
A formal [4 + 2] cycloaddition of sulfur-containing alkylidene heterocycles with allenic compounds†
Vojtěch
Dočekal‡
 a,
Bedřich
Formánek‡
a,
Ivana
Císařová
b and
Jan
Veselý
*a
a,
Bedřich
Formánek‡
a,
Ivana
Císařová
b and
Jan
Veselý
*a
aDepartment of Organic Chemistry, Faculty of Science, Charles University, Hlavova 2030/8, 128 43 Prague 2, Czech Republic. E-mail: jan.vesely@natur.cuni.cz; Web: http://orgchem.cz/vesely/
bDepartment of Inorganic Chemistry, Faculty of Science, Charles University, Hlavova 2030/8, 128 43 Prague 2, Czech Republic
First published on 2nd August 2019
Abstract
In this study, we report an enantioselective synthesis of sulfur heterocycles containing the dihydro-2H-pyran moiety. The synthesis strategy is based on quinidine catalyzed formal [4 + 2] cycloaddition between 3-alkylidene benzo[b]thiophenes and allenoates, affording the corresponding cycloadducts in high yields (up to 95%) with high enantioselectivities (up to 99% ee). Moreover, cycloadducts can be isolated by simple filtration from the reaction media.
Introduction
Benzo[b]thiophene motifs can be found in many naturally occurring and synthetic compounds and have applications in diverse areas.1 Many of them exhibit interesting biological and pharmacological activities, and several representatives are available on the pharmaceutical market. For example, raloxifene is used as a selective estrogen modulator,2 zileuton is known as a 5-lipoxygenase inhibitor,3 and benocyclidine is one of the most potent known dopamine reuptake inhibitors (Fig. 1).4 As the benzo[b]thiophene scaffold is considered as a privileged structure in drug discovery,5 the development of efficient synthetic methods affording such molecules is of high interest.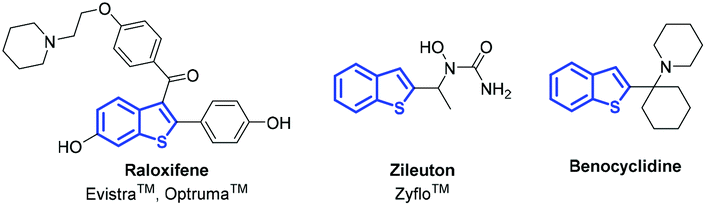 | ||
| Fig. 1 Selected examples of biologically relevant compounds. | ||
Over the past few decades, Lewis base promoted cycloaddition reaction has been extensively explored. Among the most studied substrates used for catalytic formal cycloadditions are allenoates. After the pioneering work of Lu,6 reporting a phosphine-catalyzed [3 + 2] cycloaddition of allenoates with electron-deficient alkenes, this methodology was adopted to investigate various substrates with polarized C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) X double bonds, such as imines, carbonyls, and other activated olefins.7 Allenoates represent C2 or C3 synthons in cycloadditions depending on the Lewis base used. According to the different stabilities of amines or phosphorus zwitterionic adducts formed between Lewis base and allenoate in the initial step, cycloadditions provide different products.8 Generally, the reaction catalyzed by tertiary phosphine prefers “1,3-dipole” formation, leading to formal [3 + 2] cycloadducts. In contrast, tertiary amine catalysis generates “1,2-dipole”, resulting in formal [4 + 2] or [2 + 2] cycloaddition depending on the characteristics of the electrophile. α,β-Unsaturated carbonyl compounds are commonly used heterodienes together with allenoates in amine-catalyzed formal [4 + 2] cycloadditions (Fig. 2).9 In sharp contrast to the well-developed cycloaddition reactions of allenoates with various alkylidene nitrogen- and oxygen-containing heterocycles,10 there is no report on tertiary amine catalyzed formal [4 + 2] cycloaddition between alkylidene sulfur-containing heterocycles and allenoates.
X double bonds, such as imines, carbonyls, and other activated olefins.7 Allenoates represent C2 or C3 synthons in cycloadditions depending on the Lewis base used. According to the different stabilities of amines or phosphorus zwitterionic adducts formed between Lewis base and allenoate in the initial step, cycloadditions provide different products.8 Generally, the reaction catalyzed by tertiary phosphine prefers “1,3-dipole” formation, leading to formal [3 + 2] cycloadducts. In contrast, tertiary amine catalysis generates “1,2-dipole”, resulting in formal [4 + 2] or [2 + 2] cycloaddition depending on the characteristics of the electrophile. α,β-Unsaturated carbonyl compounds are commonly used heterodienes together with allenoates in amine-catalyzed formal [4 + 2] cycloadditions (Fig. 2).9 In sharp contrast to the well-developed cycloaddition reactions of allenoates with various alkylidene nitrogen- and oxygen-containing heterocycles,10 there is no report on tertiary amine catalyzed formal [4 + 2] cycloaddition between alkylidene sulfur-containing heterocycles and allenoates.
 | ||
| Fig. 2 Cycloaddition reaction of allenoates. | ||
Results and discussion
We initiated our study with readily accessible 3-benzylidenebenzo[b]thiophen-2(3H)-one (1a) and benzyl buta-2,3-dienoate (2a) in the presence of quinidine (QD) as a chiral catalyst. To our delight, the reaction provided the desired cyclization product 3a in high yield and with very good enantioselectivity (entry 1, Table 1). Surprisingly, cinchonine (CN) without the methoxy-group on the quinoline part showed low catalytic activity, giving products 3a and 4a with decreased selectivity, and moreover, 3a with poor level of enantioselectivity (entry 2, Table 1). Introduction of the protecting group (TMS) of hydroxyl on QD had a positive effect on the yield of 3a but resulted in moderate enantioselectivity. The same observation was obtained with β-ICD (entry 4, Table 1). Widely used Sharpless dimeric bases containing quinidine units were also investigated. As expected, all the selected catalysts promoted the reaction with good enantiocontrol (entries 5–8); however, in the case of (DHQD)2PHAL and (DHQD)2PYR, an increased amount of product 4a, with an endocyclic double bond, was obtained.|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entrya | Catalyst | Solvent | Time (h) | Convers.b (%) |
3a/4a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) b b |
Yieldc (3a,%) | eed (3a, %) |
| a The reactions were conducted with 0.01 mmol of 1a, 0.12 mmol of 2a, and 20 mol% QD in 0.5 mL of solvent at rt. b Determined by 1H NMR of the crude product. c Isolated yields. d Determined by chiral HPLC analysis. e The reaction was conducted with 10 mol% 2,4-dinitrobenzoic acid as an additive. f The reaction was conducted with 5 mol% QD and 2.5 mol% 2,4-dinitrobenzoic acid as an additive. g Product precipitated from the reaction mixture. | |||||||
| 1 | QD | CHCl3 | 15 | 100 | 20:1 |
83 | 73 |
| 2 | CN | CHCl3 | 65 | 75 | 15:1 |
72 | 39 |
| 3 | C1 | CHCl3 | 15 | 100 | >20:1 |
91 | 66 |
| 4 | β-lCD | CHCl3 | 15 | 100 | >20:1 |
94 | 48 |
| 5 | (DHQD)2AQN | CHCl3 | 15 | 100 | 20:1 |
83 | 63 |
| 6 | (DHQD)2PHAL | CHCl3 | 65 | 70 | 8:1 |
47 | 64 |
| 7 | (DHQD)2PYR | CHCl3 | 15 | 90 | 11:1 |
43 | 71 |
| 8 | (DHQ)2PYR | CHCl3 | 15 | 100 | 20:1 |
86 | −68 |
| 9 | QD | Benzene | 15 | 100 | 20:1 |
93 | 79 |
| 10 | QD | THF | 15 | 100 | 19:1 |
90 | 78 |
| 11 | QD | DMF | 15 | 100 | 4:1 |
62 | 86 |
| 12 | QD | iPrOH | 15 | 100 | >20:1 |
77g | 67 |
| 13 | QD | MeOH | 15 | 100 | >20:1 |
82g | 89 |
| 14e | QD | MeOH | 15 | 97 | >20:1 |
92g | 90 |
| 15f | QD | MeOH | 72 | 90 | >20:1 |
75g | 86 |

Conversely, (DHQD)2AQN was identified to be most selective for the production of exo-olefinic product 3a, but unfortunately, with the lowest enantiocontrol from these types of catalyst. Further catalyst screening conducted with pseudoenantiomers of the abovementioned Cinchona alkaloid derivatives revealed preferential formation of the product with opposite enantiocontrol, ent-3a. For example, the model reaction of 1a with 2a catalyzed with (DHQ)2PYR afforded ent-3a in 86% yield with moderate enantioselectivity (68% ee, entry 8). For more details, see Table S1 in the ESI.†
According to our observations (Table 1), the model reaction tolerates a wide range of polar and non-polar solvents, such as chlorinated, aromatic, ethereal solvent as well as alcohols and DMF. Performing the model reaction in most of these solvents led to the formation of product 3a in high yields with moderate to good enantioselectivities (entries 1 and 9–13). The highest yields and enantioselectivities of 3a were achieved in alcohols (entries 12 and 13), where the highly enantiomerically enriched product 3a precipitated directly from the reaction mixtures. For example, the reaction in methanol afforded 3a in 82% yield with high enantioselectivity (89% ee, entry 13). For more details, see Table S2 in the ESI.† Subsequently, the presence of an additional Brønsted acid had a substantial effect on the reaction yield. When 2,4-dinitrobenzoic acid was used, product 3a was obtained in higher yield (92%, entry 14, for other additives see Table S3 in the ESI†). On the other hand, the reaction temperature and concentration did not have a marked effect on reaction efficiency and enantioselectivity (for details see Tables S4 and S5 in the ESI†). A slight decrease of reaction efficiency was observed upon reduction of catalyst loading and additives (entry 15, for details see Table S6 in the ESI†). The abovementioned results indicated that the most suitable conditions for formal [4 + 2] cycloaddition between 1a and 2a were achieved with quinidine and 2,4-dinitrobenzoic acid in methanol at room temperature (entry 13, Table 1).
After optimizing the reaction conditions, we began to explore the scope of organocatalytic cycloaddition reactions by varying sulfur-containing alkylidene 1 (Scheme 1). Cycloadducts 3b, c, and e derived from alkylidenes 1b, c, and e containing electron-withdrawing groups (EWGs) at position 5 of the benzo[b]thiophene ring were produced in high yields (72–87%) with a high degree of enantioselectivity (80–89% ee). Similarly, alkylidenes 1f–g substituted at position 6 or 7 of the benzo[b]thiophene ring showed comparable reactivity and similar enantiocontrol under the reported reaction conditions. Significantly reduced reactivity was observed, when the electron-donating group (EDG) was present at the benzo[b]thiophene ring. The observed low conversion of 1d may be explained by the electron-rich behavior of the starting material.
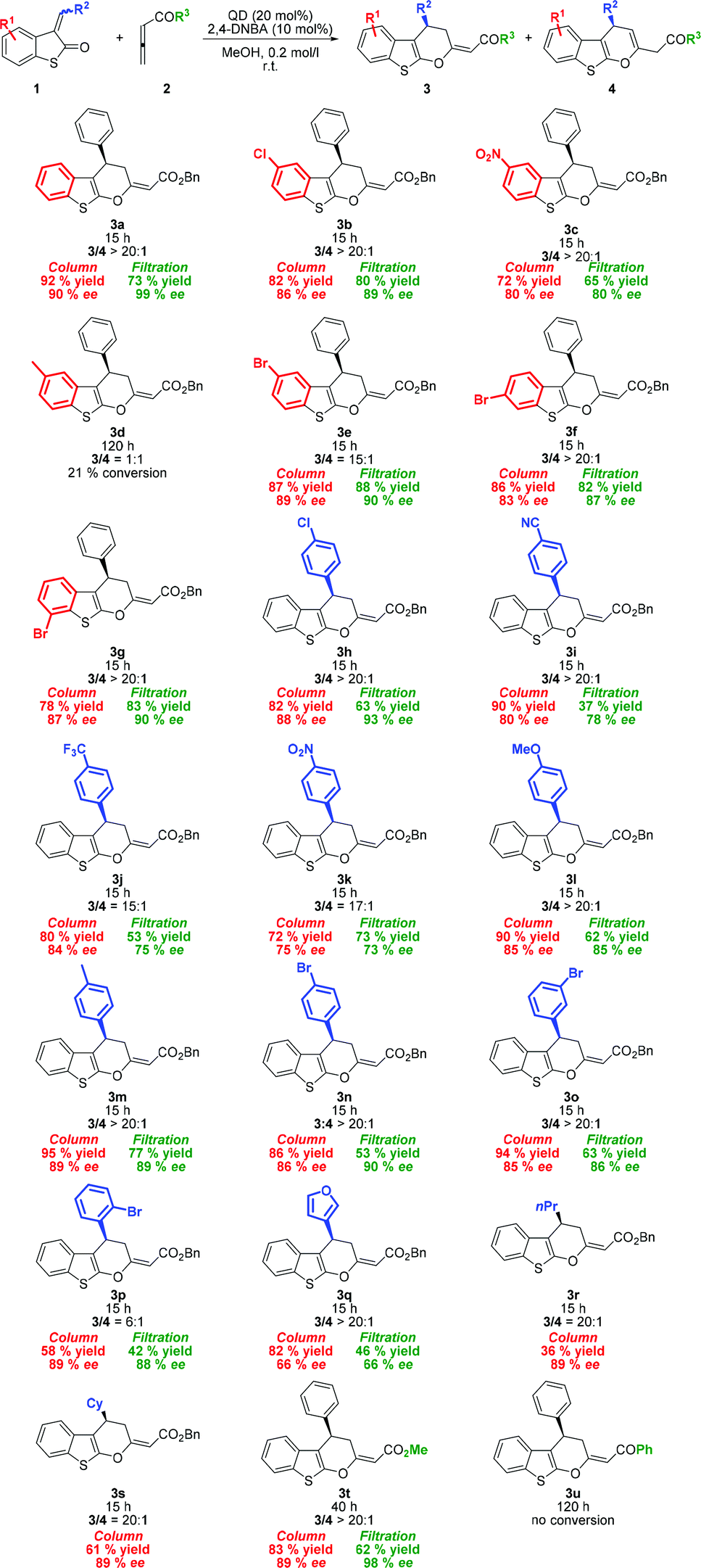 | ||
| Scheme 1 Substrate scope of the formal cycloaddition reaction. | ||
Next, we surveyed the scope of the reaction with various 3-subsituted vinyl benzothiopen-2-ones. As shown in Scheme 1, a variety of aryl vinyl derivatives bearing either EWG or EDG at the para-position are tolerated giving the desired products 3h–n in high yields (72–95%). 3-Benzylidene benzothiopen-2-ones with EWG substituents produced the corresponding products (3h–j) with a good level of enantiomeric purity (83–85% ee). When the strongly electron-withdrawing nitro group was present, enantioselectivity significantly dropped to 75% ee. Conversely, substrates bearing EDG (3l–m) were obtained in excellent yields with high enantiocontrol. For example, the reaction between p-methylbenzylidene benzothiopen-2-one 1m and 2a gave 3m in 95% yield with the enantioselectivity of 89% ee. Whereas the meta-substituted aryl derivative (1o) did not show any change in reactivity and selectivity from para-substituted analogues, ortho-substituted aryl vinyl benzothiophen-3-one (1p) produced cyclic products with diminished exo/endo selectivity (3p/4p = 6:1). Nevertheless, product 3p was isolated in acceptable yield (58%) with high enantiomeric purity (89% ee). Furthermore, the developed protocol was also applied to benzothiopen-2-ones bearing the heterocyclic moiety (1q) and alkyl groups (1r–s) affording cycloadducts 3q–s in good to acceptable yields. To our disappointment, the reaction with n-propyl derivative 1r did not reach full conversion even with a prolonged reaction time, and the corresponding product 3r was obtained in low yield.
To extend the substrate scope, other allenic substrates 2b–c were also examined (Scheme 1). As expected, methyl ester derivate 3t was obtained in high yield and with high enantioselectivity. On the other hand, the reaction with allenic ketones failed. No conversion to cycloadduct 3u was observed, probably due to increased tendency to be protonated prior to the formation of zwitterionic intermediates.
The developed organocatalytic procedure was also applied to alkylidenes derived from other sulfur-containing heterocycles 5a–c. Regioisomeric 2-benzylidenebenzo[b]thiophen-3(2H)-one (5a) exhibited lower reactivity, but the desired cycloadduct 6a was obtained after the prolonged reaction time in good yield with excellent selectivity (98% ee, Scheme 2).
 | ||
| Scheme 2 Substrate scope of the formal cycloaddition reaction with diverse sulfur containing alkylidenes 5. | ||
5-Membered heteroaromatic derivatives containing sulfur and nitrogen atoms can also be accommodated in the cycloaddition process. 5-Benzylidene-2-phenylthiazol-4(5H)-one 5b, as well as its isomeric analogue 5c, provided the corresponding products 6b and 6c, respectively, in good yields with high to excellent enantioselectivity. Noteworthily, cycloadducts 3 and 6 can also be isolated by simple filtration from the reaction performed in methanol, which supports the practical use of the developed process (Schemes 1 and 2). Using this separation technique, the corresponding products were obtained in high yields (up to 88%) with high enantioselectivities (up to 98% ee).
The applicability of the prepared cycloadducts was demonstrated on the following set of transformations. Cycloadducts 3 can be easily converted into the corresponding 4H-pyran derivatives 4. For example, compound 3a was converted by the treatment of DBU with pyran 4a in good yield (77%) without losing the enantiomeric purity (99% ee, Scheme 3). Similar results were obtained, when 3a was oxidized to sulfone 8 using MCPBA or selectively reduced to 9 under catalytic hydrogenation conditions. Furthermore, bromoderivative 3e was used in Suzuki coupling with aryl boronic acid, affording biphenyl compound 10 without losing the enantiomeric purity (Scheme 3).
 | ||
| Scheme 3 Gram-scale experiment and other transformations of cycloadducts 3. | ||
The absolute configuration of cycloadducts 3 and 6 was assigned (3R) on the dihydropyran ring using X-ray diffraction analysis of compounds 3a and 6b (Fig. 3, for details see the ESI†).11
 | ||
| Fig. 3 X-ray structure of cycloadducts 3a and 6b.11 | ||
Based on the determined absolute configuration of 3 and the previous mechanistic studies,8,12 a reaction mechanism for an asymmetric formal [4 + 2] cycloaddition can be proposed (Scheme 4). Initially, zwitterionic intermediate II is formed via an addition of a tertiary amine catalyst to allenoate 2. Subsequently, an intermolecular attack of II to electron-deficient β carbon of 1 results in the formation of enolic intermediate III. The origin of a high stereocontrol of the attack in the case of arylidene derivatives can be explained by possible aromatic stacking stabilization between the aryl moiety of 1 and the quinoline ring of II. In the final step, enolic intermediate III undergoes 6-endo-trig ring-closure and the elimination of the catalyst affording the final product 3.
 | ||
| Scheme 4 Plausible reaction mechanism. | ||
Conclusion
In summary, we have developed an asymmetric formal [4 + 2] cycloaddition between readily available sulfur-containing alkylidenes and allenoates. The reaction is efficiently catalyzed by quinidine, generating chiral cycloadducts in excellent yields (up to 95%) with high enantioselectivity (up to 99% ee). Moreover, the robustness of the developed method is demonstrated in a broad range of heterocyclic alkylidenes, and the synthetic utility of the protocol is exemplified by the user-friendly purification approach based on simple filtration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Jan Veselý gratefully acknowledges the Czech Science Foundation (No. 18-20645S) for financial support. Vojtěch Dočekal thanks the Charles University Grant Agency (No. 1504217) for financial support.Notes and references
- A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Comprehensive Heterocyclic Chemistry II, Elsevier (Perganom), Oxford, 1996 Search PubMed.
- (a) D. B. Muchmore, Oncologist, 2000, 5, 388 CrossRef CAS PubMed; (b) V. C. Jordan, J. Med. Chem., 2003, 46, 1081 CrossRef CAS PubMed; (c) C. D. Jones, M. G. Jevnikar, A. J. Pike, M. K. Peters, L. J. Black, A. R. Thompson, J. F. Falcone and J. A. Clemens, J. Med. Chem., 1984, 27, 1057 CrossRef CAS PubMed.
- (a) E. Israel, R. Dermarkarian, M. Rosenberg, R. Sperling, G. Taylor, P. Rubin and J. M. Drazen, N. Engl. J. Med., 1990, 323, 1740 CrossRef CAS PubMed; (b) P. Lu, M. L. Schrag, D. E. Slaughter, C. E. Raab, M. Shou and A. D. Rodrigues, Drug Metab. Dispos., 2003, 31, 1352 CrossRef CAS PubMed.
- J. Vignon, V. Pinet, C. Cerruti, J. M. Kamenka and R. Chicheportiche, Eur. J. Pharmacol., 1988, 148, 427 CrossRef CAS PubMed.
- R. S. Keri, K. Chand, S. Budagumpi, S. B. Somappa, S. A. Patil and B. M. Nagaraja, Eur. J. Med. Chem., 2017, 138, 1002 CrossRef CAS PubMed.
- C. Zhang and X. Lu, J. Org. Chem., 1995, 60, 2906 CrossRef CAS.
- For comprehensive book and reviews about cycloadditions of allenoates, see: (a) M. Shi, Y. Wei, M.-X. Zhao and J. Zhang, Organocatalytic Cycloadditions for Synthesis of Carbo- and Heterocycles, Wiley-VCH, Weinheim, 2018 CrossRef; (b) B. J. Cowen and S. J. Miller, Chem. Soc. Rev., 2009, 38, 3102 RSC; (c) F. López and J. L. Mascareñas, Chem. Soc. Rev., 2014, 43, 2904 RSC; (d) S. E. Denmark and G. L. Beutner, Angew. Chem., Int. Ed., 2008, 47, 1560 CrossRef CAS PubMed; (e) Z. Wang, X. Xub and O. Kwon, Chem. Soc. Rev., 2014, 43, 2927 RSC; (f) X. Lu, C. Zhang and Z. Xu, Acc. Chem. Res., 2001, 34, 535 CrossRef CAS PubMed; (g) J. L. Methot and W. R. Roush, Adv. Synth. Catal., 2004, 346, 1035 CrossRef CAS; (h) L.-W. Ye, J. Zhou and Y. Tang, Chem. Soc. Rev., 2008, 37, 1140 RSC; (i) A. Marinetti and A. Voituriez, Synlett, 2010, 174 CrossRef CAS; (j) Q.-Y. Zhao, Z. Lian, Y. Wei and M. Shi, Chem. Commun., 2012, 48, 1724 RSC; (k) Z. Wang, X. Xu and O. Kwon, Chem. Soc. Rev., 2014, 43, 2927 RSC; (l) Y. Xiao, H. Guo and O. Kwon, Aldrichimica Acta, 2016, 49, 3 CAS; (m) T. Wang, X. Han, F. Zhong, W. Yao and Y. Lu, Acc. Chem. Res., 2016, 49, 1369 CrossRef CAS PubMed; (n) Y. Wei and M. Shi, Org. Chem. Front., 2017, 4, 1876 RSC.
- (a) G.-T. Huang, T. Lankau and C.-H. Yu, J. Org. Chem., 2014, 79, 1700 CrossRef CAS PubMed; (b) Q. Shi, W. Wang, Y. Wang, Y. Lan, C. Yao and D. Wei, Org. Chem. Front., 2019, 6, 2692 RSC.
- For recent examples of amine catalyzed formal [4 + 2] cycloaddition reactions of unsaturated carbonyls with allenoates, see: (a) K. D. Ashtekar, R. J. Staples and B. Borhan, Org. Lett., 2011, 13, 5732 CrossRef CAS PubMed; (b) Y. Wang, Z.-H. Yu, H.-F. Zheng and D.-Q. Shi, Org. Biomol. Chem., 2012, 10, 7739 RSC; (c) C.-K. Pei, Y. Jiang and M. Shi, Org. Biomol. Chem., 2012, 10, 4355 RSC; (d) C.-K. Pei and M. Shi, Chem. – Eur. J., 2012, 18, 6712 CrossRef CAS PubMed; (e) D.-H. Zhang and M. Shi, ChemistryOpen, 2012, 1, 215 CrossRef CAS PubMed; (f) C.-K. Pei, Y. Jiang, Y. Wei and M. Shi, Angew. Chem., Int. Ed., 2012, 51, 11328 CrossRef CAS PubMed; (g) H.-B. Yang, Y.-Z. Zhao, R. Sang, Y. Wei and M. Shi, Adv. Synth. Catal., 2014, 356, 3799 CrossRef CAS; (h) C. Ni, Y. Yuan, Y. Zhang, J. Chen, D. Wang and X. Tong, Org. Biomol. Chem., 2017, 15, 4807 RSC; (i) K. Kasten, D. B. Cordes, A. M. Z. Slawin and A. D. Smith, Eur. J. Org. Chem., 2016, 3619 CrossRef CAS; (j) X. Wang, T. Fang and X. Tong, Angew. Chem., Int. Ed., 2011, 50, 5361 CrossRef CAS PubMed; (k) X.-S. Meng, S. Jiang, X.-Y. Xu, Q.-X. Wu, Y.-C. Gu and D.-Q. Shi, RSC Adv., 2016, 6, 66630 RSC; (l) A. L. S. Kumari and K. C. K. Swamy, J. Org. Chem., 2015, 80, 4084 CrossRef CAS PubMed; (m) T.-T.-D. Ngo, K. Kishi, M. Sako, M. Shigenobu, C. Bournaud, M. Toffano, R. Guillot, J.-P. Baltaze, S. Takizawa, H. Sasai and G. Vo-Thanh, ChemistrySelect, 2016, 1, 5414 CrossRef CAS.
- For recent examples of amine catalyzed formal [4 + 2] cycloaddition of alkylidenes with allenoates, see: (a) X.-C. Zhang, S.-H. Cao, Y. Wei and M. Shi, Org. Lett., 2011, 13, 1142 CrossRef CAS PubMed; (b) X.-Y. Chen, M.-W. Wen, S. Ye and Z.-X. Wang, Org. Lett., 2011, 13, 1138 CrossRef CAS PubMed; (c) F. Wang, Z. Li, J. Wang, X. Li and J.-P. Cheng, J. Org. Chem., 2015, 80, 5279 CrossRef CAS PubMed; (d) F. Wang, C. Luo, Y.-Y. Shen, Z.-D. Wang, X. Li and J.-P. Cheng, Org. Lett., 2015, 17, 338 CrossRef CAS PubMed; (e) F. Wang, C. Yang, X.-S. Xue, X. Li and J.-P. Cheng, Chem. – Eur. J., 2015, 21, 10443 CrossRef CAS PubMed; (f) Q.-Z. Xi, Z.-J. Gan, E.-Q. Li and Z. Duan, Eur. J. Org. Chem., 2018, 4917 CrossRef CAS; (g) S. Zhang, Y.-C. Luo, X.-Q. Hu, Z.-Y. Wang, Y.-M. Liang and P.-F. Xu, J. Org. Chem., 2015, 80, 7288 CrossRef CAS PubMed; (h) Y.-H. Deng, W.-D. Chu, X.-Z. Zhang, X. Yan, K.-Y. Yu, L.-L. Yang, H. Huang and C.-A. Fan, J. Org. Chem., 2017, 82, 5433 CrossRef CAS PubMed; (i) Z. Wang, T. Wang, W. Yao and Y. Lu, Org. Lett., 2017, 19, 4126 CrossRef CAS PubMed; (j) C. Cheng, X. Sun, Z. Wu, Q. Liu, L. Xiong and Z. Miao, Org. Biomol. Chem., 2019, 17, 3232 RSC; (k) R. Mutyalaa, V. R. Reddya, R. Donthia, V. S. R. Kallaganti and R. Chandra, Tetrahedron Lett., 2019, 60, 703 CrossRef.
- CCDC 1916993 (3a), CCDC 1916994 (6b) contains the supplementary crystallographic data for this paper.†.
- K. D. Ashtekar, X. Ding, E. Toma, W. Sheng, H. Gholami, C. Rahn, P. Reed and B. Borhan, Org. Lett., 2016, 18, 3976 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1916993 and 1916994. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qo00886a |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2019 |