
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTrioxotriangulene with carbazole: a donor–acceptor molecule showing strong near-infrared absorption exceeding 1000 nm†
Tsuyoshi
Murata
 a,
Kazuki
Kariyazono
b,
Shusaku
Ukai
a,
Akira
Ueda
c,
Yuki
Kanzaki
d,
Daisuke
Shiomi
d,
Kazunobu
Sato
d,
Takeji
Takui
d and
Yasushi
Morita
*a
a,
Kazuki
Kariyazono
b,
Shusaku
Ukai
a,
Akira
Ueda
c,
Yuki
Kanzaki
d,
Daisuke
Shiomi
d,
Kazunobu
Sato
d,
Takeji
Takui
d and
Yasushi
Morita
*a
aDepartment of Applied Chemistry, Faculty of Engineering, Aichi Institute of Technology, Toyota, Aichi 470-0392, Japan. E-mail: moritay@aitech.ac.jp
bDepartment of Chemistry, Graduate School of Science, Osaka University, Toyonaka, Osaka 560-0043, Japan
cDepartment of Chemistry, Kumamoto University, 2-39-1 Kurokami, Chuo-ku, Kumamoto 860-8555, Japan
dDepartment of Chemistry and Molecular Materials Science, Graduate School of Science, Osaka City University, Sumiyoshi-ku, Osaka, 558-8585, Japan
First published on 22nd July 2019
Abstract
A donor–acceptor type trioxotriangulene (TOT) neutral radical derivative having three carbazolyl groups as electron-donors was newly synthesized. The ESR spectrum showed that the electronic spin delocalized over both the TOT skeleton and three carbazolyl groups, causing high stability of the neutral radical species. In the cyclic voltammetry measurement, the adduct exhibited multiple redox waves from the redox processes of TOT from the monocation to radical tetraanion species and also from the oxidation of the carbazolyl groups. The UV-vis spectra showed a strong near-infrared photoabsorption band with an absorption maximum of 1028 nm, which was characterized as an intramolecular charge-transfer from the carbazolyl group to the TOT neutral radical core. In both solution and solid states, the neutral radical formed a π-dimer stabilized by the strong two-electron-multicenter bonding on the TOT skeleton.
Introduction
Organic molecules that contain electron–donor (D) and electron–acceptor (A) units (D–A) are of interest in the development of organic electronics due to the intramolecular electron transfer (IET) processes which have various applications such as non-linear optics,1 photovoltaics,2 photoconductivity,3 ambipolar charge-transport4etc. In particular, a strong IET absorption band with a large bathochromic shift extending into the near-infrared (NIR) region is the most characteristic feature of D–A molecules.5 The wavelength of the IET absorption band can be controlled by the strength of D and A units (HOMO and LUMO energies, respectively), and many D–A molecules having an absorption maximum λmax of 500–900 nm have been reported.5 On the other hand, D–A type small molecules exhibiting a IET band with λmax > 1000 nm are rare.6,7 When the D or A unit possesses an unpaired electron, additional intriguing functions can be found, originating from the electronic-spin and open-shell electronic structure. D–A molecules with the perchlorotriphenylmethyl (PTM) neutral radical as an A unit and tetrathiafulvalene (TTF) as a D unit exhibit a high electrical conductivity induced by high pressure (Fig. 1).8 A TTF derivative having a nitronyl nitroxide unit shows a high electrical conductivity of σRT = 9 × 10−4 S cm−1 as a single component under ambient conditions, and also exhibits a magnetoresistance response upon the application of a magnetic field.9 The D–A molecules of the 6-oxophenalenoxyl (6OPO, A unit) neutral radical with TTF exhibit a spin-center transfer phenomenon controlled by the external stimuli, where the spin-center (unpaired electron) moves from 6OPO to TTF units by the IET process.10 A similar transformation of the electronic-spin structure based on the bistability of open-shell D–A molecules has also been observed in PTM neutral radicals with TTF and ferrocene (Fc) units.11,12 In addition to the number of substituted D units, the interaction of the π-electronic system between D and A units would also be important for the intensity of the IET bands. These open-shell D–A molecules also exhibit IET absorption bands in the NIR region with λmax > 1000 nm; however, their intensities are usually very weak (εmax < 103 M−1 cm−1).10–12 The triphenylamine substituted PTM derivatives linked through an ethynyl spacer exhibited a strong IET band at λmax = 800–900 nm, where the intensity of the band increases with the number of substituted D units, from εmax = 5–6 × 103 M−1 cm−1 for the mono-substituted derivative to εmax = 12–17 × 103 M−1 cm−1 for the tri-substituted one.13 | ||
| Fig. 1 Molecules in text. R represents the substituent groups, and the asterisk (*) represents the electronic states of the TOT skeleton. | ||
Trioxotriangulene (TOT, Fig. 1) is a stable organic neutral radical with an electronic-spin structure delocalizing on the whole three-fold symmetric 25π-electronic system.14 The TOT neutral radical possesses a strong electron-accepting ability with the redox potential of −0.05 V vs. Fc/Fc+, which is close to that of tetracyanoquinodimethane (Fig. 1, TCNQ, E = −0.11 V). The strong self-assembling ability of TOT forming one-dimensional π-stacking columns induced high electrical conductivities as single component neutral radicals and mixed-valence salts composed of neutral radicals and monoanion species.15 We have also disclosed that the π-stacking columns of the TOT neutral radical in the crystalline state exhibit a strong NIR absorption with λmax > 1000 nm through intermolecular interactions of the SOMO.16
Here we have newly synthesized a D–A type TOT neutral radical having three carbazolyl groups as D units, 1 (Fig. 1). Carbazole is a heterocyclic chromophore, and has been widely studied as a building block of organic electronic materials due to its intrinsic photophysical and redox properties.17 In particular, the electron-donating and charge-transporting abilities of carbazole derivatives have been utilized in the development of organic light-emitting diodes (OLEDs)18 and photoconductive materials.19 Open-shell D–A compounds of the tris(2,4,6-trichlorophenyl)methyl (TTM, Fig. 1) neutral radical with carbazolyl groups as D units have demonstrated various intriguing functions such as low-energy photoabsorption/emission,19 paramagnetic liquid crystallinity20 and ambipolar charge-transport.21 In this paper, we disclose the electronic effects of the carbazolyl groups on the electronic spin structure, redox properties and π-stacking structure of 1. In particular, we emphasize that the introduction of electron-donating groups to the TOT neutral radical as substituents gave rise to a strong NIR photoabsorption with λmax > 1000 nm with εmax > 104 M−1 cm−1 due to the IET process.
Results and discussion
Synthesis and stability of 1
The synthetic method of 1 is shown in Scheme 1. The carbazolyl groups were introduced into the TOT skeleton by the cross-coupling reaction of tribromo TOT derivative 2 with corresponding 3,6-di-tert-butylcarbazole under the presence of the Pd catalyst.22 After removing the pivaloyl protecting group with a base, the product was acidified with aqueous HCl to afford the radical precursor 3. The treatment of 3 with tetra-n-butylammonium hydroxide ((n-Bu)4N+OH−) gave the monoanion salt ((n-Bu4N+)(1−)), and the oxidation of 3 with PbO2 yielded the neutral radical 1. Similarly to the pristine TOT4, both (n-Bu4N+)(1−) and 1 were stable under air both in solution and in the solid state. Neutral radical 1 did not show any decomposition even at 300 °C under air, and the decomposition point under a N2 atmosphere was ∼400 °C (Fig. S1†).14d Due to the bulky tert-butyl groups around the carbazolyl groups, 1 was highly soluble in toluene, CH2Cl2 and THF to give a green solution; however, it was insoluble in non-polar hexane and strongly polar DMSO and alcohols. | ||
| Scheme 1 Synthesis of neutral radical 1 and monoanion salt 1−. | ||
Electronic spin structure
The electronic-spin structure of neutral radical 1 was investigated by ESR and 1H-ENDOR/TRIPLE spectroscopy in toluene solution. The ESR spectrum of 1 showed a broad signal with hyperfine structures owing to the contributions from small hyperfine splitting (Fig. 2a and b). The observed g-value was 2.0040, which is very similar to that of the pristine TOT4 (g = 2.0041).14d The 1H-ENDOR spectrum (Fig. 2c) showed three kinds of proton hyperfine couplings, and their relative signs were unequivocally determined from the 1H-TRIPLE spectrum (Fig. 2d). Considering the molecular symmetry, the observed hyperfine couplings of protons were assigned with the help of DFT calculations (Fig. 3): the largest splitting is assignable to the six α-protons in the TOT skeleton (a(H1) = +2.16 MHz), whereas the smaller ones are ascribable to the protons of the carbazolyl groups (a(H2) = −0.47 MHz and a(H2) = a(H3) = +0.15 MHz). The experimental and calculated hyperfine coupling constants are in good agreement. These results show that the electronic spin delocalizes over the carbazolyl groups. The delocalization of the electronic spin to the carbazolyl groups is also suggested experimentally from the smaller hyper fine coupling constant of the α-proton on the TOT skeleton compared to that of 4 (a(H1) = 2.57 MHz),14d implying the reduction of the electronic-spin density at the α-carbon atoms. In the DFT calculations, 22% of the total spin densities of 1 is located at the three carbazolyl groups. The intensity of the ESR signal of 1 gradually decreased upon cooling due to the formation of a non-magnetic π-dimer as observed in other TOT derivatives (Fig. S2†), indicating the strong multicenter bonding nature in the π-dimer of the TOT neutral radical.14d The enthalpy and entropy changes for the π-dimerization were determined to be −12.9 kcal mol−1 and −17.5 eu, respectively, from which the dimerization constant Kdimer was determined as 3.7 × 105 M−1.23 These parameters were similar to those for the tri-tert-butyl substituted TOT5, where Kdimer is 1.2 × 105 M−1.16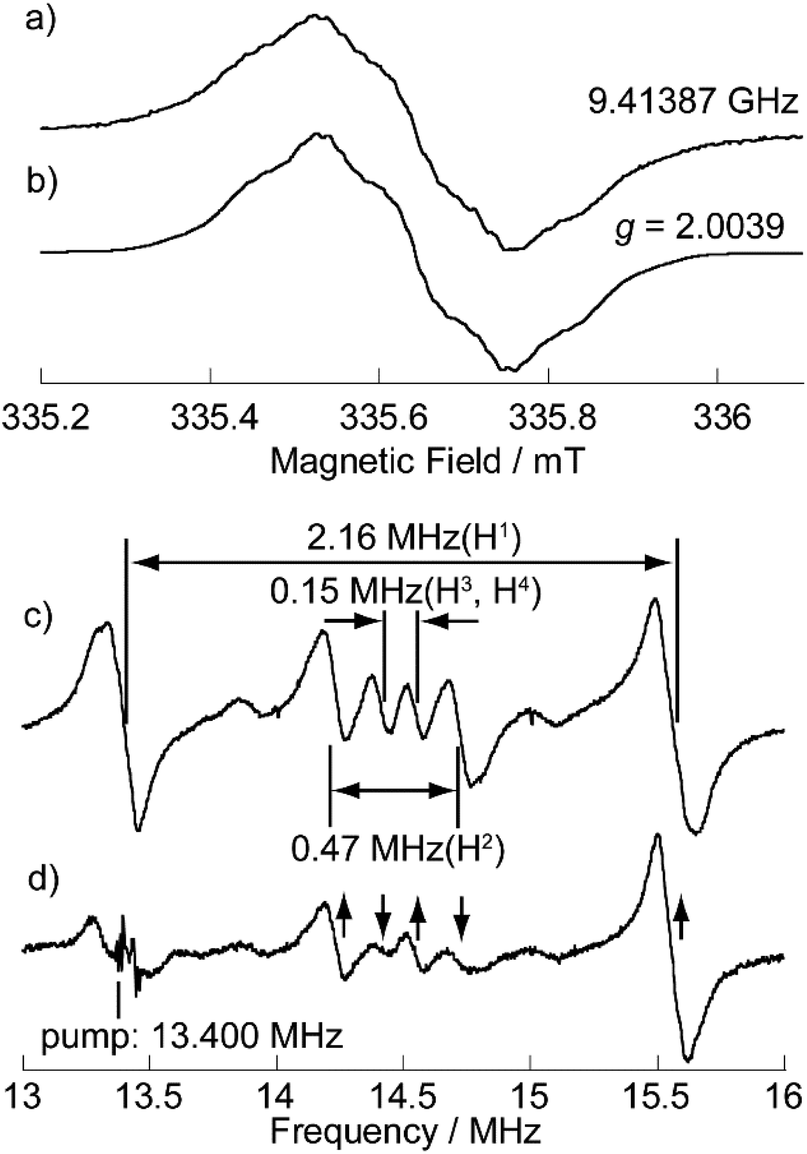 | ||
| Fig. 2 (a) Observed at 273 K and (b) simulated ESR spectra, and (c) 1H-ENDOR and (d) 1H-TRIPLE spectra of 1 in degassed toluene (9 × 10−5 M) at 260 K. The ESR simulation was achieved by optimizing the hyperfine coupling constant (hfcc) of the nitrogen nucleus and line width (Lorentzian line shape) with the proton hfcc's (a's) fixed which are experimentally determined by 1H ENDOR spectroscopy: a(H1) = 2.16 MHz, a(H2) = 0.47 MHz, a(H3) = a(H4) = 0.15 MHz, a(N) = 0.58 MHz and the line width = 0.011 mT (see Fig. S3† for detailed analysis). The upward and downward arrows in (d) denote an increase and decrease in the intensity of an ENDOR signal, respectively, when the pump frequency at 13.400 MHz is applied. Note that the two weak signals appearing at around 13.9 and 15.0 MHz in (c) and (d) are attributable to the unidentified species in extremely small amounts. These signals are free from the TRIPLE effect which is intramolecular, as shown in (d). | ||
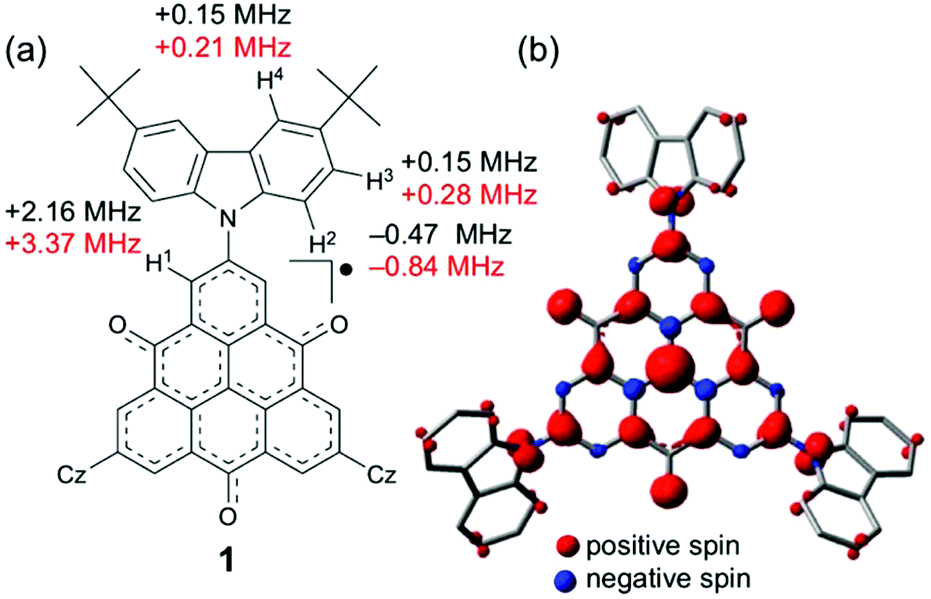 | ||
| Fig. 3 (a) Observed hyperfine coupling constants (in black) and calculated values (in red) of 1. Considering the molecular symmetry, the calculated hyperfine coupling constants for the equivalent nuclei were averaged. Cz indicates the 3,6-di-tert-butylcarbazolyl groups. (b) Calculated spin-density distribution of 1. The calculations were performed by using the DFT method at the UB3LYP/6-31G(d,p) level of theory. The tert-butyl groups were omitted during the calculation. | ||
Cyclic voltammograms
The electronic effect of the carbazolyl groups on the multistage redox ability was evaluated by means of electrochemical measurements of the salts (n-Bu4N+)(1−) in CH2Cl2 (Fig. 4 and Table 1). Due to the poor solubility of the neutral radicals of 4 and 5, the (n-Bu)4N+ salts of the monoanion species were used for the measurement. In the cyclic voltammetry (CV) measurements in CH2Cl2 (Fig. 4 and Fig. S4†), redox processes of the monoanion 1− to radical dianion (12−) and further diradical trianion (13−) were observed at E21/2 = −1.87 V and E31/2 = −2.12 V vs. Fc/Fc+, respectively, as reversible ones. On the other hand, the redox wave between monoanion 1− and neutral radical 1 at E1 ∼−0.2 V was not simple, where both oxidation and reduction proceeded in two steps probably due to the association of 1− and 1 in the solution state. The redox potential of E1 was much higher than those of TTM (−0.89 V)19a and PTM (ca. −0.7 V),13 indicating the stronger electron-accepting ability of 1. At a further higher potential, a reversible wave assignable to the redox between neutral radical 1 and monocation species 1+ was observed at E01/2 = +0.61 V. Compared to the oxidation potential of 9-phenylcarbazole at +0.80 V, the strong redox wave at the highest potential at +1.02 V originates from three carbazolyl groups (ECZp). These redox waves were observed reproducibly. In the CV measurements, in a highly polar DMF solution having a wider window to the negative potential, similar redox properties were observed, and the reduction of 13− to radical tetraanion (14−) can be observed around E4p = −2.75 V as an irreversible peak (Fig. S5†). Redox waves corresponding to E0 to E4 were also observed in 4 and 5, and therefore these waves are assignable to the redox processes of the TOT skeleton.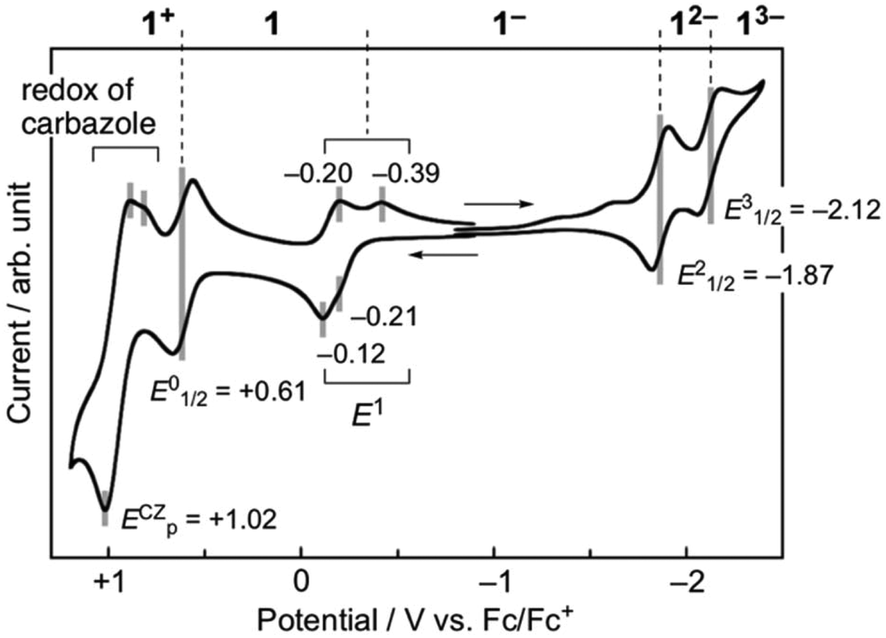 | ||
| Fig. 4 Cyclic voltammograms of 1 measured using the n-Bu4N+ salt of monoanion species (1 mM) in CH2Cl2. The result was calibrated with an Fc/Fc+ couple. | ||
| 1 | 4 | 5 | ||||
|---|---|---|---|---|---|---|
| CH2Cl2 | DMF | CH2Cl2 | DMF | CH2Cl2 | DMF | |
| a Potentials were calibrated with an Fc/Fc+ couple = 0 V. b Calculated by the DFT method at the ROB3LYP/6-31G(d,p) level of theory. c Peak potentials were listed due to the irreversibility. d Calculated with an optimized structure where the dihedral angle between TOT and carbazolyl groups is 47°. | ||||||
| E 4 | — | −2.75c | — | −3.09c | — | −3.18c |
| E 3 | −2.12 | −2.14 | — | −2.39 | −2.53c | −2.48 |
| E 2 | −1.87 | −1.79 | −2.18c | −2.03 | −2.18c | −2.10 |
| E 1 | −0.21c | −0.13c | +0.07c | −0.05c | −0.35 | −0.26c |
| E 0 | +0.61 | +0.70c | — | — | +0.79c | — |
| E cz | +1.02c | +0.97c | — | — | — | — |
| LUMO | −2.63d | −2.53 | −2.29 | |||
| SOMO | −3.19d | −3.33 | −3.11 | |||
According to the molecular orbital (MO) calculations of 1 and 4 (Fig. S6†), the E1 and E0 waves correspond to the SOMO and the E2–E4 waves correspond to the doubly degenerate LUMOs.14d The redox events E2–E4 of 1 in DMF are positively shifted by 0.2–0.3 V compared to those of 4 and 5 due to the effective π-extension to the carbazolyl groups (Fig. S5†). The redox potential of E1 of 1 in CH2Cl2 was slightly higher than that of 5, and was lower than that of 4 (Fig. S4†). These changes in the redox potentials originating from the substituent effect showed a good agreement with the MO calculation (Table 1 and Fig. S6†). It is known that the electronic effects of the substituent groups are strongly influenced by relative conformation (stereoelectronic chameleon).24 We also calculated the MO energies of 1 with various dihedral angles between TOT and the carbazole moieties (Fig. S7†). The calculation indicates that the increasing dihedral angle causes the weakening of the delocalization of the SOMO and the lowering of the SOMO and LUMO energies. When TOT and the carbazolyl groups are co-planar (θ = 0°), the SOMO energy becomes higher, and redox of E1 of 1 will occur at a lower potential compared to that of 5. On the other hand, when TOT and the carbazolyl groups are vertical to each other (θ = 90°), the carbazolyl group acts as an electron-withdrawing group, and the redox potential of E1 of 1 will become higher than that of 4. The steric repulsion between the hydrogen atoms at the α-positions of TOT and 1-positions of the carbazole moieties (H1 and H2 in Fig. 3a) causes a twisting between TOT and the carbazole moieties. The CV measurements imply that the molecular structure of 1 in the solution state has a twist angle close to that of the optimized structure (θ = 47°).
Absorption spectra
The effects of the introduction of carbazolyl groups into TOT neutral radical were further studied by the UV/Vis measurement in CH2Cl2 (Fig. 5). The spectrum of 1 at a shorter wavelength than 700 nm can be explained by the superimposition of the carbazole and 4. At the longer wavelength region, 1 showed a new strong and broad absorption band with λmax of 1028 nm and εmax of 1.3 × 104 M−1 cm−1. In the previous work, we revealed that TOT neutral radicals are in an equilibrium between the monomer and π-dimer, and the π-dimer exhibits a low energy absorption band around 800–900 nm due to the strong intermolecular interaction of two SOMOs (indicated by an arrow in Fig. 5).16 In the temperature dependent measurements from 293–213 K, the intensity of low energy absorption in 1 was almost kept constant at room temperature (Fig. S6†). A similar behavior can be seen in the concentration variable UV/Vis measurement of 1 in CH2Cl2, where the concentration had a negligible effect on the intensity of the low energy absorption band (Fig. S8†). Such a behavior was quite different from those of other TOT derivatives, where an increase in temperature and decrease in concentration caused a significant weakening of the band due to the dissociation of the π-dimer (Fig. S9†).16 These experimental results suggest that the low energy absorption band is caused by the IET from the electron-donating carbazolyl groups to the electron-accepting TOT neutral radical.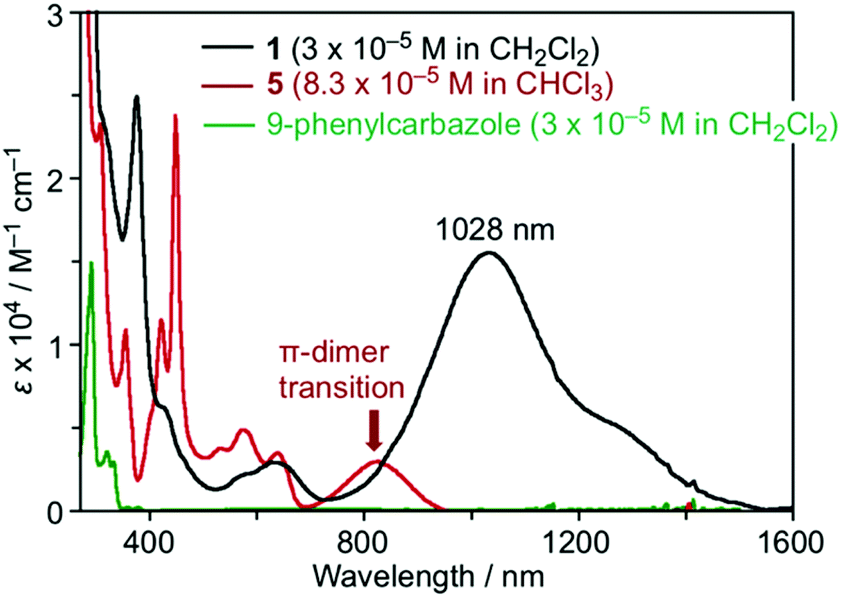 | ||
| Fig. 5 UV-Vis spectra of 1 (black line), tri-tert-butyl TOT5 (brown line), and 9-phenylcarbazole (green line) at room temperature. | ||
In order to obtain an insight into the electronic transition, we performed TD-DFT calculations of 1 (Fig. S10†). The calculation reproduced the IET band around 1180 nm originating from the electronic transition from the doubly degenerate HOMOs to SOMO (Fig. S6†). The HOMO and SOMO coefficients reside on the carbazole and TOT parts, respectively. Thus, the calculation also indicates that the low energy absorption band of 1 is an optically induced IET from the carbazolyl group to the TOT moiety. The difference between the oxidation potentials of the TOT skeleton (E1 ∼−0.2 V) and carbazolyl groups (ECZp = +1.02 V) in the CV measurements was ca. 1.2 V (Fig. 4), corresponding to ca. 1000 nm. This value is close to the experimental and calculated IET excitation energy.
A similar absorption band was also observed in the carbazole substituted TTM radicals at around λmax = 600–700 nm with εmax = 2–5 × 103 M−1 cm−1.19–21 Since TOT possesses a much higher electron-accepting ability than TTM, the transition energy became much smaller. Among the numerous open- and close-shell D–A molecules reported previously, those showing absorption bands at λmax > 1000 nm are rare, and further these bands are weak with ε ∼103 M−1 cm−1. In the TTF–TCNQ dyad linked through a σ-bonding linker, a low energy IET band was observed at λmax of 1630 nm with εmax ∼3 × 103 M−1 cm−1.25 The open-shell D–A molecule composed of TTF and 6OPO which has an electron-accepting ability close to that of p-chloranil exhibits a very weak IET band at λmax = 1350 nm with εmax ∼6 × 102 M−1 cm−1.10a
In the low temperature measurements in toluene (3.0 × 10−5 M), a weak shoulder band was observed at around 1260 nm, in which the intensity decreased with increasing temperature and almost disappeared at 353 K (Fig. 6). Considering the π-dimerization observed in the temperature dependence of the ESR measurement, this low energy absorption band can be characterized as the intermolecular transition within the π-dimer. Such a behavior is very similar to the π-dimerization of 5, where the intermolecular transition was observed at λmax = 834 nm.16 The significant red-shift of 1 in comparison with 5 is caused by the delocalization of the π-electronic system within the molecule. In the solid-state spectrum, the low energy absorption was observed as a single band around 800–1400 nm, where IET and intermolecular transition bands were gathered by broadening (Fig. S11†).
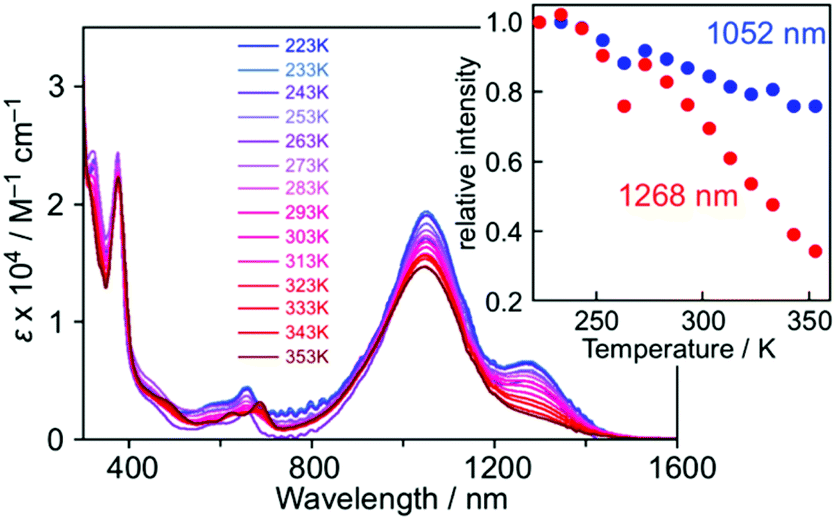 | ||
| Fig. 6 Temperature-dependent UV-Vis spectra of 1 in toluene (3.0 × 10−5 M). The inset shows the plots of the intensities at 1052 nm (blue) and 1268 nm (red), where the values were normalized with those at 223 K. | ||
The UV-Vis spectra of 1 were measured in various solvents, where both λmax and intensity of the IET band became lower with increasing the solvent polarity from toluene (1049 nm) to acetonitrile (874 nm) (Fig. 7). The hypsochromic effect of the IET band suggests an increase in the energy gap with the solvent polarity. A similar observation was also found in the PTM radical having a carbazolyl moiety; however, the change in λmax and intensity in carbazole-PTM was remarkably smaller than those of 1.26 The difference in the solvent effects between 1 and carbazole-PTM would be the consequence of the stronger electron-accepting ability of the TOT moiety compared to that of PTM and/or the extended π-system of TOT that enables strong interaction with solvent molecules.
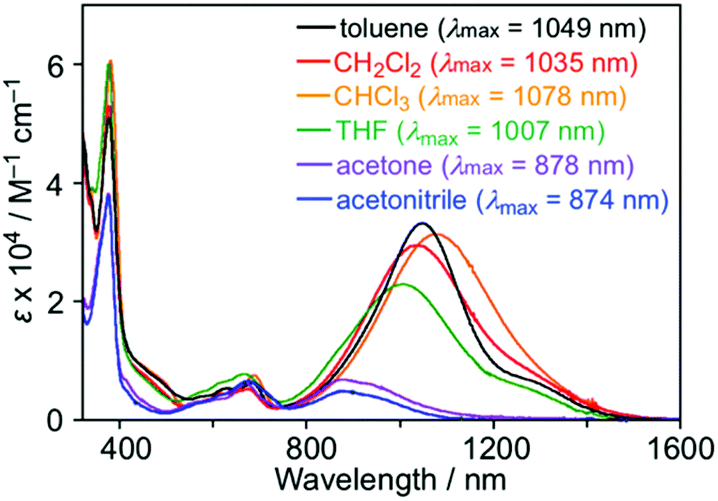 | ||
| Fig. 7 UV-Vis spectra of 1 in various solvents (∼1.0 × 10−5 M) at room temperature. Numerical values in parentheses are λmax of the IET band. | ||
Crystal structure and solid-state properties
A single crystal of 1 was obtained by vapor diffusion using CHCl3 and EtOH, and the quality of the structural analysis was poor due to the efflorescence of CHCl3 solvent and disorder of tert-butyl groups (R = 0.15). The TOT skeleton of 1 was nearly planar, and the dihedral angle between the TOT and three carbazolyl groups were 42–47° (Fig. 8a). In the crystal packing, 1 formed a π-dimer in a staggered manner with a face-to-face distance of 3.28 Å, similarly to 3 and 4 (stack-A, Fig. 8b).14d,16 The π-dimers further stacked on the TOT and carbazole moieties (stack-B, Fig. 8c, ca. 3.2–3.8 Å), forming a one-dimensional chain structure along the c-axis (Fig. 8d). Such a stacking pattern is in a sharp contrast with the π-dimer-based columns in the crystal structures of 4 and 5.14d,16 This structural feature is probably due to the steric repulsion of the bulky tert-butyl groups on the carbazole moiety.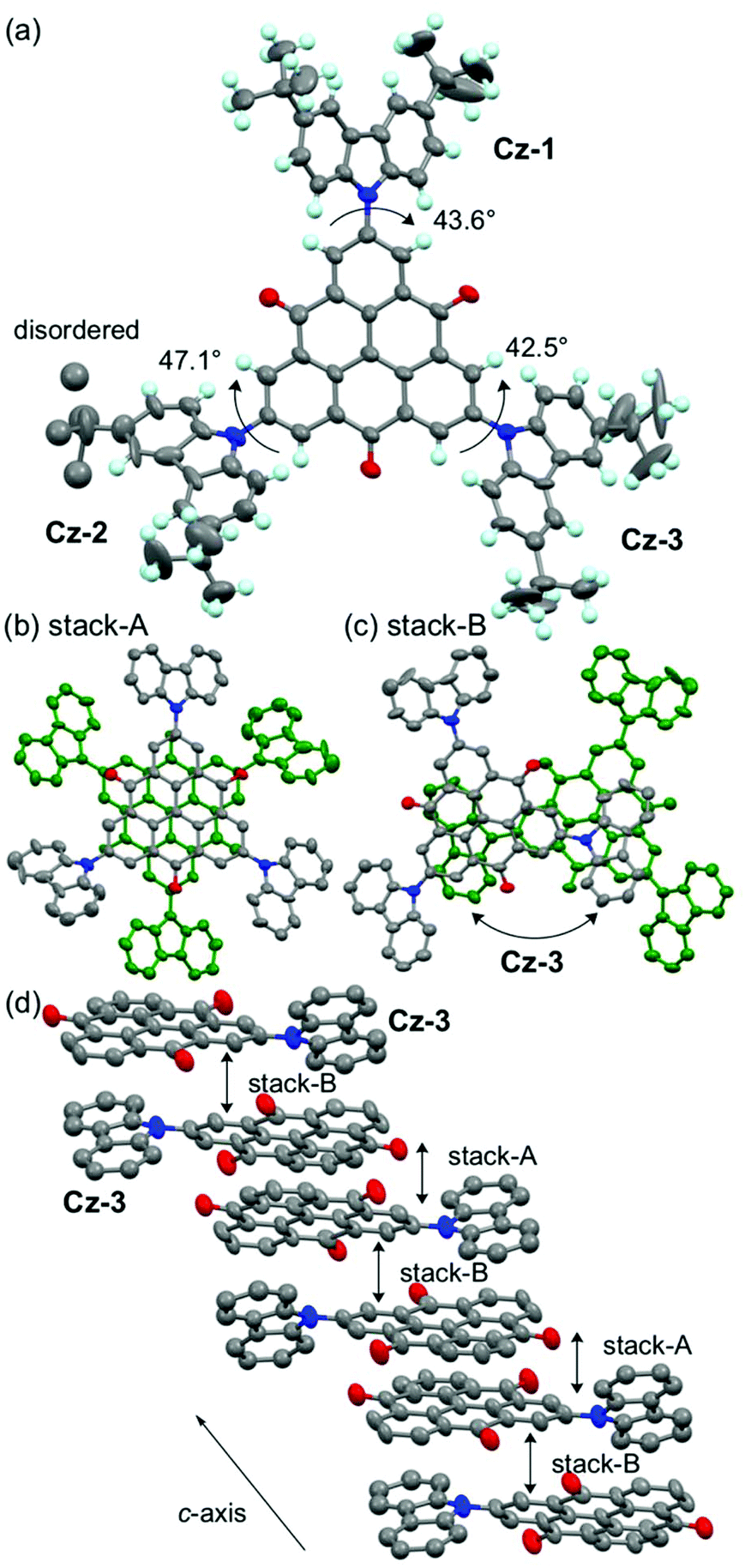 | ||
| Fig. 8 Crystal structure of 1. (a) Top view of 1. Ellipsoids are drawn at the 25% probability level, and labels Cz-A to Cz-C indicate the carbazole moieties. (b, c) Overlap patterns in stacks-A and -B. Hydrogen atoms and tert-butyl groups are omitted. (d) 1D π-stacking column along the c-axis. Cz-A and -B moieties which do not participate in the π-stacking are omitted. | ||
In the temperature dependence of magnetic susceptibility (χp, Fig. S12†), almost all spins of 1 were quenched due to the strong antiferromagnetic interaction within the π-dimers shown in the crystal structure (Fig. 8b). The analysis based on the singlet-triplet model27 resulted in the 2J/kB > −2000 K of antiferromagnetic interaction, which is similar to those in π-dimers of other TOT derivatives.14d The 2J/kB values of the π-dimer of 1 was also estimated by the DFT method.28 At the UB3LYP/3-21G level of theory, the calculations showed a 2J/kB value of −2273 K, which is very close to the experimental value. On the other hand, the calculated magnetic interaction between the π-dimers was 2J/kB = −6 K, much smaller than that within the π-dimer.
Conclusions
In conclusion, we have designed, synthesized and characterized an open-shell D–A molecule, 1, where three carbazolyl groups were introduced into the TOT neutral radical preserving its threefold symmetry. ESR spectroscopy and DFT calculations showed that a large amount of the electronic-spin delocalized over the carbazolyl groups. In the electrochemical measurement, 1 exhibited six-stage redox processes consisting of five steps of TOT and one step of the carbazolyl group. Furthermore, 1 exhibited a self-assembling ability to form a π-dimer inherent in TOT neutral radicals through strong two-electron-multicentered bonding in both solution and crystalline states. Most importantly, 1 exhibited a strong NIR photoabsorption band both in solution and in the solid state, which was formed by the IET from the carbazolyl groups to the TOT neutral radical. The high absorbance of the IET band in the NIR region exceeding 1000 nm is a substantial electronic feature among many D–A molecules.π-Conjugated molecules showing NIR photoabsorption have been noticed in the research studies not only of organic electronic materials5,29 but also of biological applications.30 One of the molecular design strategies for NIR dyes is the extension of the π-conjugated systems as demonstrated in the polymeric fused porphyrin array (3500 nm, dodecamer)31 and the giant hydrocarbon sheet (C222H42, 250–1400 nm).32 The π-stacking assemblies of open-shell molecules also exhibit NIR absorption bands at 1000–2000 nm as shown in radical-ion salts of TTF and TCNQ33 and also in TOT neutral radicals of our previous studies.14,15 In both cases, the construction of large π-electronic system through the assembly of many π-units is the key for lowering the absorption energy. The open-shell D–A molecules 1 in the present work realized a strong NIR photoabsorption without extension of the π-electronic system.
Recently, interesting properties and functions of D–A molecules with an open-shell electronic structure have been disclosed. Furthermore, the electronic structures of these systems are sensitive to temperature and solvent, and cause a drastic response on their electronic-spin structures.10–12 The development of the functionalities of 1 based on the responses to the external stimuli is currently underway. It should also be noted that the one-dimensional π-stacking column of TOT neutral radicals exhibits a high electron-transport ability to show a high conductivity in the crystalline state.15 Our current interest is aimed at the exploration of pressure or NIR photoinduced electrical conductivity as a single component neutral radical crystal or thin film, where IET locally generates TOT− species within the one-dimensional column providing a conduction carrier.
Experimental
Materials and methods
The synthesis of compound 2 is described in our previous work.14c,16 Toluene used for the synthesis and ESR measurement was purified by distillation from CaH2. Rf values on TLC were acquired on E. Merck precoated (0.25 mm) silica gel 60 F254 plates. Melting and decomposition points were measured using hot-stage apparatus with a Yanako MP-J3, and were uncorrected. Melting and/or decomposition were detected by eye observation. Elemental analyses were performed at the Graduate School of Science, Osaka University. 1H and 13C NMR spectra were obtained on a JEOL ECA-500 with DMSO-d6 using Me4Si as an internal standard. The infrared spectrum was recorded on a JASCO FT/IR-660 Plus spectrometer using a KBr plate (resolution 4 cm−1).Synthetic procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ethyl acetate/MeOH).
:1 ethyl acetate/MeOH).
In a 50 mL round-bottomed flask, 3 (80.0 mg, 0.069 mmol) was suspended in acetone (10 mL), and then n-Bu4NOH (1 M MeOH solution, 4.0 mL, 4.0 mmol) was added. After stirring at room temperature for 1 h, the mixture was concentrated under reduced pressure. The residue was washed with hot water, and then the resulting precipitate was collected by filtration, to give (n-Bu4N+)(1−) (94 mg, 97%) as a deep blue powder. M.p. ∼280 °C (in air); TLC Rf 0.60 (10:1 ethyl acetate/MeOH); 1H NMR (500 MHz, DMSO-d6, 80 °C) δ 0.94 (m, 12H), 1.28–1.36 (m, 8H), 1.46 (s, 54H), 1.54–1.62 (m, 8H), 3.12–3.18 (m, 8H), 7.50 (s, 12H), 8.32 (s, 6H) 8.90 (s, 6H) ppm; 13C NMR (500 MHz, DMSO-d6, 80 °C) δ 13.83, 19.76, 23.78, 32.41, 35.05, 58.58, 109.59, 117.07, 123.64, 124.33, 128.84, 130.19, 131.21, 131.65, 139.81, 143.35, 180.31 ppm; IR (KBr) ν 3049, 2961, 2871, 1614, 1548, 1496, 1297 cm−1; UV (CH2Cl2) λmax (ε) 239 (6.0 × 104), 291 (4.8 × 104), 382 (2.7 × 104), 625 (sh, 4.9 × 103), 860 (7.2 × 103) nm; MALDI-TOFMS (negative-mode) m/z 1152.6 ([M − (n-Bu)4N]−) (Fig. S13–S15†).
:1 hexane/ethyl acetate); IR (KBr) ν 2959, 1639, 1577, 1480, 1372, 1320, 1298 cm−1; UV (CH2Cl2) λmax (ε) 294 (3.7 × 104), 376 (2.2 × 104), 660 (2.2 × 103), 1034 (1.3 × 104) nm; UV (KBr) λmax 290, 382, 690, 1064 nm; Anal. Calcd for (C82H78N3O3)(H2O)0.7: C, 85.38; H, 6.82; N, 3.64. Found: C, 84.42; H, 6.96; N, 3.55.
Magnetic measurements
Magnetic resonance measurements were performed on a Bruker ELEXSYS E500 spectrometer for X-band liquid-phase ESR spectra and on Bruker ESR/ENDOR spectrometers ESP 300/350 for 1H-ENDOR, and 1H-TRIPLE spectra equipped with a wide-band 500 W radiofrequency amplifier. The solution of the radical was degassed by the freeze–pump–thaw method before the measurements were recorded. Magnetic susceptibility was measured on a Quantum Design SQUID magnetometer MPMS-XL at 2–300 K. The magnetic responses were corrected with diamagnetic blank data of the sample holder obtained separately. The diamagnetic contribution of the sample itself was estimated from Pascal's constants.Electrochemical measurements
Electrochemical measurements were made with an ALS Electrochemical Analyzer model 630A. Cyclic voltammograms were recorded with a 3 mm-diameter carbon plate and Pt wire counter electrodes in dry CH2CH2 or DMF containing 0.1 M n-Bu4NClO4 as the supporting electrolyte at room temperature. The experiment employed a Ag/AgNO3 reference electrode, and the final results were calibrated with an Fc/Fc+ couple.UV-Vis absorption spectroscopy
Electronic spectra were measured for KBr pellets or solutions on a Shimadzu UV-vis scanning spectrophotometer UV-3100PC. Temperature-dependent measurements were performed using an Oxford ITC502 temperature controller and an Oxford OptistatDN valuable temperature liquid nitrogen cryostat.X-ray crystallography
X-ray crystallographic measurements were made on a Rigaku Raxis-Rapid imaging plate with graphite monochromated Cu Kα (λ = 1.54187 Å). The structure was determined by a direct method using SHELXS program,34 and refinement was performed by a full-matrix least-squares on F2 using SHELXL-2014.35 All non-hydrogen atoms were refined anisotropically, and all hydrogen atoms were included but not refined. Empirical absorption correction was applied.568(1) Å3, T = 200 K, Z = 8, dcalcd = 1.270 g cm−3, μ = 2.552 mm−1, 2θmax = 136.5°, λ(CuKα) = 1.54187 Å, ω scan mode, 172468 reflections, of which 13292 were unique and 3126 were included in the refinement [I > 2.00σ(I)], data corrected for Lorentzian and polarization effects; an empirical absorption correction resulted in transmission factors ranging from 0.605 to 0.880. The final values R1 = 0.150, wR2 = 0.474, GOF = 1.077, maximum positive and negative peaks in ΔF map were ρmax = 0.506 e−3 and ρmin = −0.580 e−3. The quality of the structural analysis was poor due to the efflorescence of the CHCl3 solvent and disorder of tert-butyl groups.
Computational details
Density functional theory (DFT) calculations were performed using the Gaussian 03 program package. The calculation of electronic-spin structures of the neutral radicals 1 and 4 was performed at the UB3LYP/6-31G** level of theory with optimization of the geometries. The MO energy levels were calculated at the ROB3LYP/6-31G** level of theory with the geometries obtained from UB3LYP/6-31G**. TD-DFT calculations were performed at the UB3LYP/6-31G** level of theory with the geometries obtained from UB3LYP/6-31G**. In the calculation of intermolecular magnetic interactions within π-stacking structures, the geometries were extracted from the crystal structure, and the UB3LYP/3-21G level of theory was applied.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was supported by the Canon Foundation, the Grants-in-Aid for Scientific Research B (No. 25288022 and 16H04114), the Elements Science and Technology Project from the Ministry of Education, Culture, Sports, Science and Technology, Japan, and the Core Research for Evolutional Science and Technology (CREST) Basic Research Program “Creation of Innovative Functions of Intelligent Materials on the Basis of Element Strategy” of the Japan Science and Technology Agency (JST). Also, this work has been partially supported by the Grants-in-Aid for Scientific Research on Innovative Areas (Quantum Cybernetics), and the work has been supported by the AOARD Scientific Project on “Quantum Properties of Molecular Nanomagnets” (Award No. FA2386-13-1-4029, 4030, 4031) and the AOARD Project on “Molecular Spins for Quantum Technologies” (Grant No. FA2386-17-1-4040), USA.Notes and references
- (a) Materials for Nonlinear Optics: Chemical Perspectives, ed. S. R. Marder, J. E. Sohn and G. D. Stucky, ACS Symposium Series 455, American Chemical Society, Washington, DC, 1991 Search PubMed; (b) R. R. Tykwinski, U. Gubler, R. E. Martin, F. Diederich, C. Bosshard and P. Günter, J. Phys. Chem. B, 1998, 102, 4451 CrossRef CAS.
- (a) D. Gendron and M. Leclerc, Energy Environ. Sci., 2011, 4, 1225 RSC; (b) A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020 CrossRef CAS PubMed; (c) W. Li, K. H. Hendriks, M. M. Wienk and R. A. J. Janssen, Acc. Chem. Res., 2016, 49, 78 CrossRef CAS PubMed; (d) K. Nakano and K. Tajima, Adv. Mater., 2017, 29, 1603269 CrossRef PubMed; (e) I. Osaka and K. Takimiya, Adv. Mater., 2017, 29, 1605218 CrossRef PubMed.
- (a) H. Fujiwara, Y. Sugishima and K. Tsujimoto, Tetrahedron Lett., 2008, 49, 7200 CrossRef CAS; (b) K. Isoda, T. Yasuda and T. Kato, Chem. – Asian J., 2009, 4, 1619 CrossRef CAS PubMed; (c) K. Tsujimoto, R. Ogasawara, Y. Kishi and H. Fujiwara, New J. Chem., 2014, 38, 406 RSC; (d) K. Tsujimoto, R. Ogasawara, T. Nakagawa and H. Fujiwara, Eur. J. Inorg. Chem., 2014, 3960 CrossRef CAS; (e) Y. Beldjoudi, M. A. Nascimento, Y. J. Cho, H. Yu, H. Aziz, D. Tonouchi, K. Eguchi, M. M. Matsushita, K. Awaga, I. Osorio-Roman, C. P. Constantinides and J. M. Rawson, J. Am. Chem. Soc., 2018, 140, 6260 CrossRef CAS PubMed.
- (a) H. Chiba, J. Nishida and Y. Yamashita, Chem. Lett., 2012, 41, 482 CrossRef CAS; (b) J. Fan, J. D. Yuen, M. Wang, J. Seifter, J.-H. Seo, A. R. Mohebbi, D. Zakhidov, A. Heeger and F. Wudl, Adv. Mater., 2012, 24, 2186 CrossRef CAS PubMed; (c) J. D. Yuen and F. Wudl, Energy Environ. Sci., 2013, 6, 392 RSC.
- (a) J. Fabian, H. Nakazumi and M. Matsuoka, Chem. Rev., 1992, 92, 1197 CrossRef CAS; (b) G. Qian and Z. Y. Wang, Chem. – Asian J., 2010, 5, 1006 CrossRef CAS PubMed; (c) Z. Y. Wang, Near-infrared organic materials and emerging applications, CRC Press, Boca Raton, FL, 2013 CrossRef.
- (a) M. Tian, S. Tatsuura, M. Furuki, Y. Sato, I. Iwasa and L. S. Pu, J. Am. Chem. Soc., 2003, 125, 348 CrossRef CAS PubMed; (b) G. Qian, B. Dai, M. Luo, D. Yu, J. Zhan, Z. Zhang, D. Ma and Z. Y. Wang, Chem. Mater., 2008, 20, 6208 CrossRef CAS; (c) M. Luo, H. Shadnia, G. Qian, X. Du, D. Yu, D. Ma, J. S. Wright and Z. Y. Wang, Chem. – Eur. J., 2009, 15, 8902 CrossRef CAS PubMed; (d) G. Qian and Z. Y. Wang, Can. J. Chem., 2010, 88, 192 CrossRef CAS.
- Several π-conjugated polymers of D–A molecules showing NIR absorption bands have been reported: (a) Y. Wang and T. Michinobu, J. Mater. Chem. C, 2016, 4, 6200 RSC; (b) T. T. Steckler, P. Henriksson, S. Mollinger, A. Lundin, A. Salleo and M. R. Andersson, J. Am. Chem. Soc., 2014, 136, 1190 CrossRef CAS PubMed.
- (a) M. Souto, H. Cui, M. Peña-Álvarez, V. G. Baonza, H. O. Jeschke, M. Tomic, R. Valentí, D. Blasi, I. Ratera, C. Rovira and J. Veciana, J. Am. Chem. Soc., 2016, 138, 11517 CrossRef CAS PubMed; (b) M. Souto, C. Rovira, I. Ratera and J. Veciana, CrystEngComm, 2017, 19, 197 RSC; (c) M. Souto, M. C. Gullo, H. Cui, N. Casati, F. Montisci, H. O. Jeschke, R. Valentí, I. Ratera, C. Rovira and J. Veciana, Chem. – Eur. J., 2018, 24, 5500 CrossRef CAS PubMed.
- H. Komatsu, M. M. Matsushita, S. Yamamura, Y. Sugawara, K. Suzuki and T. Sugawara, J. Am. Chem. Soc., 2010, 132, 4528 CrossRef CAS PubMed.
- (a) S. Nishida, Y. Morita, K. Fukui, K. Sato, D. Shiomi, T. Takui and K. Nakasuji, Angew. Chem., Int. Ed., 2005, 44, 7277 CrossRef CAS PubMed; (b) S. Nishida, K. Fukui and Y. Morita, Chem. – Asian J., 2014, 9, 500 CrossRef CAS PubMed.
- (a) J. Guasch, L. Grisanti, V. Lloveras, J. Vidal-Gancedo, M. Souto, D. C. Morales, M. Vilaseca, C. Sissa, A. Painelli, I. Ratera, C. Rovira and J. Veciana, Angew. Chem., Int. Ed., 2012, 51, 11024 CrossRef CAS PubMed; (b) J. Guasch, L. Grisanti, M. Souto, V. Lloveras, J. Vidal-Gancedo, I. Ratera, A. Painelli, C. Rovira and J. Veciana, J. Am. Chem. Soc., 2013, 135, 6958 CrossRef CAS PubMed.
- (a) I. Ratera, D. Ruiz-Molina, F. Renz, J. Ensling, K. Wurst, C. Rovira, P. Gütlich and J. Veciana, J. Am. Chem. Soc., 2003, 125, 1462 CrossRef CAS PubMed; (b) J. Guasch, L. Grisanti, S. Jung, D. Morales, G. D'Avino, M. Souto, X. Fontrodona, A. Painelli, F. Renz, I. Ratera and J. Veciana, Chem. Mater., 2013, 25, 808 CrossRef CAS.
- X. Wu, J. O. Kim, S. Medina, F. J. Ramírez, P. M. Burrezo, S. Wu, Z. L. Lim, C. Lambert, J. Casado, D. Kim and J. Wu, Chem. – Eur. J., 2017, 23, 7698 CrossRef CAS PubMed.
- (a) Y. Morita, S. Suzuki, K. Sato and T. Takui, Nat. Chem., 2011, 3, 197 CrossRef CAS PubMed; (b) Y. Morita, S. Nishida, T. Murata, M. Moriguchi, A. Ueda, M. Satoh, K. Arifuku, K. Sato and T. Takui, Nat. Mater., 2011, 10, 947 CrossRef CAS PubMed; (c) T. Takui, K. Sato and Y. Morita, WO. Pat, WO2013042706A1, 2013 Search PubMed; (d) Y. Morita, T. Murata, A. Ueda, C. Yamada, Y. Kanzaki, D. Shiomi, K. Sato and T. Takui, Bull. Chem. Soc. Jpn., 2018, 91, 922 CrossRef CAS; (e) T. Murata, N. Asakura, S. Ukai, A. Ueda, Y. Kanzaki, K. Sato, T. Takui and Y. Morita, ChemPlusChem, 2019, 84, 680 CrossRef CAS; (f) H. Enozawa, S. Ukai, H. Ito, T. Murata and Y. Morita, Org. Lett., 2019, 21, 2161 CrossRef CAS PubMed; (g) T. Murata, K. Kotsuki, H. Murayama, R. Tsuji and Y. Morita, Commun. Chem., 2019, 2, 46 CrossRef.
- T. Murata, C. Yamada, K. Furukawa and Y. Morita, Commun. Chem., 2018, 1, 47 CrossRef.
- Y. Ikabata, Q. Wang, T. Yoshikawa, A. Ueda, T. Murata, K. Kariyazono, M. Moriguchi, H. Okamoto, Y. Morita and H. Nakai, npj Quantum Mater., 2017, 2, 27 CrossRef.
- (a) J. F. Ambrose and R. F. Nelson, J. Electrochem. Soc., 1968, 115, 1159 CrossRef CAS; (b) D. R. Prudhomme, Z. Wang and C. J. Rizzo, J. Org. Chem., 1997, 62, 8257 CrossRef CAS PubMed; (c) Z. Gomurashvili, Y. Hua and J. V. Crivello, Macromol. Chem. Phys., 2001, 202, 2133 CrossRef CAS; (d) M. Sangermano, G. Malucelli, A. Priola, S. Lengvinaite, J. Simokaitiene and J. V. Grazulevicius, Eur. Polym. J., 2005, 41, 475 CrossRef CAS; (e) S.-K. Chiu, Y.-C. Chung, G.-S. Liou and Y. O. Su, J. Chin. Chem. Soc., 2012, 59, 1 CrossRef.
- (a) V. I. Adamovich, S. R. Cordero, P. I. Djurovich, A. Tamayo, M. E. Thompson, B. W. D'Andrade and S. R. Forrest, Org. Electron., 2003, 4, 77 CrossRef CAS; (b) A. van Dijken, J. J. A. M. Bastiaansen, N. M. M. Kiggen, B. M. W. Langeveld, C. Rothe, A. Monkman, I. Bach, P. Stössel and K. Brunner, J. Am. Chem. Soc., 2004, 126, 7718 CrossRef CAS PubMed; (c) J. F. Morin, N. Drolet, Y. Tao and M. Leclerc, Chem. Mater., 2004, 16, 4619 CrossRef CAS; (d) K. R. Justin Thomas, M. Velusamy, J. T. Lin, Y. T. Tao and C. H. Chuen, Adv. Funct. Mater., 2004, 14, 387 CrossRef; (e) K. Brunner, A. V. van Dijken, H. Borner, J. J. A. M. Bastiaansen, N. M. M. Kiggen and B. M. W. Langeveld, J. Am. Chem. Soc., 2004, 126, 6035 CrossRef CAS.
- (a) V. Gamero, D. Velasco, S. Latorre, F. López-Calahorra, E. Brillas and L. Juliá, Tetrahedron Lett., 2006, 47, 2305 CrossRef CAS; (b) J. V. Grazulevicius, P. Strohriegl, J. Pielichowski and K. Pielichowski, Prog. Polym. Sci., 2003, 28, 1297 CrossRef CAS; (c) K. Parimal, K. R. Justin Thomas, T. L. Jiann, Y. T. Tao and C. H. Chien, Adv. Funct. Mater., 2003, 13, 445 CrossRef; (d) B. Akira, O. Ken, K. Wolfgang and C. A. J. Rigoberto, Phys. Chem. B, 2004, 108, 18949 CrossRef.
- (a) S. Castellanos, D. Velasco, F. López-Calahorra, E. Brillas and L. Julia, J. Org. Chem., 2008, 73, 3759 CrossRef CAS PubMed; (b) S. Castellanos, L. López-Calahorra, E. Brillas, L. Julia and D. Velasco, Angew. Chem., Int. Ed., 2009, 48, 6516 CrossRef CAS PubMed.
- (a) S. Castellanos, V. Gaidelis, V. Jankauskas, J. V. Grazulevicius, E. Brillas, F. López-Calahorra, L. Juliá and D. Velasco, Chem. Commun., 2010, 46, 5130 RSC; (b) M. Reig, C. Gozalvez, V. Jankauskas, V. Gaidelis, J. V. Grazulevicius, L. Fajarí, L. Juliá and D. Velasco, Chem. – Eur. J., 2016, 22, 18551 CrossRef CAS PubMed.
- J. F. Hartwig, M. Kawatsura, S. I. Hauck, K. H. Shaughnessy and L. M. Alcazar-Roman, J. Org. Chem., 1999, 64, 5575 CrossRef CAS PubMed.
- D. Small, V. Zaitsev, Y. Jung, S. V. Rosokha, M. Head-Gordon and J. K. Kochi, J. Am. Chem. Soc., 2004, 126, 13850 CrossRef CAS.
- S. Z. Vatsadze, Y. D. Loginova, G. dos Passos Gomes and I. V. Alabugin, Chem. – Eur. J., 2017, 23, 3225 CrossRef CAS.
- D. F. Perepichka, M. R. Bryce, C. Pearson, M. C. Petty, E. J. L. McInnes and J. P. Zhao, Angew. Chem., Int. Ed., 2003, 42, 4636 CrossRef CAS PubMed.
- A. Gilabert, L. Fajarí, I. Sirés, M. Reig, E. Brillas, D. Velasco, J. M. Anglada and L. Juliá, New J. Chem., 2017, 41, 8422 RSC.
- B. Bleaney and K. D. Bowers, Proc. R. Soc. London, Ser. A, 1952, 214, 451 CrossRef CAS.
- Y. Takano, T. Taniguchi, H. Isobe, T. Kubo, Y. Morita, K. Yamamoto, K. Nakasuji, T. Takui and K. Yamaguchi, J. Am. Chem. Soc., 2002, 124, 11122 CrossRef CAS PubMed.
- Z. Sun, Q. Ye, C. Chi and J. Wu, Chem. Soc. Rev., 2012, 41, 7857 RSC.
- A. Alabugin, Photochem. Photobiol., 2019, 95, 722 CrossRef CAS PubMed.
- (a) A. Tsuda and A. Osuka, Science, 2001, 293, 79 CrossRef CAS PubMed; (b) H. Mori, T. Tanaka and A. Osuka, J. Mater. Chem. C, 2013, 1, 2500 RSC.
- C. D. Simpson, J. D. Brand, A. J. Berresheim, L. Przybilla, H. J. Räder and K. Müllen, Chem. – Eur. J., 2002, 8, 1424 CrossRef CAS.
- (a) J. B. Torrance, B. A. Scott, B. Welber, F. B. Kaufman and P. E. Seiden, Phys. Rev. B, 1979, 19, 730 CrossRef CAS; (b) J. B. Torrance, Acc. Chem. Res., 1979, 12, 79 CrossRef CAS; (c) S. Yamaguchi, Y. Moritomo and Y. Tokura, Phys. Rev. B, 1993, 48, 6654 CrossRef CAS; (d) M. Meneghetti, Phys. Rev. B, 1994, 50, 16899 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A, 2008, 64, 112 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C, 2015, 71, 3 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures of the intermediates, and additional experimental data and molecular orbital energy calculations. CCDC 1894816. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qo00663j |
| This journal is © the Partner Organisations 2019 |