81 (–CH3). HR-MS (m/z): calc. for C69H55N6 (M + H)+: 967.4488; found, 967.4488. Anal. calc. for C69H54N6: C, 85.68; H, 5.63; N, 8.69 found: C, 85.49; H, 5.67, N, 8.58.
1,3,5-Tris(2-(1,1′-biphenyl-4-yl)benzimidazol-1-ylmethyl)-2,4,6-triethylbenzene (2b). White solid product (661 mg, 96%) was prepared from L2 (551 mg, 2.04 mmol), NaH (60 mg, 2.5 mmol) and 1,3,5-tris(bromomethyl)-2,4,6-triethylbenzene (300 mg, 0.68 mmol). 1H NMR (500 MHz, d6-DMSO, ppm): δ 7.90 (6H, d, JHH = 8.0 Hz, Ha), 7.84–7.80 (6H, m, Hb), 7.76 (6H, d, JHH = 7.6 Hz, Hc), 7.65 (3H, d, JHH = 8.0 Hz, H4), 7.51 (6H, t, JHH = 7.5 Hz, Hd), 7.44–7.41 (3H, m, He), 7.14 (3H, t, JHH = 7.5 Hz, H5), 6.50 (3H, t, JHH = 6.0 Hz, H6), 6.37 (3H, d, JHH = 8.0 Hz, H7), 5.55 (6H, s, –CH2–), 2.71–2.63 (6H, m, –CH2–CH3) and 0.47 (9H, s, –CH2–CH3). 13C NMR (100 MHz, CDCl3, ppm): δ 153.74, 145.40, 143.24, 142.74, 139.95, 134.65, 130.69, 130.14, 129.18, 129.01, 128.04, 127.45, 127.38, 127.16, 123.30, 122.23, 120.11, 111.62 (aromatic and biphenyl), 44.97 (–CH2–), 23.76 (–CH2–CH3) and 14.48 (–CH2–CH3). HR-MS (m/z): calc. for C72H61N6 (M + H)+: 1009.4958; found, 1009.4961. Anal. calc. for C72H60N6: C, 85.68; H, 8.33; N, 8.33 found: C, 85.76; H, 5.99, N, 8.33.
1,3,5-Tris(2-(5-hexylthiophen-2-yl)benzimidazol-1-ylmethyl)-2,4,6-trimethylbenzene (3a). Yellow solid product (209 mg, 83%) was prepared from L3 (213 mg, 0.75 mmol), NaH (22 mg, 0.9 mmol) and 1,3,5-tris(bromomethyl)mesitylene (100 mg, 0.25 mmol). 1H NMR (500 MHz, d6-DMSO, ppm): δ 7.62 (3H, d, JHH = 3.6 Hz, Ha), 7.55 (3H, d, JHH = 8.1 Hz, H4), 7.10 (3H, t, JHH = 7.6 Hz, H5), 6.97 (3H, d, JHH = 3.4 Hz, Hb), 6.63 (3H, t, JHH = 7.6 Hz, H6), 6.29 (3H, d, JHH = 8.2 Hz, H7), 5.70 (6H, s, –CH2–), 2.85 (6H, t, JHH = 7.4 Hz, thio–CH2–), 1.99 (9H, s, –CH3), 1.66 (6H, quint, JHH = 7.4 Hz, –C–CH2–), 1.35–1.27 (18H, m, –(CH2)3–), 0.85 (9H, t, JHH = 6.8 Hz, –C–CH3). 13C NMR (100 MHz, CDCl3, ppm): δ 150.41, 148.58, 143.26, 138.80, 134.80, 131.19, 129.23, 128.90, 124.96, 122.96, 122.94, 122.27, 119.97, 110.83 (aromatic and thiophene), 46.29 (–CH2–), 17.01 (–CH3), 31.54, 31.48, 30.18, 28.77, 22.57 and 14.09 (hexyl). HR-MS (m/z): calc. for C63H73N6S3 (M + H)+: 1009.5059; found, 1009.5054. Anal. calc. for C63H72N6S3: C, 74.96; H, 7.19; N, 8.33; S, 9.53 found C, 74.89; H, 7.15; N, 8.26; S, 9.70.
1,3,5-Tris(2-(5-hexylthiophen-2-yl)benzimidazol-1-ylmethyl)-2,4,6-triethylbenzene (3b). Yellow solid product (421 mg, 88%) was prepared from L3 (387 mg, 1.36 mmol), NaH (40 mg, 1.7 mmol) and 1,3,5-tris(bromomethyl)-2,4,6-triethylbenzene (200 mg, 0.45 mmol). 1H NMR (500 MHz, d6-DMSO, ppm): δ 7.69 (3H, d, JHH = 3.2 Hz, Ha), 7.56 (3H, t, JHH = 6.8 Hz, H4), 7.08 (3H, quint, JHH = 6.9 Hz, H5), 7.00 (3H, s, Hb), 6.45–6.39 (3H, m, H6), 6.24–6.20 (3H, m, H7), 5.67 (6H, s, –CH2–), 2.87 (6H, t, JHH = 7.2 Hz, thio–CH2–), 2.46 (6H, d, JHH = 1.8 Hz, –CH2–CH3), 1.72–1.63 (6H, m, –C–CH2–), 1.39–1.27 (18H, m, –(CH2)3–) and 0.86–0.83 (18H, m, –CH2–CH3 and –C–CH3). 13C NMR (100 MHz, CDCl3, ppm): δ 150.30, 148.26, 145.77, 143.28, 134.91, 130.62, 129.08, 128.92, 124.92, 123.16, 122.21, 119.94, 114.54 (aromatic and thiophene), 44.83 (–CH2–), 23.75 (–CH2–CH3), 14.59 (–CH2–CH3), 31.53, 30.16, 28.6, 22.57, and 14.08 (hexyl). HR-MS (m/z): calc. for C66H79N6S3 (M + H)+: 1051.5528; found, 1051.5525. Anal. calc. for C66H78N6S3: C, 75.38; H, 7.48; N, 7.99; S, 9.15 found C, 75.29; H, 7.56; N, 7.91; S, 9.24.
1,3,5-Tris(2-(5-octylthiophen-2-yl)benzimidazol-1-ylmethyl)-2,4,6-trimethylbenzene (4a). Yellow solid product (486 mg, 89%) was prepared from L4 (470 mg, 1.504 mmol), NaH (43 mg, 1.8 mmol) and 1,3,5-tris(bromomethyl)mesitylene (200 mg, 0.501 mmol). 1H NMR (500 MHz, d6-DMSO, ppm): δ 7.60 (3H, s, Ha), 7.55 (3H, d, JHH = 7.6 Hz, H4), 7.09 (3H, t, JHH = 6.8 Hz, H5), 6.95 (3H, s, Hb), 6.63 (3H, t, JHH = 7.6 Hz, H6), 6.29 (3H, d, JHH = 7.6 Hz, H7), 5.69 (6H, s, –CH2–), 2.83 (6H, s, thio–CH2–), 1.98 (9H, s, –CH3), 1.65 (6H, s, –C–CH2–), 1.29–1.22 (30H, m, –(CH2)5–), 0.83 (9H, s, –CH3). 13C NMR (100 MHz, d6-DMSO, ppm): δ 149.52, 148.39, 143.09, 138.76, 135.16, 131.16, 131.44, 129.78, 129.70, 125.79, 122.81, 122.06, 119.35, 111.44 (aromatic and thiophene), 46.37 (–CH2–), 16.78 (–CH3), 31.74, 31.49, 29.85, 29.17, 29.11, 28.96, 22.56 and 14.36 (octyl). HR-MS (m/z): calc. for C69H85N6S3 (M + H)+: 1093.5998; found, 1093.5991. Anal. calc. for C69H84N6S3: C, 75.78; H, 7.74; N, 7.68; S, 8.79 found C, 75.86; H, 7.68; N, 7.75; S, 8.71.
1,3,5-Tris(2-(5-octylthiophen-2-yl)benzimidazol-1-ylmethyl)-2,4,6-triethylbenzene (4b). Yellow solid product (427 mg, 83%) was prepared from L4 (425 mg, 1.36 mmol), NaH (40 mg, 1.7 mmol) and 1,3,5-tris(bromomethyl)-2,4,6-triethylbenzene (200 mg, 0.45 mmol). 1H NMR (500 MHz, d6-DMSO, ppm): δ 7.67 (3H, d, JHH = 3.4 Hz, Ha), 7.57 (3H, d, JHH = 8.0 Hz, H4), 7.09 (3H, t, JHH = 7.5 Hz, H5), 6.99 (3H, s, Hb), 6.40–6.35 (3H, br s, H6), 6.21 (3H, s, H7), 5.67 (6H, s, –CH2–), 2.87 (6H, t, JHH = 7.1 Hz, thio–CH2–), 2.49 (6H, s, –CH2–CH3), 1.67 (6H, t, JHH = 6.7 Hz, –C–CH2–), 1.30–1.23 (30H, m, –(CH2)5–), 0.82 (9H, t, JHH = 5.5 Hz, –CH3), 0.79–0.72 (9H, m, –CH2–CH3). 13C NMR (100 MHz, CDCl3, ppm): δ 150.31, 148.26, 145.76, 143.76, 143.28, 134.91, 130.63, 129.09, 128.91, 124.92, 123.16, 122.21, 119.94, 111.94 (aromatic and thiophene), 44.83 (–CH2–), 23.78 (–CH2–CH3), 14.59 (–CH2–CH3), 31.85, 31.58, 30.17, 29.22, 29.05, 22.66, and 14.11 (octyl). HR-MS (m/z): calc. for C72H91N6S3 (M + H)+: 1135.6467, found 1135.6458. Anal. calc. for C72H90N6S3: C, 76.14; H, 7.99; N, 7.40; S, 8.47 found C, 76.24; H, 7.89; N, 7.32; S, 8.55.
DFT calculations. Geometry optimization of 1a–1b and 2a–2b structures were performed with Gaussian 09 suite of packages18 at MPW1PW91/6-311G(d,p) level of theory and basis set. The stationary points were characterized by the absence of imaginary frequencies in the harmonic normal vibrational mode analysis. The optimized structures at this level of theory and basis are furnished in the Tables S1–S4 in ESI.† C–H⋯π intramolecular interaction energy calculations were performed at MPW1PW91 and M06-2X levels of theory with 6-311+G(d,p) basis function. The relative magnitude of the NMR chemical shift in the presence of DMSO solvent was calculated relative to ligand (L1–L2) evaluated with solvation model based on density (SMD) solvent model20 and MPW1PW91 level of theory with 6-311+G(d,p) basis function in Gaussian 09 suite of packages.18 MESP of the 1a–2b molecules were evaluated with Gaussian 09 suite of packages at MPW1PW91/6-311+G(d,p) level of theory and basis set.24
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
CKK thank UGC-DSKPDF, SDS thank UGC-NRC, and KVJJ thank UGC-FRPS for funding.
References
- E. Lanzarotti, R. R. Biekofsky, D. A. Estrin, M. A. Marti and A. G. Turjanski, J. Chem. Inf. Model., 2011, 51, 1623 CrossRef CAS PubMed and references therein.
-
(a) T. P. Sauer and C. D. Sherrill, J. Phys. Chem. A, 2005, 109, 10475 CrossRef PubMed;
(b) S. R. Gadre, S. D. Yeole and N. Sahu, Chem. Rev., 2014, 114, 12132 CrossRef CAS PubMed;
(c) A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2016, 116, 2775 CrossRef CAS PubMed;
(d) C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885 RSC;
(e) M. Nishio, Phys. Chem. Chem. Phys., 2011, 13, 13873 RSC;
(f) O. Takahashi, Y. Kohno and M. Nishio, Chem. Rev., 2010, 110, 6049 CrossRef CAS PubMed.
-
(a) S. Sarkhel and G. R. Desiraju, Proteins: Struct., Funct., Bioinf., 2004, 54, 247 CrossRef CAS PubMed;
(b) R. N. V. K. Deepak and R. Sankararamakrishnan, Biophys. J., 2016, 110, 1967 CrossRef CAS PubMed.
-
(a) C. A. Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310 CrossRef CAS PubMed;
(b) J. Singh and J. M. Thornton, FEBS Lett., 1985, 191, 1 CrossRef CAS.
-
(a) T. Morimoto, H. Uno and H. Furuta, Angew. Chem., Int. Ed., 2007, 46, 3672 CrossRef CAS PubMed;
(b) S. Janich, R. Frohlich, A. Wakamiya, S. Yamaguchi and E. U. Wurthwein, Chem.–Eur. J., 2009, 15, 10457 CrossRef CAS PubMed.
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2016, 116, 2775 CrossRef CAS PubMed.
-
(a) K. Funatsu, A. Kimura, T. Imamura, A. Ichimura and Y. Sasaki, Inorg. Chem., 1997, 36, 1625 CrossRef CAS PubMed;
(b) M. Sathiyendiran, J. Y. Wu, M. Velayudham, G. H. Lee, S. M. Peng and K. L. Lu, Chem. Commun., 2009, 3795 RSC;
(c) S. H. Lin, C. I. Yang, T. S. Kuo, M. H. Chiang, K. C. Hsu and K. L. Lu, Dalton Trans., 2012, 41, 1448 RSC;
(d) J. Zaho, K. Peng, Y. Guo, J. Zhang, S. Chen and J. Hu, New J. Chem., 2015, 39, 6016 RSC.
-
(a) P. Elumalai, P. Rajakannu, F. Hussain and M. Sathiyendiran, RSC Adv., 2013, 3, 2171 RSC;
(b) S. D. Sathiyashivan, B. Shankar, P. Rajakannu, P. Vishnoi, D. T. Masram and M. Sathiyendiran, RSC Adv., 2015, 5, 74705 RSC;
(c) S. D. Sathiyashivan, C. K. Kumar, B. Shankar, M. Sathiyendiran and D. T. Masram, RSC Adv., 2017, 7, 17297 RSC.
- R. Ludwig, F. Weinhold and T. C. Farrar, J. Phys. Chem. A, 1997, 101, 8861 CrossRef CAS.
- A. S. Mahadevi, A. Rahalkar, S. R. Gadre and G. N. Sastry, J. Chem. Phys., 2010, 133, 164308 CrossRef PubMed.
- R. Wieczorek and J. J. Dannenberg, J. Am. Chem. Soc., 2004, 126, 14198 CrossRef CAS PubMed.
- T. van Mourik and A. J. Dingley, J. Phys. Chem. A, 2007, 111, 11350 CrossRef CAS PubMed.
- R. D. Parra, S. Bulusu and X. C. Zeng, J. Chem. Phys., 2005, 122, 184325 CrossRef PubMed.
- P. Salvador, N. Kobko, R. Wieczorek and J. J. Dannenberg, J. Am. Chem. Soc., 2004, 126, 14190 CrossRef CAS PubMed.
- K. Ohno, M. Okimura, N. Akai and Y. Katsumoto, Phys. Chem. Chem. Phys., 2005, 7, 3005 RSC.
- S. S. Xantheas, Chem. Phys., 2000, 258, 225 CrossRef CAS.
- S. Shimizu and H. S. Chan, Proteins: Struct., Funct., Bioinf., 2002, 48, 15 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision E.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- M. M. Deshmukh and S. R. Gadre, J. Phys. Chem. A, 2009, 113, 7927 CrossRef CAS PubMed.
- S. R. Gadre, S. A. Kulkarni and I. H. Shrivastava, J. Chem. Phys., 1992, 96, 5253 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- S. R. Gadre and I. H. Shrivastava, J. Chem. Phys., 1991, 94, 4384 CrossRef CAS.
- C. H. Suresh and S. R. Gadre, J. Am. Chem. Soc., 1998, 120, 7049 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
-
(a) Y. Wei, Y. Yang and J.-M. Yeh, Chem. Mater., 1996, 8, 2659 CrossRef CAS;
(b) C. Zheng, S. Pu, J. Xu, M. Luo, D. Huang and L. Shen, Tetrahedron, 2007, 63, 5437 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR spectra and theoretical data of L1–L4 and 1a–4b. See DOI: 10.1039/c8ra06647g |
| ‡ Both authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Shankar Deval Sathiyashivan‡b,
Dhanraj T. Masram*b,
K. V. Jovan Jose*a and
Malaichamy Sathiyendiran
a,
Shankar Deval Sathiyashivan‡b,
Dhanraj T. Masram*b,
K. V. Jovan Jose*a and
Malaichamy Sathiyendiran