
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNeglected sulfur(VI) pharmacophores in drug discovery: exploration of novel chemical space by the interplay of drug design and method development†
U.
Lücking

Bayer AG, Pharmaceuticals Division, Drug Discovery, Müllerstr. 178, 13353 Berlin, Germany. E-mail: ulrich.luecking@bayer.com
First published on 22nd March 2019
Abstract
Historically, sulfoximines, sulfondiimines and sulfonimidamides have been neglected pharmacophores in drug discovery even though they offer very interesting properties. This highlight shares the key learnings of various lead optimization approaches at Bayer AG that have successfully utilized these neglected sulfur(VI) functional groups to deliver multiple clinical candidates. In this context, the key synthetic methods utilized for the synthetic preparation of these unusual compounds will be outlined.
Sulfones 1 and sulfonamides 2 are important pharmacophores found in many drugs on the market (Scheme 1). In contrast, the corresponding aza analogues, sulfoximines13, sulfondiimines 4 and sulfonimidamides 5, have received little interest in medicinal chemistry until recently. The infrequent take-up of these sulfur(VI) functional groups in drug discovery is surprising since they offer very interesting properties, such as high stability, favorable physicochemical properties, multiple hydrogen-bond acceptor/donor functionalities and structural diversity. Possible reasons for the neglected use of these functional groups are the lack of commercial availability, limited synthetic methods for their preparation and an incomplete understanding of their properties relevant to medicinal chemistry.
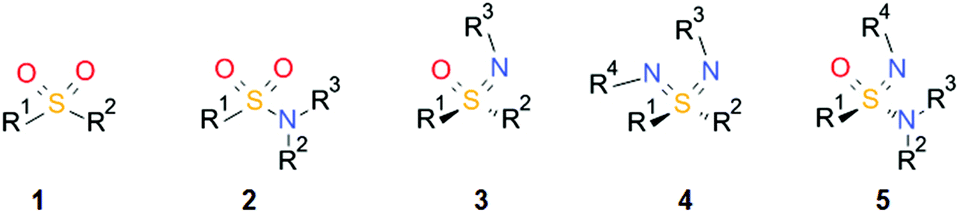 | ||
| Scheme 1 General structures of sulfones 1, sulfonamides 2, and aza analogues sulfoximines 3, sulfondiimines 4 and sulfonimidamides 5. | ||
The long-standing interest at Bayer AG in these neglected sulfur(VI) pharmacophores stems from the pan-CDK inhibitor project from the oncology section. In this approach, the potent sulfonamide ZK 304709 was selected as the clinical candidate due to its promising preclinical overall profile (Scheme 2). However, a dose-escalation study in patients was terminated due to dose-limited absorption at high doses of ZK 304709 which was mainly attributed to its limited aqueous solubility of only 8 mg L−1. Moreover, ZK 304709 was found to accumulate in the erythrocytes of patients due to an off-target activity against carbonic anhydrases (CAs), a finding which has been discussed as a parameter contributing to the observed interpatient variability of exposure.2 The revised project aims therefore focused on two key aspects in the follow-up program: first, the limited absorption at high dose was to be addressed by reducing dose size via significant improvement of antitumor potency, as well as increased aqueous solubility; second, the follow-up compound was to be devoid of CA inhibitory properties.3
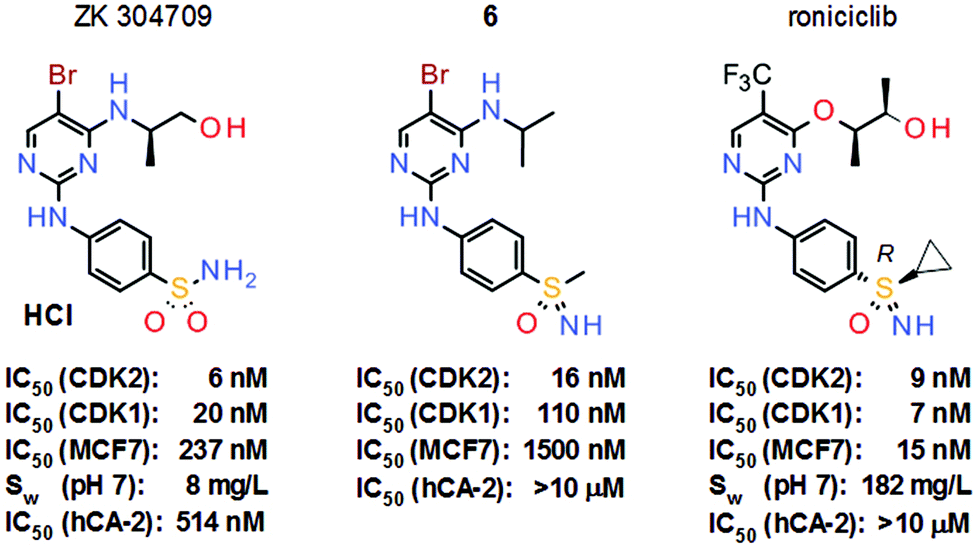 | ||
| Scheme 2 Structures and key properties of the pan-CDK inhibitors ZK 304709, sulfoximine model compound 6 and roniciclib. | ||
With respect to the off-target activity, it was established that N-unsubstituted arenesulfonamides can interact with a zinc cation in the active center of the CA binding pocket. In-house modelling studies supported the hypothesis that the sulfonamide group of ZK 304709 is crucial for the activity against CAs, and various structural modifications of ZK 304709 were evaluated with the goal of eliminating the off-target activity without compromising the activity against CDKs. One idea was to exchange the sulfonamide group, which mediates the interaction with CAs, for a new functional group. In this context, the coincidental finding of a review article on the sulfoximine group4 aroused an interest in this unusual pharmacophore. Sulfoximines are isoelectronic with sulfones but the introduction of the nitrogen creates asymmetry (Scheme 3). Sulfoximines are stable compounds and the nitrogen offers an additional point for substitution. The nitrogen is also basic enough to allow metal ion coordination or salt formation. The heteroatoms bound to the sulfur are hydrogen-bond acceptors and, in the case of NH sulfoximines (R3 = H), the group has dual hydrogen-bond donor/acceptor functionality. Moreover, structurally simple sulfoximines are readily soluble in protic solvents.5 Due to these promising properties, the sulfoximine group was introduced into the lead structure, even though there was hardly any precedence for use of the sulfoximine group in medicinal chemistry at that time.
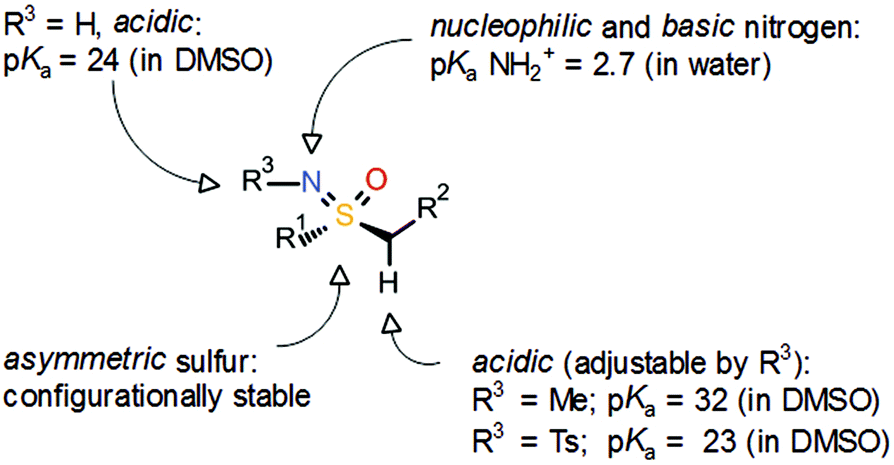 | ||
| Scheme 3 General properties of the sulfoximine group.4,6 | ||
Sulfoximine model compound 6 was prepared by imination of the corresponding sulfoxide with hydrazoic acid, generated in situ from sodium azide and concentrated sulfuric acid. This traditional method for the preparation of sulfoximines7 has yielded the desired products in many literature examples; however, since hydrazoic acid is not only toxic but also potentially explosive, the preparation of model compound 6 was only performed on a very small scale. Nevertheless, sulfoximine 6 indeed revealed activity against CDKs in vitro, but no activity against CAs (Scheme 2). Moreover, model compound 6 also demonstrated high hydrolytic and metabolic stability in vitro and was used as a starting point for further lead optimization. These efforts ultimately led to the discovery of roniciclib, which was the most potent pan-CDK inhibitor of the project (Scheme 2). However, the synthesis of the sulfoximine roniciclib on a larger scale remained an issue since the traditional hydrazoic acid method could not be utilized due to safety concerns. Fortunately, a then novel synthetic method developed by Okamura and Bolm8 could be readily applied. This rhodium-catalyzed process relies on commercially available chemicals and was used to convert sulfoxide 8 into sulfoximine 9 in good yield without safety concerns on a multigram scale (Scheme 4). Notably, Bolm's imination method8 was an important factor in the decision to continue the sulfoxmine project at Bayer AG since this key advance would allow the safe, scaled up production of potential sulfoximine clinical candidates.
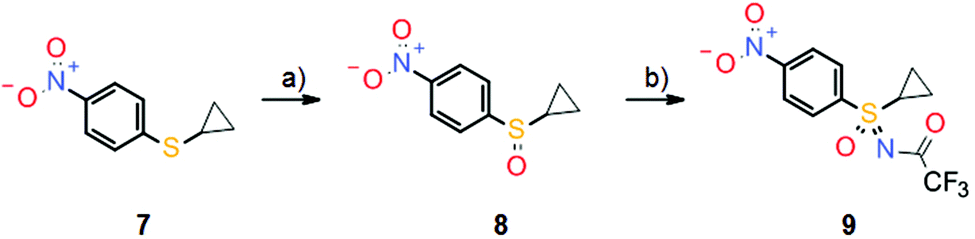 | ||
| Scheme 4 Synthesis of key sulfoximine building block 9 by rhodium-catalyzed imination of sulfoxide 8. Reagents and conditions: (a) H5IO6, FeCl3, MeCN, rt, 76%; (b) CF3C(O)NH2, PhI(OAc)2, MgO, [Rh2(OAc)4], DCM, rt, 78%. | ||
With this new synthetic approach to prepare larger quantities at hand, roniciclib was finally evaluated in various xenograft models and revealed high antitumor efficacy with good tolerability.9 The required therapeutic dose of roniciclib in mice is about 50-fold lower than that of ZK 304709. Moreover, the aqueous solubility of roniciclib of 182 mg L−1 is more than 20-fold higher than that of the former clinical candidate. Furthermore, roniciclib does not inhibit CAs and does not accumulate in erythrocytes. Roniciclib also revealed excellent PK in humans in phase I trials but phase II studies were terminated due to a safety signal.10
Nonetheless, selective inhibition of exclusively transcription-regulating CDK9 is a promising new approach in cancer therapy. Selective CDK9 inhibition results in rapid depletion of short-lived mRNA transcripts of important survival proteins, such as Mcl-1. Lead structure BAY-958 (LDC 526) revealed good in vitro activity against CDK9, along with very high kinase selectivity, surprisingly even within the CDK family (Scheme 5).11 BAY-958 also exhibited good antiproliferative activity and metabolic stability in vitro. On the other hand, BAY-958 has limited aqueous solubility, moderate permeability and high efflux which results in low oral bioavailability (e.g., in rats). Lead optimization efforts finally led to the discovery of the benzyl sulfoximine atuveciclib, which shows similar potency and selectivity as BAY-958 in vitro, but much improved aqueous solubility, improved permeability and reduced efflux, resulting in a much improved bioavailability in animals. Moreover, the switch from the sulfonamide to the sulfoximine removed a potential CYP induction liability.
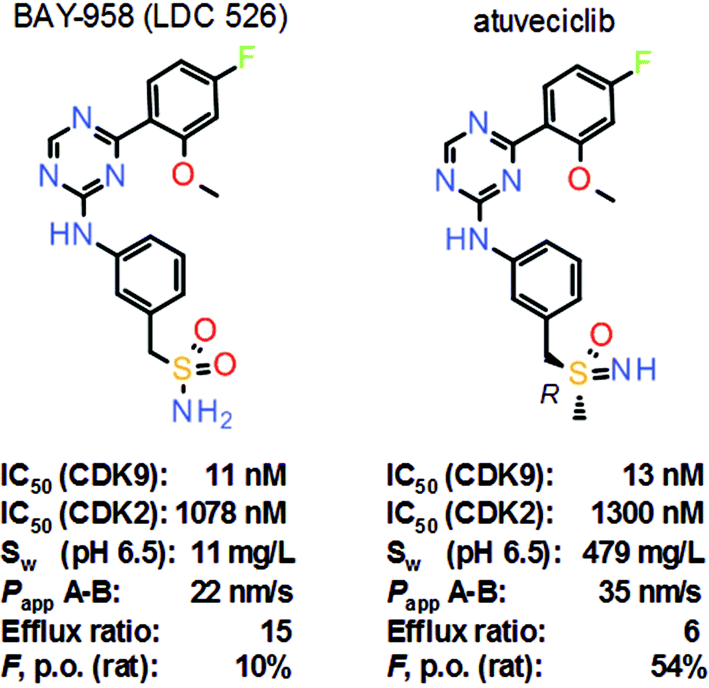 | ||
| Scheme 5 Structures and key properties of the selective CDK9 inhibitors BAY-958 (LDC 526) and atuveciclib. | ||
The initial synthesis of atuveciclib11 also relied on the sequential oxidation/imination of a suitable thioether, which gave good yields (Scheme 6).
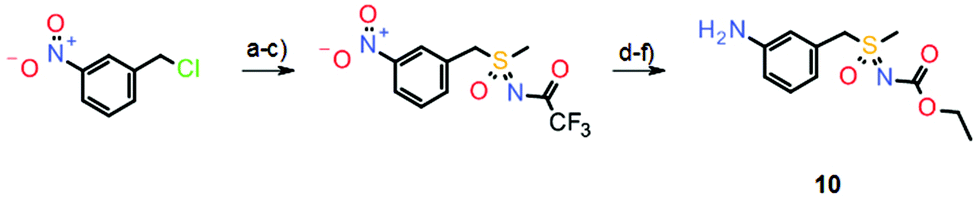 | ||
| Scheme 6 Synthesis of key sulfoximine building block 10 in the initial preparation of atuveciclib. Reagents and conditions: (a) NaSMe, EtOH, −15 °C to rt, crude; (b) H5IO6, FeCl3, MeCN, rt, 70%; (c) CF3C(O)NH2, PhI(OAc)2, MgO, [Rh2(OAc)4], DCM, rt, 99%; (d) K2CO3, MeOH, rt, 79%; (e) ClC(O)OEt, pyridine, 0 °C to rt, crude; (f) TiCl3, THF, rt, crude. | ||
However, such a sequential oxidation/imination strategy can have two drawbacks. First, the syntheses tend to be quite long since they often require the preparation of the initial thioether and then the employment of a suitable protecting group at the sulfoximine nitrogen. Second, if more complex thioethers are used, for instance those containing additional heteroatoms, oxidation/imination procedures can result in low yields or even fail completely.12 Therefore, a new synthetic approach was envisaged based on a pre-formed sulfoximine building block, avoiding oxidation/imination reactions. Subsequently, the first examples of intermolecular α-arylations of dimethyl sulfoximine were achieved by employing the p-methoxybenzyl (PMB)-protected derivative 11 in a palladium-catalyzed cross-coupling reaction with aryl halides.12,13 As an application, a much shorter synthesis of atuveciclib was developed which relies on this palladium-catalyzed α-arylation reaction as a key step (Scheme 7).
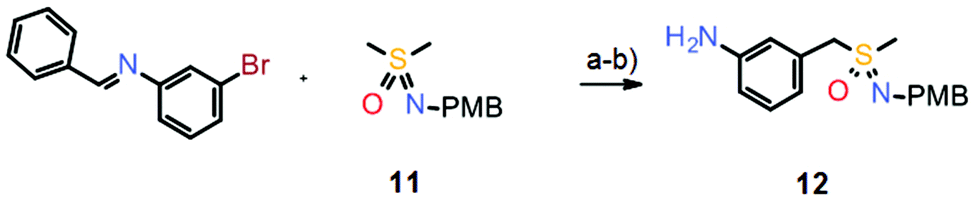 | ||
| Scheme 7 Synthesis of key sulfoximine building block 12 in the improved preparation of atuveciclib, by palladium-catalyzed α-arylation of PMB-protected dimethyl sulfoximine 11. Reagents and conditions: (a) Pd(OAc)2 (5 mol%), Xantphos (10 mol%), LiOtBu, CPME, 110 °C; (b) HCl, rt, 33% yield over two steps. | ||
Phase 1 trials of atuveciclib were recently terminated due to a strategic decision. However, daily oral administration of atuveciclib also resulted in neutropenia as a dose-limiting toxicity in patients. To improve tolerability, the aim then became the identification of a selective CDK9 inhibitor suitable for intermittent i.v. application. The lead structure of the follow-up project, BAY-332, exhibited good CDK9 inhibitory activity and selectivity against CDK2 in vitro (Scheme 8). However, aqueous solubility of BAY-332 was not sufficient to enable formulation of the predicted therapeutic dose in humans for i.v. application in patients. Since solubility was a key optimization parameter in this approach, which proved to be difficult to address, the properties of the corresponding sulfondiimine analogue 13 were then considered, with the hope that the introduction of an additional nitrogen at the sulfur could further improve solubility. Moreover, no chiral separation of enantiomers would be required in the case of achiral sulfondiimine 13. Matched analogue 13 revealed reduced activity against CDK9 in vitro by a factor of three, but similar selectivity against CDK2, and significantly improved aqueous solubility.14 Moreover, sulfoximine BAY-332 and matched sulfondiimine 13 displayed comparable metabolic stability in rat hepatocytes in vitro. In a subsequent rat PK study in vivo, an increased volume of distribution (Vss) and increased blood clearance (CLb) of analogue 13 were recorded. Increased CLb was not considered to be an issue in this approach, since the target profile were compounds with a short half-life to evaluate the effects on tolerability.
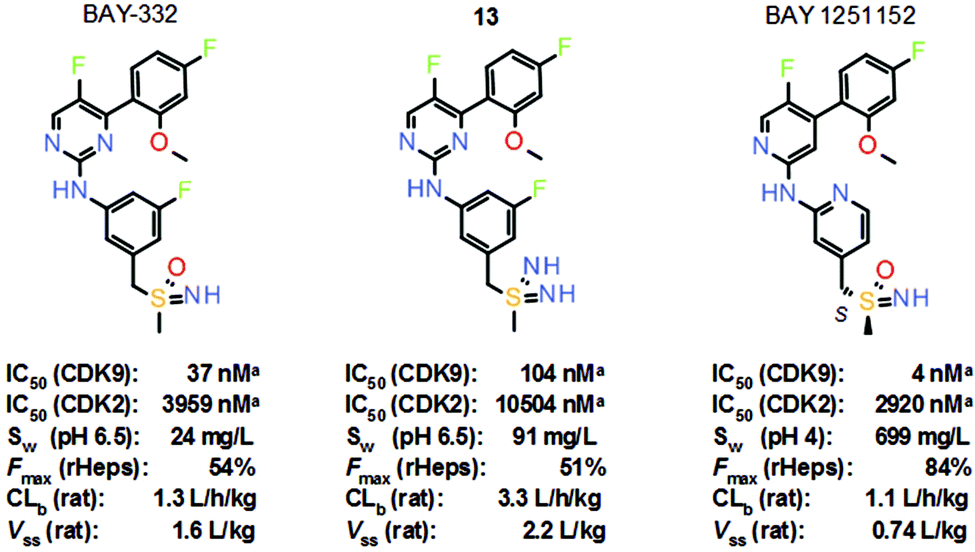 | ||
Scheme 8 Structures and key properties of the selective CDK9 inhibitors BAY-332, sulfondiimine 13 and BAY 1251152. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) High ATP conditions. High ATP conditions. | ||
The major drawback in the sulfondiimine compound series was the synthesis of the test compounds. Again, a then novel procedure reported by Bolm and co-workers15 was successfully applied to prepare small quantities of test compounds (Scheme 9).14 However, the initial imination of the thioethers (e.g., compound 14) was achieved by the action of O-(2,4,6-trimethylbenzenesulfonyl)hydroxylamine (MSH), which is also potentially explosive, and therefore large-scale synthesis of sulfondiimines for extended in vivo testing remains an issue. Whilst the sulfondiimine group is a very interesting pharmacophore from the perspective of medicinal chemistry, new and safe synthetic methods are needed to further explore its potential.
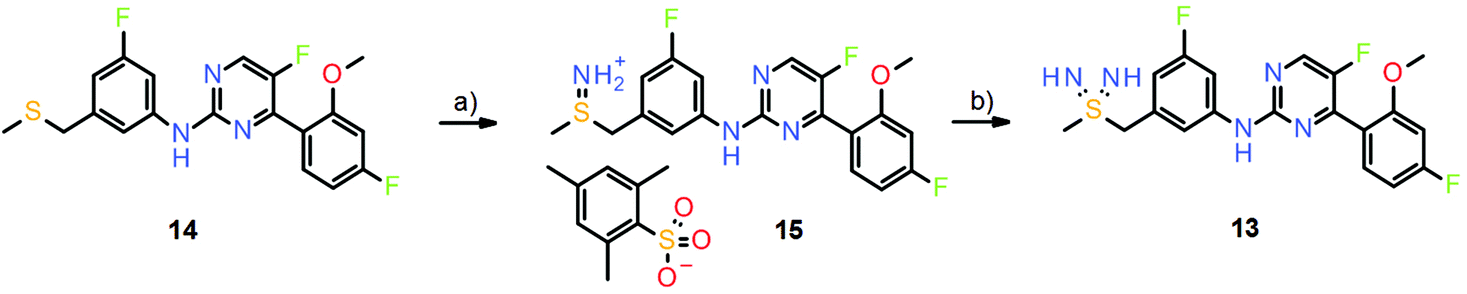 | ||
| Scheme 9 Formation of the sulfondiimine group in the preparation of sulfondiimine 13. Reagents and conditions: (a) MSH, DCM rt, 78%; (b) NaHDMS, NCS, Na2CO3, DMF, rt, 19%. | ||
Nevertheless, extensive scaffold modifications finally led to the discovery of the modified benzyl sulfoximine BAY 1251152 (Scheme 8).16 Based on a (pyridin-2-ylamino)pyridine scaffold, BAY 1251152 showed significantly improved in vitro potency and kinase selectivity, even within the CDK family. In various xenograft models, BAY 1251152 displayed high antitumor efficacy after once weekly i.v. administration. High, pH-dependent aqueous solubility of BAY 1251152 along with its low predicted therapeutic dose in humans finally enabled the formulation of this highly selective CDK9 inhibitor for i.v. administration in patients. BAY 1251152 is currently being evaluated in clinical phase I trials of once weekly i.v. administration in patients (NCT02635672; NCT02745743).
Interest in the sulfoximine group as a versatile pharmacophore in drug discovery has increased substantially in recent years, as evidenced by a significant increase in life science patent applications incorporating sulfoximine compounds and novel sulfoximines reported as clinical candidates.17 The sulfoximine group has been evaluated as a pharmacophore in a variety of molecular settings and indications with diverse rationales. In particular, the switch from sulfones and sulfonamides to sulfoximines has elicited much interest. In a recent study, a series of matched sulfoximine analogues of marketed drugs and advanced clinical candidates were prepared to investigate the effects of the replacement of suitable non-sulfur-based functional groups, such as amines, by sulfoximines.5c Based on the rationale that under physiological conditions, most amines, which are ubiquitous in life science approaches, are predominantly protonated, it was thought that the tetrahedral sulfoximine group could be utilized as a structural alternative with a differentiated pharmacological profile. Sulfoximine analogue 16 of the marketed PDE5 inhibitor vardenafil, for instance, was shown to be basically equipotent in an in vitro PDE5 enzyme assay (Scheme 10). The thermodynamic aqueous solubility of vardenafil at pH 6.5 is higher than that of analogue 16, which displayed a significantly reduced logD. Sulfoximine 16 also showed improved in vitro stability in rat hepatocytes, but in the Caco2 screening assay, analogue 16 exhibited a very low permeability coefficient (Papp A–B) of <1 nm s−1 and a high efflux ratio of >200. Overall, however, very promising results were recorded in this study of sulfoximine analogues of marketed drugs and advanced clinical candidates, suggesting that the sulfoximine moiety should be added to the medicinal chemist's toolbox.5c
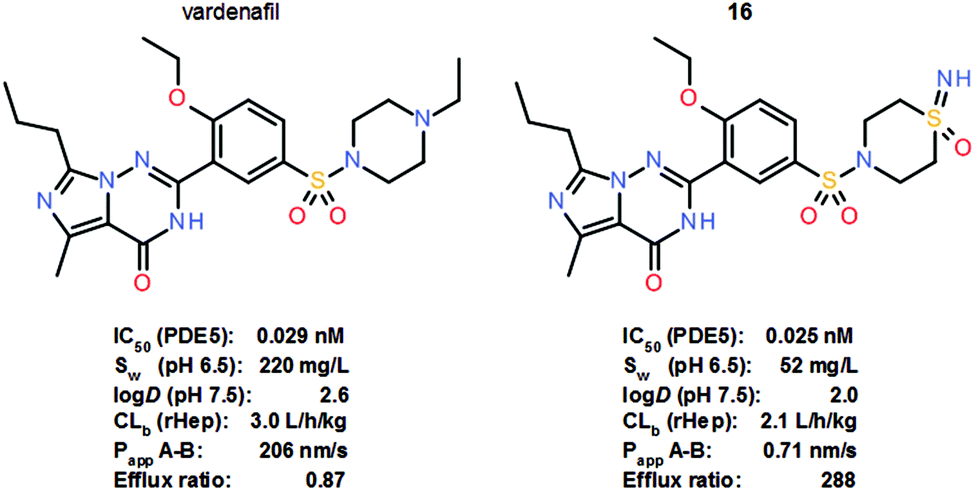 | ||
| Scheme 10 Structures and key properties of the PDE5 inhibitors vardenafil and sulfoximine analogue 16. | ||
The sulfonamide group 2 has proven a very important pharmacophore which is found in many approved drugs and clinical candidates. In contrast, the corresponding aza analogues, known as sulfonimidamides 5, have been neglected even though they also seem to offer very interesting properties. Similar to sulfoximines 3, the introduction of an additional nitrogen atom to the sulfonamide group 2 induces asymmetry and offers an additional point for substitution. Moreover, the sulfonimidamide group 5 offers multiple possibilities of hydrogen-bond donor/acceptor functionalities (Scheme 11). So far, there are only very few examples of sulfonimidamides in drug discovery, as recently highlighted by Arvidsson and co-workers.18
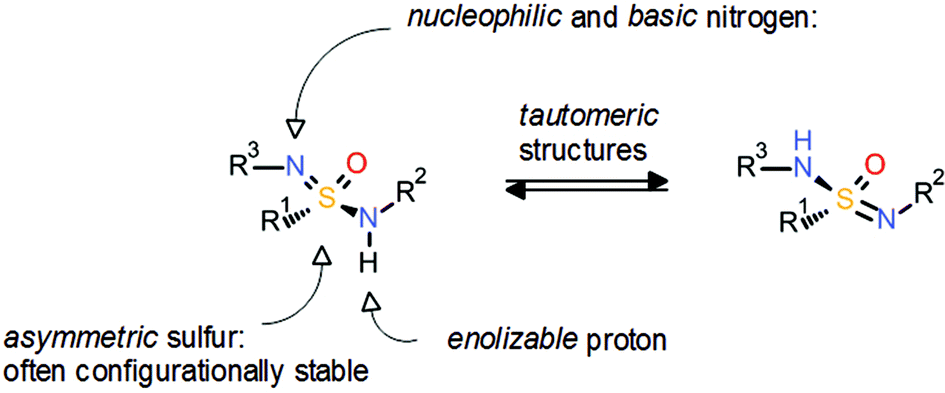 | ||
| Scheme 11 General properties of the sulfonimidamide group.18a | ||
Similar to sulfoximines 3 about 15 years ago, it can be speculated that the use of the sulfonimidamide group 5 has been hampered by limited synthetic methods, limited commercial availability and an incomplete understanding of the properties of sulfonimidamides relevant to medicinal chemistry. Since the synthetic methodology for the preparation of sulfoximines 3 has progressed significantly over the years, it was thought that perhaps these methods could be applied to the synthesis of sulfonimidamides 5. Against this background, a then new publication from Bull, Luisi and co-workers19 on the synthesis of NH sulfoximines 3 (R3 = H) by NH transfer to sulfoxides using ammonium carbamate and (diacetoxyiodo)benzene provided inspiration. The reported method is easy to use, has a broad scope and no safety concerns. Thus, it was speculated whether or not these reaction conditions could also be utilized for the synthesis of sulfonimidamides 5. Indeed, a series of unprotected tertiary sulfonimidamides was prepared in good to excellent yield in a one-pot transformation from tertiary sulfinamides 17via NH transfer using ammonium carbamate and (diacetoxyiodo)benzene in methanol (Scheme 12). A wide range of functional groups was tolerated and initial results indicated that the NH transfer is stereospecific.20
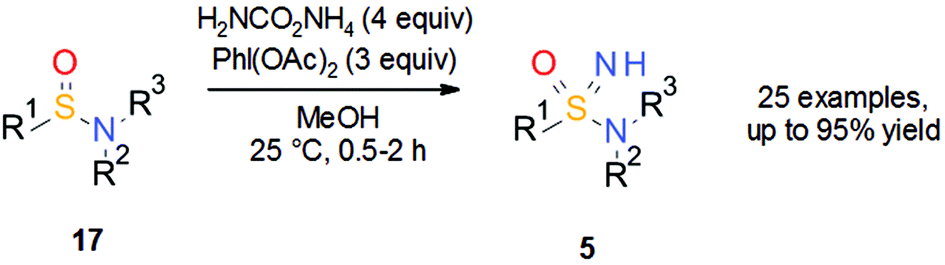 | ||
| Scheme 12 One-pot synthesis of NH sulfonimidamides 5 by NH transfer to sulfinamides 17. | ||
With a new method to prepare NH sulfonimidamides 5 in hand, it was thought to utilize the NH group as an additional point for substitution. Again, the idea was to make use of the prior experience gained from the synthesis of sulfoximines. It was assumed that the reactivity of NH sulfonimidamides 5 in cross-coupling reactions should be similar to that of NH sulfoximines 3, and thus reaction conditions that had been successfully used for the decoration of sulfoximines 3 were applied to NH sulfonimidamide model compound 5a (Scheme 13) to prepare an unprecedented set of structurally diverse sulfonimidamides.21 The N-functionalization reactions of model compound 5a included arylation, alkylation, trifluoromethylation, cyanation, sulfonylation, alkoxycarbonylation and aminocarbonylation. Subsequent in vitro studies of selected, structurally diverse N-functionalized sulfonimidamides 5b did not reveal any intrinsic flaw of the sulfonimidamide group with respect to its application as a versatile pharmacophore in drug discovery.
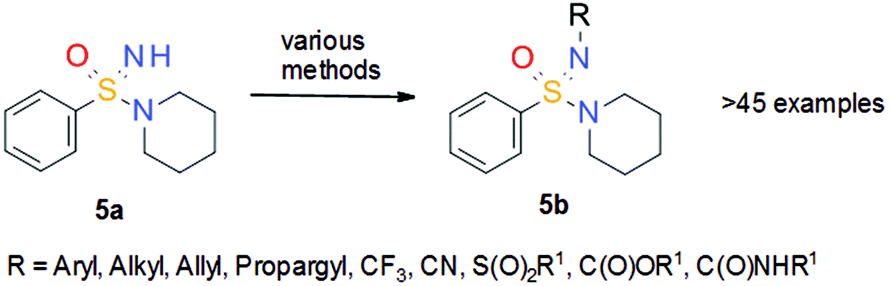 | ||
| Scheme 13 N-Functionalization of NH sulfonimidamide model compound 5a. | ||
In conclusion, the neglected sulfur(VI) pharmacophores sulfoximines 3, sulfondiimines 4 and sulfonimidamides 5 offer very interesting properties and drug design options to the medicinal chemist, who today is confronted with biological targets of increased complexity. In this class of sulfur-based functional groups, the sulfoximine group 3 has had a pioneering role, exemplified by at least four sulfoximine compounds (roniciclib, atuveciclib, BAY 1251152, AZD6738) selected for clinical evaluation in recent years. Moreover, a significant and ever-increasing number of sulfoximines featured in scientific articles and life science patent applications, as well as commercially available sulfoximine building blocks, serve to highlight the increased acceptance of the sulfoximine group by the drug discovery community. Notably, the development of significantly improved methodologies for the synthesis of sulfoximines 3 was an important trigger for this encouraging development. In contrast, scattered literature reports do suggest that the sulfondiimine group 4 could be another versatile pharmacophore in drug discovery; however, the development of safe synthetic methods for sulfondiimine preparation is needed to progress this neglected compound class. The sulfonimidamide group 5 has been the subject of a growing interest in medicinal chemistry recently. In the last two years, a few other groups have outlined interesting, novel synthetic approaches to sulfonimidamides18b,22 but, as far as I am aware, a sulfonimidamide candidate for clinical testing has yet to be disclosed. Recent in vitro studies have not revealed any intrinsic flaw of the sulfonimidamide group regarding properties relevant to medicinal chemistry and the Bayer AG group has thus started to utilize the highly complex and structurally diverse sulfonimidamide group 5 in their drug discovery efforts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
I would like to especially thank the core team members of the lead optimization phase of the pan-CDK inhibitor approaches (G. Siemeister, R. Jautelat, M. Krüger, T. Brumby, H. Briem, A. Hillisch, P. Lienau, D. Mumberg, M. Brands) and the selective CDK9 inhibitor approaches (A. Scholz, D. Kosemund, R. Bohlmann, L. Zorn, G. Siemeister, P. Lienau, D. Mumberg, F. von Nussbaum) at Bayer AG. Thanks are also due to J. A. Sirvent for his postdoctoral work on the sulfoximines, and to F. Izzo and R. Stockman from the University of Nottingham for a successful collaboration on the sulfonimidamides. Finally, I would like to thank M. Bergmann and K. Greenfield for valuable support with the manuscript.Notes and references
- Whilst the official IUPAC name is sulfoximide, this designation is seldom used. Sulfoximine is synonymous (see: https://goldbook.iupac.org/html/S/S06125.html; accessed December 11, 2018), and is the generally accepted terminology. Such compounds are also indexed by Chemical Abstracts Service under the heading sulfoximine. Similarly, the term sulfondiimine rather than sulfondiimide is more frequently used by researchers in this field.
- (a) J. S. Graham, R. Plummer, C. McCoy, K. Kowal, H. Wiesinger, K. Detjen, H. Calvert, B. Wiedenmann and J. Cassidy, Eur. J. Cancer, 2008, 44, 2162 CrossRef CAS PubMed; (b) E. N. Scott, A. L. Thomas, L. R. Molife, S. Ahmed, S. Blagden, P. C. Fong, K. Kowal, C. McCoy, H. Wiesinger, W. Steward and J. De Bono, Cancer Chemother. Pharmacol., 2009, 64, 425 CrossRef CAS PubMed.
- U. Lücking, R. Jautelat, M. Krüger, T. Brumby, P. Lienau, M. Schäfer, H. Briem, J. Schulze, A. Hillisch, A. Reichel, A. M. Wengner and G. Siemeister, ChemMedChem, 2013, 8, 1067 CrossRef PubMed.
- M. Reggelin and C. Zur, Synthesis, 2000, 1 CrossRef CAS.
- (a) U. Lücking, Angew. Chem., Int. Ed., 2013, 52, 9399 CrossRef PubMed; (b) M. Frings, C. Bolm, A. Blum and C. Gnamm, Eur. J. Med. Chem., 2017, 126, 225 CrossRef CAS PubMed; (c) J. A. Sirvent and U. Lücking, ChemMedChem, 2017, 12, 487 CrossRef CAS PubMed.
- N. A. Meanwell, J. Med. Chem., 2011, 54, 2529 CrossRef CAS PubMed.
- C. R. Johnson, M. Haake and C. W. Schroeck, J. Am. Chem. Soc., 1970, 92, 6594 CrossRef CAS.
- H. Okamura and C. Bolm, Org. Lett., 2004, 6, 1305 CrossRef CAS PubMed.
- G. Siemeister, U. Luecking, A. M. Wengner, P. Lienau, W. Steinke, C. Schatz, D. Mumberg and K. Ziegelbauer, Mol. Cancer Ther., 2012, 11, 2265 CrossRef CAS PubMed.
- B. C. Cho, G. K. Dy, R. Govindan, D.-W. Kim, N. A. Pennell, G. Zalcman, B. Besse, J.-H. Kim, G. Koca, P. Rajagopalan, S. Langer, M. Ocker, H. Nogai and F. Barlesi, Lung Cancer, 2018, 123, 14 CrossRef PubMed.
- U. Lücking, A. Scholz, P. Lienau, G. Siemeister, D. Kosemund, R. Bohlmann, H. Briem, I. Terebesi, K. Meyer, K. Prelle, K. Denner, U. Bömer, M. Schäfer, K. Eis, R. Valencia, S. Ince, F. von Nussbaum, D. Mumberg, K. Ziegelbauer, B. Klebl, A. Choidas, P. Nussbaumer, M. Baumann, C. Schultz-Fademrecht, G. Rühter, J. Eickhoff and M. Brands, ChemMedChem, 2017, 12, 1776 CrossRef PubMed.
- J. A. Sirvent, D. Bierer, R. Webster and U. Lücking, Synthesis, 2017, 49, 1024 CAS.
- For an earlier intermolecular Pd-catalyzed α-arylation of an activated N-benzoyl sulfoximine ethyl ester, see: G. Y. Cho and C. Bolm, Org. Lett., 2005, 7, 1351 CrossRef CAS PubMed.
- U. Lücking, A. Scholz, P. Lienau, G. Siemeister, R. Bohlmann and U. Bömer, (Bayer Pharma AG), WO2015/150273, 2015 Search PubMed.
- M. Candy, C. Guyon, S. Mersmann, J.-R. Chen and C. Bolm, Angew. Chem., Int. Ed., 2012, 51, 4440 CrossRef CAS PubMed.
- U. T. Luecking, A. Scholz, D. Kosemund, R. Bohlmann, H. Briem, P. Lienau, G. Siemeister, I. Terebesi, K. Meyer, K. Prelle, R. Valencia, S. Ince, F. von Nussbaum, D. Mumberg, K. Ziegelbauer and M. Brands, AACR Annual Meeting, Washington, D.C., April, 2017, MS.CH01.01 #984.
- See, for example: (a) K. M. Foote, A. Lau and J. W. M. Nissink, Future Med. Chem., 2015, 7, 873 CrossRef CAS PubMed; (b) G. Ouvry, F. Bihl, C. Bouix-Peter, O. Christin, C. Defoin-Platel, S. Deret, C. Feret, D. Froude, F. Hacini-Rachinel, C. S. Harris, C. Hervouet, G. Lafitte, A.-P. Luzy, B. Musicki, D. Orfila, V. Parnet, C. Pascau, J. Pascau, R. Pierre, C. Raffin, P. Rossio, D. Spiesse, N. Taquet, E. Thoreau, R. Vatinel, E. Vial and L. F. Hennequin, Bioorg. Med. Chem. Lett., 2018, 28, 1269 CrossRef CAS PubMed.
- (a) P. K. Chinthakindi, T. Naicker, N. Thota, T. Govender, H. G. Kruger and P. I. Arvidsson, Angew. Chem., Int. Ed., 2017, 56, 4100 CrossRef CAS PubMed; (b) G. C. Nandi and P. I. Arvidsson, Adv. Synth. Catal., 2018, 360, 2976 CrossRef CAS.
- M. Zenzola, R. Doran, L. Degennaro, R. Luisi and J. Bull, Angew. Chem., Int. Ed., 2016, 55, 7203 CrossRef CAS PubMed.
- F. Izzo, M. Schäfer, R. Stockman and U. Lücking, Chem. – Eur. J., 2017, 23, 15189 CrossRef CAS PubMed.
- F. Izzo, M. Schäfer, P. Lienau, U. Ganzer, R. Stockman and U. Lücking, Chem. – Eur. J., 2018, 24, 9295 CrossRef CAS PubMed.
- See, for example: (a) J. Wen, H. Cheng, S. Dong and C. Bolm, Chem. – Eur. J., 2016, 22, 5547 CrossRef CAS PubMed; (b) T. Q. Davies, A. Hall and M. C. Willis, Angew. Chem., Int. Ed., 2017, 56, 14937 CrossRef CAS PubMed; (c) B. Gao, S. Li, P. Wu, J. E. Moses and K. B. Sharpless, Angew. Chem., Int. Ed., 2018, 57, 1939 CrossRef CAS PubMed; (d) H. Yu, Z. Li and C. Bolm, Angew. Chem., Int. Ed., 2018, 57, 15602 CrossRef CAS PubMed.
Footnote |
| † Dedicated to Professor Julius Rebek on the occasion of his 75th birthday. |
| This journal is © the Partner Organisations 2019 |