
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Progress in the synthesis of perylene bisimide dyes
Agnieszka
Nowak-Król
 and
Frank
Würthner
*
and
Frank
Würthner
*
Universität Würzburg, Institut für Organische Chemie and Center for Nanosystems Chemistry, Am Hubland, 97074 Würzburg, Germany. E-mail: wuerthner@uni-wuerzburg.de
First published on 7th March 2019
Abstract
With their versatile absorption, fluorescence, n-type semiconducting and (photo-)stability properties, perylene bisimides have evolved as the most investigated compounds among polycyclic aromatic hydrocarbons during the last decade. In this review we collect the results from about 200 original publications, reporting a plethora of new perylene bisimide derivatives whose properties widely enrich the possibility for the application of these dyes beyond traditional fields. While some applications are highlighted, different from other recent reviews, our focus here is on the advances in the synthetic methodologies that have afforded new bay functionalizations, recently addressed functionalizations at the ortho-positions to the carbonyl groups, and annulation of carbo- and heterocyclic units. An impressive number of perylene bisimide oligomers are highlighted as well which are connected by single bonds or spiro linkage or in a fused manner, leading to arrays with fascinating optical and electronic properties.
 Agnieszka Nowak-Król | Agnieszka Nowak-Król graduated with honors from Rzeszów University of Technology in Poland. She earned her doctorate at the Polish Academy of Sciences in Warsaw with Prof. D. Gryko in 2013 and continued her career as an Alexander von Humboldt postdoctoral fellow with Prof. F. Würthner at the University of Würzburg. In 2016, she started her independent career as a group leader at the Center for Nanosystems Chemistry. She has published >30 papers on porphyrins, corroles, and perylene bisimides and their applications in nonlinear optics, organic electronics and photovoltaics, and supramolecular photosystems. Her current activities focus on chiral π-conjugated organoboron compounds. |
 Frank Würthner | Frank Würthner received his education at the University of Stuttgart, Germany and at MIT in Cambridge, MA, USA. In 1995 he started his independent research on functional dyes and supramolecular dye chemistry at BASF in Ludwigshafen and the University of Ulm. In 2002, he became a professor at the University of Würzburg. His research interests include the synthesis of novel π-scaffolds and their application in organic electronics, the construction of complex supramolecular architectures, the mechanistic elucidation of self-assembly processes, and the investigation of light-induced processes in dye-based nanosystems. He has published >400 papers and has been listed since 2014 as a highly cited researcher. He is also an elected member of the National Academy of Sciences Leopoldina, Germany. |
Introduction
About a hundred years after their discovery, perylene-3,4:9,10-bis(dicarboximide)s, commonly abbreviated as PDIs or PBIs, have emerged as one of the most important classes of functional dyes. Similar to porphyrins and phthalocyanines that are the leading examples of the class of tetrapyrrole dyes, PBIs are the most important compounds of the family of polycyclic aromatic hydrocarbons. This outstanding role of PBIs has evolved not only due to their properties, but also due to the incredible development of the synthetic chemistry of this class of dyes, in particular during the last decade.Initially used exclusively as red colorants in the vat dyeing process and as industrial pigments,1 nowadays PBIs are intensively explored in the field of functional organic materials. Accordingly, while the first seventy years of research on PBIs focused on the variation of imide substituents2 to control their solubility and packing arrangement in the solid state, as is required to receive the desired pigment color,1 functionalization of the electron-deficient aromatic core was addressed only rather late to tune the fluorescence and electronic properties. Early examples include bay functionalization by chlorination under rather drastic conditions3 and a few examples of chlorine exchange by a limited number of nucleophiles during the 1980s, e.g. phenolates,4 which were shown to alter the absorption and emission properties of PBIs to afford interesting red light emitters. It took another two decades until synthetic methods became available that enable the functionalization of a desired position of the PBI aromatic core. This research was strongly motivated by the use of PBIs in electronics, photovoltaics and photonics and as building blocks for supramolecular photosystems (Fig. 1).
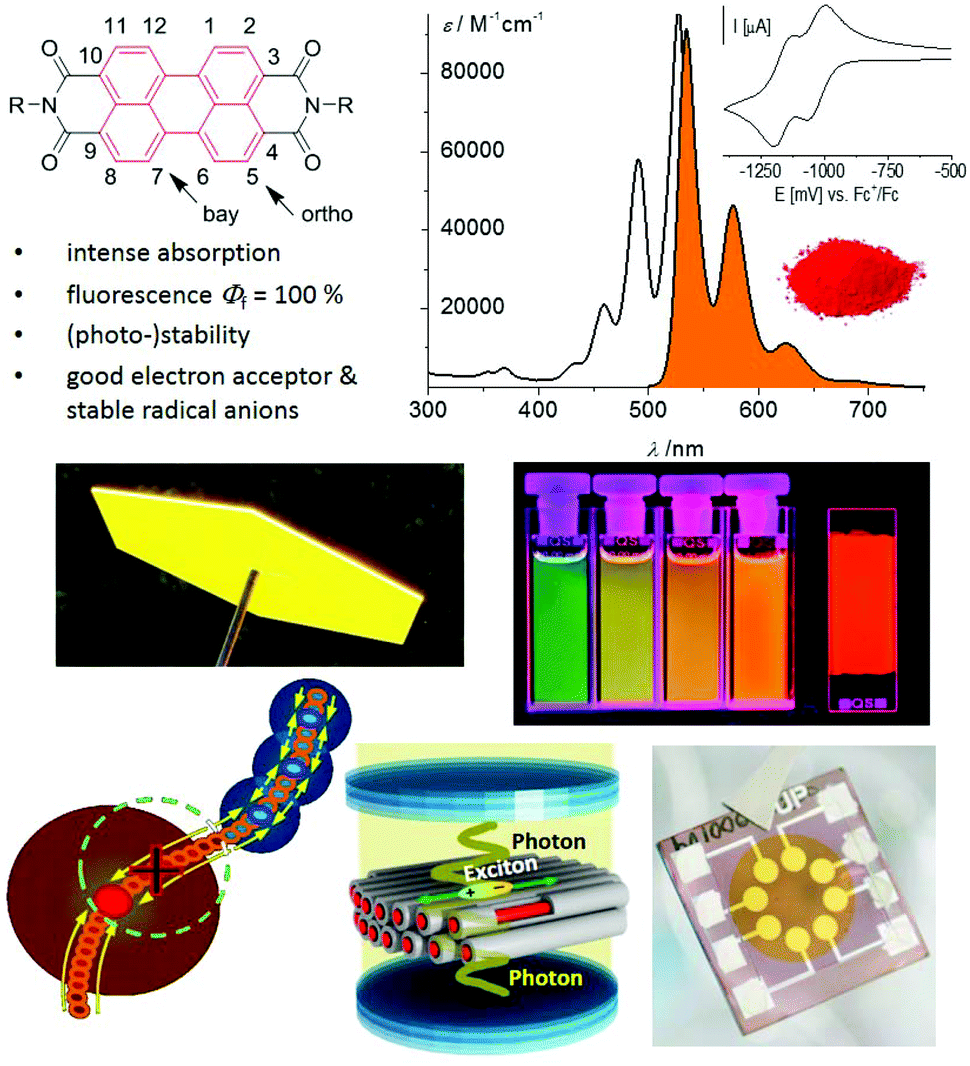 | ||
| Fig. 1 Perylene bisimides and their structural and functional properties for application as fluorophores, color pigments, solar light concentrators, thin film emitters, supramolecular photosystems, in optical microcavities and as electron acceptors in organic transistors and solar cells. | ||
As illustrated in Fig. 1, PBIs constitute indeed a highly versatile class of dyes. The popularity of these compounds is due to their excellent functional properties in combination with their application-relevant features of photo- and thermal stability and chemical robustness. From a functional point of view, the optical properties, in particular fluorescence, are probably still the most appreciated motivation to work with PBIs. Only a few dyes can match PBIs in terms of fluorescence quantum yields, which are close to unity for the parent PBI and many core-functionalized PBI derivatives.5 This feature makes PBIs interesting for various applications including single molecule spectroscopy,6–8 biomolecular imaging,9 and control of light–matter interactions in photonics devices.10 Second, their electron-poor character with the first reduction potential for core-unsubstituted PBIs of around −1.0 V vs. the ferrocenium/ferrocene (Fc+/Fc) redox couple makes PBIs equally suited to fullerenes as electron transport materials.11 Different from fullerenes, however, ambient stable organic transistor devices featuring high electron mobility could be achieved by controlling the packing arrangement and adjusting the position of the LUMO level. The latter was achieved thanks to the ease of manipulation of the perylene chromophore via substitution at the ortho- or bay-positions of the PBI core.5,12
Appropriate LUMO levels, high mobility, and exceptional optical properties were also the reasons why PBIs were recognized as attractive candidates for organic solar cells (OSCs). Within the last decade, a small number of researchers compared to those working with fullerenes succeeded in advancing the PBI-based organic solar cell from a power conversion efficiency of around 3%13,14 up to more than 10%.15,16
Third, strong π–π-interactions between PBIs and the possibility for hydrogen-bond-directed self-assembly via the self-complementary imide functional groups enabled a plethora of supramolecular structures with distinct photophysical properties, which provided considerable insights into fundamental processes such as exciton migration,17 relaxation into excimer states,18 symmetry-breaking charge separation,19 and singlet fission.20 More comprehensive insights into the use of PBIs for supramolecular photosystems as well as organic electronics and photovoltaics can be found in several recent review articles focusing mainly on design principles to control packing and to understand the relationship between a molecular structure and morphology, and their implications for the operation of organic transistor and organic photovoltaic (OPV) devices.21,22
While the judicious selection of imide and core-substituents is crucial for tuning the PBIs’ functional properties for the desired applications, these reviews do not cover the important progress in synthetic methodologies that enabled this progress. Therefore, almost a decade after the review by Marder and co-workers,12 here, we address the amazing more recent progress in the synthesis of PBIs. As we will show, the field of PBI dyes benefited greatly from the recent advances in general organic synthetic methodologies, particularly in C–H activation and metal-catalyzed coupling reactions. There is a clear trend in the implementation of new methods developed for simple aromatic molecules or other classes of dyes, in addition to reactions specific only for a PBI scaffold. A large number of these reactions were unknown until fairly recently. Thus, we provide an overview of the currently existing synthetic tools and introduce the reader to the available methods and strategies for the preparation of a desired PBI in the most suitable, convenient and efficient way.
The review is organized according to the methodology in the synthetic route towards desired molecules in order to facilitate the design of the synthetic sequences. We highlight the relevance of some new protocols and the utilization of various building blocks and show differences between particular approaches. We also put emphasis on the smart strategies that either facilitate purification of key building blocks or take advantage of the intrinsic reactivity of particular perylene molecules in order to achieve high selectivity. The recently reported procedures are contrasted with older methods, and the benefits and limitations of the new protocols are discussed.
As the reader will notice, the vast majority of the discussed PBIs are equipped with certain types of imide residues (Fig. 2). Their choice is not coincidental. Apart from bulky aryl groups, among which 2,6-diisopropylphenyl is the most ubiquitous, linear and branched alkyl groups are very common to improve the solubility.23 The so-called swallow tails introduced by Langhals are particularly suited because their preferential conformation at the imide bond with the two alkyl groups protruding away from the PBI plane leads to a similarly strong attenuation of PBI aggregation as observed for the 2,6-diisopropylphenyl group (Fig. 2).24,25 Crystallographic data for the latter indeed confirm the perfectly orthogonal arrangement of the PBI scaffold and the phenyl planes of the imide substituents.26
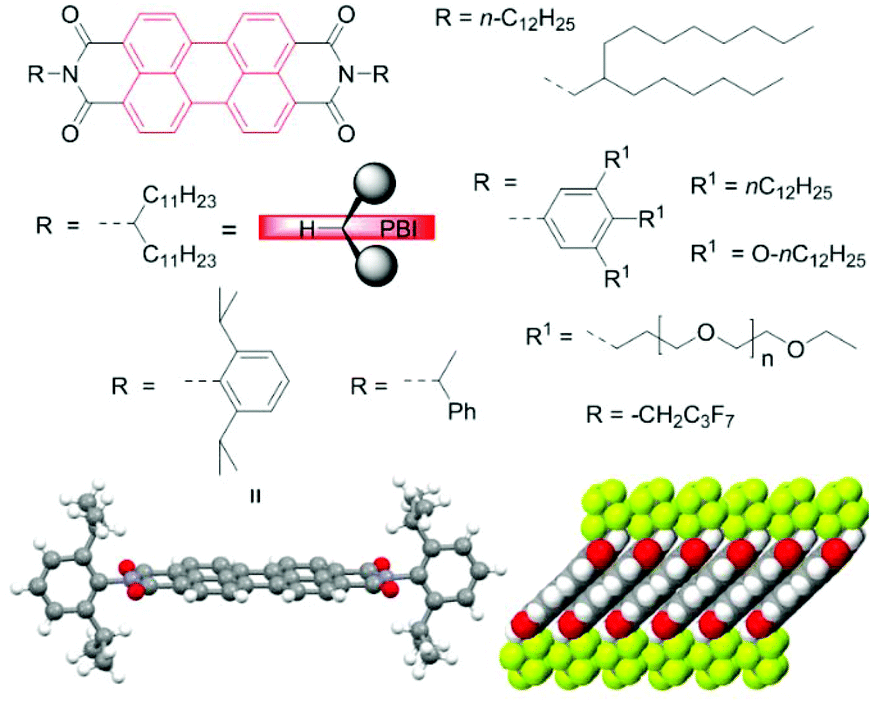 | ||
| Fig. 2 Commonly utilized imide substituents. Long and branched alkyl chains as well as sterically demanding substituents and oligoethylene glycol chains are the most common choices to reach high solubility.23 Specific imide substituents may direct specific packing arrangements as depicted for CH2C3F7 substituents which support the formation of densely packed perylene core and form a water-repellent interlayer.25 | ||
These structural manipulations were inevitable for the utilization of PBIs as fluorescence dyes4,24 but also more recently for the exploration of the synthetic potential of perylene bisimide compounds covered in this review. The judicious selection of imide substituent groups has also become a critical issue for specific applications in more recent years. Thus, PBIs were equipped with per- or polyfluoroalkyl chains to increase the electron affinity, modify the packing arrangement and protect PBI radical anions against humidity in n-channel organic transistors (Fig. 2),25 whilst specific branched alkyl chains of various lengths are needed to tune the morphology of the blend film in OSCs by alkyl chain engineering. The type of imide groups is also of paramount importance in the field of supramolecular chemistry, for instance, to direct hydrogen bond or metallosupramolecular polymer formation.5 In this context the simple removal of solubilizing tert-butyloxycarbonyl (BOC)27 or N-methylbenzyl protecting groups28 by heating or acid treatment is noteworthy. The role of imide substituents in self-assembly of PBIs was addressed more broadly in a recent review article.29 Here, we focus on the core modification rather than on this aspect of PBI chemistry, although it is important to emphasize that solubility affects not only purification but also the reaction outcome.
A very attractive alternative to the already discussed bulky aryl or branched alkyl chains is the utilization of perylene tetraesters (PTEs) as relevant intermediates in the synthesis of the desired PBIs.30–33 These compounds offer a potential advantage over PBIs owing to their enhanced solubility in a wide range of solvent media. PTEs and other perylene derivatives also differ from PBIs in chemical reactivity because of a reduced electron-withdrawing effect of the out-of-plane rotated carboxylic ester groups, which can assist in the execution of the synthesis of some molecules which would otherwise be hardly conceivable for PBIs. In this regard, a Diels–Alder reaction executed by Bock and co-workers between perylene tetracarboxylic acid tetraester and N-alkyl maleimide serves as a good example.34
Halogenation, borylation and nitration at the bay and ortho-positions
Efficient halogenation and borylation procedures of the perylene core afford building blocks of high value for further functionalization. As a consequence of the inherent reactivity of the 3,4,9,10-tetracarboxylic acid functionalized perylene dyes, electrophilic substitution occurs preferentially at bay positions of the PBI and perylene bisanhydride (PBA) cores. It is noteworthy that the sterical congestion imparted in the bay area by non-hydrogen atoms always leads to the distortion of the originally planar PBI core.35 According to studies by Osswald and Würthner, a propeller-type distortion is favored and the dihedral angles in the bay area increase with the number of halogen atoms from two to four and with the size of a given substituent from ∼5° for 1,7-difluoro-PBI to ∼37° for 1,6,7,12-tetrabromo-PBI (Fig. 3).36 For the more distorted PBIs it is even possible to isolate the enantiopure atropisomers by chiral high-performance liquid chromatography (HPLC) at room temperature.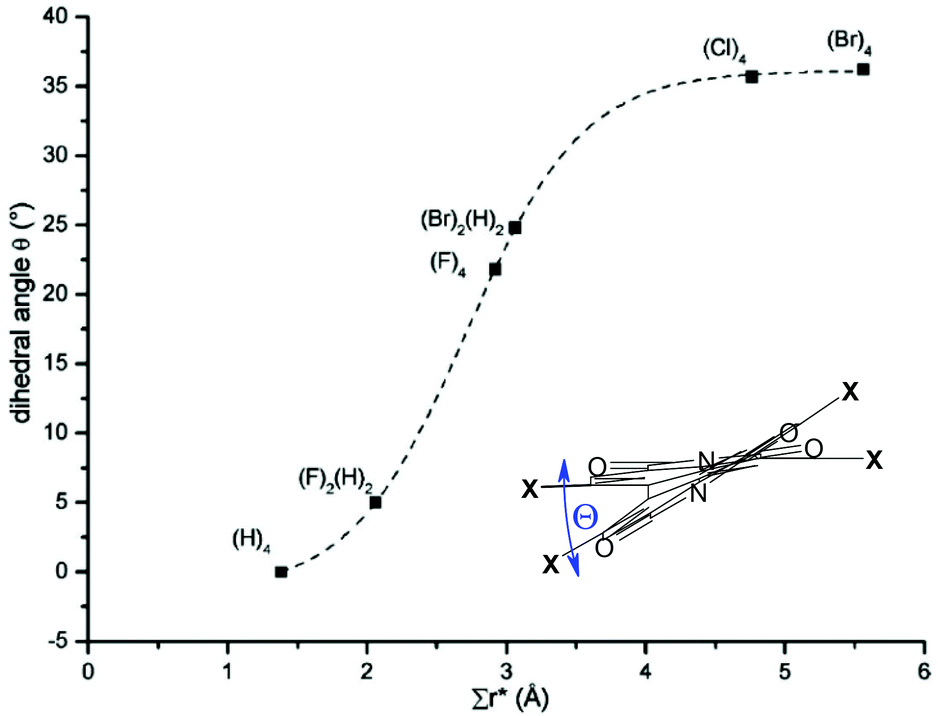 | ||
| Fig. 3 Dependence of the dihedral angle and the apparent overlap for different halogen-substituted perylene bisimides (substituents with numbers are given). Reprinted with permission from ref. 36. Copyright 2007 American Chemical Society. | ||
Initially formed all-bay-halogenated perylene dyes can be further reacted with electrophiles to produce perhalogenated dyes. An effective strategy to impede the inherent reactivity of perylene dyes and promote the formation of other regioisomers is directed ortho-metalation (DoM), followed by a halogen or boron quench. This approach gives highly selective access to valuable ortho-substituted building blocks with regiochemistry complementary to those obtained in metal-uncatalyzed reactions. In this section, general strategies for the preparation of the halogenated and borylated perylene molecules are discussed. Special attention is given to the recent advances in the traditional electrophilic substitution determined by the electronic properties of these classes of substrates, DoM processes supported by the carbonyl function and halogen exchange reactions.
Bromination
Dibromo-PBIs are typically prepared according to the procedure developed by BASF, which includes bromination of perylene bisanhydride 1 with bromine in fuming sulfuric acid at high temperature, and subsequent imidization of brominated bisanhydrates.37 As revealed by Würthner et al.,38 the reaction under these conditions yields a regioisomeric mixture of 1,7- and 1,6-dibrominated PBAs 1,7-2 and 1,6-2 along with the tribrominated product 3 in a ratio of ca. 76![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:4 (Scheme 1).
:20:4 (Scheme 1).
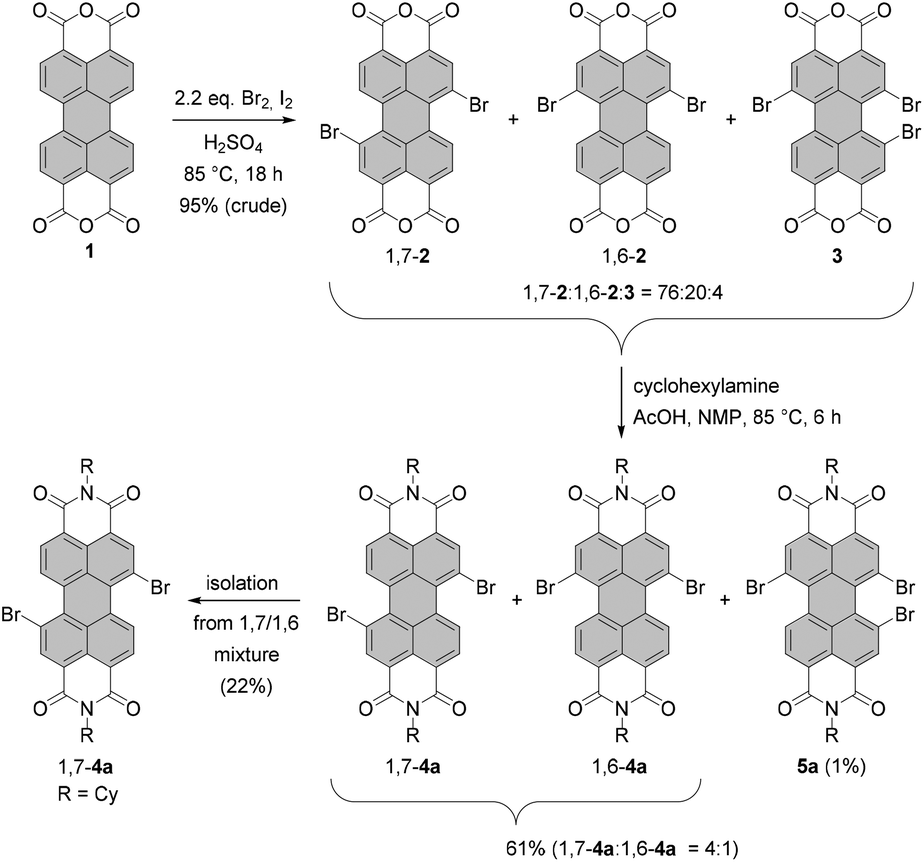 | ||
| Scheme 1 Preparation of isomerically pure 1,7-dibromo-PBIs by bromination of PBA and subsequent imidization. | ||
These compounds are virtually insoluble in any organic solvent. For this reason, separation of the mixture is carried out at a later stage for the corresponding brominated PBIs. Imidization of the mixture of PBAs with cyclohexylamine in N-methyl-2-pyrrolidone (NMP) with the addition of AcOH afforded the 1,7- and 1,6-dibrominated PBIs 1,7-4a and 1,6-4a in 61% yield accompanied by a small amount of tribrominated PBI 5a. The latter could be removed by column chromatography and the 1,7- and 1,6-regioisomers were separated by repetitive recrystallization to afford gram quantities of the isomerically pure 1,7-dibrominated PBI.
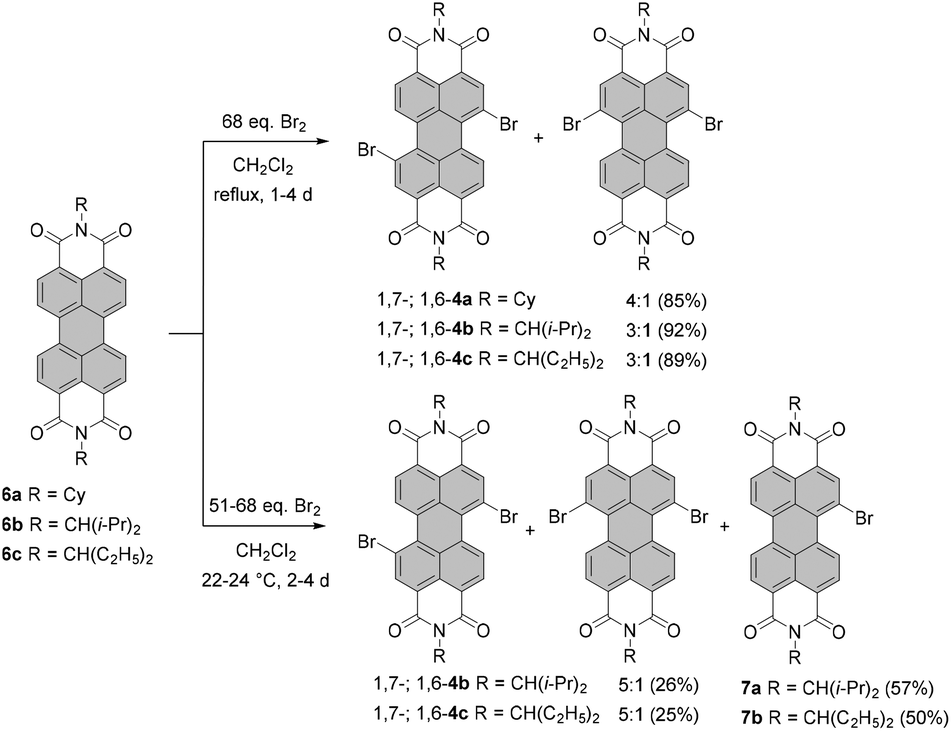
Milder bromination conditions were introduced by Rybtchinski and co-workers.39 Accordingly, core-unsubstituted PBIs 6a–c were reacted in dichloromethane (DCM) with 68 eq. of bromine to exclusively produce 1,7- and 1,6-dibrominated PBIs 4a–c in a 3:1 or 4:1 ratio in 85–92% yield. Isolation of 1,7-isomers 4b and 4c was achieved by repetitive crystallization (Scheme 2).
 | ||
| Scheme 2 Synthesis of monobromo- and dibromo-PBIs via bromination of PBIs. | ||
Monobromination of perylene dyes is also challenging as the reaction affords typically a mixture of monobromo and dibromo products along with the unreacted starting material. An analogous reaction to exclusive dibromination performed at the decreased temperature of 22–24 °C yielded monobromo 7a in 57% yield along with 26% of regioisomeric dibromo-PBIs and 15% of the unreacted starting material.39 For ethylpropyl imide substituents, the reaction time had to be extended from two to four days (in this case ca. 51-fold excess of Br2 was used) to afford monobromo-PBI 7b in 50% yield. Due to low solubility, monobromination of 6a could not be executed. Bromination of 6c could also be expedited under the conditions applied by Xiao. Refluxing PBI 6c with 50 eq. of bromine in the presence of K2CO3 in chloroform for only 4 h produced compound 7b in 41% yield. Nevertheless, the formation of the regioisomeric mixture of 1,6- and 1,7-dibromo-PBIs (42% yield) could not be avoided.40
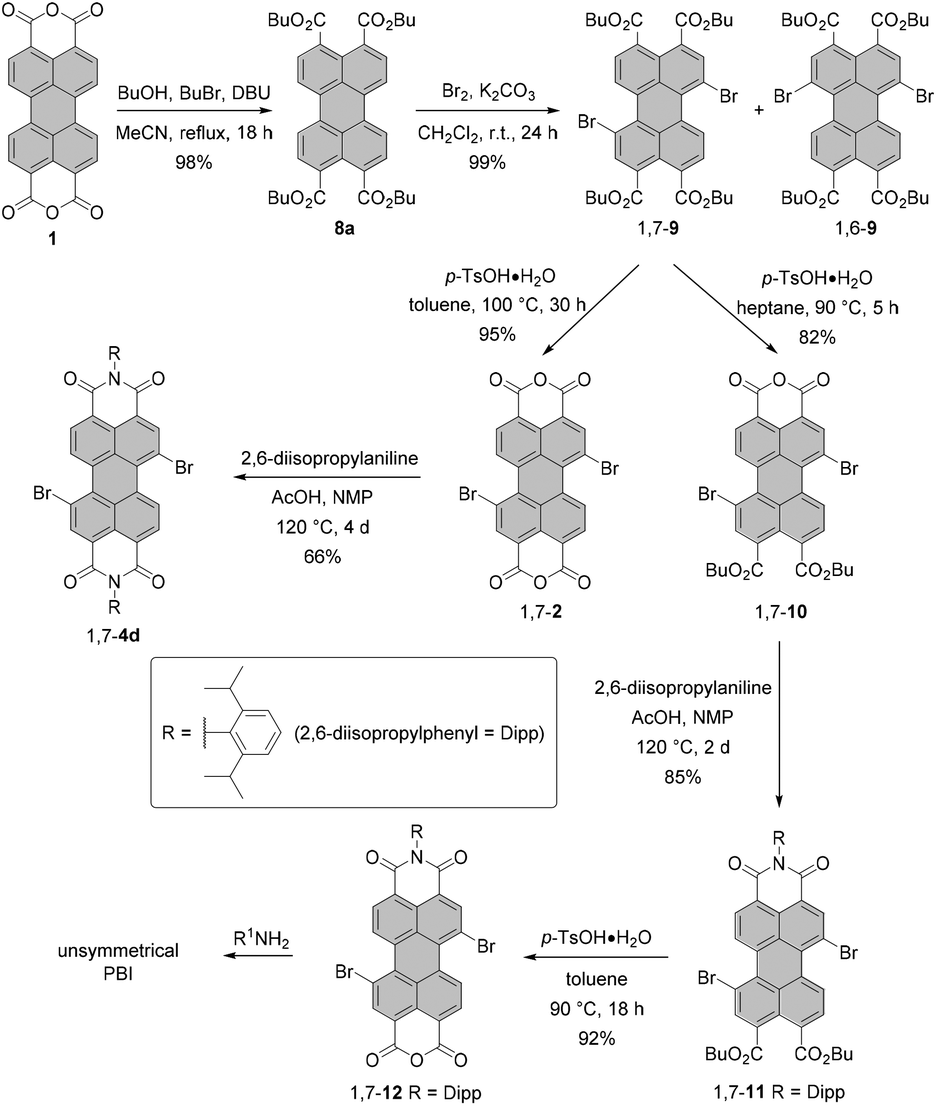
During the last few years, several groups have explored the more elaborate pathway via perylene tetracarboxylic acid esters (PTEs),31–33 which are better soluble and more reactive in electrophilic aromatic substitution. This approach was demonstrated indeed to be advantageous for a number of situations. One is the synthesis of halogen-substituted perylene monoimide monoanhydrides. These compounds cannot be accessed by the cleavage of one imide functionality of a PBI, as the treatment of a PBI with a concentrated solution of KOH under harsh conditions would lead to unwanted reactions involving halogen atoms. Here, Shi and coworkers and Sengupta et al. showed how to apply perylene tetraesters 8a for the rational synthesis of unsymmetrical PBIs from bisanhydride 1 to obtain either monobromo-33 or dibromo-substituted31 PBIs. Scheme 3 illustrates the approach of Sengupta.31
 | ||
| Scheme 3 Strategy for the synthesis of isomerically pure dibromo-PTE and unsymmetrical dibromo-PBIs via bromination of a PTE. | ||
After the formation of the PTE derivative from PBA, dibrominated PTE is obtained under mild conditions and in excellent yield by employing bromine and K2CO3. As in the case of bromination of PBI or PBA, the 1,7-dibromo PTE is contaminated with the 1,6-isomer. The desired 1,7-compound can, however, be more easily isolated by a sequence of two crystallizations in 62% yield. Work-up of the mother liquor increases the efficiency of the process up to 80%. Importantly, the reaction can be easily scaled up due to a facile purification protocol. Thus, isomerically pure 1,7-9 is available in multigram quantities after a single reaction. This compound can be then converted either into bisanhydride 1,7-2 or diester monoanhydride 1,7-10 in the presence of p-toluenesulfonic acid. The key factor determining the major product is the solubility of 1,7-10 in the reaction medium at a given temperature. When the solubility is too high, the reaction may proceed further to produce bisanhydride 1,7-2. The same holds true for reactions carried out at temperatures above 90 °C. Accordingly, the reaction towards 1,7-10 was performed in heptane at 90 °C, whereas PBA 1,7-2 was formed in a high yield in toluene at 100 °C. Anhydrides 1,7-2 and 1,7-10 can be easily reacted with amines in NMP and acetic acid to give the corresponding imides, e.g. 1,7-4d and 1,7-11. Monoimide 1,7-11 bearing two ester groups serves as a precursor of monoimide monoanhydride 1,7-12, which is an excellent starting material for the preparation of a variety of unsymmetrical PBIs.
Utilization of PTEs in the synthesis of PBIs may become even more advantageous with the development of new methods for the conversion of ester functionalities into anhydride. In 2018, Achalkumar reported a versatile microwave-assisted synthesis of bay-unsubstituted perylene tetracarboxylic diester monoimides (PEIs) from PTE. The reactions thereof with aromatic or aliphatic amines under microwave irradiation produced the desired molecules in yields above 50%. The method is also suitable for the preparation of bay-annulated PEIs and unsymmetrical PBIs.41
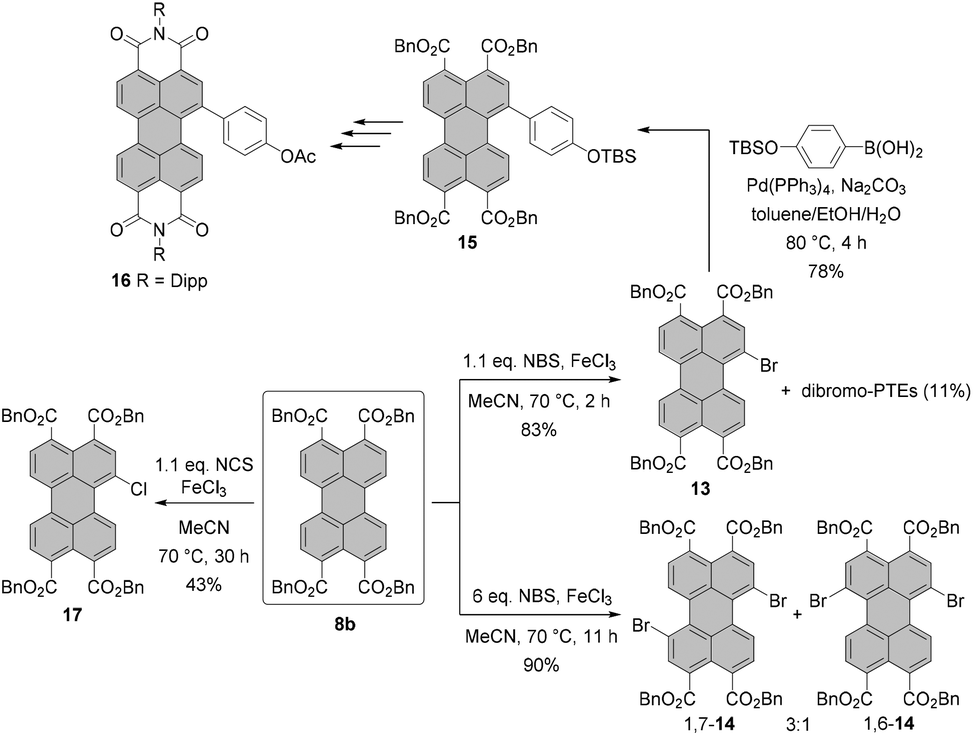
Another remarkable example of utilization of PTE as a key intermediate in the synthesis of PBI is the highly efficient monobromination of PTE reported recently by Takahashi, Yoda and co-workers.32 Their protocol employs 1.1 eq. NBS as a brominating agent in combination with a catalytic amount of FeCl3. Stirring of PTE 8b in MeCN at 70 °C for 2 h affords mono-bromo compound 13 in a high yield of 83%, while only small amounts of the dibromo product and the unreacted starting material were observed. Compound 13 could be next functionalized and readily converted into the desired PBI 16 in several steps (Scheme 4).
 | ||
| Scheme 4 Synthesis of monobromo- and monochloro-PTEs. TBS = tert-butyldimethylsilyl. | ||
Thus far, this is the most effective method for the synthesis of bay-monobrominated perylenes. The method has, however, some limitations. It was shown that the reaction proceeded smoothly only for tetrabenzyl perylene-3,4,9,10-tetracarboxylate, whereas for n-butyl tetraester, commonly used as an intermediate in the synthesis of PBIs, the conversion was poor, and no reaction was observed for core-unsubstituted PBIs bearing either 2-ethylhexyl or 2,6-diisopropylphenyl imide substituents.
Analogous conditions were also tested for chlorination and iodination with N-chlorosuccinimide (NCS) and N-iodosuccinimide (NIS) as halogenation agents, respectively. However, none of these reactions matched the success of bromination. These conditions were also adapted to the preparation of dibromo-PTE. The reaction was carried out with 6 eq. of NBS and over an extended reaction time providing PTEs 1,7-14 and 1,6-14 in 90% yield as a mixture in a 3:1 ratio.32
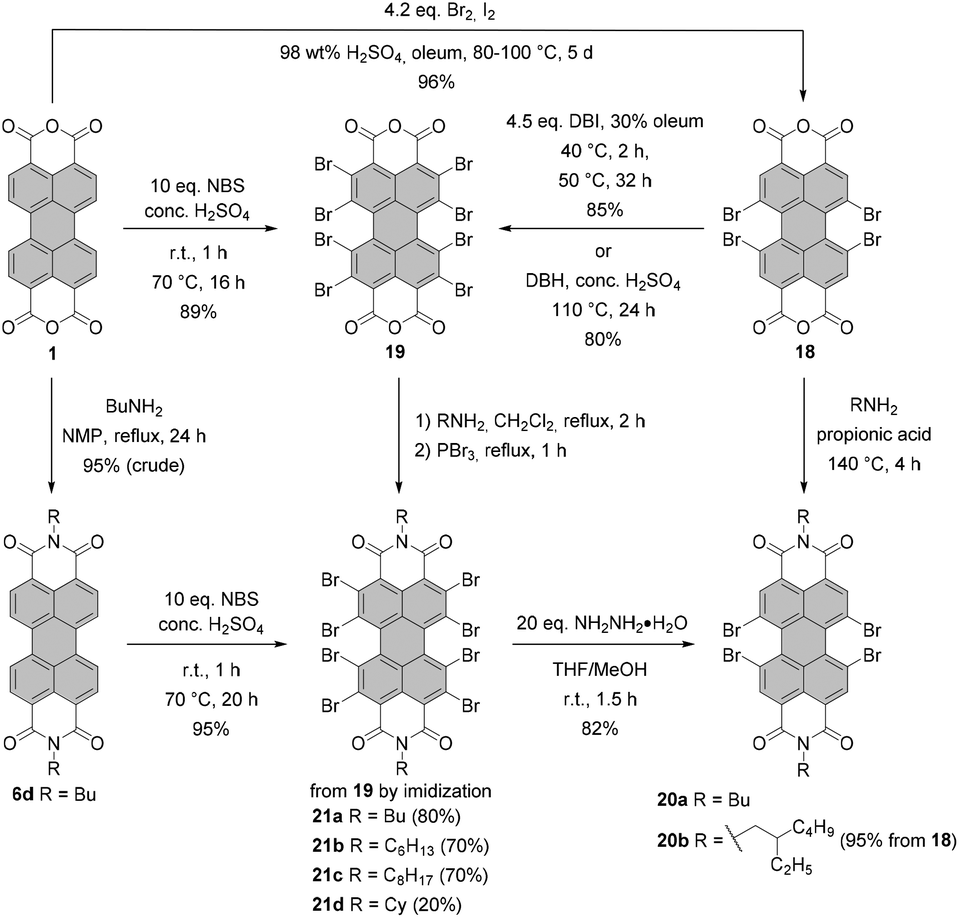
Conventionally, tetrabromo-substituted PBIs are prepared via imidization of tetrabromo-PBA. The latter compound is obtained by bromination of core-unsubstituted PBA 1 with bromine in the mixture of sulfuric acid and oleum according to the procedure published by Liu and Zhu.42 The reaction requires stirring at elevated temperature over an extended reaction time, which gives rise to the desired compound 18 contaminated with tri- and pentabrominated derivatives (Scheme 5). This inseparable mixture is next subjected to the imidization reaction. Introduction of the imide substituent increases solubility, hence allowing the separation of a fourfold brominated PBI from other multi-brominated products. Imidization of the brominated PBA with various amines can be carried out in propionic acid. These conditions were applied to obtain PBI 20b bearing 2-ethylhexyl substituent in excellent yield.42 PBAs are also readily reacted with amines under microwave irradiation (see Scheme 7).43 Product 18 can be converted into octabromo-PBA 19 upon treatment with dibromoisocyanuric acid (DBI) in oleum44 or 1,3-dibromo-5,5-dimethylhydantoin (DBH) in concentrated sulfuric acid (Scheme 5).45
 | ||
| Scheme 5 Synthesis of tetrabromo- and octabromo-PBIs. | ||
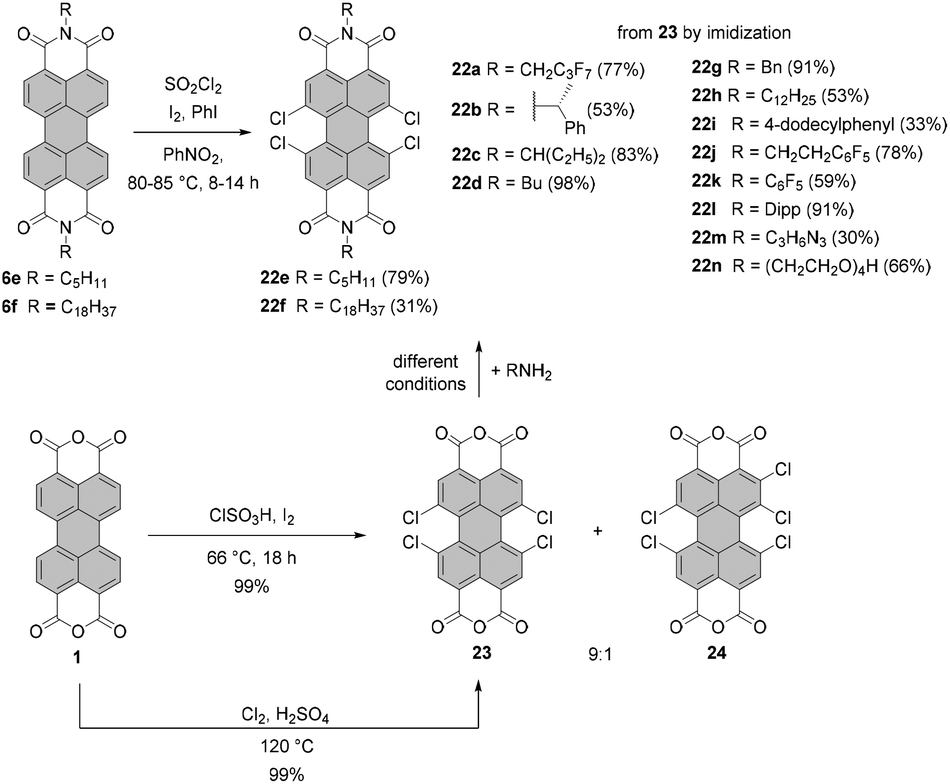 | ||
| Scheme 6 Synthesis of tetrachloro-PBIs by chlorination of PBIs or imidization of tetrachloro-PBA 23 under different conditions. For the reaction conditions of imidization of 23 see the respective ref. 25, 54–56 and 58–61. | ||
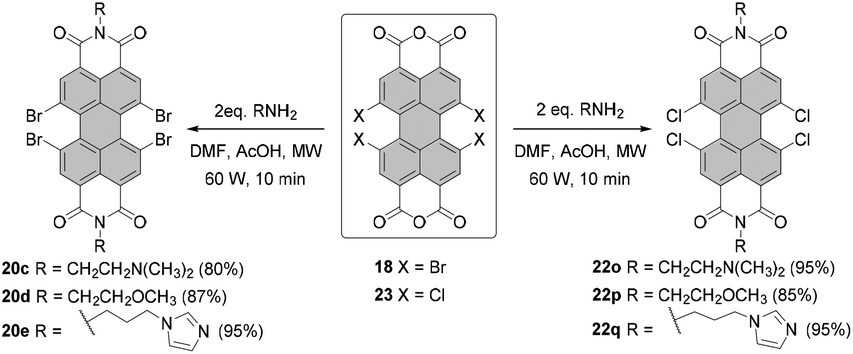 | ||
| Scheme 7 Microwave-assisted imidization of bay-tetrahalogenated PBAs. | ||
An attractive alternative to these two-step bromination procedures was reported by Orentas and co-workers.46 A facile protocol employing NBS as a brominating agent in concentrated H2SO4 afforded perbrominated 21a in one step from core-unsubstituted PBI 6d in 95% yield. Not only does it offer milder conditions, but also it shortens significantly the reaction time from around one week to ca. 20 h. Since at room temperature a small amount of heptabrominated PBI was detected in addition to 21a, the reaction was stirred at 70 °C to ensure full bromination. The method was also successfully applied for the synthesis of 19 from 1, which extends the scope of the imide substituents for octabrominated PBIs and is particularly important for N-aryl substituents that are prone to SNAr. Treatment of octabromo-PBI 21a with hydrazine induces the cleavage of ortho-bromine atoms and gives access to pure tetrabromo-PBI 20a in a yield of 82%. Most probably, the reaction is initiated by the electron transfer from hydrazine to electron-poor 21a. Likewise, pure 20a can be obtained by a slight overbromination of 6d with 6 eq. of NBS, followed by removal of the excessive bromine atoms by hydrazine hydrate.46 This method is superior to direct bromination of a core-unsubstituted precursor, which always leads to a mixture of tetrabromo-PBA with other polyhalogenated derivatives.
Under standard imidization conditions, such as heating with amines in propionic acid or NMP, the introduction of imide substituents into 19 could not be achieved. Imidization studies for core-unsubstituted naphthalene bisanhydrides revealed that the reaction may proceed via a number of intermediates, including carbamoyl derivatives.47 Indeed, the tetrabromonaphthalene derivative bearing two dicarbamoyl functions and two carboxyl groups was observed by Liu and Zhu in the MALDI spectrum.48 Endeavors to accomplish dehydration of this compound by stirring the reaction mixture under reflux in high-boiling solvents, organic acids or quinoline with addition of a zinc catalyst were fruitless. The reason for this could be the presence of four electron-withdrawing bromine atoms, which converts this naphthalene intermediate into a strong organic acid and entails the formation of the internal salt. On the other hand, heating the reaction mixture over an extended time period could not be applied, as it resulted in the replacement of bromine substituents by amine nucleophiles.48–50 These findings can be extrapolated to structurally related perylene derivatives. Accordingly, it is reasonable to assume that the reactivity of the octabromo perylene derivative can be shut down by the formation of the internal salt. Extension of the reaction time is no option, as more reactive ortho-bromine atoms are prone to displacement reactions with amines. The problem of low reactivity of the carbamoyl naphthalene intermediate was tackled by converting the carboxylic group into more reactive acyl bromide with phosphorus tribromide (PBr3) to support intramolecular condensation.48 Imidization of octabromo-PBA 19 was approached in a similar way. Owing to the improved solubility of 19 when compared to tetrabromo-PBA 18, the reaction with amines could be carried out in boiling methylene chloride in the presence of PBr3 to furnish PBIs 21a–d in good yields. Most importantly, the addition of phosphorus tribromide helped to avoid ortho-amination.45
Chlorination
Tetrachloro-PBIs can be synthesized by two possible routes: (1) chlorination of PBA followed by imidization or (2) chlorination of PBI. The latter approach employs chlorination with sulfuryl chloride in the presence of iodine and iodobenzene in nitrobenzene.4,51 The reaction of PBIs 6e and 6f with sulfuryl chloride performed at 80–85 °C over 8–14 h yielded desired tetrachloro-derivatives 22e and 22f in 79% and 31% yields, respectively.52 Utilization of chlorosulfonic acid in the presence of iodine was also described.53 Nevertheless, a considerably more common approach for the synthesis of tetrachloro-PBIs involves tetrachloro-PBA 23. Under the standard laboratory conditions, the synthesis of 23 is executed by treatment of core-unsubstituted PBA 1 with gaseous chlorine in sulfuric acid. Chlorine gas can be generated in situ, for instance by the addition of concentrated sulfuric acid to a NaClO solution and venting the liberated elemental chlorine into the reaction mixture.54 More convenient and straightforward conditions involve treatment of PBA with chlorosulfonic acid and a catalytic amount of iodine. Typically the process was performed at 70 °C for 20 h.55–57 It was observed that this protocol, applied either for PBA or PBI,53,56,58 gives rise to the tetrachloro-product contaminated with the pentachloro side-product. In addition to these impurities a hexachlorinated derivative was detected.58 Optimization of the conditions identified the reaction time as a critical parameter determining the composition of the crude product. Since the poor solubility of this material precluded a proper analysis of its purity, PBA 23 was converted in two steps into a soluble PBI derivative in each case and the sample composition was analyzed by mass spectrometry, 1H NMR spectroscopy and/or elemental analysis. As revealed by the combined techniques, the formation of pentachloro and hexachloro impurities could be avoided when the reaction time was shortened to 5 h. The crude product was purified by Soxhlet extraction to give 23 in ca. 62% yield. Under these conditions, the trichlorinated compound was also observed. Yet, the removal of this impurity by column chromatography was possible after its conversion into the corresponding PBI. A study by Dubey and Jager revealed that the formation of pentachloro-PBI is strongly dependent on the reaction temperature.30,56 Thus, the ratio of 23 to 24 was 8:2 for chlorination with chlorosulfonic acid and a catalytic amount of iodine at a temperature of 70 °C over 20 h. The amount of the pentachloro side-product was reduced to less than 10% when the reaction was carried out at 66 °C for 18 h (Scheme 6). This temperature was identified as optimal since a further decrease in the reaction temperature resulted in the formation of a trichloro-PBI, while a pentachloro product was still observed.30 Subsequent condensation with aliphatic or aromatic primary amines under varied reaction conditions afforded the desired tetrachloro-PBIs 22a–d,g–n in moderate to excellent yields depending on the amine and the synthetic protocol.25,54–56,58–61 In general, the reaction with aromatic amines requires harsher conditions, such as higher temperatures and prolonged time (e.g. synthesis of 22jversus22k). The most commonly applied protocols employ NMP with the addition of acetic acid or propionic acid as the reaction media.
The preparation of tetrachloro- and tetrabromo-PBIs is supported by the progress in imidization of PBAs. In 2015, Syrgiannis and Prato published an efficient microwave-assisted method for the synthesis of PBIs from the core-unsubstituted and halogenated PBAs (Scheme 7).43 The procedures differed in some details, e.g. power, additive, reaction time, and work-up, for various types of amines and PBAs. Essentially, the new method proved to be a versatile tool for the synthesis of PBIs. A protocol that tolerates both chloride and bromide functionalities in bay areas included the addition of acetic acid, similarly to conventional imidization conditions. The reactivity of bay-halogenated PBIs is markedly higher than that of core-unsubstituted congeners under these conditions. To achieve full conversion of tetrabromo- or tetrachloro-PBAs 18 and 23, a single reaction cycle including microwave irradiation for 10 min sufficed for all the tested aliphatic amines such as N,N-dimethylethylenediamine, (2-methoxyethyl)amine and amine appended with an imidazole moiety. The new synthetic protocol allowed the preparation of corresponding tetrabrominated PBIs 20c–e and tetrachlorinated PBIs 21o–q in high yields of 80–95% (Scheme 7).
Generally, a conversion of the crude PBA 23 contaminated with pentachloro-PBA 24 into PBIs does not allow the isolation of a pure fourfold chlorinated product. Pure 1,6,7,12-substituted PBIs may be obtained upon further derivatization and a subsequent purification.53,56,59,60 Dubey and Jager proposed a very simple, but very effective solution to this problem via the highly soluble PTEs 25 and 26 (Scheme 8).30,56 Since the retention factors of the compounds differ substantially, their separation was achieved by column chromatography to obtain pure tetrachloro-PTE 25 in yields as high as 85%. Isolation of 25 can also be executed by recrystallization from an acetonitrile/dichloromethane mixture, albeit in a lower yield of 60%. Acid-catalyzed removal of ester groups in refluxing chlorobenzene (bp 131 °C) provided pure bisanhydride 23 in 95% yield. On the other hand, treatment of 25 with p-toluenesulfonic acid in low-boiling heptane (bp ∼98.4 °C) afforded monoanhydride 27 with two preserved ester moieties in 65% yield. The following sequence of steps echoes the synthesis of unsymmetrical tetrabromo-PBIs and involves imidization of 27 in refluxing propionic acid, the formation of the monoanhydride 29 and the second imidization with a different amine to furnish unsymmetrical PBI 30 bearing diisopropylphenyl and 4-methoxyphenyl imide substituents in an overall yield of 69% after three steps (Scheme 8).30
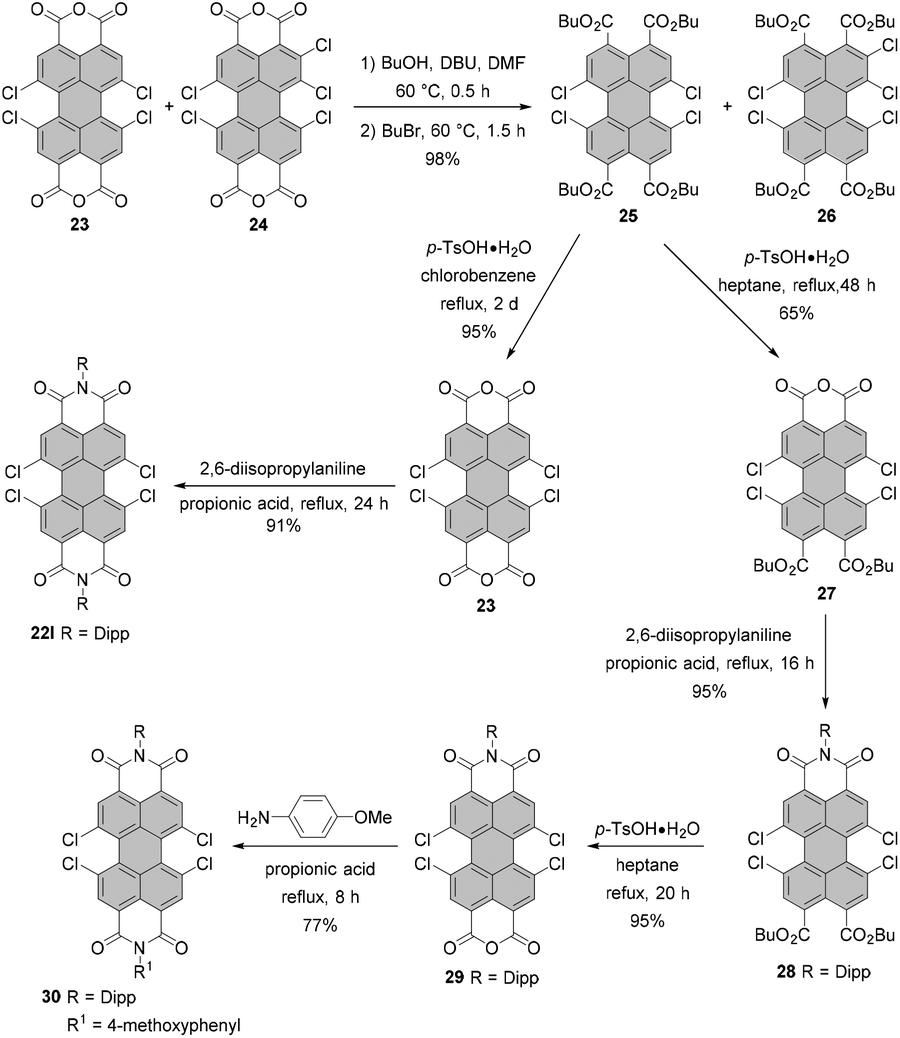 | ||
| Scheme 8 Strategy for the synthesis of pure tetrachloro-PBA and unsymmetrical tetrachloro-PBIs. | ||
Tetrachloro-PBI can also be converted into PBI bearing two chlorine atoms in one bay area (Scheme 9). The first example of the monobay-dichloro-PBI was reported by Wang in 2009. Accordingly, the cleavage of two chlorine atoms was assisted by copper iodide in the presence of L-proline in DMSO. Dichloro-PBI 31a was isolated in 48% along with 50% of the recovered starting material.62 Later, Hoffmann optimized the reaction conditions increasing the yield to 79%, while nearly full conversion of the starting material was observed.63 Using the same approach, the cleavage of two bromine atoms from one bay area was executed to yield monobay-dibromo product 32.64
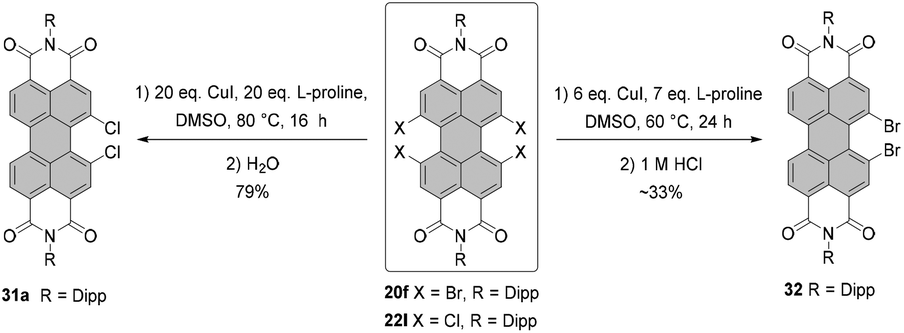 | ||
| Scheme 9 Synthesis of dihalogenated monobay-substituted PBIs. | ||
Würthner and co-workers reported on the synthesis of octachloro-PBI 34a by chlorination of parent PBI 35 in chlorosulfonic acid at 80 °C with excess chlorine and iodine as catalyst (Scheme 10).65 The compound showed excellent properties as an ambient-stable n-channel semiconductor with a mobility of 0.82 cm2 V−1 s−1, which was attributed to the dense brickstone packing arrangement governed by the hydrogen-bonding interactions. Thus, the presence of free NH imide functionalities was beneficial for the performance of the devices but unfavorable for its solubility and purification. It is noteworthy that the charge carrier mobility and stability of octachloro-PBI were better than those of the NH tetrachloro-derivative.66 As opposed to octabromo-PBIs 21a–d (Scheme 5), which exhibit superior solubility in chlorinated solvents and THF,4534a is not soluble in common organic solvents. The isolation of this compound was challenging and required two successive recrystallizations from NMP and acetic acid, followed by gradient sublimation. Similar conditions were used by Sadrai for the preparation of N-methyl PBI.67N-Aryl octachloro-PBIs can be synthesized by efficient perchlorination of core-unsubstituted PBA, followed by imidization. For instance, the synthesis of 34b was accomplished via intermediate 33. Treatment thereof with 2,6-diisopropylaniline in propionic acid furnished PBI in 40% yield (Scheme 10).68 Attempts at fourfold chlorination of tetrachloro-PBIs led either to overchlorination or low yields of the desired octa-substituted target PBIs.68
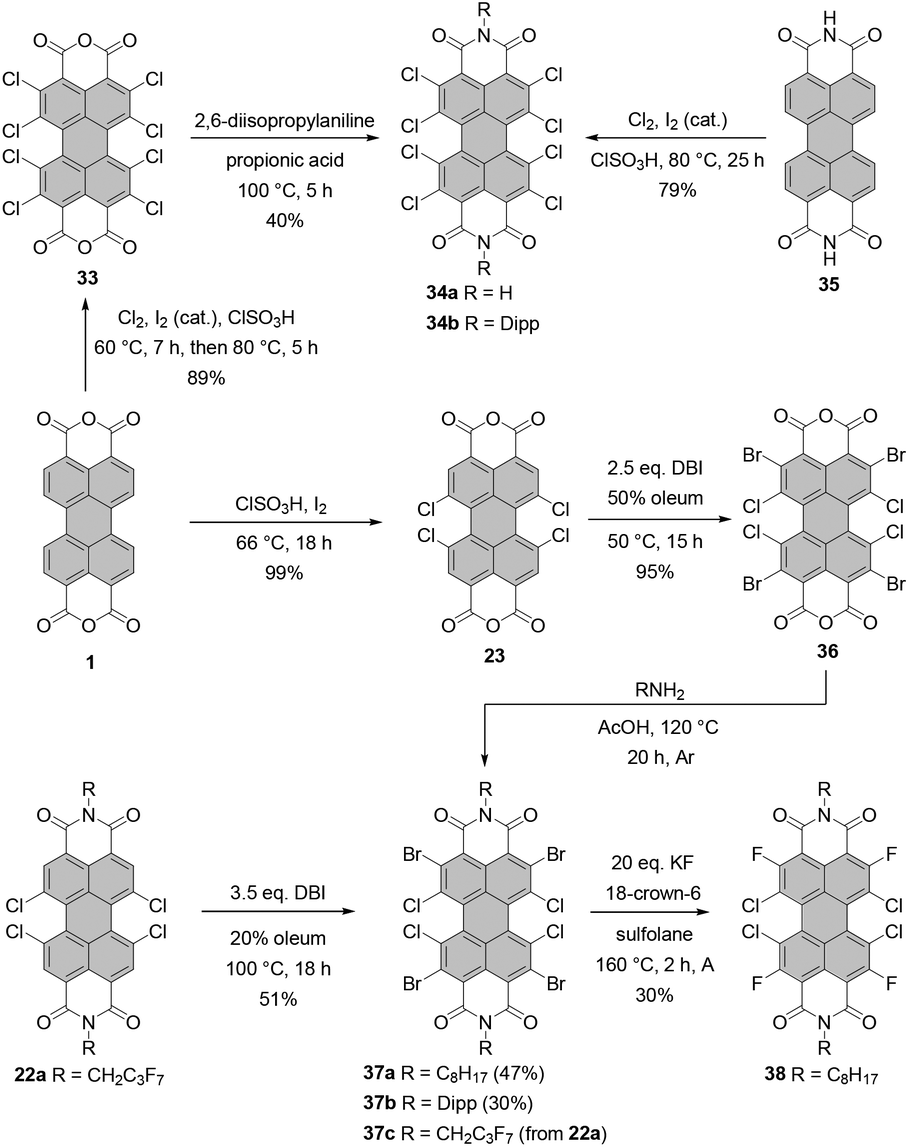 | ||
| Scheme 10 Synthesis of perchlorinated and mixed perhalogenated PBIs. | ||
Core-perhalogenated PBIs bearing different halogen atoms at the ortho and bay positions can be prepared by stepwise halogenation either of PBA or directly PBI. Tetrabromotetrachloro-PBA 36 was obtained by bromination of tetrachloro-precursor with 2.5 eq. of dibromoisocyanuric acid in 50% oleum at 50 °C.69 Subsequent treatment of 36 with n-octylamine or diisopropylaniline in glacial acetic acid under reflux afforded desired PBIs 37a and 37b in rather moderate yields of 47% and 30%, respectively, probably due to the undesired bromide displacement with amines (Scheme 10). These compounds served as starting materials for nucleophilic aromatic substitutions with various nucleophiles. In addition, 37a was converted into tetrachlorotetrafluoro-PBI 38 by the nucleophilic halogen exchange of bromine atoms (Scheme 10) under conditions developed for the synthesis of PBI bearing four fluoride substituents at bay positions (vide infra).70 Compound 37c was synthesized by ortho-bromination of PBI 22a with DBI in 20% oleum in 51% yield (Scheme 10).71
Fluorination
Halogen exchange with fluoride anions, the so-called Halex process, is next to the reaction via diazotization of anilines in hydrogen fluoride or thermal decomposition of either arenediazonium tetrafluoroborates or hexafluorophosphates (Balz–Schiemann reaction), the most common method for the synthesis of aromatic fluorides. This approach was also applied for the synthesis of bay and ortho-substituted PBI dyes. Bay-substituted difluorinated PBIs are accessible via two routes. The first approach involves a halogen-exchange reaction of dibromo-PBA and subsequent imidization, whereas in the second the order of events is reversed, that is, the initial imidization of dibromo-PBA is followed by the Halex reaction of PBIs (Scheme 11).70,72 The latter method is superior, as it eliminates the risk of the undesired replacement of fluoride atoms with amine during the imidization reaction, which was observed for the first method. Tetrafluoro-PBIs are synthesized from the respective tetrachloro-PBIs (Scheme 11). | ||
| Scheme 11 Synthesis and reactivity of bay-substituted fluorinated PBIs. | ||
Nucleophilic substitution of halogenated precursors with KF as a common fluorinating agent was carried out in the presence of either (N,N-dimethylimidazolidino)tetramethylguanidinium (CNC+) chloride or 18-crown-6 as a catalyst. CNC+ proved more effective at the conversion of the tetrachloro-PBIs into the corresponding fluorides, while 18-crown-6 was favored, in most cases, for dibromo-PBI starting materials. Sulfolane was chosen as a suitable reaction medium because of its low cost, high thermal stability up to 250 °C and high boiling point. The protocol employing KF and 18-crown-ether furnished difluoro-PBIs 39a–g from the respective dibromo-precursors 4a,d and 4e–i in modest yields (5–63%).70,72,73 However, cyclohexyl derivative 39a was obtained in significantly better yield, when CNC+Cl− was used as a catalyst.70 Fourfold substitution of chloride atoms with fluoride in the presence of CNC+Cl− proved even more challenging. Tetrafluoro-PBIs 40a–e were formed in low yields (up to 25% but typically lower) from the corresponding tetrachloro-PBIs 22a,b, 22l and 22r,s.70,72,73 It is noteworthy that the efficiency of the halogen exchange is strongly dependent on the solubility of the starting material in sulfolane. In addition, the lower yields for the tetrachloro-PBIs can be attributed to the higher steric encumbrance in the bay areas. Treatment of 40b with BBr3 induces cleavage of α-methylbenzyl groups to yield pigment 41 with NH imide functionalities in nearly quantitative yield. Saponification of 40e with KOH was also successful in leading to bisanhydride 42, although the material was contaminated with a product in which one fluoride was replaced with a hydroxyl group. Some of the bay-fluorinated derivatives were utilized in vapor-deposited thin film transistors that could be operated under ambient conditions. The almost planar PBI 39e appended with fluorocarbon chains showed a good electron mobility of 0.34 cm2 V−1 s−1 in air, which was attributed to both a better stability of the radical anion (the first reduction potential at −0.92 V vs. Fc+/Fc), closely stacked π-cores, and the water-repellent effect of the fluoroalkyl layer. In contrast, the mobility of the even more electron-poor tetrafluoro-substituted PBI 40a (the first reduction potential at −0.87 V vs. Fc+/Fc) was one order of magnitude lower due to less ideal arrangement of these more strongly core-distorted molecules.73 As opposed to bay-substitution, fourfold nucleophilic displacement of bromine atoms with fluoride at ortho positions proceeds more smoothly. Accordingly, heating of 37a with KF in sulfolane in the presence of 18-crown-6 afforded 38 in 30% yield (Scheme 10).69
Iodination
Due to the high steric encumbrance in the bay areas, introduction of four bulky iodide substituents has not been accomplished for monomeric PBI so far. Tetraiodinated di(perylene bisimide) 43 represents the only example of the successful iodination of bay positions.74 The compound was synthesized from tetrabromo PBI 20f by a cascade of several reactions including the Ullmann coupling, C–H transformation and halogen exchange reactions in 15% yield (Scheme 12). The halogen exchange was facilitated by the initial formation of the fused bis-PBI. This molecule features a distorted molecular structure due to the steric repulsion of the neighboring imide rings, which in turn led to the release of strain energy and supported the incorporation of the iodine atoms.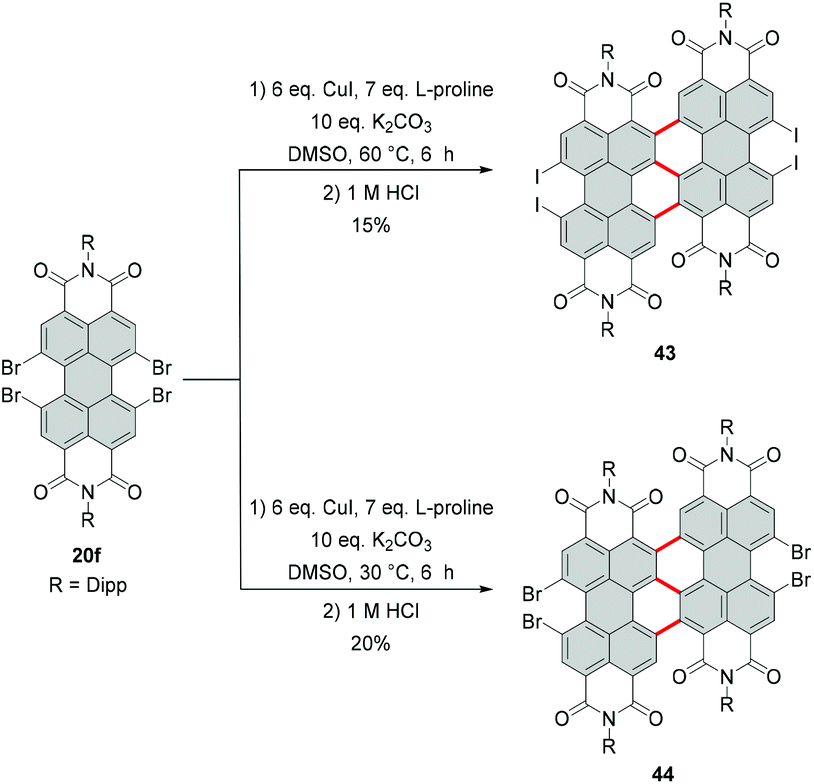 | ||
| Scheme 12 Introduction of iodide into bay areas of a dimeric PBI compound. | ||
The reaction was conducted with an excess of CuI and L-proline as a ligand. CuI played a dual role: firstly as a catalyst and secondly as a source of iodide, while the amino acid was necessary to decrease the temperature of the bromide–iodide exchange. The accelerating effect of α-amino acids bearing hydrophobic side chains was previously recognized.75,76 The outcome of the reaction of 20f with CuI depends largely on the applied temperature. A decrease from 60 °C to 30 °C produced tetrabromo derivative 44 (Scheme 12),74 which suggests that CuI acted here only as a catalyst for the C–C coupling reaction, while the halogen-exchange reaction did not proceed at lower temperature.
Nitration
Nitro compounds of PBIs, despite being non-fluorescent, constitute a class of valuable building blocks for derivatization. Nevertheless, they are sporadically utilized in the field of PBI chemistry. In contrast to bromination or chlorination, the introduction of only one group is facile owing to the strongly electron-withdrawing character of the nitro group, which impedes the second electrophilic aromatic substitution. Nitration of PBI can be carried out by treatment of PBI with cerium(IV) ammonium nitrate (CAN) and HNO3 in dichloromethane at room temperature. The preference towards mono- or dinitro was controlled by the reaction time and an excess of the nitrating agent. Mononitro-PBIs 45a and 45b were synthesized in >90% yields, when the reaction mixture was stirred for 2 h (Scheme 13, conditions A).77 This is a clear advantage over bromination of PBIs or PBAs, as the latter reaction cannot be so easily controlled and produces typically a mixture of mono- and dibromo compounds along with an unreacted starting material (vide supra). The protocol employing CAN and nitric acid solved the problems previously encountered during nitration of PBIs with fuming nitric acid, which produced a mixture of mono- and dinitro-PBIs.78,79 Thus, the CAN/HNO3 method offers access to mononitrated PBIs with high selectivity and under mild conditions. The synthesis of mononitro-PBIs 45c,e,f in almost quantitative yields was also accomplished with (toxic!) N2O4 gas upon stirring at room temperature for 4–7 h in dichloromethane (Scheme 13, conditions B). Preparation of a N2O4 solution is, however, cumbersome and requires thermal decomposition of Pb(NO3)2. A catalytic amount of methanesulfonic acid considerably accelerated the reaction and increased the yields of nitro-PBIs. Despite its low selectivity at room temperature, nitric acid can be successfully applied for the nitration of PBIs with one bay area being already blocked, that is, in the cases where selectivity is not a critical issue.80 Furthermore, a high selectivity for mononitration with nitric acid was achieved when the temperature was decreased to 0 °C (Scheme 13, conditions C). Specifically, core-unsusbtituted PBI 6i was treated with concentrated nitric acid for 2 h to give 45d in quantitative yield.81 Extension of the reaction time of CAN/nitric acid with core-unsubstituted PBIs to 48 h and utilization of a higher excess of a nitrating agent or subsequent nitration of mononitro-PBIs 45a under these conditions afforded mixtures of 1,7- and-1,6-dinitrated PBI 46a in ca. 60% yields in the ratio of 3:1 based on 1H NMR (Scheme 13).77,82,83 Regioisomeric compounds 1,7-46a and 1,6-46a were successfully separated by HPLC.82 Moreover, isomerically pure 1,7-dinitroisomers bearing cyclohexyl imide residues or other substituents such as octyl or butyl could be separated by repetitive crystallization,77,83 similarly to dibromo-PBIs. It is important to note that nitration of PBIs always provides a mixture of 1,6 and 1,7-dinitro compounds whose 1H NMR spectra do not differ significantly in terms of chemical shifts of aromatic protons.82 For this reason, similar to 1,6- and 1,7-dibromo-PBIs, a mixture can be erroneously taken to be a single isomer.
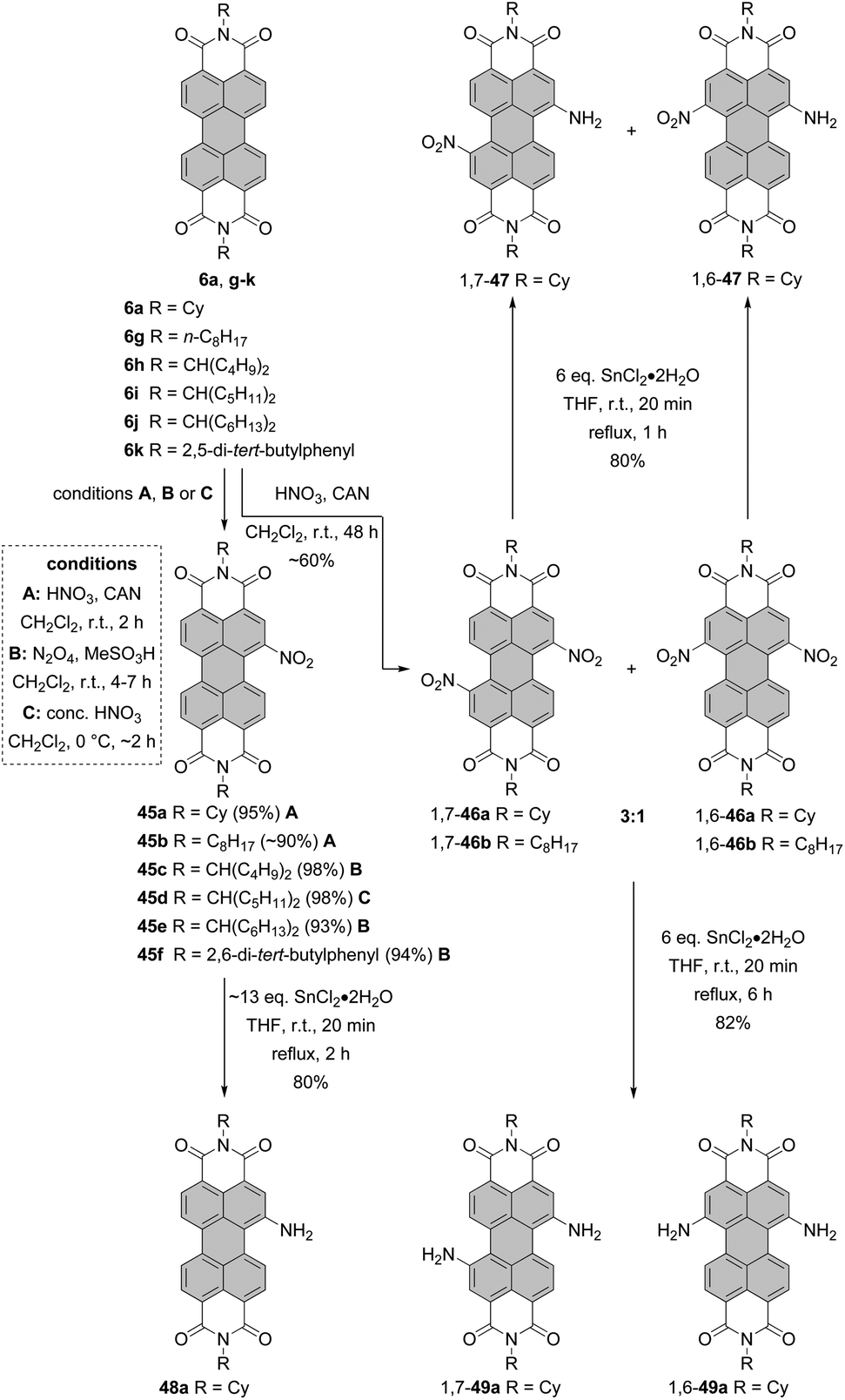 | ||
| Scheme 13 Nitration and subsequent reduction of PBIs. | ||
Nitration of PTEs leads, in general, to similar results. Mononitration of PTE 8a in fuming nitric acid was straightforward and afforded 50 in a yield of 80%. Likewise, introduction of two NO2 groups with double the amount of the nitrating agent was high-yielding (86%) but produced a regioisomeric mixture of 1,7-51 and 1,6-51, which could not be separated by column chromatography.84 High selectivity and even higher yield were achieved when the tetraester was nitrated with sodium nitrite in nitric acid at 0 °C (Scheme 14).85 These conditions also proved effective for various PTEs, i.e. hexyl, octyl, decyl and dodecyl tetraesters. The yields of these compounds were in the range of 80–90%.86
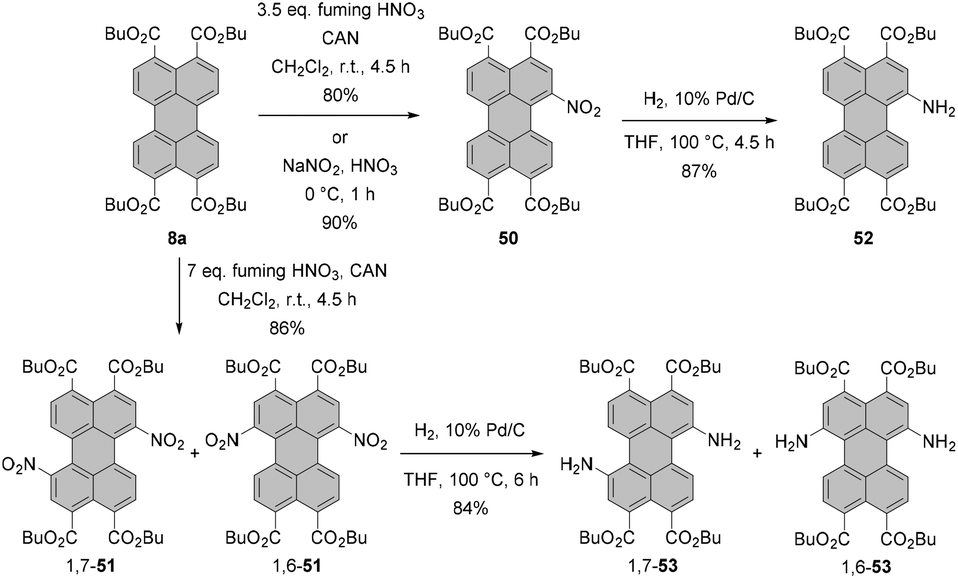 | ||
| Scheme 14 Nitration and subsequent reduction of PTEs. | ||
The nitro group can be replaced by various nucleophiles or reduced to give amino-PBIs. Reduction is carried out either using Pd/C-catalyzed hydrogenation,87 iron powder and hydrochloric acid, or triethyl ammonium formate and Pd/C,79 or SnCl2. Stirring the solution of mononitro-PBI 45a with ∼13 eq. of tin chloride dihydrate for 2 h afforded amino-product 48a in 80% yield (Scheme 13).88
In another report, it was shown that treatment of isomerically pure 1,7- or 1,6 dinitro-PBI 46a with 6 eq. of SnCl2·2H2O for 6 h provided diamino products 1,7-49a and 1,6-49a in 82% yields. Reducing the reaction time to 1 h produced 1-amino-7-nitro or 1-amino-6-nitro PBIs 1,7-47 and 1,6-47 in 80% yields (Scheme 13).89 Likewise, reduction of PTEs is straightforward. Hydrogenation of the regioisomeric mixture 1,7-51 and 1,6-51 afforded the corresponding mixture of 1,7- and 1,6-diamino PTEs 1,7-53 and 1,6-53 in 84% yield, comparable to the yield of reduction of mononitro-PTE 50 to 52 under these conditions.84
Stepwise functionalization as a strategy for the selective synthesis of 1,7-disubstituted PBIs
The synthesis of isomerically pure 1,6- or 1,7-substituted PBIs is indeed a challenge and designing new and convenient ways to access these molecules are of high interest to the community. Thus far, there are only a few reported methods for the preparation of isomerically pure 1,7-dibromo- or dinitro-PBIs involving either time-consuming repetitive recrystallization or separation by HPLC. As was demonstrated, the separation of 1,7- and 1,6-isomers can be executed upon functionalization of dibromo or dinitro compounds with amines or ethers. However, this approach depends largely on the nature of both bay and imide substituents and does not guarantee success in each single case. This limits significantly the scope of PBI chromophores available for various applications. An elegant approach to tackle this problem is by taking advantage of the modulation of electron density via stepwise functionalization. Introduction of the first substituent changes the distribution of the electron density of the perylene core, which favors substitution at one site over another. Thus, selective 1,7-bay-functionalization of perylene dyes can be accomplished by utilization of the directing effect of the first bay-substituent. In principle, this strategy offers the most convenient access to the respective PBIs. Following this idea, Zhan, Yao and co-workers90 studied the impact of the 2-methoxyethoxyl group on bromination of PBIs with bromine in DCM under reflux. The electron-donating group (EDG) at the 1 position of 54a selectively directed the Br substituent into the 7 position to give 55a in 96% yield. The cooperative effect of these two groups allowed selective bromination of the 12 position versus the 2-methoxyethoxy group to obtain compound 56a in 95% yield (Scheme 15). The effect of the positions of EDG and Br substituents was also evident for bromination of the mixture of 1,6 and 1,7-bis(2-methoxyethoxy)-PBIs. The reaction was considerably faster when EDG groups were in 1,6 mutual relationship, since such a constellation allowed the activation of the 7 and 12 positions by EDGs in the same and in the second bay areas. On the other hand, a strongly electron-withdrawing cyano group deactivated the PBI aromatic scaffold towards electrophilic aromatic substitution and prevented the introduction of any bromine atom under these conditions.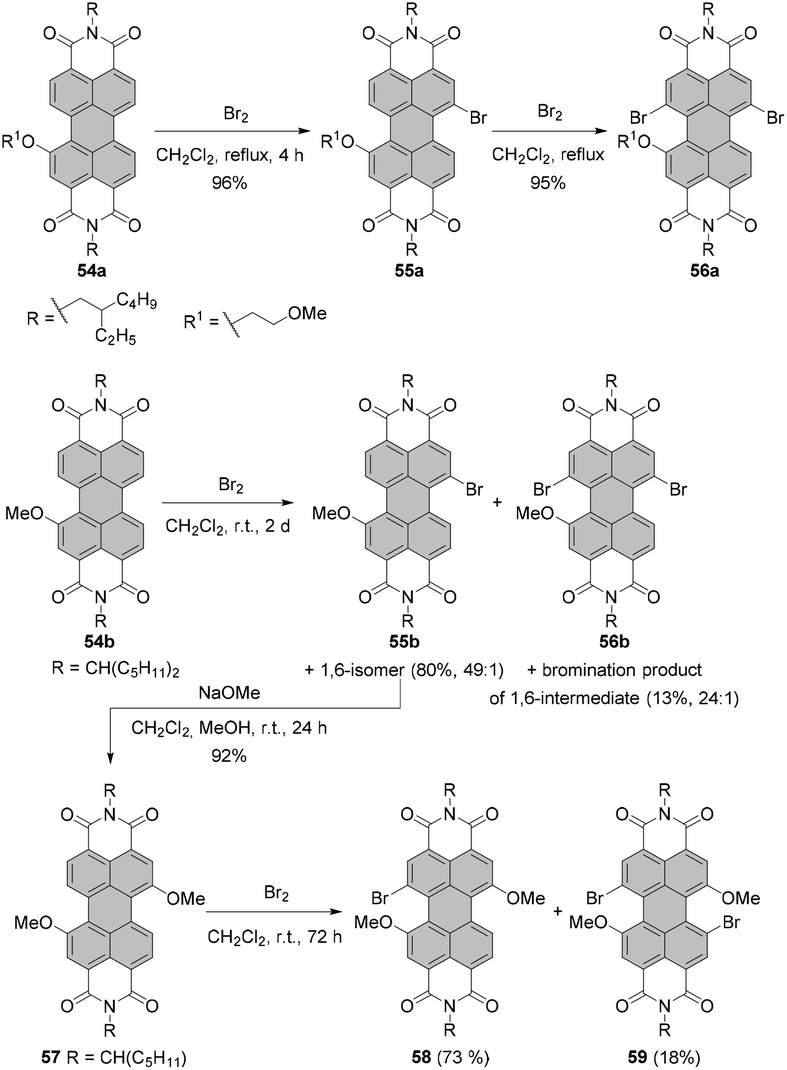 | ||
| Scheme 15 Stepwise regioselective functionalization of PBIs using directing effect of bay-substituents. | ||
Recently, Nuckolls utilized the directing effect of the electron-donating methoxy group to introduce a bromine atom at the 7 position versus the OMe substituent with high regioselectivity (Scheme 15). Bromination of 54b carried out at room temperature over an extended period of time afforded 1,7-substituted-PBI 55b in a high yield of 80%, along with a trace amount of 1,6-isomer and dibrominated products in 13% yield. The efficiency of dibromination could be enhanced using higher excess of bromine and extended reaction time. Compound 56b is analogous to 56a, which was obtained via sequential bromination of the alkoxylated precursor. PBI 55b was converted into dimethoxy-PBI 57. A subsequent bromination provided tri- and tetra-substituted PBIs 58 and 59 of unique substitution patterns. Alternatively, compound 58 could be obtained in 89% yield directly from 56b by methoxylation thereof.91
ortho-Borylation and halogenation
The first syntheses of tetraborylated PBIs at 2,5,8,11-positions were published in 2011 by Shinokubo92 and independently by Li and Müllen.93 In the latter work, 6c and 6l were treated with bis(pinacolato)diboron (B2pin2) under ruthenium catalysis with RuH2(CO)(PPh3)3 (conditions B, Scheme 16) to afford 62a and 62c in good yields. RuH2(CO)(PPh3)3 was previously used for ortho-alkylation94 and ortho-arylation95 (first ortho-functionalization reactions). Ru-catalyzed borylation was unsuccessful for the diisopropylphenyl imide substituents. As suggested by Kakiuchi for the arylation of aromatic ketones96 and by Müllen for the borylation of PBIs,93 the high level of regioselectivity of the Ru-catalyzed reaction may be attributed to the precoordination of the catalyst to the Lewis basic directing group of the PBI substrate, which promotes cyclometallation by insertion into the ortho C–H bond to render a ruthenacycle intermediate.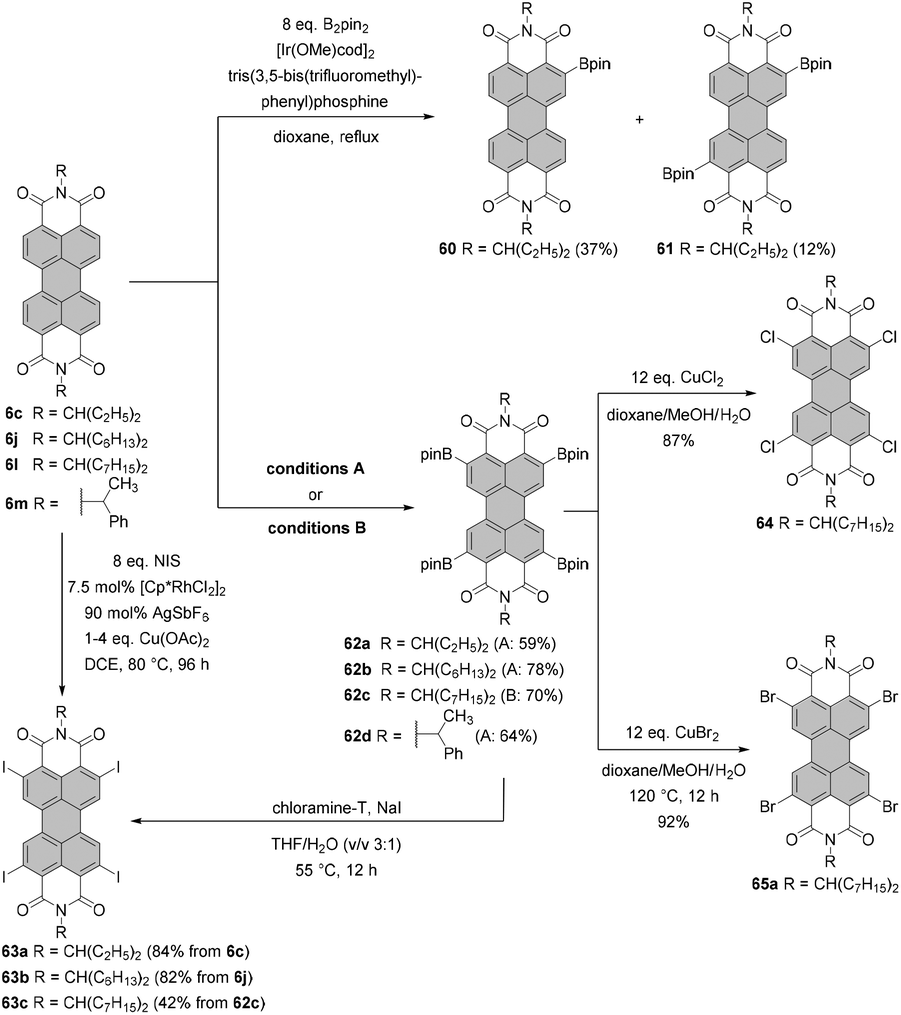 | ||
| Scheme 16 ortho-Borylation and ortho-halogenation of PBIs. Conditions A: 8 eq. B2pin2, 3–4 mol% [Ir(OMe)cod]2, P(C6F5)3, dioxane, reflux, 2–4 d. Conditions B: 7.9–8.3 eq. B2pin2, ∼50 mol% [RuH2(CO)(PPh3)3], mesitylene/pinacolone, 140 °C, 24–30 h. | ||
Shinokubo employed an iridium catalyst to accomplish perfectly regioselective borylation of PBIs 6c, 6j and 6m with B2pin2 at proximal positions of the directing imide carbonyl groups (conditions A, Scheme 16). The corresponding products were obtained in yields ranging from ca. 60 to 80%. When Ir-catalyzed borylation was carried out in the presence of 4,4′-di-tert-butyl-2,2′-bipyridine or tris(3,5-bis(trifluoromethyl)phenyl)phosphine as a ligand instead of tris(pentafluorophenyl)phosphine, only mono- or disubstituted products 60 and 61, respectively, were observed in low to moderate yields.92
Tetraborylated PBIs serve as key intermediates in the synthesis of tetrahalo- and tetracyano-PBIs, and PBIs with four heteroatoms at ortho-positions. Accordingly, treatment of 62c with in situ generated iodine monochloride (ICl) from chloramine-T and sodium iodide in a THF/water mixture at 55 °C produces 63c in 42% yield (Scheme 16).93 A notable improvement was presented in the report by Lin and Zhang, who demonstrated high yielding ortho-selective C–H iodination of core unsubstituted PBIs under Rh-catalysis.97 The straightforward protocol employing N-iodosuccinimide (NIS) as a source of iodine, a Rh(III) catalyst, and AgSbF6 and Cu(OAc)2 as additives furnished ortho-tetraiodo-PBIs 63a and 63b in excellent yields above 80% directly from 6c and 6j (Scheme 16). These conditions were also successfully applied for the multigram (up to 10 g) chromatography-free synthesis of 63a and 63b. The reactions were carried out with a substantial loading of Ag(I) salt (80–90 mol%), which was required for an effective activation of NIS.98 The presence of Cu(OAc)2 was also indispensable to the positive reaction outcome. As reported by Glorius for reactions of heterocyclic compounds under similar conditions, the metal catalyst may play a dual role. It determines the reaction site at ortho positions vs. directing groups, while suppressing electrophilic substitution resulting from the inherent reactivity of the heterocycles. The latter was explicitly validated for bromination with NBS.99 This holds true for the majority of starting materials, excepting highly reactive electron-rich heterocycles. However, bromination with NBS analogous to the reaction with NIS yielded a mixture of ortho- and bay-substituted PBIs.97 The excellent selectivity of iodination may be also partially explained by the high steric requirements of the iodide substituents. 2,5,8,11-Tetrachloro- and tetrabromo-PBIs 64 and 65a can be readily obtained by copper(II)-mediated halogenation of PBI boronate 62c (Scheme 16).100
The conditions developed by Lin and Zhang for the synthesis of ortho-tetraiodo-PBIs can be modified accordingly to produce partially iodinated PBIs.97 More specifically, a lower loading of Ag(I) salt and Cu(OAc)2 additives along with a decrease in the excess of NIS to 1.2 eq. furnishes the 5-monoiodo PBI in 50% yield and the addition of a bit larger amount of the iodinating agent (3.0 eq.) gives rise to a mixture of three diiodo-PBIs (2,5-, 2,8-, and 2,11-derivatives) in an overall yield of 45%, out of which only one regioisomer was isolated successfully. ortho-Monobrominated PBIs are also readily accessible by two-step reactions employing ruthenium-catalyzed functionalization with pinacolatoboron, followed by the exchange for bromide with CuBr2 to give bromo-PBI 7c from 6n in 36% yield (Scheme 17).101 Intermediate monoborylated PBI could be isolated in 38% yield. Likewise, Shinokubo reported the preparation of the Bpin derivative via an iridium-promoted reaction. Treatment of 6c with an equimolar amount of B2pin2 in the presence of [Ir(OMe)cod]2 and P(C6F5)3 afforded compound 60 in 57% yield.102
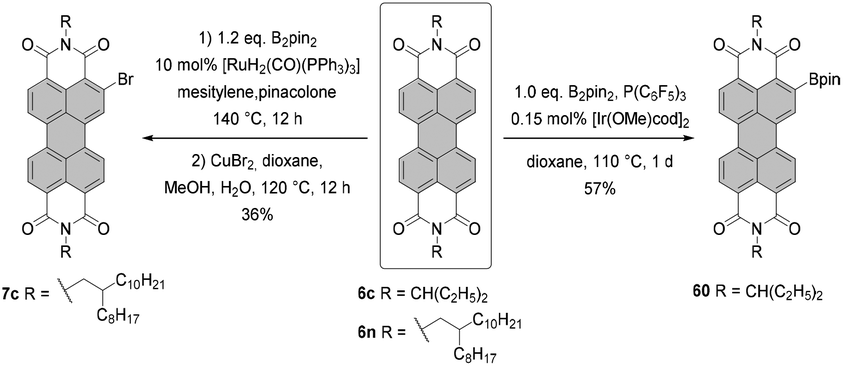 | ||
| Scheme 17 Synthesis of mono-functionalized PBIs at the ortho position. | ||
Substitution reactions at bay areas
Halogenated and borylated PBIs are the most valuable starting materials which upon subsequent substitution afford derivatives with desired optical, redox and packing properties. These properties can be tailored by functionalization of a PBI core at the ortho or bay positions. In contrast, modification of the imide substituents has a rather small impact on the optical and electronic properties of these chromophores due to the nodes in the HOMO and LUMO orbitals at the imide nitrogen atoms. The most effective strategy to tune the absorption and emission wavelengths, as well as positions of the frontier molecular orbital levels of PBIs, is bay substitution but it is also far more difficult to perform due to the steric encumbrance in bay areas. In particular, fourfold substitution is highly challenging. Therefore, the diversity of nucleophiles which can be reacted successfully with fourfold bay-halogenated PBIs is rather limited. In contrast, twofold bromine exchange is more straightforward with traditional nucleophilic substitution reactions as well as a variety of metal-catalyzed coupling reactions, such as Suzuki, Stille, Sonogashira, and Ullmann couplings. However, as discussed before, the separation of 1,7- and 1,6-dibromo regioisomeric mixtures is cumbersome. Consequently, there are numerous publications reporting, de facto, the properties of 1,7- and 1,6-disubstituted PBI derivatives as a mixture rather than isomerically pure compounds.38,103 This section gives an overview of the diversity of bay and ortho-substituted perylene dyes with a strong emphasis on the recent achievements in the synthesis of these molecules.Replacement of halogen atoms by amines
The two most common derivatizations are those with amines and alcohols/phenols. PBIs bearing amino functional groups in the bay position constitute an entirely different class of chromophores compared to the parent PBI as they are characterized by a long-wavelength charge transfer band, which originates from the HOMO localized on the amine to the LUMO located on the PBI scaffold. As a consequence, the absorption band exhibits notable solvatochromism, the fluorescence is weak and the (photo-)stability is reduced. Nucleophilic displacement of bromine atoms in 1,7 and 1,6 regioisomeric mixtures of 4a,d–f by piperidine and pyrrolidine gives rise to dipiperidinyl-PBIs 66a–c104 and dipyrrolidinyl-PBIs 67a,105 respectively, as 1,7 and 1,6 mixtures which could be separated by column chromatography on silica (Scheme 18). | ||
| Scheme 18 Synthesis of 1,7- and 1,6-diamino-PBIs from the regioisomeric mixture of dibromo-PBIs. | ||
It should be noted that nucleophilic substitution with secondary amines gives typically much better results for the more nucleophilic cyclic amines. Accordingly, there are numerous reports on the synthesis and investigation of the properties of PBI substituted with piperidine, pyrrolidine and morpholine.103–107 While other dialkylamines suffer from lower nucleophilicity, at least dimethylamino-PBI 70 was obtained in an excellent yield of 93% by substitution of 1,7-4a with gaseous dimethylamine in NMP/pyridine at 80 °C (Scheme 19).108
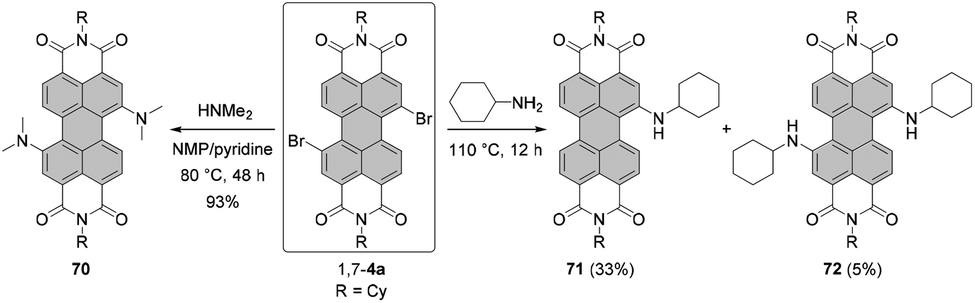 | ||
| Scheme 19 Synthesis of dimethylamino- and cyclohexylamino-PBIs. | ||
The yields with primary alkyl amines are usually much lower.109 In line with these findings, the reaction of 1,7-4a with cyclohexylamine produced primarily mono-amino PBI 71 in 33% yield and only a small amount of diamino compound 72 (Scheme 19),110 which points out that dehalogenation is a competing process to amination. Better results were obtained for amination with n-octylamine. The reaction of the 1,7 and 1,6 regioisomeric mixture of 4e in neat amine provided the corresponding mixture of diamino products 1,7-68 and 1,6-68 in 60% yield, but was also accompanied by the formation of a significant amount of mono-substituted product 69 (Scheme 18). Separation of the isomers was executed afterwards by HPLC, as conventional column chromatography proved ineffective.111 It is noteworthy that the latter reaction could be carried out at higher temperature than amination with cyclohexylamine due to the higher boiling point of n-octylamine.
Alternatively, dialkylamino-PBIs can be synthesized from amino-substituted precursors by the Eschweiler–Clarke reaction. Mainly, treatment of PBI 48c with formaldehyde produced tertiary amine 73 in around 60% yield (the yield varied greatly). Better results were obtained by methylation of 48c with methyl iodide under phase transfer catalysis (PTC) conditions with KOH and benzyltriethyl ammonium chloride (TEBA) in toluene (Scheme 20).79
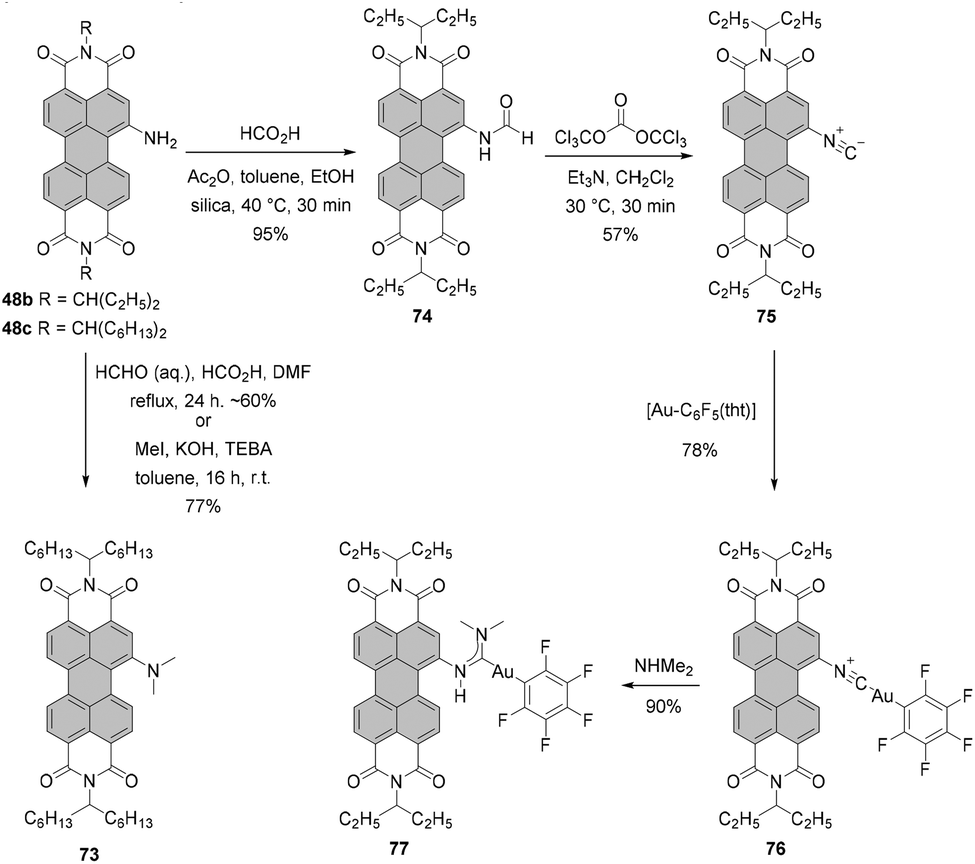 | ||
| Scheme 20 Reactivity of amino-PBIs. Tht = tetrahydrothiophene. | ||
Amino-PBI is also an intermediate in the synthesis of isocyano-substituted PBI 75. Accordingly, compound 48b was first formylated and subsequently dehydrated with bis(trichloromethyl) carbonate (triphosgene) to give isocyanide 75. This compound was used to form complexes with gold. First, a neutral gold isocyanide complex, such as 76, was prepared from the respective gold(I) precursor and PBI 75. A subsequent nucleophilic attack of dimethylamine on the coordinated isocyanide yielded the gold carbene compound 77. The isonitrile derivative features absorption and emission spectra with well-resolved vibronic fine structures and an impressive fluorescence quantum yield of 50%. The optical properties of the gold complexes are similar to those of the parent isonitrile, whereas for carbene complexes the emission bands are structureless and fluorescence is markedly quenched (Φfl = ∼10–20% in DCM).112
Replacement of halogen atoms by alcohols and phenols
In contrast to amino-functionalized PBIs, the typical properties of PBIs such as intense fluorescence and good (photo-)stability are preserved upon bay-functionalization with alkoxy and aryloxy substituents. Accordingly, these functionalizations are most suitable to shift the PBI emission color from the green-yellow (cp. Fig. 1) to the red spectral range. Some diaryloxy-PBIs may be prepared in a pure form from the regioisomeric mixture of 1,7- and 1,6-dibromo PBIs. For instance, PBIs 1,7-78 and 1,6-78 bearing 2,4-di-tert-butylphenoxy groups could be separated by repetitive crystallization from the corresponding 7:3 mixture of diaryloxy-PBIs owing to a marked difference in solubility in toluene of both isomers (Scheme 21).105 This approach is, however, not applicable to most of the aryloxy substituents.
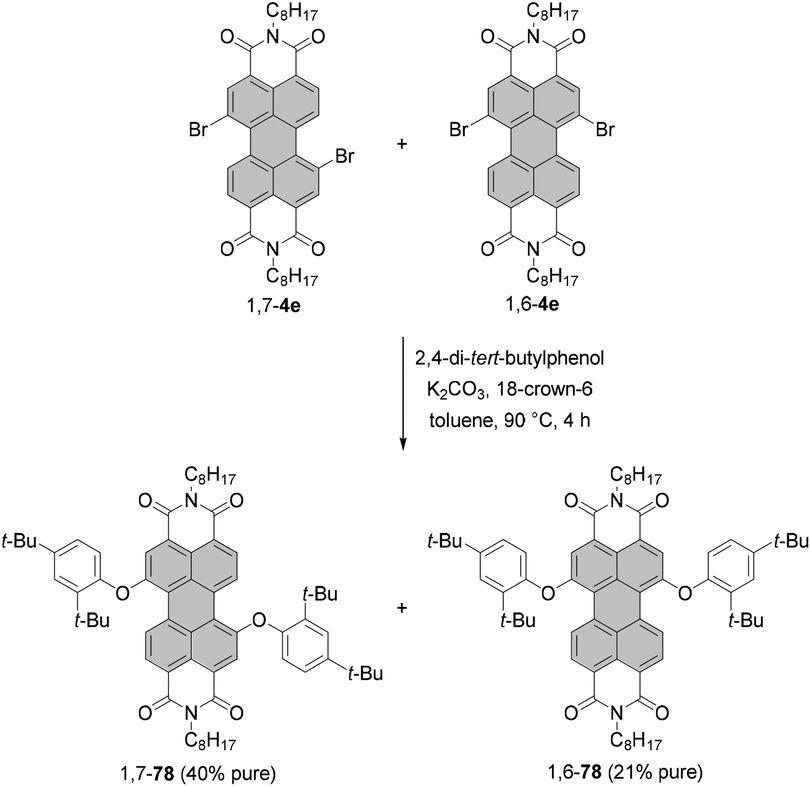 | ||
| Scheme 21 Synthesis of diaryloxy-PBIs from the regioisomeric mixture of dibromo-PBIs. | ||
The introduction of aryloxy substituents is, in general, a very efficient process. Standard preparation of 1,7-diaryloxy-PBIs includes the reaction of the corresponding dibromo derivatives with aryl alcohols. The attachment of even bulky 2,6-diphenylphenoxy and 2,6-diisopropylphenoxy substituents at 1,7 positions was high-yielding. Compounds 81a and 81b were obtained in 79% and 88% yields, respectively, by reacting dibromo-PBI 1,7-4a with the respective phenols in the presence of K2CO3 in NMP (Scheme 22).35,113 As indicated by single crystal X-ray analysis, compound 81a features an essentially planar perylene π-scaffold. The torsion angle derived from the positions of bay carbon atoms is only 1.5(2)°.35 As a consequence of the effective shielding of the PBI core by the sterically demanding groups and reduced conformational flexibility, 81a displays remarkable properties in solution and in the solid state. More specifically, (1) sharp vibronic progressions were observed in the absorption spectrum of 81a in solution due to the small conformational space imparted by the bulky substituents, which is rather unusual for bay-substituted PBIs, (2) the Stokes shift was smaller than even that of the parent unsubstituted PBI owing to the high rigidity of the aromatic scaffold, and (3) the compound displayed a high fluorescence quantum yield (Φfl = 37%) in the solid state.35,113 It should be noted that 81b also revealed well-defined vibronic progressions and a small Stokes shift originating from the enhanced rigidity of the molecule. However, fluorescence in the solid state was almost entirely quenched.35
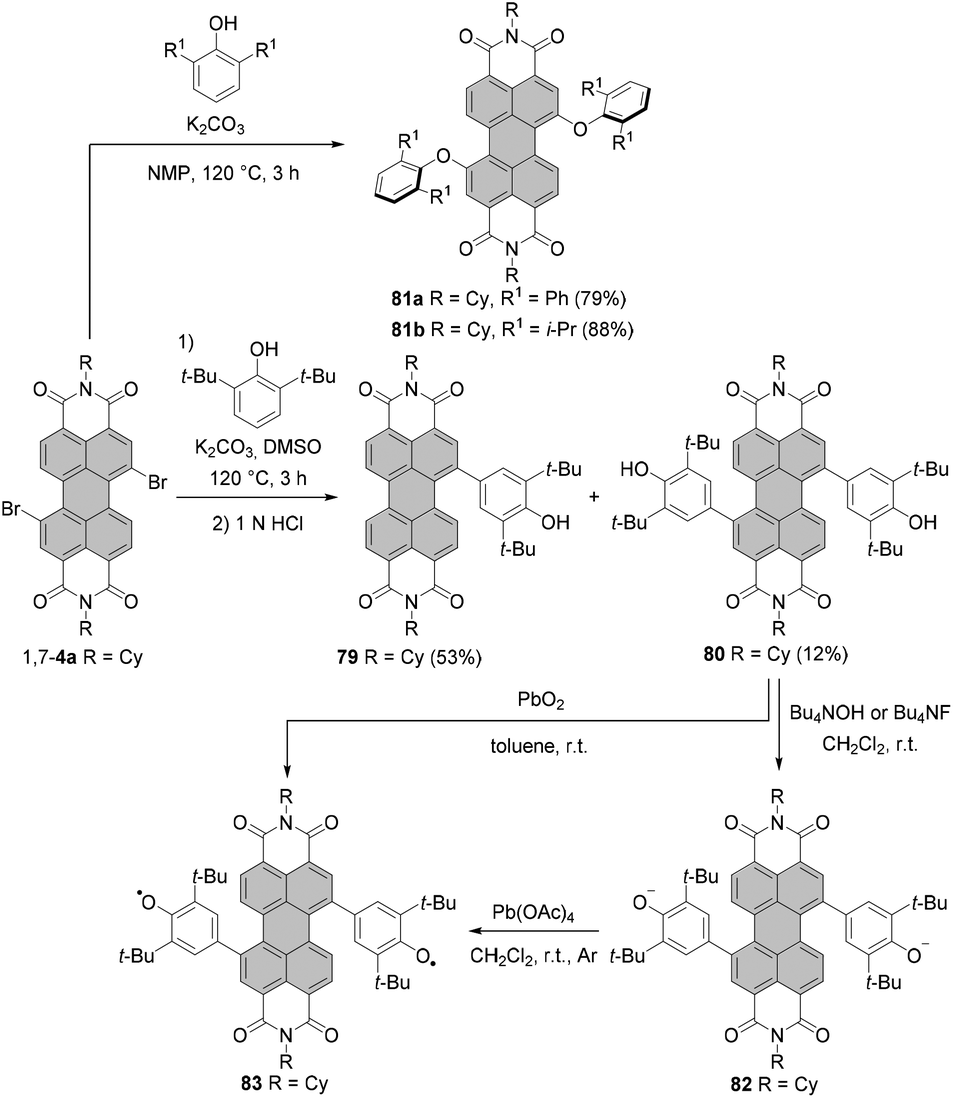 | ||
| Scheme 22 Substitution of a 1,7-dibromo-PBI with sterically demanding phenols. | ||
The reaction of 1,7-dibromo-PBI 1,7-4a with 2,6-di-tert-butylphenol, in which the hydroxyl group is effectively protected by two bulky ortho-substituents, furnished C–C rather than C–O coupled products 79 and 80 in 53% and 12% yields, respectively. The type of the product reflects the tendency of 2,6-di-tert-butylphenol to undergo oxidative coupling with the formation of the carbon–carbon bonds (Scheme 22).114
While the formation of compounds 81a and 81b is, in general, attributed to a nucleophilic aromatic substitution (SNAr), the radical nucleophilic aromatic substitution (SRN1) mechanism was suggested for the reaction of 1,7-4a with 2,6-di-tert-butylphenol. Here, initially an electron is transferred from the phenoxide to the electron-poor PBI to give the corresponding PBI radical anion. In the next step, this species loses bromide and the neutral PBI radical may react with phenoxide to give a hydroxyphenyl-substituted-PBI radical anion. Subsequent bromide elimination, followed by the second reaction with phenoxide and an electron transfer to the starting material, affords finally the disubstituted product 80. Monofunctionalized PBI 79 is formed upon abstraction of hydrogen from the solvent by the neutral PBI radical. 79 and 80 showed halochromic properties. Deprotonation with tetrabutylammonium hydroxide (TBAH)114 or tetrabutylammonium fluoride (TBAF)115 in dichloromethane to give the corresponding monoanion or dianion 82 (Scheme 22) entailed substantial and unprecedented changes in absorption spectra of both compounds. More specifically, new bands at 1179 and 1185 nm arose in absorption spectra of these species, which could be attributed to the pronounced charge transfer between the exceptionally strong electron-donating phenoxide substituents and the electron-poor PBI core.114 Chemical oxidation of 82 with lead(IV) tetraacetate in dichloromethane or the direct treatment of 80 with PbO2 in toluene furnished remarkably stable open-shell singlet biradical 83 (Scheme 22). The singlet biradical character y of 83 was estimated to be 0.72.115
Tetraaryloxy PBIs are routinely obtained from tetrachloro-PBIs by aromatic nucleophilic substitution of chlorides with aromatic alcohols. The fact that these reactions only work for phenols rather than alcohols may again be taken as an indication of the radical nucleophilic substitution pathway. For many phenols including heteroaromatic 3-hydroxypyridines,116 despite the steric encumbrance in bay areas, the reactions typically provide the desired products in good to high yields. However, to achieve acceptable conversion of the starting material, extended stirring at high temperatures may be inevitable. Dubey and Jager reported on the improved reaction conditions for the replacement of chlorine atoms by phenoxy substituents via utilization of Cs2CO3 as a base and DMF as a reaction medium in place of the conventionally used K2CO3/NMP. By means of this, fourfold chlorine substitution by 4-tert-butylphenol could be achieved in 81% yield within only 3 h.30
The synthesis of alkoxylated PBIs is far more challenging. These molecules may be obtained by treatment of brominated PBIs with alcohols or via alkylation of hydroxylated PBIs. The latter method requires the efficient preparation of intermediate hydroxy-PBIs, which is, however, not trivial. Li reported the synthesis of monobay-substituted dihydroxy-PBI 86 starting from PBIs bearing only one bromine atom in a given bay area.117 The reaction under basic conditions followed by aqueous work-up produced hydroxy-PBI 84 along with furan-annulated-PBI 85 with an intact bromine atom in the second bay area. Treatment of the latter compound with trifluoroacetic acid (TFA) affords 1,12-dihydroxy derivative 86 (Scheme 23).
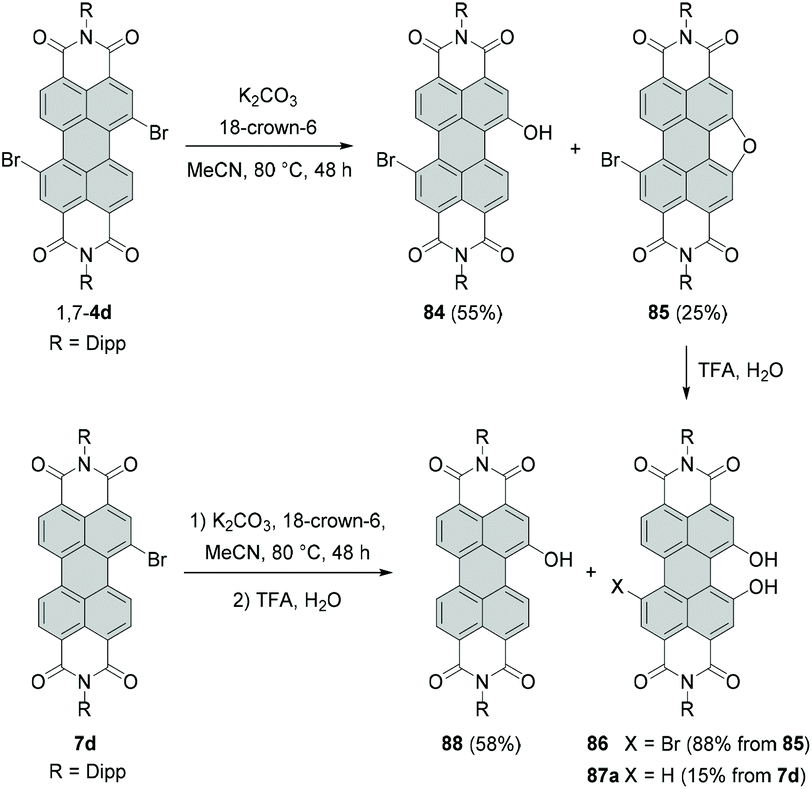 | ||
| Scheme 23 Synthesis of bay-hydroxylated PBIs from bay-halogenated PBIs. | ||
When monobromo-PBI 7d is used as a starting material, the reaction gives access to the monohydroxy-PBI 88 and dihydroxy-PBI 87a (Scheme 23). Thus, to synthesize a PBI bearing two hydroxy groups in one bay area, only one bromine atom in the bay area is necessary. However, these compounds are formed in only low to moderate yields, whereas the main products are monohydroxy derivatives 84 and 88. The reaction may proceed via the radical pathway (SRN1-type reaction mechanism) by analogy to the synthesis of 4-hydroxyphenyl-substituted PBIs from dibromo precursors.114 The proposed mechanism involves the electron transfer from a hydroxyl anion to the PBI, followed by elimination of bromide and recombination with the hydroxyl radical anion to give a monohydroxy intermediate. Subsequently, the OH group of the PBI is deprotonated and the consecutive electron transfer to another PBI may occur, while the perylenoxy radical undergoes intramolecular cyclization to afford a furan-annulated type product. Finally, acidic work-up affords 1,12-dihydroxy-PBI.
It is only recently that Schenning and Brunsveld published a one-pot high-yielding procedure for the preparation of a monobay dihydroxylated PBI directly from a core-unsubstituted precursor (see the section “Direct hydroxylation reactions”, Scheme 56).118 Matile applied another approach to bay-hydroxylated PBIs. Initially, 1,7-dibromo PBI 4a was reacted with allyl alcohol in the presence of NaH to give 89 in 55% yield. Subsequent Pd-catalyzed deallylation gave access to dihydroxy-PBI 90 with the 1,7-substitution pattern in 79% yield.119 Alkoxylation of 1,7-4a carried out in neat dodecyl alcohol in the presence of NaH produced a mixture of mono derivative 91 as a major component and disubstituted product 92 which was formed in barely 17% yield (Scheme 24).110 Likewise, the synthesis of 1,7-dioctyloxy PBI 93 from the corresponding 1,7-dibromo precursor was not efficient. The reaction of 1,7-4d with n-octylbromide in the presence of K2CO3 in DMF furnished 93 in only 28% yield (Scheme 24).118
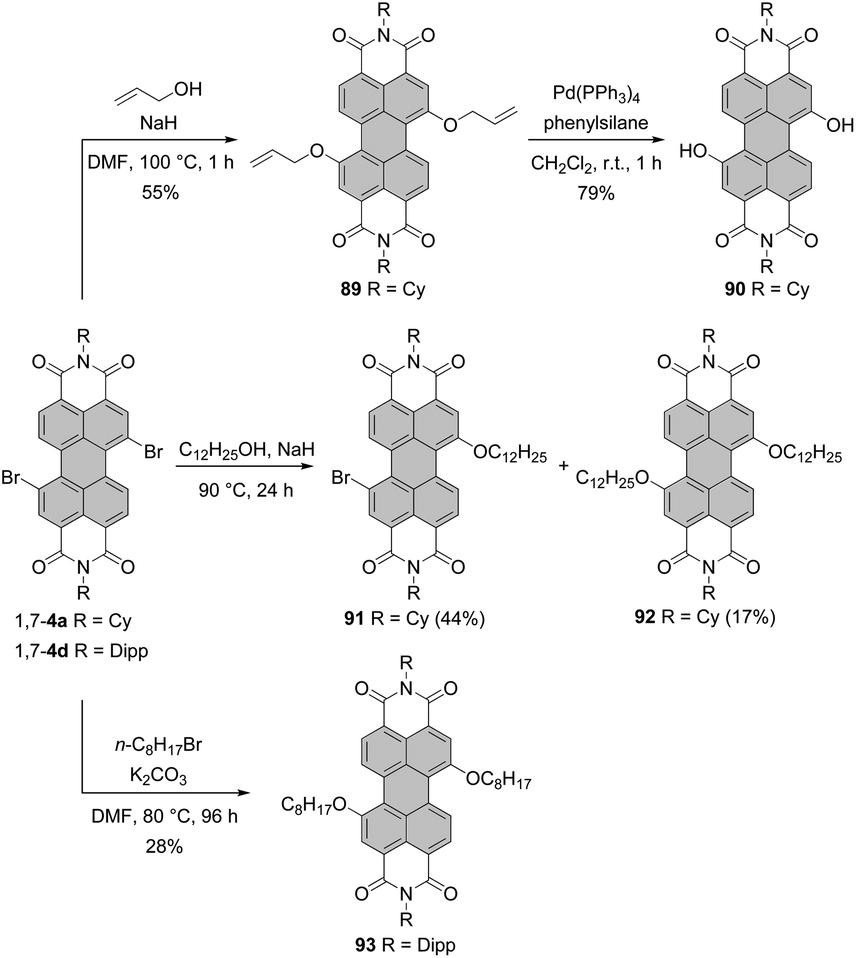 | ||
| Scheme 24 Synthesis of 1,7-dihydroxy and dialkoxy PBIs. | ||
Fernández-Lázaro and co-workers proposed another way to access alkoxylated PBIs.120 The method based on the fluoride-assisted reaction of halogenated PBIs with S or O reagents was an extension of their studies of fluoride-mediated functionalization of core-unsusbtituted PBIs (vide infra). Depending on the type of starting material, i.e. either alcohol or thiol, a combination of CsF/18-crown-6 or KF/18-crown-6 was identified as the most appropriate to facilitate the functionalization of a PBI compound. The reaction of a regioisomeric mixture of dibromo PBI 1,7-4j and 1,6-4j with butanol in the presence of CsF and crown ether proved highly efficient. Dialkoxy compounds 1,7-94 and 1,6-94 were isolated in yields as high as 95% (Scheme 25), which constitutes a tremendous improvement when compared to previously described alkoxylation reactions.
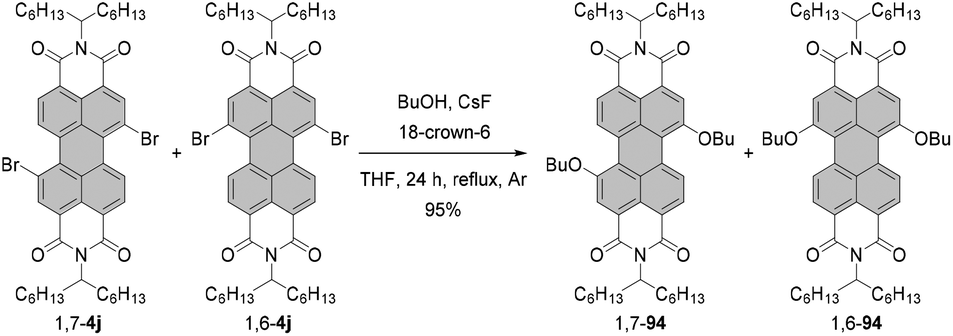 | ||
| Scheme 25 Fluoride-mediated synthesis of dialkoxy-PBIs. | ||
For many years, the almost quantitative reactions of tetrachloro-PBIs (bearing various imide substituents) with phenols leading to the beautifully red emissive tetraphenoxy-PBIs remained the unique example of a successful chlorine exchange. Variations of this chemistry included even substitution with 2,2′-biphenol.121,122 The reaction of tetrachlorinated PBI 22d with 1 or 2.1 equivalents of 2,2′-biphenol under identical reaction conditions afforded in one or both bay areas biphenol-bridged PBIs 99 and 100, respectively. Interestingly, the mono-bridged PBI 99 is chiral and conformationally stable. Thus, the (P)- and (M)-enantiomers could be resolved at room temperature by HPLC on a chiral stationary phase.121 Excellent yields of fourfold substitution also may be achieved by means of fluoride-mediated reactions, as shown for the conversion of PBI 22t to tetraphenoxy-PBI 95 in Scheme 26. Despite the success of substitution with phenols, treatment of tetrachlorinated PBI with aliphatic alcohols such as n-butanol did not lead to the desired tetraalkoxy products. The reaction of 22t afforded only tributoxychloro-PBI 96 in 32% yield along with the dibutoxydichloro-PBIs 97 and 98 in 63% yield, whereas no product of fourfold substitution was observed (Scheme 26).120
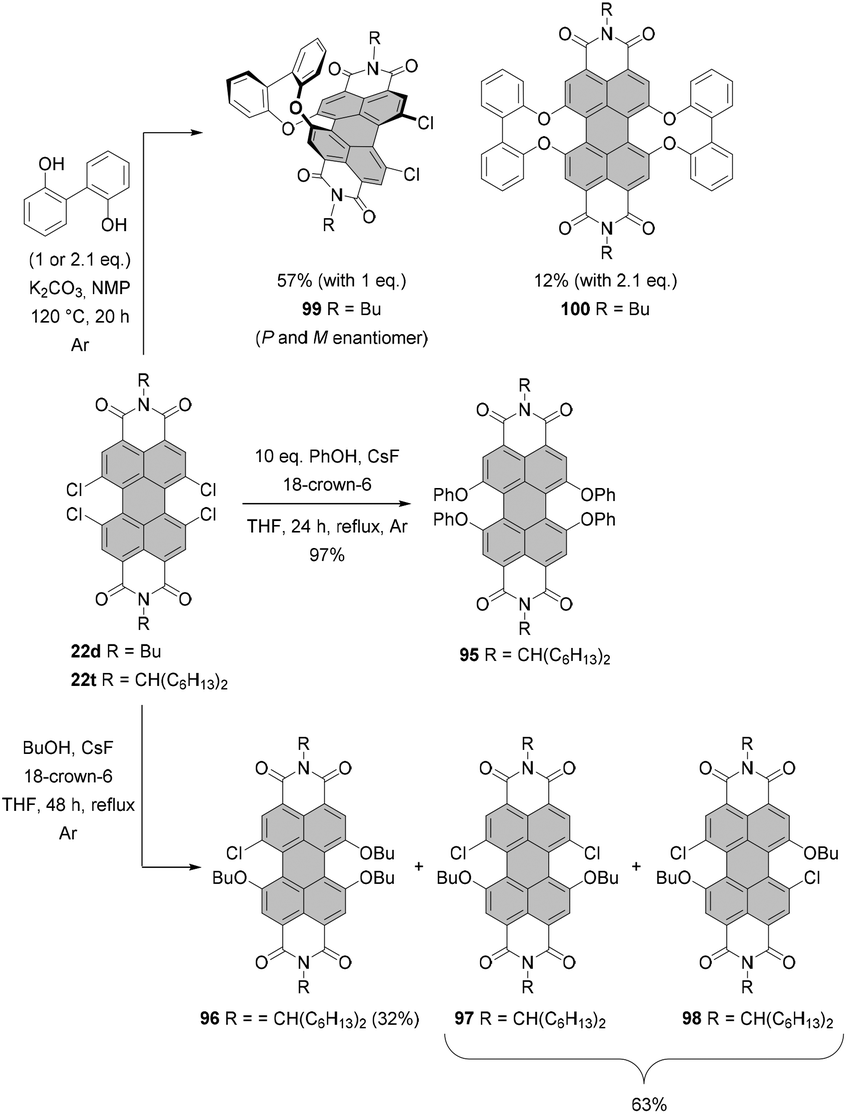 | ||
| Scheme 26 Synthesis of fourfold alkoxy- and aryloxy-substituted PBIs. | ||
Tetraalkoxy-bay-substituted PBIs were reported for the first time by Würthner and co-workers.123 The introduction of four methoxy groups was executed via CuBr-mediated cross-coupling of the tetrabromo-substituted perylene precursor with sodium methoxide. In general, these compounds can be accessed via two routes. That is, methoxy groups can be attached either directly to perylene bisimide, e.g.20g (route A) or to perylene tetracarboxylic ester 101 (route B) whose common precursor is bisanhydride 18. Both routes afforded the target tetramethoxy-perylene derivatives 102a and 103 in moderate yields, 20% and 33%, respectively (Scheme 27). In route B, tetramethoxy-PTE 103 was further transformed into perylene bisanhydride 104 in a quantitative yield, which was reacted with amines in propionic acid to give target PBIs 102a–d in yields ranging from 50% for the labile phenylethyl substituent to ∼70–90% for aromatic and aliphatic groups. The method of choice depends largely on the imide substituents. Firstly, particularly labile imide substituents should be introduced at a possibly late step of the synthesis. Thus, it is recommended to perform the Ullmann-type reaction for the PTE starting material rather than tetrabromo-PBI. Secondly, if the imide substituent does not provide sufficient solubility to the PBI, separation of the target tetramethoxy-PBI from the trimethoxy side-product, which is always formed in the course of the reaction, may impose significant difficulties. On the other hand, isolation of the desired tetramethoxy-PTE benefits from the superior solubility of tetraesters. In addition, separation of PTE 103 from the corresponding trimethoxy derivative is well-established and can be routinely carried out, whereas the purification conditions of tetramethoxy-PBIs have to be optimized for each imide substituent independently. Interestingly, whereas the copper-mediated reaction of tetrabromo perylene derivatives with sodium methoxide provided the corresponding bay-substituted methoxylated products, treatment of tetrachloro-PBI 22l with NaOMe in DMF induced rearrangement to give oxazine imide 105 (Scheme 28).124 Neither did fluoride-mediated reaction of tetrachloro precursor 22t with butanol give rise to tetraalkoxy-PBI (see Scheme 26).120
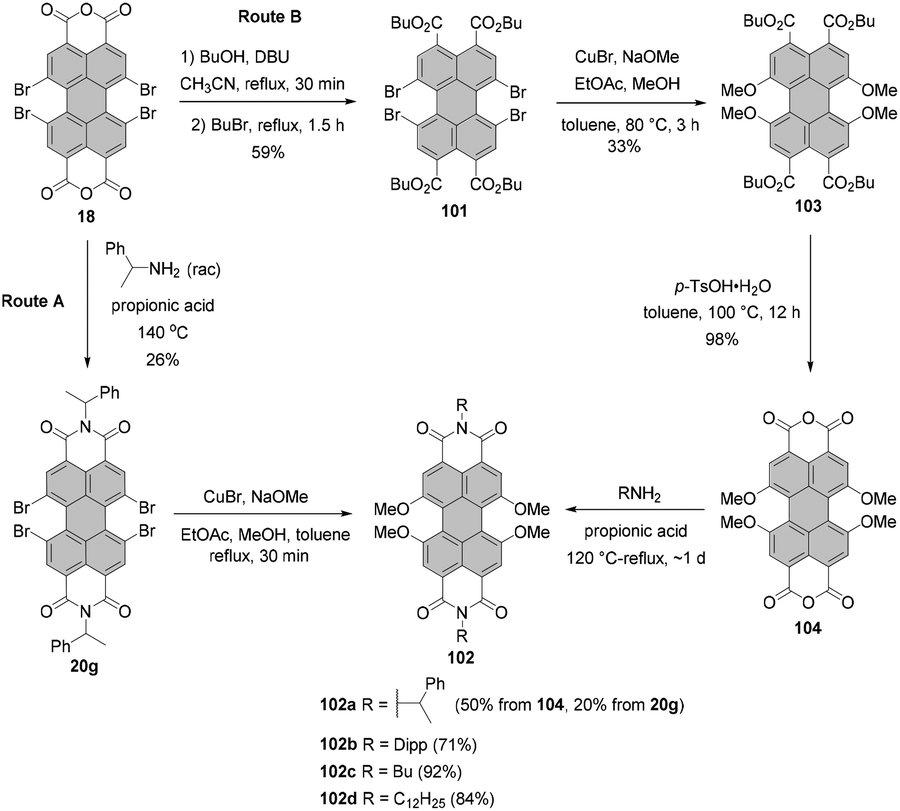 | ||
| Scheme 27 Synthesis of tetramethoxy-PBIs via Ullmann-type coupling of PBI or PTE with NaOMe. | ||
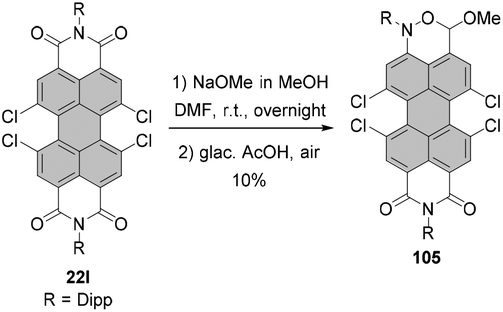 | ||
| Scheme 28 Base-induced rearrangement of tetrachloro-PBI to oxazine imide. | ||
Tetramethoxy PBI 102d bearing dodecyl imide substituents showed an unprecedented crystal packing arrangement (Fig. 4). The uniqueness of this crystal lies in the presence of three crystallographically independent molecules: saddle-shaped achiral and two pairs of propeller-like twisted (P)- and (M)-enantiomeric PBI frameworks. These five distinguishable PBI scaffolds are organized within a single regular π-stack showing an inversion of chirality from P to M via achiral conformation in the middle of the stack.125
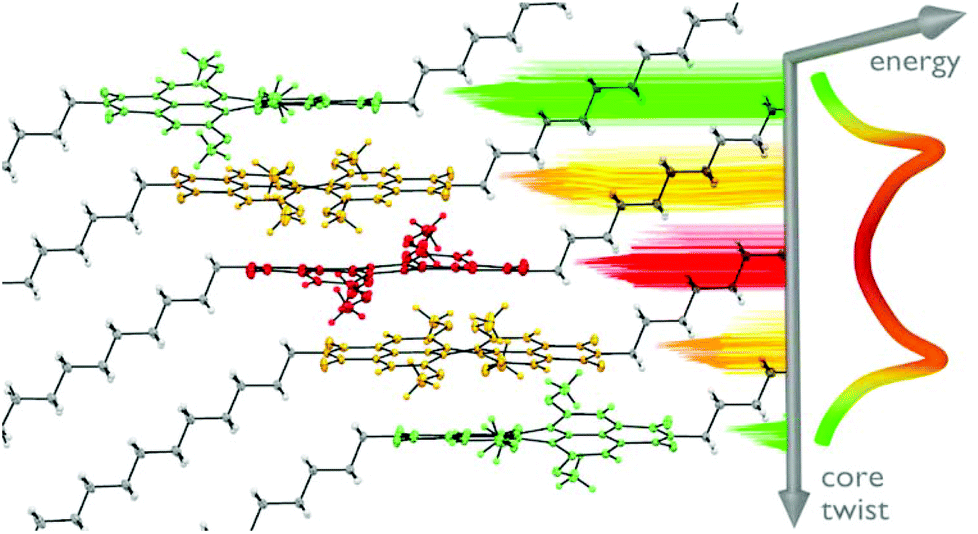 | ||
| Fig. 4 Columnar stacking of tetramethoxy-PBI 102d in a single crystal, revealing a unique transfer of chirality from (P)- to (M)-atropisomers (yellow, green) via an achiral saddle-shaped molecule (red). Reprinted with permission from ref. 125. Copyright 2017, John Wiley and Sons, Inc. | ||
As opposed to 1,7-dipyrrolidino-PBIs, e.g. 1,7-67, compounds 102a–d feature high (photo-)stability. This can be attributed to the fact that lifting the HOMO level by only two pyrrolidino substituents5 is more pronounced than that by four methoxy groups. The absorption and emission maxima of tetramethoxy-PBIs are shifted beyond 600 nm, which reflects substantial interactions between the electron-poor PBI core and electron-donating methoxy groups. Nevertheless, high fluorescence quantum yields (of around 70% in DCM) could be retained for these dyes. For comparison, the Φfl of 1,7-dipyrrolidino-PBI bearing 2,6-diisopropylphenyl imide substituents was decreased to only 19% in DCM as a consequence of the deactivation pathway via charge transfer between amino groups and the PBI scaffold.126 The increased electron-density of methoxy-substituted PBIs versus parent molecules is also reflected by cathodically shifted redox potentials. The first oxidation processes were observed at potentials of around +0.6 V. In contrast to the challenging introduction of four methoxy group in bay areas, the synthesis of 1,7-dimethoxy-PBIs is straightforward and can be carried out under milder reaction conditions (methanol in the presence of K2CO3 in DMF) to give dimethoxy-PBIs in good yields.127
Replacement of halogen atoms by thiols
In comparison with alkoxylation, substitution of dibromo-PBI 1,7-4a with alkylthiols proceeds more smoothly due to the higher nucleophilicity of alkylthiolates than that of alkoxides. Under phase transfer catalysis (PTC) conditions with cetyltrimethylammonium bromide (CTAB) as a PTC catalyst, K2CO3 as a base in xylene compound 106 was obtained in 61% yield. An even better yield, as high as 94%, was achieved when dihalogenated PBI was reacted with thiophenol to afford 107 as a consequence of the highly nucleophilic character of aromatic thiols (Scheme 29).110 Analogous conditions applied to tetrachlorinated PBIs proved ineffective.128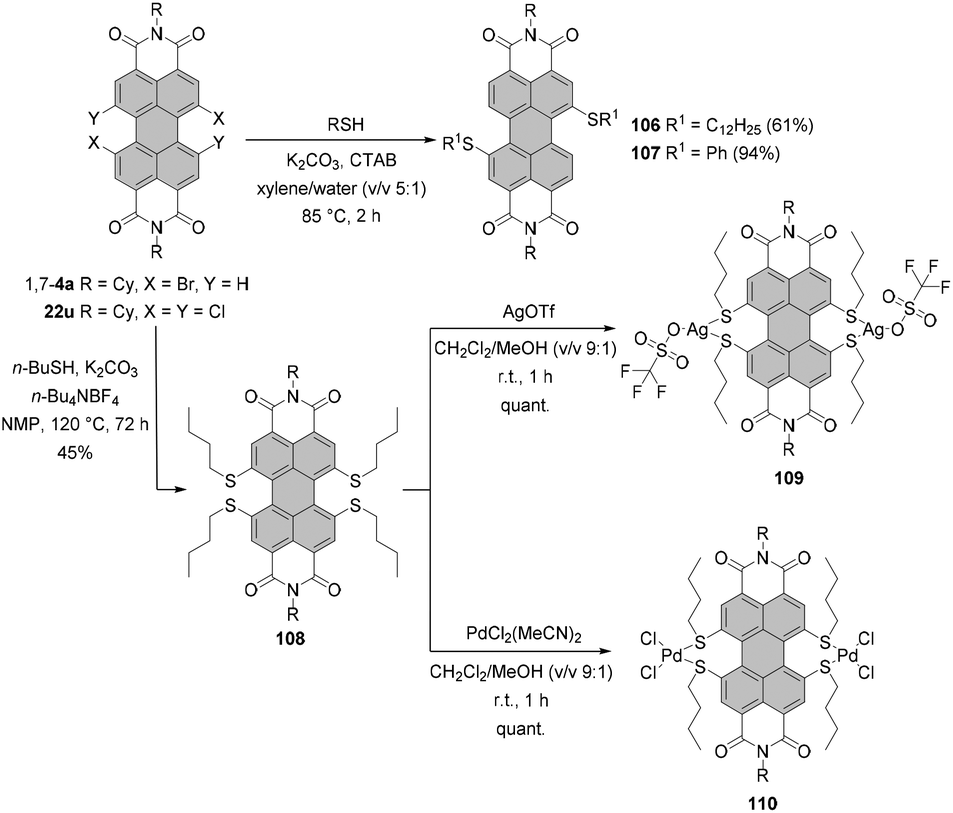 | ||
| Scheme 29 Thiolation of halogenated PBIs. | ||
In order to accomplish the fourfold replacement of chlorine atoms, the reaction of n-butanethiolate with 22u was carried out at a much higher concentration in NMP in the presence of n-Bu4NBF4 as a PTC catalyst. These endeavors resulted in the successful preparation of bay-tetrathiolated PBI 108 in good yield as for the attachment of four sterically demanding groups at bay positions (Scheme 29) entailing a substantial twist of the perylene core. The compound was obtained as a racemic mixture of (P)- and (M)-enantiomers, which were successfully resolved by HPLC on a chiral stationary phase.128 Due to the high van-der-Waals radius of sulfur (1.80 Å), these entities featured a high activation barrier for racemization (ΔG‡ = 119 kJ mol−1), comparable to the bay-substituted tetrabromo-PBI (ΔG‡ = 118 kJ mol−1).36 Compound 108 was utilized to prepare unique silver and palladium complexes 109 and 110, in which transition metal ions were coordinated to sulfur atoms in both bay areas (Scheme 29). As revealed by single-crystal X-ray analysis, 110 formed a 1-D supramolecular polymer owing to weak Pd–Pd interactions.128
Like for alkoxylation, Fernández-Lázaro's method was also effective for thiolation. Under similar conditions, except that CsF was replaced with KF, 22t was reacted with hexanethiol.120 The reaction outcome could be controlled by varying the concentration and the excess of alkanethiol to produce monothio- 111, trithio- 112a or tetrathio-PBIs 113a in yields of 99%, 98% and 96%, respectively (Scheme 30). Such a high thiolation efficiency was not observed for classical bay-substitution reactions. Remarkably, the conditions were suitable for the synthesis of tetra-tert-butylthio derivative 113b. Even though tert-butylthio groups have significant steric requirements, compound 113b could be isolated in yields as high as 54% in addition to trisubstituted compound 112b. Excellent results were also obtained for substitution of 22t with ethane-1,2-dithiol. The reaction proceeded smoothly towards diannulated product 114 (Scheme 30).
 | ||
| Scheme 30 Fluoride-mediated thiolation of chlorinated PBIs. | ||
Despite the broad scope of the fluoride-assisted functionalization of PBIs, an analogous reaction of p-tolylthiophenol with tetrachloro-PBI produced only low yields of dichlorodithio- and chlorotrithio-PBIs together with dehalogenated twofold thiolated derivatives. Thus, contrary to the classical nucleophilic aromatic substitution by sulfur reagents, the fluoride-assisted reaction led to superior results for alkylmercaptan reagents. Both approaches may therefore be considered to be complementary.
The mechanism of these fluoride-assisted transformations remains controversial. Some researchers considered indeed fluoride oxidation, which was, however, recently objected for the related naphthalene bisimide by Gabbaï.129 Oxidation of fluoride is a highly endergonic process and it is therefore rather unlikely that fluoride acts as a single electron donor transferring an electron to the electron-poor perylene bisimide. Nevertheless, the exact role of fluoride in this reaction is not clear and the reaction itself may proceed via the radical pathway.
Replacement of the nitro group
Contrary to the substitution of halogens, the replacement of nitro groups has not received much attention in PBI chemistry. As a consequence of their strong electron-withdrawing character, nitro derivatives are more reactive than their halogenated congeners in nucleophilic aromatic substitution. Replacement reactions with ethanol and n-propyl mercaptan in the presence of K2CO3 of mononitro-PBI 45a were carried out in boiling chloroform to give the corresponding alkoxy and thio compounds 54c and 115 in 45% and 35% yields, respectively. Yet, displacement of a nitro group with more reactive phenols, such as 4-hydroxybenzaldehyde and 4-tert-butylphenol, could be carried out at room temperature in NMP to provide products 54d and 54e in excellent yields (Scheme 31).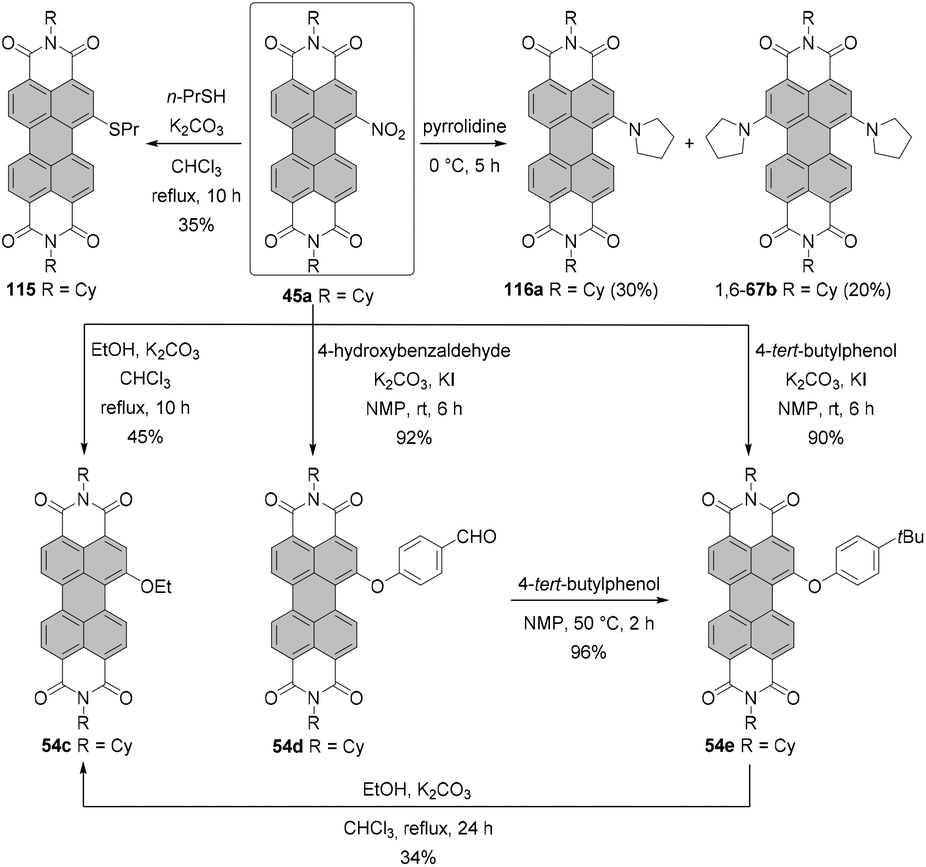 | ||
| Scheme 31 Substitution of nitro group and ether exchange in PBIs. | ||
Thus, substitution of nitro-compounds with phenols has an advantage over analogous reactions of bromine-functionalized starting materials, as the latter require elevated temperatures. It is noteworthy that, due to the high reactivity of mononitro-PBI 45a, the reaction with highly nucleophilic pyrrolidine afforded mono-substituted product 116a in 30% yield along with a substantial amount of 1,6-diamino derivative 1,6-67b (20% yield), even though the temperature was lowered to 0 °C (Scheme 31). At higher temperatures the ratio of mono- and di-substituted products changed in favor of 1,6-67b, and above 25 °C diamino-PBI was formed as the major product.130
Nitro-PBIs are also relevant starting materials or intermediates in the synthesis of bay-annulated PBIs. For instance, incorporation of the 2H-pyran into bay areas of a PBI core involves probably displacement of the nitro group,80 whereas the formation of a 1,2-dihydro-1,2-azaborine ring requires the conversion of nitro-PBI 45a into amino-PBI 48a (Scheme 32).87 Compound 48a was used as-prepared for the next step due to air instability. The azaborine ring was constructed by intramolecular electrophilic borylation of amino-PBI 48a with dichlorophenylborane in 81% yield. The reaction was carried out in refluxing toluene for 24 h in the presence of triethylamine as a base. Azaborine PBI 117 featured remarkable stability. As revealed by the thermogravimetric analysis, the decomposition temperature was as high as 402 °C.
 | ||
| Scheme 32 Synthesis of a 1,2-dihydro-1,2-azaborine PBI derivative. | ||
Ether exchange
Interestingly, some ether derivatives may also serve as starting materials for nucleophilic aromatic substitution. In these reactions, a weaker nucleophile is replaced by the stronger nucleophile to form a new C–O bond. Accordingly, replacement of 4-formylphenoxy precursor 54d with 4-tert-butanol gave rise to the corresponding derivative 54e in 96% yield. As expected, a much lower yield was obtained for a more demanding substitution of this compound with ethanol (see Scheme 31). Since the reactivity of phenoxy-PBI towards SNAr is lower than that of a nitro derivative, both reactions were carried out at elevated temperature. Attempts to replace a stronger nucleophile, e.g. a reaction of 54e with 4-hydroxybenzaldehyde, were unsuccessful.130The synthesis of unsymmetrical PBIs is more challenging. High selectivity was achieved for PBI 118 bearing three 4-tert-butylphenoxy groups in bay areas. The utilization of this compound as a starting material constitutes another alternative to halogen replacement. Ether-exchange reactions were carried out with alcohols and alkylthiol in the presence of NaH in boiling THF, affording products 119–121 in 71–78% yields (Scheme 33). These conditions proved inert to the terminal ethylene moiety in 119. In contrast to triaryloxy PBI, analogous reactions were unsuccessful for tetraaryloxy PBI, which could be attributed to the higher steric hindrance in the bay area and the higher electron density of the latter compound.131
 | ||
| Scheme 33 Synthesis of bay-substituted O-alkyl and S-alkyl PBI derivatives via replacement of ether functionality. | ||
C–C coupling reactions at bay positions
As in any other branch of organic chemistry, palladium-catalyzed reactions have come to the fore in the field of perylene dyes as simple synthetic tools providing access to a wellspring of molecular scaffolds, including those highly elaborated for applications, e.g. in organic electronics and photovoltaics. The latter compounds are briefly discussed in the section “Core-extended and annulated PBIs”, in some cases together with the synthesis of their precursors. Pd-Catalyzed reactions which are commonly used for the preparation of substituted PBIs include the Suzuki coupling, Sonogashira reaction and Stille coupling. In the following, we discuss the implementation of these reactions for the synthesis of bay-substituted PBIs. We also show utilization of other metals for the construction of carbon–carbon single bonds.Replacement of halogen atoms by cyano groups
Introduction of two electron-withdrawing cyano groups into the PBI bay area proved to be highly beneficial to stabilize the PBI radical anions for applications in n-channel organic transistors. A suitable protocol for these materials was developed by Wasielewski and co-workers132via treatment of bromo-PBIs 4a and 4g (1,7 and 1,6 regioisomeric mixtures) with zinc cyanide in the presence of tris(dibenzylideneacetone)dipalladium(0) (Pd2dba3) and 1,1′-bis(diphenylphosphino)ferrocene (dppf) to give compounds 122a and 122b (Scheme 34). While these conditions were high-yielding, a simple Rosenmund–von Braun reaction failed to produce any bay-substituted cyano-PBIs.133 As revealed by the crystal structure of 122b, the core twist is only ∼5°. Thus, introduction of the nitrile group did not induce a significant distortion of the PBI scaffold from planarity, which is beneficial for co-facial packing of the dyes. In contrast, cyanation considerably affects redox potentials. The first reduction potential of 122b was recorded at a high value of −0.48 V vs. Fc+/Fc redox couple, which is about 0.5 V higher than that for the parent PBI. Both compounds were successfully applied as n-type semiconductors in organic field-effect transistor (OFET) devices with mobilities of 0.10 and 0.64 cm2 V−1 s−1, respectively.132 | ||
| Scheme 34 Synthesis of dicyano-PBIs from dibromo-PBI precursors. | ||
Replacement of halogen atoms by fluoroalkyl groups
It was recognized already in 2000 that CH2(CF2)nF substituents at imide positions endow naphthalene bisimides with ambient stability for n-channel transistor operation.134 In the following years it became clear that, indeed, many naphthalene as well as perylene bisimides can take advantage of particular packing arrangements where the electron-transporting rylene cores are densely packed and protected against humidity by the protruding perfluoroalkyl chains as illustrated for the crystal structure of CH2(CF2)3F-substituted PBI (Fig. 2).25 Inspired by this work, also perfluoroalkylation of PBI bay positions was considered as an interesting option to both lower the PBI LUMO to add further stability to the PBI radical anion and to repel water.Fluoroalkyl-substituted compounds 123a–d were synthesized by Wang and co-workers135 in excellent yields (80–90%) by copper-mediated coupling of 1,7-dibromo-PBIs 4d,f,g with perfluoroalkyl iodides (Scheme 35). The temperature was kept below 135 °C during the whole process. The reaction tolerated various imide substituents, e.g. aryl, fluoroalkyl and simple n-alkyl, and proceeded smoothly for perfluoroalkyl iodides of different length. As expected, attachment of electron-withdrawing substituents entailed anodic shifts of reduction potentials in comparison with core-unsubstituted PBIs and the target compounds could be applied as n-type semiconducting materials to fabricate air-stable OFETs. A transistor device employing 123c as an active layer reached an electron mobility of 0.052 cm2 V−1 s−1. Installation of trifluoromethyl groups onto PBI 123e and several other rylene bisimides could be carried out by Wasielewski and co-workers136 under mild conditions by the method of Hartwig for the trifluoromethylation of aryl iodides and bromides (Scheme 35).137 These mild conditions can be contrasted with protocols for trifluoromethylation of core-unsubstituted PTEs138 or PBAs.139 Perfluoroalkyl PBIs 123a–d exhibit high fluorescence quantum yields above 90% in chloroform. In contrast, emission of bis(trifluoromethyl)-PBI 123e (Φfl = 64% in DCM) was not stronger than that of tetramethoxy derivatives 102a–d (see Scheme 27).
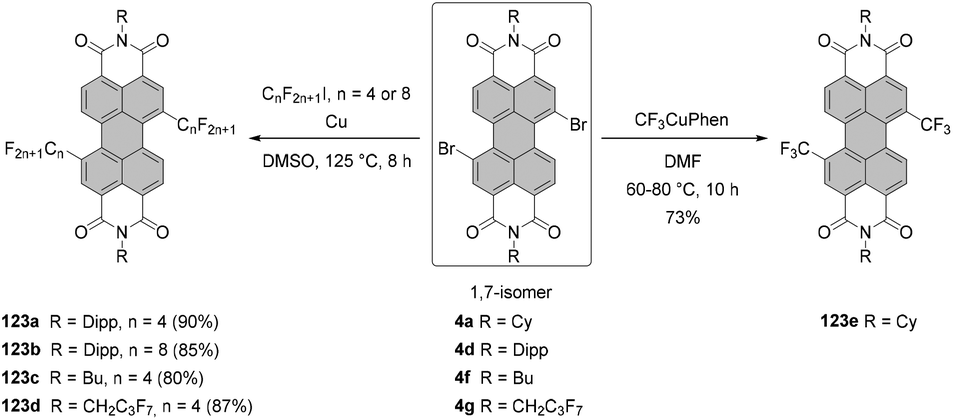 | ||
| Scheme 35 Copper-mediated perfluoroalkylation of dibromo-PBIs. Phen = phenanthroline. | ||
Replacement of halogen atoms by Suzuki coupling
Fourfold arylation of bay positions was demonstrated for the first time by Liu and Zhu in 2006. The Suzuki coupling of tetrabromo-PBI 20b with phenylboronic acid catalyzed by Pd(PPh3)4 yielded tetraphenyl-PBI 124a in 47% yield (Scheme 36). The success of this reaction is strongly dependent on the applied base. While the utilization of the commonly used K2CO3 ended in failure, the application of weaker bases such as KF or even better CsF afforded the desired product in moderate yields.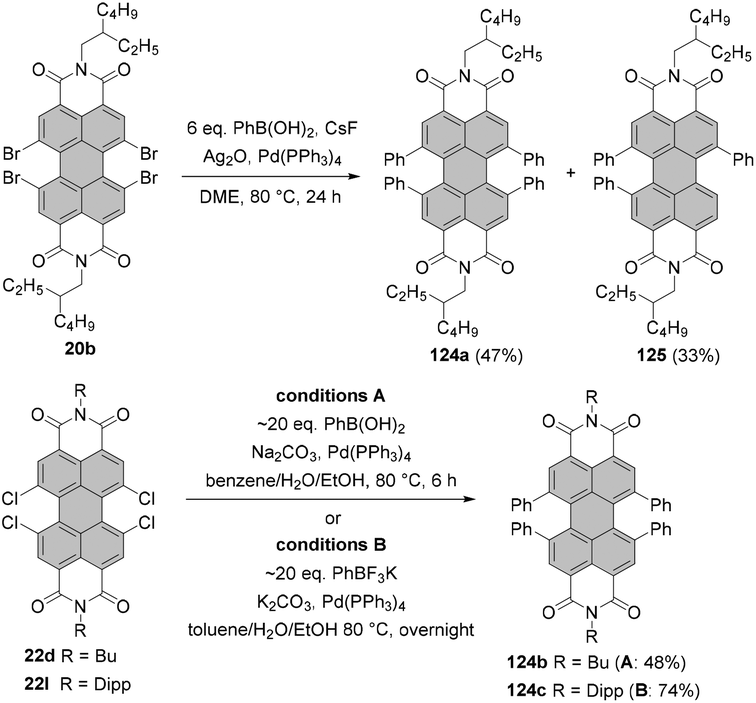 | ||
| Scheme 36 Suzuki coupling of tetrahalogenated PBIs. | ||
Moreover, the presence of Ag2O increased the efficiency of the transformation. Although optimization of the reaction conditions allowed the reduction of side-reactions, the threefold substituted side-product 125 was formed each time in high quantities as a consequence of the great steric encumbrance in the bay areas.42 In 2010, Hoffmann reported a successful transformation of tetrachlorinated precursors 22d and 22l into tetraphenyl PBI 124b and 124c. The palladium-catalyzed reaction of 22d with the high excess of phenylboronic acid in the presence of Na2CO3 produced the desired product 124b in a yield of 48% (Scheme 36).55 A comparable yield was obtained for the Suzuki coupling between 22l and phenylboronic acid under similar conditions employing K2CO3 as a base. A considerable improvement was achieved when phenyl boronic acid was replaced with potassium phenyltrifluoroborate.63 This modification gave rise to PBI 124c in yields as high as 74% (Scheme 36).
The synthesis of the corresponding only diarylated PBIs from 1,12-dichloro derivative 31a was surprisingly not more efficient, although the reaction scope in terms of the electronic nature of substituents was broader. Accordingly, diaryl-PBIs could be formed even for boronic acids bearing electron-withdrawing substituents (see PBIs 1,12-126a–c, Scheme 37), whereas the reactions of electron-poor arylboronic acids with tetrachloro precursors, e.g.22l, were unsuccessful. However, the efficiency of this process was substantially reduced due to hydrodehalogenation, which can be considered as the main competing process for substrates with strong crowding in the bay area. Accordingly, the reaction of 31a with methoxyphenylboronic acid afforded 1,12-126b in 53% yield in addition to a nearly equimolar amount of dehalogenated compound 127a. The latter compound could not be isolated due to spontaneous photocyclization to fused product 128a (Scheme 37). Moreover, as opposed to the Suzuki coupling of tetrachloro-PBI, the utilization of potassium phenyltrifluoroborate or phenylboronic acid pinacol ester led preferably to the product of mono-hydrodehalogenation 127b. Conversely, the reaction of 1,7-dibromo-PBIs 4d and 4e proceeded smoothly to give diaryl products 1,7-126a–d in good to very high yields for boronic acids with both electron-withdrawing and -donating end groups (Scheme 37).63
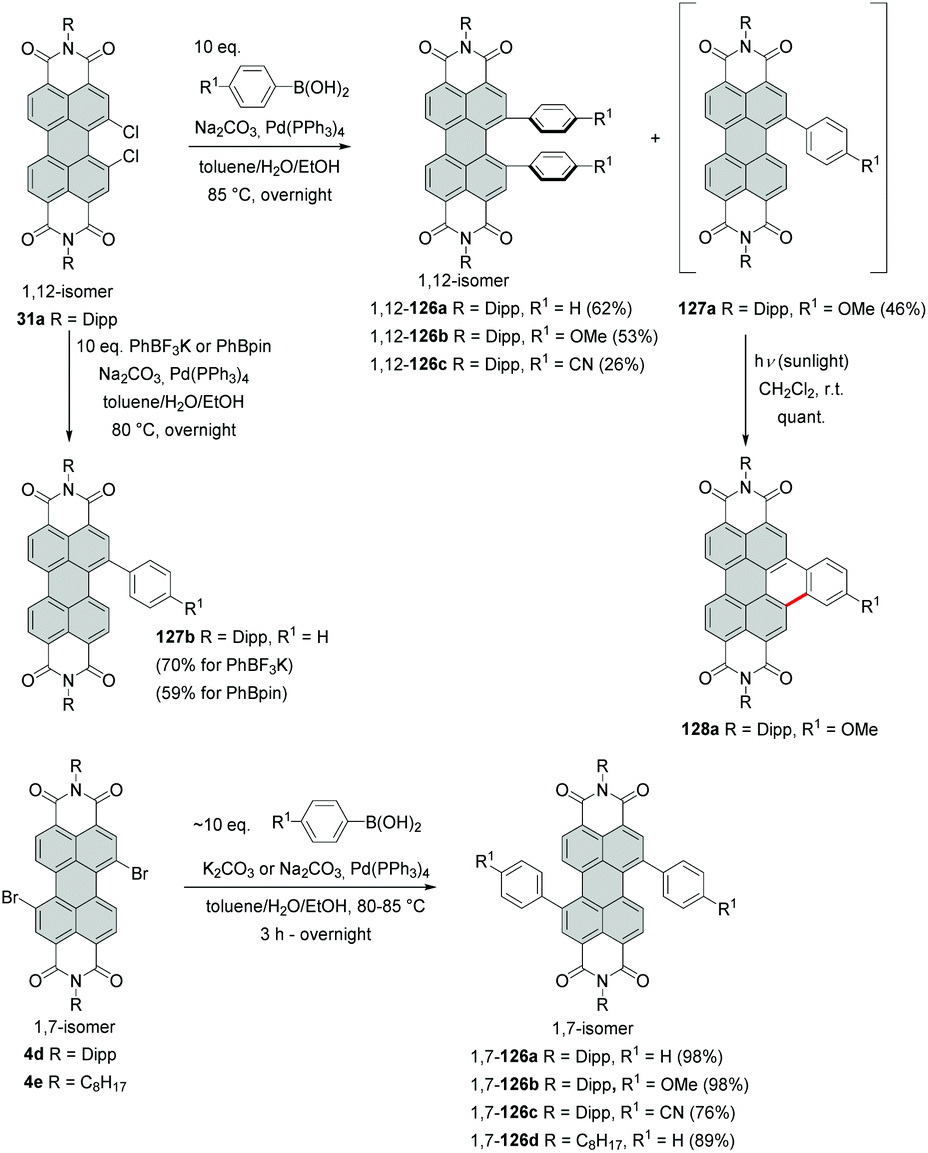 | ||
| Scheme 37 Suzuki coupling of 1,12- and 1,7-dihalogenated PBIs. | ||
The application of the Suzuki coupling is not limited to single-core functionalized PBIs. The reaction became one of the key methods to couple PBIs with aromatic, typically carbocyclic, cores and is routinely used to produce multichromophoric PBI arrays. Some of the examples are discussed in the section “PBI arrays by fusion of carbocyclic and heterocyclic rings”.
Replacement of halogen atoms by Stille, Sonoghasira and Glaser coupling
The Stille coupling was utilized to form C–C bonds between a bay-carbon atom of PBI and a squaric acid derivative. The reaction of 1,7-dibromo-PBI 4a with the stannyl cyclobutenedione reagent in the presence of Pd(PPh3)4 and CuI afforded desired PBI 129 in 70%. Subsequent cleavage of the isopropyl group produced PBI 130 with the free hydroxyl group (Scheme 38). The acidity of this compound is significantly higher than that of 80 (see Scheme 22), leading to the interesting properties of 130. Due to high acidity of the squaric acid moiety, not only is the compound halochromic, but also it exhibits hydrochromism, which can be used for colorimetric sensing of humidity.140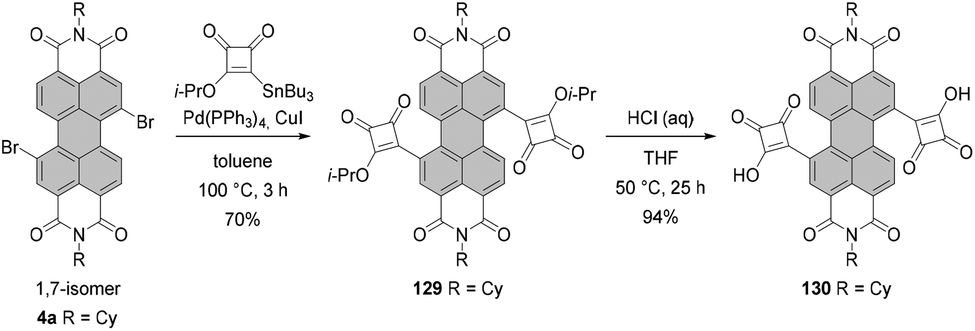 | ||
| Scheme 38 Synthesis of a squaric acid PBI derivative by Stille coupling. | ||
The importance of the Stille coupling is evident in the field of organic semiconductors. Next to the Suzuki coupling, it is the most commonly utilized reaction to couple aromatic, in particular, thiophene-based cores with PBI units. It was used to prepare a plethora of aryl-bridged systems consisting of two or more PBI chromophores with linear,141,142 spiro143 or other twisted linkers.
The Sonogashira and Glaser couplings are commonly used to prepare oligomeric PBIs linked by either ethynyl or butadiynyl bridges. The selected examples are depicted in Scheme 39. Accordingly, monobromo-PBI 7e was reacted with the ethynyl coupling partner 131a in the presence of PdCl2(PPh3)2, CuI and triethylamine to afford 132a in 76% yield. The Glaser coupling was carried out under similar conditions with the addition of p-benzoquinone as an oxidant. The corresponding butadyinyl-linked bis-PBI 133 was isolated in 83% yield. Analogous reactions carried out with N,N-diisopropylamine (DIPA) as a secondary amine base gave inferior results. The authors suggested nucleophilic displacement of the bromine atom as a plausible reason for decreasing the reaction yields.144 As pointed out recently by Takai and Takeuchi it was rather due to the addition of amine to the terminal alkyne (vide infra).145 Studies of the optical and electrochemical properties of these compounds revealed that the molecules are effectively conjugated, leading to bathochromic shifts of absorption maxima and decreases of the LUMO levels versus the parent PBI. The shift for the ethynyl-PBI was more pronounced than that for the butadyinyl derivative.144 The latter molecule showed conformational heterogeneity due to the longer linker.
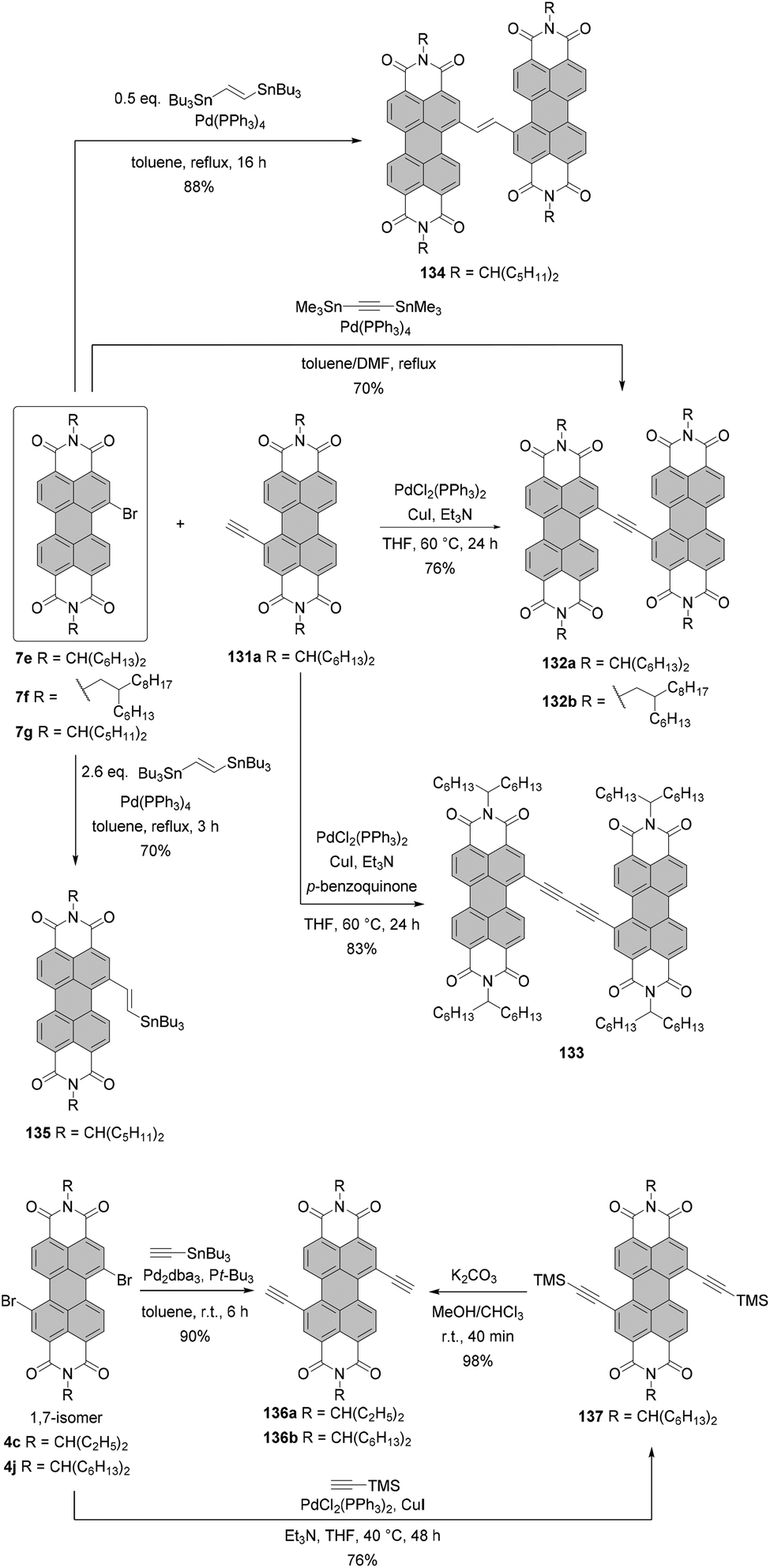 | ||
| Scheme 39 Synthesis of vinyl-, ethynyl- and butadiynyl-bridged bis-PBIs and building blocks for the synthesis of oligomeric PBIs with these linkers. | ||
The number of building blocks required for the synthesis of alkynyl-linked PBI 132b and consequently the number of synthetic steps were reduced by applying the Stille coupling between bis(trimethylstannyl)acetylene and bromo-PBI 7f (Scheme 39).146 The yield of 132b was comparable to the yield of 132a by the Sonogashira coupling. The reaction was carried out in the toluene/DMF solvent mixture under reflux, but analogous transformations with bis(tributylstannyl)acetylene can also be carried out at room temperature with the catalytic system Pd2dba3/tri-tert-butylphosphine (Pt-Bu3) to produce bis-PBIs in excellent yields.147,148 The Stille coupling between trans-1,2-bis(tributylstannyl)ethylene and bromo-substituted PBIs is of even greater synthetic importance as it gives access to vinylene-bridged oligomeric PBIs. The latter compounds represent the key building blocks in the synthesis of fused dimeric PBIs and PBI nanoribbons,149 in addition to ethylene-fused polymers,150,151 although it should be pointed that the synthetic route for the preparation of the first ethynylene-fused dimeric PBI involved cyclization of alkyne-bridged PTE.152 Ethenyl-bridged dimeric PBI 134 was synthesized in 88% yield by coupling of bromo-PBI 7g with 0.5 equivalent of 1,2-bis(tributylstannyl)ethylene, using Pd(PPh3)4 as a catalyst (Scheme 39).149
These methods can be extended to the synthesis of higher oligomers. The essential building blocks are 2-(tributylstannyl)vinyl-substituted PBIs, e.g.135,149 and PBIs bearing two terminal alkyne substituents, such as 136a and 136b. The latter compounds were prepared either by a two-step sequence including the Sonogashira coupling of a bromo-precursor with a silylated acetylene, followed by deprotection of the alkyne,153 or by the Stille reaction between bromide and tributylstannylacetylene (Scheme 39).148 Specifically, the Sonogashira coupling of PBI 1,7-4j with trimethylsilylacetylene under standard conditions produced alkyne-functionalized PBI 137 in 76% yield. Quantitative cleavage of the TMS groups with K2CO3 afforded the building block 136b.153 PBI 136a was synthesized directly from 1,7-4c in 90% yield by the Stille coupling with tributylstannylacetylene under mild conditions.148 An analogous approach was applied for the preparation of PBI bearing the 2-(tributylstannyl)vinyl group. To avoid the formation of a dimeric PBI, an excess of stannane reagent was applied. The reaction was carried out in the presence of Pd(PPh3)4 in toluene under reflux to give 135 from 7g in 70% yield.149
PBIs substituted with only one arylethynyl group, such as 138a–e, were synthesized via the Sonogashira coupling from monobromo-precursor 7a and the corresponding aryl alkyne.154,155 The reactions were carried out in the presence of PdCl2(PPh3)2, copper(I) iodide and DIPA in THF under reflux. The yields were excellent, typically above 90%, for a broad scope of phenylethynyls bearing both electron-donating and electron-withdrawing terminal groups, in addition to heteroaryl alkynes, such as 5-ethynyl-1-methyl-1H-imidazole (Scheme 40). The only exception was phenylethynyl PBI 138c with the N,N-dimethylamino group. In the latter case the yield was reduced to 64% due to the tedious purification by column chromatography. The distortion of the phenylethynyl-PBIs in the bay area is negligible. Accordingly, for the PBI 138a C1–C12b–C12a–C12 bay area a dihedral angle of only 1.8° was measured, which means that introduction of the alkynyl group exerts minimal steric strain.154 Ethynyl-substituted single-core PBIs also exhibit interesting properties and functions. For instance, imidazolylethynyl-PBI 138e was used as a specific fluorochrome for the labelling of fat bodies in Drosophila and adipocytes in mammals. Emission of this compound was quenched in polar solvents (reducing the background emission noise) and was restored upon binding to these cells or fat bodies.155
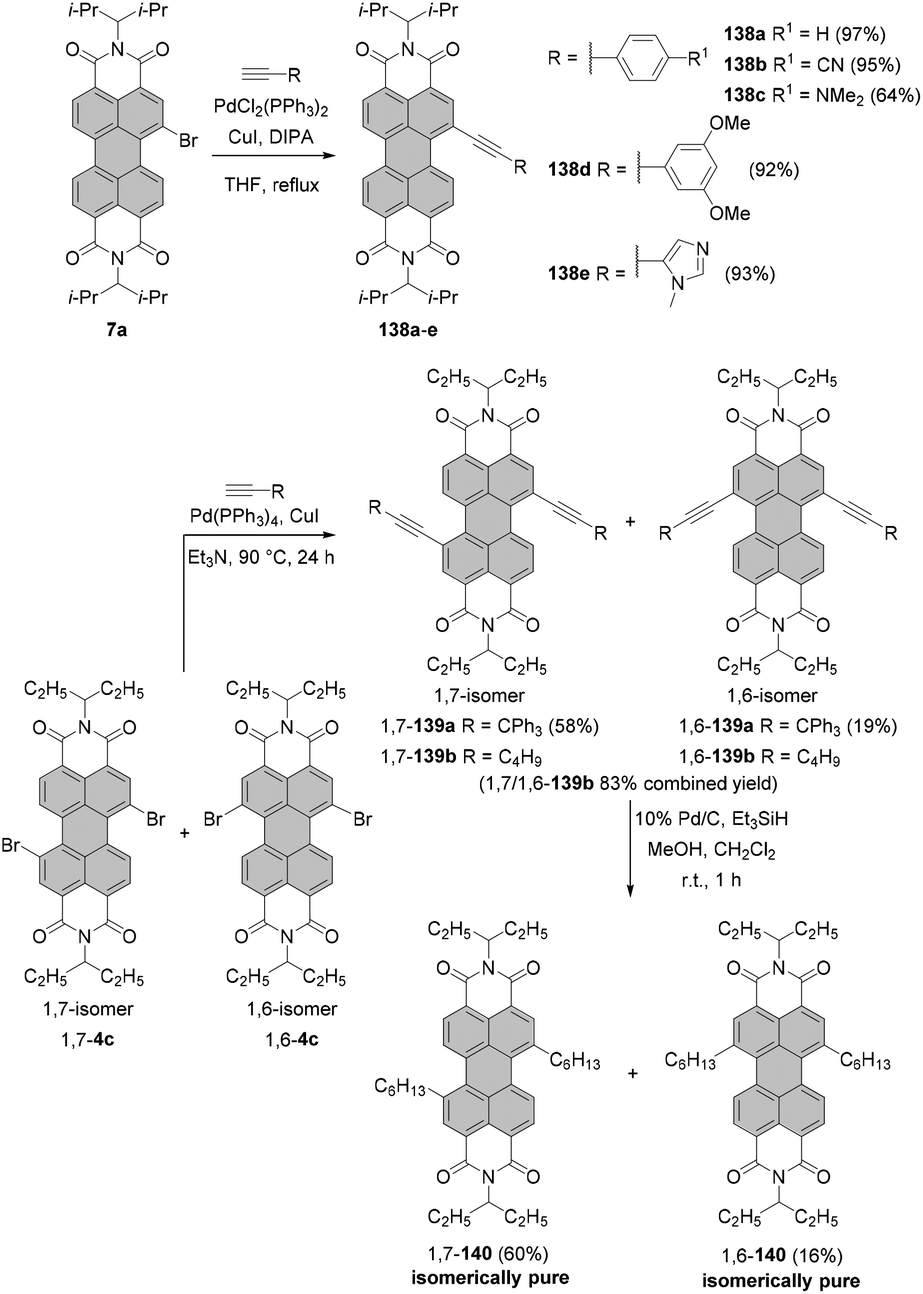 | ||
| Scheme 40 Synthesis of dialkynyl-PBIs and their hydrogenation. | ||
Shirtcliff disclosed the preparation of dialkynyl PBIs 1,7-139a and 1,6-139a bearing bulky triphenylmethyl terminal groups from a 1,7 and 1,6 regioisomeric mixture of PBI 4c (Scheme 40).156 The mixture was enriched with the latter compound after a fraction of a pure 1,7-isomer was isolated by crystallization. Introduction of these substituents allowed the differentiation of both isomers on silica and accordingly 1,6- and 1,7-isomers could be isolated by column chromatography. In contrast, a reaction of 1,7-4c/1,6-4c with 1-hexyne produced a 1,7/1,6 mixture that was indistinguishable by TLC and thus inseparable by column chromatography. Interestingly, though, when hexynyl PBIs 1,7-139b and 1,6-139b were converted to 1,7-140 and 1,6-140 by Pd-catalyzed hydrogenation, both regioisomers were readily separated by column chromatography. In view of the eventual derivatization by exchange for imide substituents, PBIs 139a,b and 140 were subjected to saponification with KOH in boiling tert-butanol. While triphenylmethylethynyl-PBIs 1,7-139a and 1,6-139a, and hexyl-substituted PBIs 1,7-140 and 1,6-140 provided the respective bisanhydrides in satisfactory yields (50–75%), hexynyl derivatives experienced decomposition under these harsh conditions. Another interesting observation was reported by Takai and Takeuchi.145 Mainly, when a PBI bearing terminal alkyne functionalities at 1,7 positions was treated with dibutylamin, a gradual color change to green via dark yellow was observed. The changes were ascribed to the formation of mono- and bisadducts by the Michael-type addition of amine to the triple bond. Analogous transformations were observed for naphthalene bisimides and diketopyrrolopyrrols.145 It was demonstrated that the reaction may occur not only in solution but also in the solid state and upon exposure of a film to amine vapor.
In addition to the omnipresent palladium-catalyzed reactions allowing the attachment of a large variety of aromatic systems to a PBI scaffold or linking two PBI units via (hetero)aromatic or aliphatic bridges, there is a handful of important reactions giving access to PBI systems directly linked by C–C bonds. These reactions include the Negishi reaction and copper-mediated coupling of halogenated PBIs. Bromo-PBIs 7d,g,h and 141a unsubstituted in the second bay area or bearing two chlorine atoms underwent the Ullmann reaction to afford singly linked products 142a–d in high yields (76–84%) (Scheme 41).64,157,158 Coupling was carried out in the presence of an excess of nanosized copper powder in dry DMSO. When 1,7-dibromo-PBI was used as a starting material, the reaction yielded a mixture of oligomeric PBIs which were composed of PBI units linked by C–C single bonds with each other.64 Se- or N-bay-annulated bis-PBIs 142e and 142f were synthesized via the Negishi coupling of monobrominated precursors 141b and 141c in the presence of zinc and palladium catalysts (Pd2dba3 or bis(dibenzylideneacetone)palladium(0) (Pddba2)).81,159 These homocoupling conditions afforded 142e and 142f in excellent yields (Scheme 41).81 Owing to the strong absorption in the visible region and their ability to act as electron transport materials, singly linked PBIs were tested as acceptor materials for OSCs. Particularly good results were obtained for bay-annulated PBIs, such as 142e and 142f. Photovoltaic devices based on these materials paired with suitable donors, PDBT-T1 and the medium band gap donor polymer P3TEA, respectively, achieved power conversion efficiencies (PCEs) of 8.4% and 7.6%, respectively.81
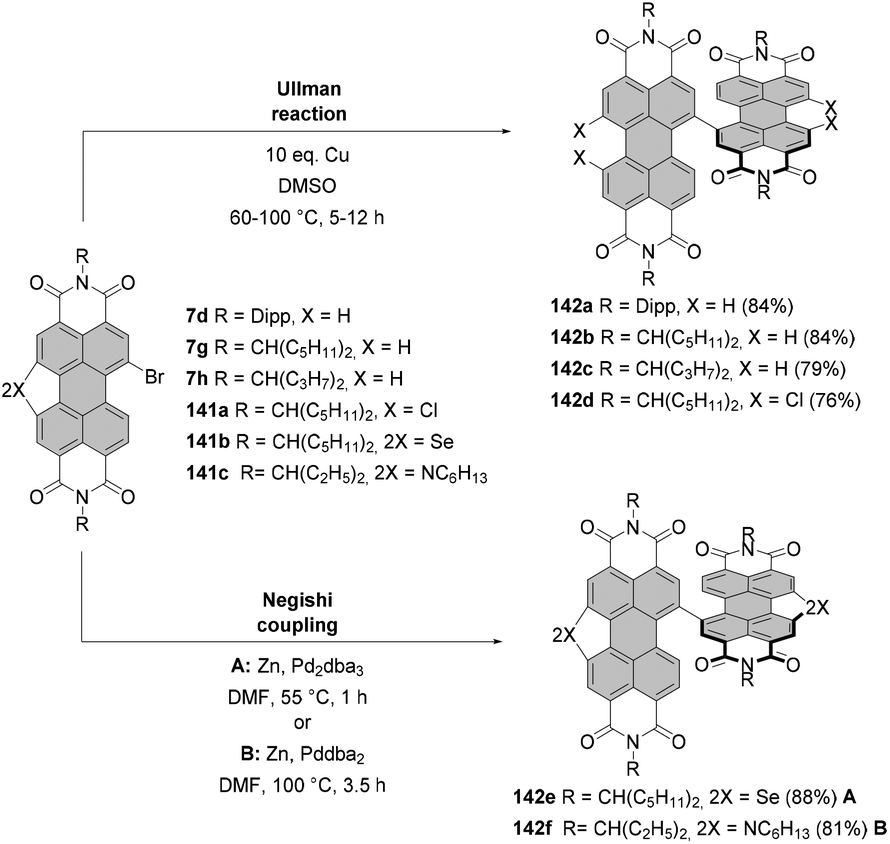 | ||
| Scheme 41 Synthesis of bis-PBIs by Ullmann reaction and Negishi coupling. | ||
Substitution reactions at ortho-positions
2,5,8,11-Functionalized PBIs have been made available only within the last decade and initiated new and intriguing research directions for PBI chemistry.92,93 Thus, the ortho-halogenated and -borylated precursors discussed before serve, like their bay-functionalized counterparts, as suitable starting materials for a number of nucleophilic substitution reactions with anilines, phenols, and thiophenols, as well as cross-coupling reactions with CuCN, terminal alkynes, and arylboronic acids.Introduction of nitrogen, oxygen and sulfur nucleophiles
Following the well-known protocols for the structurally related halogenated naphthalene bisimides,49,50,160 heating of tetraiodo-PBI 63c in neat n-octylamine at 130 °C under an argon atmosphere produced 143 in almost quantitative yield.93 Likewise, introduction of aniline or thiophenol was straightforward, giving products 144 and 145a from 63b in excellent yields, while the reaction with phenol was less effective as the corresponding product 146a was obtained only in 64% yield (Scheme 42).97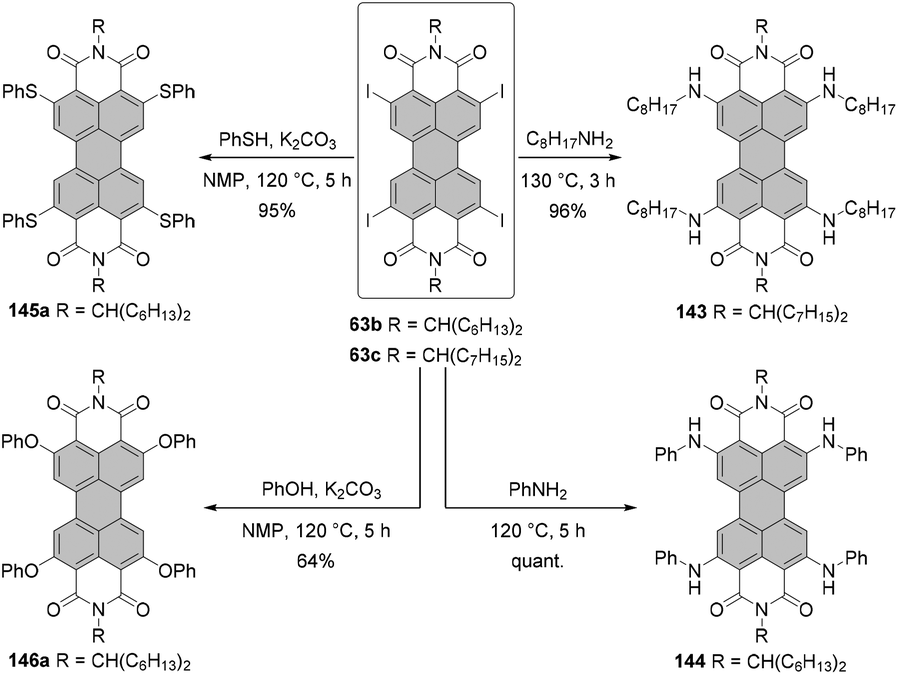 | ||
| Scheme 42 Nucleophilic aromatic substitution of 2,5,8,11-tetraiodo-PBIs. | ||
Analogous transformations were also successfully conducted for mixed perhalogenated PBI 37a (Scheme 43).69 Importantly, the reactions performed in neat aromatic amines, such as aniline and 3,5-dimethylaniline, proceeded selectively at ortho positions despite the high excess of nucleophilic reagents (∼160 eq.) and the temperature of 120 °C.
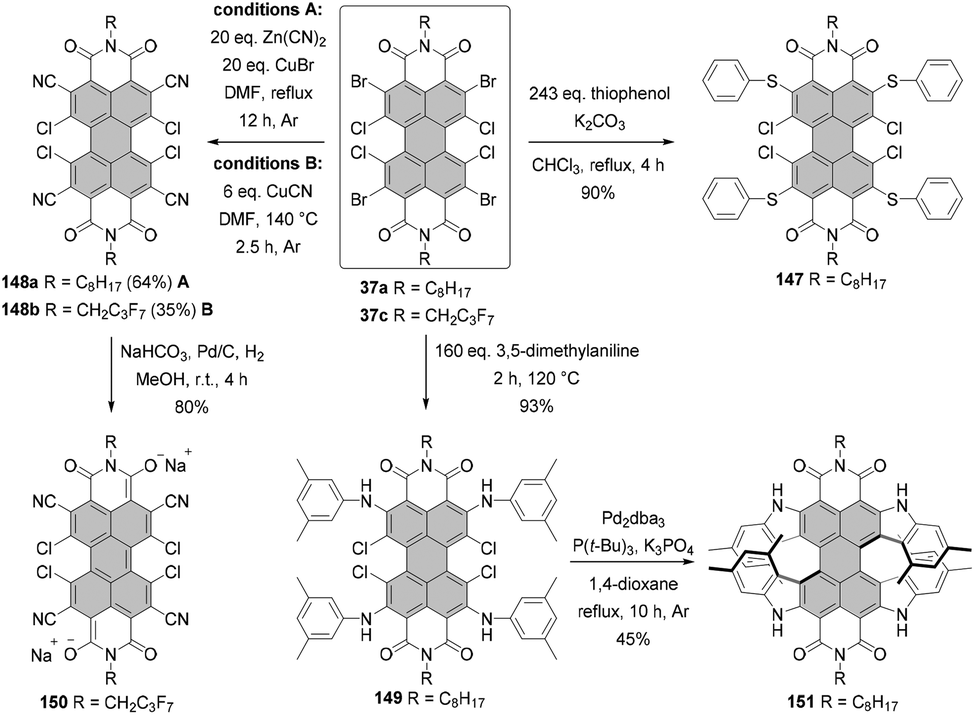 | ||
| Scheme 43 Substitution reactions of mixed perhalogenated PBIs. | ||
Even higher excess of the reagent was used for the substitution of 37a with thiophenol, yielding selectively ortho-substituted product 147, although in this case the reaction mixture was stirred at lower temperature in chloroform.
2,5,8,11-Tetrahydroxy-PBI 152 was obtained by oxidation of borylated PBI 62b with hydroxylamine hydrochloride as an oxidant in the presence of NaOH in a yield of 82%. PBI 152 was further converted into methoxy derivative 102e upon treatment of 152 with methyl iodide in the presence of Ag2CO3 as a base (Scheme 44).92 Interestingly, for ortho-hydroxy and ortho-amino functional groups, the formation of intramolecular hydrogen bonds to the neighboring carbonyl groups was observed. Different from the related ortho-functionalized naphthalene bisimides,161 so far no detailed spectroscopic studies have been conducted to elucidate the impact of these hydrogen bonds on the photophysical properties.
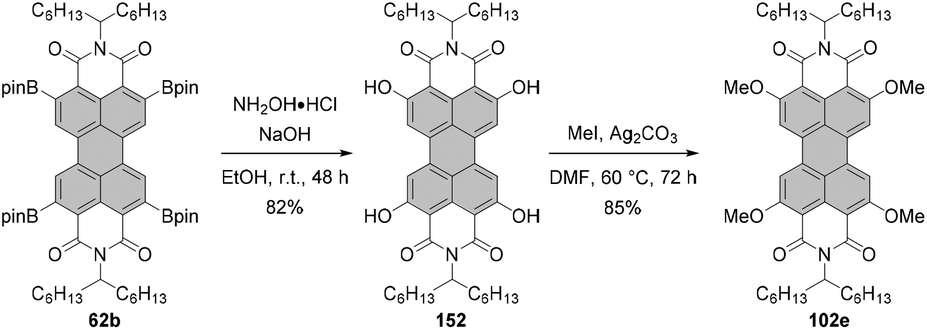 | ||
| Scheme 44 Synthesis of 2,5,8,11-tetrahydroxy- and tetramethoxy-PBIs. | ||
As recently described by Orentas and co-workers, octabromo-PBI 21a can be reacted with various nucleophiles in a regioselective fashion to afford ortho-substituted PBIs due to a substantially higher reactivity of ortho-bromine atoms located in the immediate proximity of imide moieties as compared to the bromine atoms in the bay areas (Scheme 45). Thus, it was possible to achieve a high level of selectivity without the need for mixed perhalogenated PBIs, such as 37a (see Scheme 43). A notable example is the introduction of four methoxy groups at ortho positions which could be executed in one step by the treatment of octabromo-derivative 21a with sodium methoxide in a THF/MeOH mixture (Scheme 45).46 As opposed to the challenging introduction of four methoxy groups in bay areas by copper-mediated exchange of chlorine,123 the reaction was straightforward, such that stirring at room temperature overnight sufficed to produce compound 153 in 72% yield, with all four bromine atoms in the bay area being retained. Likewise, aromatic nucleophilic substitution with aliphatic amine at room temperature only for 2 h provided tetraamino product 154 in excellent yield of 92%. The reaction proceeded smoothly also for 4-n-butylaniline, although for less reactive aromatic amine refluxing of the reaction mixture and extension of the reaction time to one day was inevitable to produce 155 in 95% yield. Thiophenol, similar to alkyl amine, could be reacted with 21a at ambient temperature. Substitution with alkyl thiols or more electron-rich thiophenols is impaired by the competing formation of the radical anion of the octabromo-PBI. To obtain the corresponding thiophenol products, the umpolung methodology was applied. Accordingly, 21a was reacted with PhSO2SNa to give intermediate benzenesulfonothioate 156. Reduction thereof with Grignard reagent gave rise to 157a in overall 83% yield. Thus, the reaction leads to satisfactory yields of the otherwise poorly accessible 157a, providing a further improvement in the methodology for the PBI synthesis.
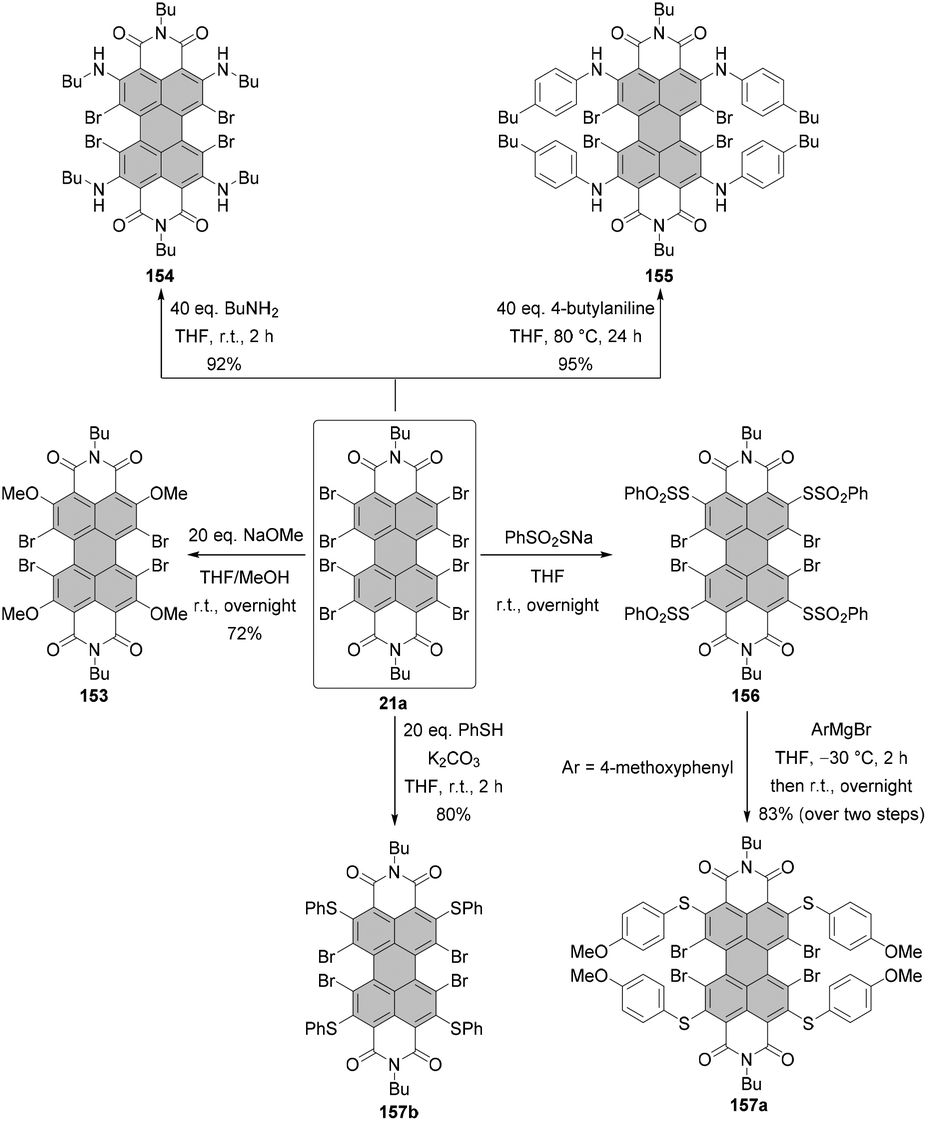 | ||
| Scheme 45 ortho-Substitution reactions of perbrominated PBI. | ||
As shown by Fernández-Lázaro and co-workers, fluoride-mediated thiolation provides fourfold ortho-substituted PBIs in excellent yields (Scheme 46) regardless of the type of sulfur reagent, whether primary or tertiary aliphatic or aromatic.120 In contrast, the differences in reactivity are clearly visible for alcohols. Whilst substitution of 65b with phenols proceeded smoothly to give tetraaryloxy PBIs, e.g. compound 146b (Scheme 46), an analogous reaction with an aliphatic alcohol afforded a mixture of PBIs with different degrees of substitution. Accordingly, tetrabutoxy PBI 146c could be isolated in 40% yield in addition to partially alkoxylated products like 158.120 The reaction of alkyl and aryl thiols with 65b is highly feasible, leading to the respective tetra-substituted products 145b–d in yields of 80–99%.
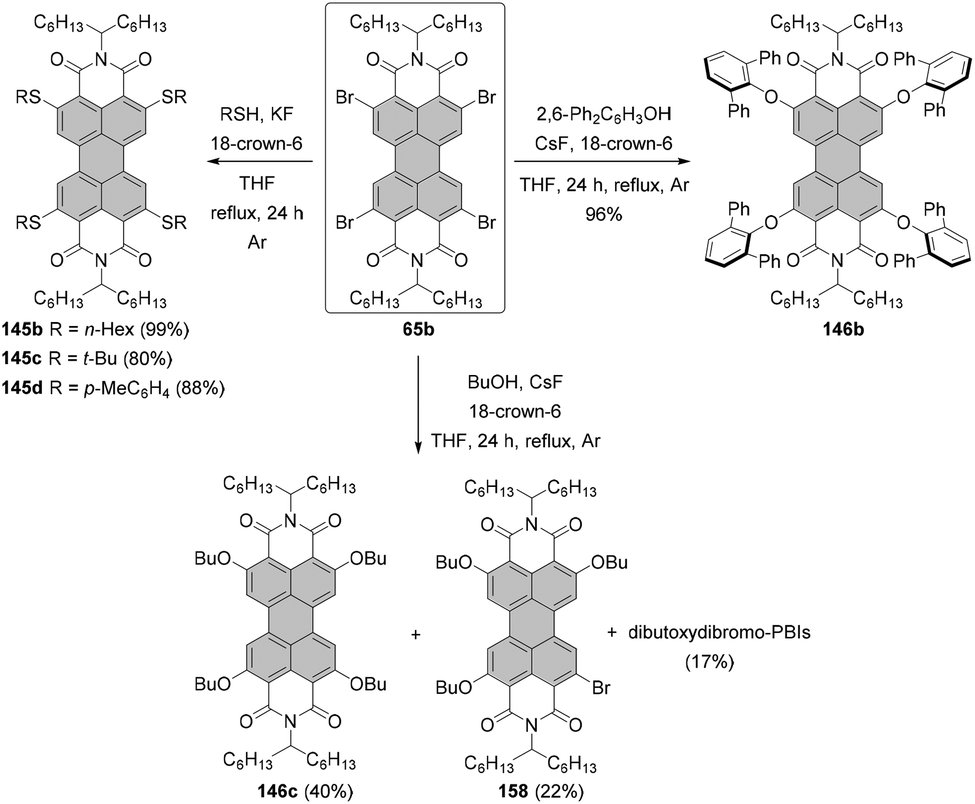 | ||
| Scheme 46 Fluoride-mediated synthesis of ortho-alkoxy-, ortho-aryloxy- and ortho-thio-substituted PBIs. | ||
Introduction of cyanide
Cyanide substituents can be introduced by the Rosenmund–von Braun reaction employing ortho-bromo-substituted PBI 37c and an over-stoichiometric amount of copper(I) cyanide to afford 148b in 35% (Scheme 43). The compound revealed intriguing behavior arising from the presence of four chloro and four cyano substituents. The first reversible reduction process was observed at −0.07 V (vs. Fc+/Fc), confirming the highly electron-deficient character of 148b. Catalytic reduction of this compound in methanol provided dianion disodium salt 150, which featured remarkable stability under ambient conditions over several months and constitutes the first example of an ambient isolable PBI dianion.71 Alternatively, ortho-cyanation can be executed via copper-mediated reaction of 37a with a high excess of both Zn(CN)2 as a cyanide source and CuBr as a copper source to provide 148a in 64% yield (Scheme 43).69 Both the reactions required heating the reagents at high temperature in anhydrous DMF.Cu(II)-Mediated oxidative cyanation was applied for a tetraboronate PBI, but with a rather moderately positive outcome. This transformation was conducted under modified conditions developed originally by Hartwig. The key modification was the utilization of dioxane as a co-solvent in order to solubilize the starting material. Accordingly, the microwave-assisted reaction of 62c with Cu(NO3)2 and Zn(CN)2 in the presence of CsF in a dioxane/methanol mixture produced 159b only in 40% yield (Scheme 47).100 The CsF base presumably activates the arylboronate ester by converting thereof into an intermediate borate compound.162 A significant improvement in the synthesis of cyanated PBIs was achieved when PBI tetraiodide was used as a starting material instead of bromo-PBI. Compound 159a was obtained from iodo derivative 63b in as high a yield as 95% via the Rosenmund–von Braun reaction97 in line with the fact that the cyanation of aryl iodides is, in general, smoother than that of aryl bromides (Scheme 47).
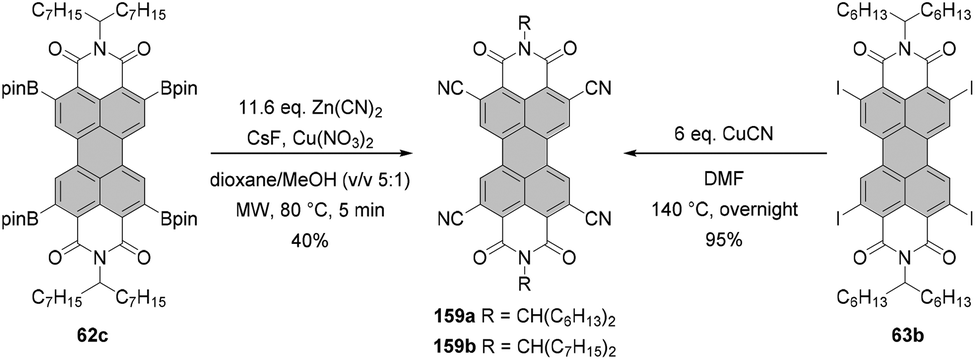 | ||
| Scheme 47 ortho-Cyanation of PBIs. | ||
C–C-coupling reactions
The utilization of ortho-functionalized PBIs proved to be advantageous for the attachment of heteroaryl and electron-deficient aryl substituents to the PBI core (compared with arylation of core-unsubstituted PBIs, vide infra). More specifically, PBI-tetraboronate 62c was readily coupled with electron-poor 4-bromobenzonitrile to give 160d in 70% yield (Scheme 48). The reaction was carried out in the presence of Pd(PPh3)4 as a catalyst.93 Pd2dba3 in combination with SPhos was identified as an optimal catalytic system to promote the introduction of heteroaryl groups, such as 2-thienyl, 3-thienyl and 3-pyridyl. The corresponding thienylated PBIs 160a and 160b were isolated in yields exceeding 80%, whereas pyridylation was somewhat less efficient (Scheme 48).92 The Suzuki coupling was also successful, when the location of functional groups on the coupling partners was reversed. That is, a Pd(PPh3)4-catalyzed reaction of tetraiodido-PBI 63b with phenyl and 2-thiophenyl boronic acids afforded 160e and 160a in very good yields.97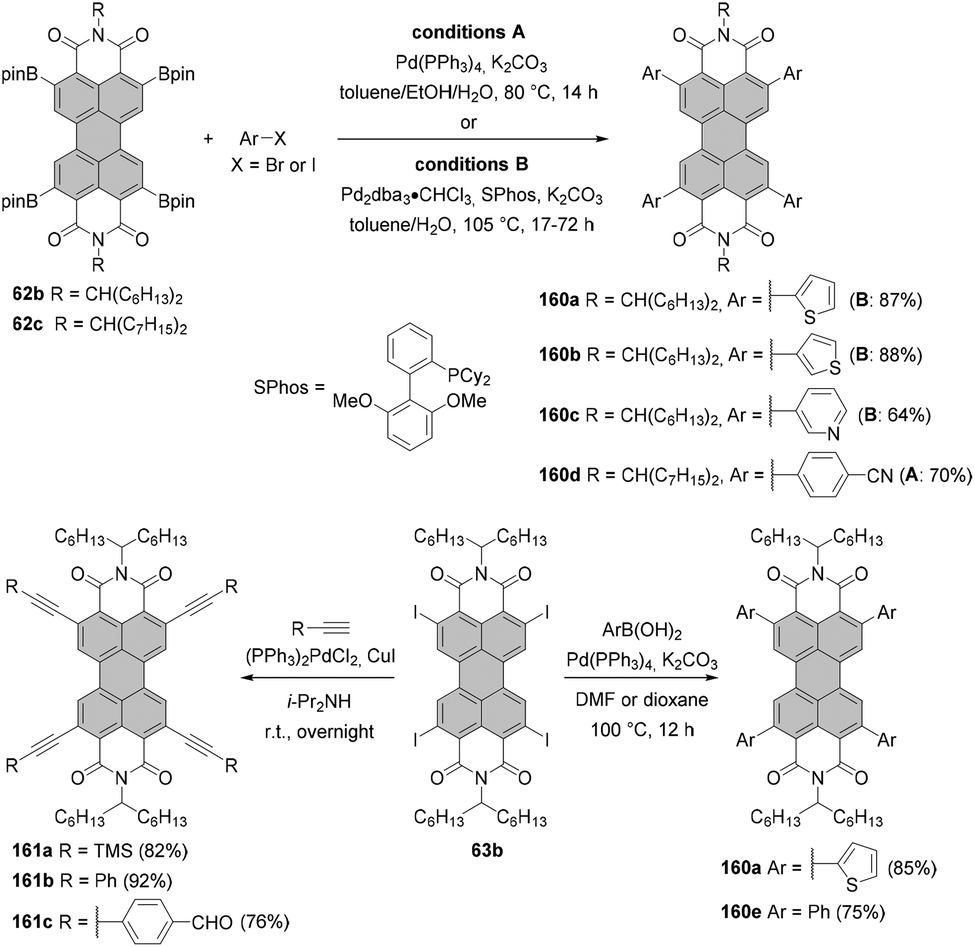 | ||
| Scheme 48 Palladium-catalyzed reactions of ortho-functionalized PBIs. | ||
It is noteworthy that compound 160a could be obtained in comparable yields by both methods. The versatility of the iodinated PBI building block was demonstrated in other palladium-catalyzed reactions such as the Sonogashira coupling. Alkynylation of ortho-positions with aryl- and silylacetylenes was smooth, affording tetra-substituted products 161a–c in high yields97 (Scheme 48). Introduction of four ethynyl substituents at ortho-positions of a PBI core was also realized by a Castro–Stephens reaction (historically a predecessor of the Sonogashira coupling in which the organocopper reagent is not generated in situ unlike the latter reaction) between 2,5,8,11-tetrabromo-PBIs and [(triisopropylsilyl)ethynyl]copper(I) in DMSO at 90 °C. The reaction afforded the corresponding tetralkynyl-PBIs in good to high yields, depending on the imide substituents.163 Monobromo PBIs are also key building blocks for the preparation of three-dimensional multichromophoric PBI arrays via the Suzuki or Stille coupling. The synthesis of these compounds was motivated by the development of non-fullerene acceptor materials for organic solar cells. For instance, solar cells utilizing PTB7-Th as a donor and an electron acceptor based on the 4,8-di(thiophen-2-yl)benzo[1,2-b:4,5-b′]dithiophene central core and four PBI units linked via ortho-positions showed PCEs over 8%.164
Direct functionalization of core-unsubstituted PBIs
As shown above, the synthesis of PBIs is typically accomplished either by the nucleophilic displacement of halogen atoms or by metal-catalyzed carbon–carbon coupling utilizing halogenated or optionally borylated starting materials. Despite being less abundant in the literature, C–H functionalization of a PBI core deserves to be given respectable consideration as it allows streamlining the synthetic strategies by reducing the number of synthetic steps.C–C-coupling reactions between PBIs and unsaturated coupling partners
The first successful method for the selective 2,5,8,11-functionalization of core-unsubstituted PBIs was published only in 2009.94 It gave access to tetraalkyl-PBIs via ruthenium-catalyzed regioselective C–H activation in which the substitution pattern was determined by the ortho-directing carbonyl imide groups. Treatment of 6c or PBI with 2,6-diisopropylphenyl imide groups with terminal alkenes in the presence of [RuH2(CO)(PPh3)3] gave rise to alkyl-substituted PBIs. The yields varied from moderate for cyclic or linear alkyl to high for silylated groups (see PBIs 162a–d, Scheme 49). The reaction of unsubstituted PBIs with terminal alkenes yielded olefination products when the process was oxidative. Accordingly, Lin and Zhang introduced two sets of conditions based on ruthenium(II) or rhodium(III) catalysis.165 Not only were these reactions regioselective, but also they showed excellent E-stereoselectivity. Activated alkenes bearing electron-accepting groups such as ester or benzenesulfinate were reacted successfully with PBI 6g and 6j in the presence of [RuCl2(p-cymene)]2, AgSbF6, or Cu(OAc)2 as an oxidant and AcOH as an additive. As proposed by Jeganmohan, the tentative reaction mechanism involves Ru(II) active species, generated in situ by the removal of chloride ligands by silver cations.166 The products were formed in 33–82% yields depending on the alkene EWG group and imide substituents of the PBI (e.g.163a–e). An analogous reaction with styrene was low-yielding. Therefore, the more reactive but several times more expensive rhodium catalyst [RhCp*Cl2]2 was applied for styryl derivatives to give the corresponding products 163f and 163g in 96% and 43% yields, respectively.165 Interestingly, these two sets of conditions provided alkenylated products rather than alkylated ones in the absence of an oxidant. | ||
| Scheme 49 ortho-Functionalization of core-unsubstituted PBIs. DCE = 1,2-dichlorethane. | ||
Arylation at ortho positions can be executed in a two-step one-pot fashion from unsubstituted PBI. The method originates from the synthetic protocol for the ruthenium-catalyzed arylation of aromatic ketones.96 According to the modified procedure, the PBI starting material and 7 eq. of arylboronic acid neopentyl glycol ester were treated with 20 mol% of the ruthenium catalyst RuH2(CO)(PPh3)3 in a solvent mixture of pinacolone and mesitylene. Addition of the latter as a co-solvent ensured higher reaction temperatures, which shortened the reaction time and gave rise to higher yields (see products 160f–j, Scheme 49). Yet, the reaction with 4-bromophenyl boronate was extremely sluggish and produced desired 160k in only 12% yield after being stirred for 91 h, while 50% of the starting compound 6c was recovered. It is also worth noting that the reaction of unsubstituted PBIs was unsuccessful with a strongly electron-deficient boronate substrate, such as p-nitrophenyl and pentafluorophenyl boronates, as well as heteroaromatic starting materials, e.g. 2-thienyl boronate.95 Conversely, the Suzuki coupling of 2,5,8,11-PBI tetraboronate with 4-bromobenzonitrile proved to be efficient, which points out the superiority of the sequential synthesis to the one-pot protocol.93
As demonstrated by Lai, Wudl and Zheng, direct introduction of heteroaryl substituents at ortho positions of the PBI core is feasible by the nucleophilic addition of the Grignard reagent to the PBIs devoid of halogen or boron functionalities at ortho positions, followed by oxidation in air (Scheme 50).167 The unusual reactivity of the Grignard reagents with PBIs can be rationalized by (a) the lower reactivity of imide carbonyl, which results in the absence of products of 1,2-addition, (b) the coordination of magnesium to the carbonyl oxygen, lower steric requirements and lower electron density at ortho carbon atoms, facilitating addition at ortho rather than at bay positions. The reaction of thienyl as well as mesityl Grignard reagents with core unsubstituted and S-annulated PBIs 6i and 164a furnished arylated products 160l,m and 165a,b though with moderate yields (30–50%). The tetrathiophene-functionalized compound 160m was obtained in a substantially lower yield than by the Suzuki coupling of either tetraborylated or tetraiodinated PBIs (compare with Scheme 48). Substitution at ortho positions combined with the formation of a five-membered S-ring is a good strategy to extend the π-conjugated system and tune FMO levels and optical properties, while retaining the planarity of the PBI core. Photovoltaic devices utilizing the S-annulated molecule 165b as an acceptor material showed PCEs up to 5%, which are high for single PBI-core compounds in OPV.
 | ||
| Scheme 50 ortho-Functionalization of core-unsubstituted and S-annulated PBIs with Grignard reagents. | ||
Direct alkylation and fluoroalkylation reactions
Introduction of trifluoromethyl and longer perfluoroalkyl substituents can be executed by cross-coupling of halogenated PBIs and alkyl halides (vide supra). Direct C–H functionalization constitutes a viable alternative, although it is more challenging for electron-poor PBI aromatic scaffolds. Wang demonstrated two effective protocols for the installation of either alkyl and oligo(ethylene glycol)168 or perfluoroalkyl substituents169 in bay areas of core unsubstituted PBIs. Simple alkyl chains were attached by Pd-catalyzed reaction of 6l and 6o with the corresponding alkyl halides (Scheme 51). The reaction was optimized in terms of ligand, catalyst loading and temperature. These efforts allowed the identification of the most effective conditions. Accordingly, coupling between PBIs and halides was carried out in the presence of Pd(OAc)2 as a catalyst, PPh3 as a ligand and Cs2CO3 as a base in o-xylene at 130 °C. The reactions afforded monoalkylation products in moderate yields up to 55%. The presence of dialkylated products in the mixture was detected by mass spectrometry but the isolation of these compounds was not reported. The type of imide substituents did not affect the efficiency of the process and compounds 166a and 166b were obtained in comparable yields, when PBIs 6l and 6o bearing aromatic and swallow-tail imide substituents, respectively, were reacted with hexyl bromide. Alkyl iodides, as exemplified by the reaction of 6o with hexyl iodide, gave slightly better results than bromides, whereas reactivity of chlorides was not sufficient under these conditions. The addition of an excess of alkyl halides due to possible β-elimination to the corresponding alkenes, and the excess of Cs2CO3 were indispensable to the success of these transformations. Nevertheless, a substantial amount of the unreacted starting material could be recovered in each case. The substrate scope was not limited only to alkyl halides. The reactions also worked for partially fluorinated alkyl and oligo(ethylene glycol) coupling partners, although with rather low efficiency. Accordingly, products 166c and 166d were obtained in yields below 30%.168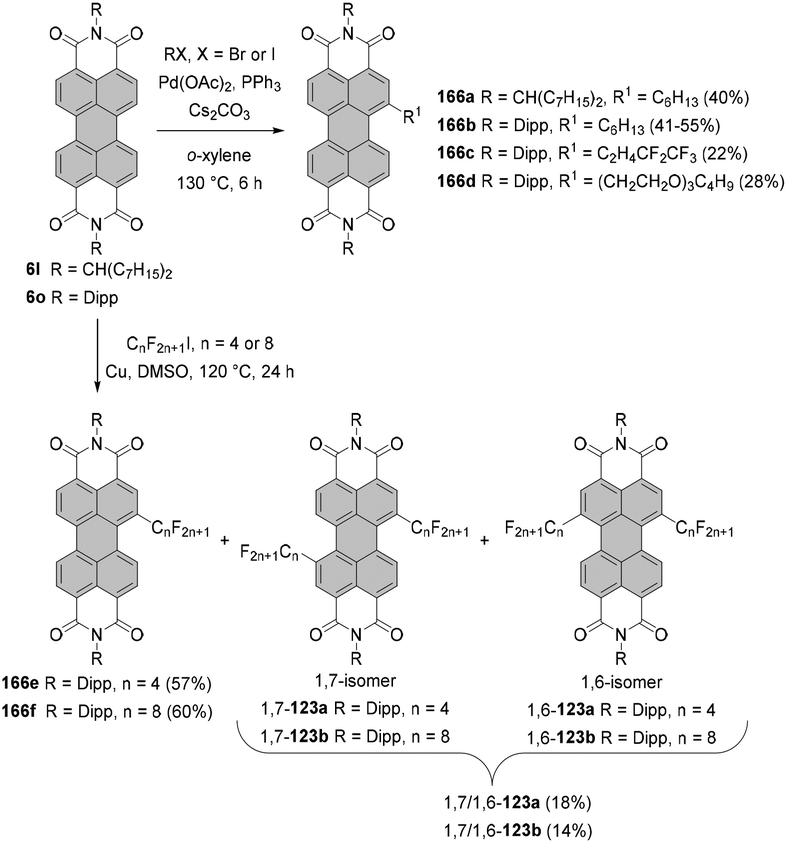 | ||
| Scheme 51 Alkylation and perfluoroalkylation of core-unsubstituted PBIs. | ||
In contrast to the copper-mediated reaction of pre-functionalized PBIs with perfluoroalkyl halides (see Scheme 35), an analogous transformation of core-unsubstituted PBIs leads preferentially to monoalkylated products. Accordingly, alkylation of 6o with 2 eq. of perfluoroalkyl iodide in the presence of 3 eq. of Cu powder afforded PBIs 166e and 166f in 57% and 60% yields, respectively (Scheme 51). The compounds were accompanied by some amount of a regioisomeric mixture of 1,7 and 1,6 bis-perfluoroalkylated PBIs 123a and 123b in a ratio of 5:1. Addition of TEMPO to the reaction mixture inhibited the reaction, which indicates the radical mechanism.169 Due to low yields of dialkylated PBIs by C–H functionalization of the PBI core, it is reasonable to consider coupling of perfluoroalkyl iodides with 1,7-dibrominated PBI precursors for the preparation of 1,7-bis(perfluoroalkylated)-PBIs. On the other hand, 1,6-regioisomers can be accessed via PTE intermediate.170
A process published by Strauss and Boltalina in 2015 gave access to PBIs with an even higher degree of substitution, though in low yields (Scheme 52). The reaction was carried out in a gradient-temperature gas–solid (GTGS) hot plate reactor at around 400 °C for 4.5 h to yield a mixture of PBAs bearing two, three or even four CF3 groups with different substitution patterns. A difference in solubility was used to separate trifluoromethyl-substituted PBAs from an essentially insoluble starting material. Subsequently, a crude product was subjected to imidization in propionic acid. Since trifluoromethylation of PBAs was not selective, the corresponding mixture of PBIs was likewise a complex composition of PBIs. Flash chromatography of the corresponding mixture of PBIs allowed the separation of PBIs bearing different numbers of CF3 from each other, whereas resolution of regioisomers was conducted by HPLC. This purification procedure afforded the regioisomers of di, tri and tetra trifluoromethyl-substituted PBIs 1,7/1,6-123f,g, 1,5,7/1,6,8-167a,b, and 1,5,7,11/1,6,8,11-167a typically in estimated yields up to 10% (Scheme 52). The only exception was 1,7-bis(trifluoromethyl)-PBI 123f bearing butyl imide groups; it was isolated in ca. 20% yield. When the reaction sequence was reversed and core-unsubstituted PBI was subjected to trifluoromethylation under these conditions, only a monoalkylated product could be detected by mass spectrometry. Due to technical requirements, this method cannot be considered at this point to be of general importance for the synthetic organic chemists. Nevertheless, a systematic study of the variety of trifluoromethylated compounds shed light on the influence of the number of CF3 groups and their redistribution on the PBI core on the properties of PBI dyes. Accordingly, attachment of each CF3 group to the core moves the first reduction by 0.14 V towards more positive values. Another observation was that the chain length of perfluoroalkyl substituents does not affect this trend and that two perfluoroalkyl imide residues exert a similar effect to one CF3 group at the perylene core on reduction potentials.
 | ||
| Scheme 52 Synthesis of core-substituted trifluoromethyl derivatives by reaction in a gradient-temperature gas-solid (GTGS) hot plate reactor. | ||
Direct amination reactions
Synthesis of bay-substituted diamino-PBIs via dibromo intermediates is cumbersome due to the tedious method for the isolation of a 1,7-dibromo compound from its 1,6-regioisomer by repetitive crystallization. Moreover, this method does not give an access to the pure dibrominated compound with a 1,6-substitution pattern. Separation of the two isomers is, indeed, possible upon introduction of amino groups. Yet, it is substituent-dependent, which does not guarantee that the products would be successfully resolved for any introduced imide substituents or amine. For this reason, methods that allow the selective synthesis of 1,6-substituted PBIs are particularly valuable. Remarkably, core-unsubstituted PBIs can be efficiently aminated in neat amines in the presence of the oxidant even at room temperature. As unambiguously proved by Höger107,171 and confirmed for similar reactions by Efimov,106 the reactions of core-unsubstituted PBIs with amines produce exclusively compounds identified as mono or diamino bay-substituted products. Notably, in the latter case, the reaction proceeds selectively at 1 and 6 positions of the PBI core. The number of the introduced amino groups depends on the imide substituents, amine and the oxidizing agent. The highest yields were obtained for cyclic amines, such as pyrrolidine and piperidine. The introduction of pyrrolidine was particularly smooth for PBIs bearing aryl imide substituents and occurred already at room temperature. Mainly, amination of 6k and 6p catalyzed by copper(II) chloride afforded dipyrrolidino-PBIs 1,6-67d and 1,6-67e in 68% and 82% yields, respectively, within only 1–3 h. An analogous transformation of PBI 6j with the hexylheptyl imide functionality into 1,6-67c (49% yield) was somewhat less efficient (Scheme 53),107 whereas the less soluble N-octyl PBI gave only a trace amount of the corresponding di-substituted product.106 Efimov considered solubility as a key parameter determining the success of the process.106 The reactions with piperidine supported by CuCl2 required prolonged stirring and were carried out at higher temperatures in some cases to provide disubstituted-PBI 1,6-66f in excellent yield or mono-piperidinyl PBIs 168a, 168c and 168d as major products accompanied by diamino-PBIs 1,6-66b, 1,6-66d and 1,6-66e.107,171 Finally, attempts to react morpholine, 1-methylpiperazine and 1-hexylamin resulted in only low to moderate yields of typically mono-amino-PBIs 169a–e under these conditions.107 | ||
| Scheme 53 Direct amination of core-unsubstituted PBIs. Method A: Zn(OTf)2, Cs2CO3, H2O, 60 °C, 6 h; method B: KMnO4, AgNO3, r.t., 24 h; method C: CuCl2, r.t.−80 °C, 6–24 h; method D: CuBr2, Cs2CO3, H2O, 60 °C, 6 h. | ||
The yield of disubstituted products could be substantially improved when CuBr2 was used as a catalyst in addition to Cs2CO3 and water as additives (method D, Scheme 53).171 Under these conditions, 1,6-66b was obtained in yields as high as 67% along with mono-piperidynyl-PBI 168c in 29% yield. Moreover, these conditions allowed the synthesis of 1,6-disubstituted products from PBI 6o and morpholine as well as 1-methylpiperazine. PBI 170b was formed as a minor product in only 19% yield, whereas morpholine derivative 170a was formed as the major product. In the latter case, the preference towards the formation of a particular product, “mono” versus “di”, was reversed, when compared to the reaction under CuCl2 catalysis. Interestingly, mono-piperidynyl-PBI 168c was favored in the absence of the additives and CuBr2. Nevertheless, markedly better results (yield 70%) were obtained for Zn(OTf)2 as a catalyst in the presence of water and Cs2CO3 (Method A, Scheme 53).171 The mechanism of this transformation is not clear.107,171 Efimov reported catalyst-free amination of core-unsubstituted PBIs.106 The number of introduced amino groups, regioselectivity of diamination (1,6 versus 1,7) and yields were strongly affected by the oxidant and temperature in addition to the solubility of the respective PBIs. High selectivity towards 1,6-diamino PBIs 67a, 67d (pyrrolidine derivatives) and 66e (piperidine derivatives) was observed for KMnO4 in combination with AgNO3, whereas the mono-substituted product 116b was preferentially synthesized in the presence of pyridinium dichromate (PDC) (Scheme 53). The driving force for the spontaneous amination of core-unsubstituted PBIs to give mono or disubstituted products under these conditions is likely to derive from the presence of air or other substances that are able to act as oxidants. The mechanism most likely proceeds via the PBI radical anion pathway, as revealed by the spectroscopic study. Both methods, uncatalyzed and catalyzed by metal complexes, provide an alternative synthesis of 1,6-diamino- or 1-monoamino-PBIs that does not rely on the use of the respective halogenated PBIs as the precursors.
Efficient conditions for direct amination of core-unfunctionalized PBIs employing CuBr2 as a catalyst (see Scheme 53) were successfully applied for diamines (Scheme 54). As opposed to the reactions with monoamines, addition of a small amount of water was inevitable to obtain a detectable amount of cyclized products. Unexpectedly, substitution with primary amines, ethylenediamine or (±)-trans-1,2-diaminocyclohexan took place exclusively at the 1 and 2 positions to give 171a and 171c in 12% and 32% yields, respectively, whereas 1,12-diamino product was not observed under these conditions. In contrast, N,N′-dimethylethylenediamine afforded a mixture of 1,2- and 1,12-cyclized products 171d and 172 in comparable yields (31% and 30%, respectively). The absorption maximum of the bay-substituted diamino compound 172 was positioned at 689 nm and was bathochromically shifted versus the absorption maximum of the 1,2-isomer (λabs = 618 nm). When the mixed primary and secondary amines were applied, the reaction proceeded towards the 1,2-cyclized isomer as depicted for 171b in Scheme 54. Thus, the reactivity of the diamine had a tremendous impact on the selectivity of the reaction. Moreover, in all cases, only singly cyclized products were isolated, despite the fact that the reactions were carried out with a high excess of diamines.171
 | ||
| Scheme 54 Synthesis of azacyclic PBI derivatives from core-unsubstituted PBIs and diamines. | ||
When the reactivity of a particular starting material or intermediate is high enough, the initial substitution of halogen is followed by direct substitution on the perylene core. This holds true for the replacement of a nitro group in mononitro-PBI 45a with pyrrolidine, which, apart from the expected mono-substituted derivative, produced diamino-derivative in a considerable yield (see Scheme 31).130 The number of amino substituents in bay areas could be increased even up to four. Already in 2014, direct amination of unactivated C–H bonds leading to triamino-PBIs was briefly mentioned by Höger, though without any experimental details.107 In 2018, Fernández-Lázaro published a systematic study of fluoride-mediated introduction of the third amino group to the bay-substituted diamino-PBIs focusing exclusively on cyclic amines.172 The reactions were carried out in refluxing THF in the presence of TBAF as a source of fluoride. Since the attachment of the third identical amino group either to 1,7-diamino or 1,6-diamino starting material leads naturally to the same product, triamino PBIs can be derived from a 1,7/1,6 regioisomeric mixture. Accordingly, PBIs 173c–f bearing either ethylpropyl or hexylheptyl imide residues were synthesized from the corresponding 1,7/1,6 isomeric mixtures of dipiperidino- or dimorpholino-PBIs and the respective amines in yields of 65–80% as illustrated in Scheme 55.
 | ||
| Scheme 55 Amination of diamino-PBIs. | ||
The reactivity of both isomers was verified by reacting regioisomerically pure diamino compounds 1,7-66a and 1,6-66a with piperidine. Both isomers yielded product 173a in essentially the same yield of ∼60% (Scheme 55). To avoid the formation of mixtures, preparation of PBIs bearing mixed amino substituents necessitated isomerically pure diamino precursors.
Thus, PBIs 173b and 174 were synthesized from 1,7-dipiperidino-PBI 66a and N-methylpiperazine or pyrrolidine in 65% and 40% yields, respectively (Scheme 55). The conditions were also successfully applied for aromatic NH reagents, although the efficiency of substitution was rather low. When the reaction of dipyrazol-N-yl-PBI 175a with pyrazole was performed, trisubstituted 176 was isolated in 11%. Surprisingly, even the tetra-substituted product 177 was formed in only 1% yield (Scheme 55). A detailed inspection of the mechanism was not carried out. In accord with the increased electron density, redox potentials of triamino-PBIs were shifted cathodically versus diamino congeners, e.g. the first oxidation and reduction processes for compound 173a were observed at +0.06 V and −1.43 V, respectively. Aromatic pyrazole substituents do not exert a strong effect on the electrochemistry of the corresponding PBIs.
Direct hydroxylation reactions
The direct hydroxylation method published by Schenning and Brunsveld's labs constitutes a more straightforward way to prepare monobay difunctionalized-PBIs than the sequence shown in Scheme 23. Thus, the reaction of 6e with activated manganese dioxide in sulfuric acid, followed by partial reduction with sodium sulfite, afforded quantitatively 87b. The NMR spectrum indicated the presence of only the dihydroxylated PBI which could be alkylated with halides or pseudohalides in the presence of N,N-diisopropylethylamine (DIPEA) or K2CO3 as a base in DMF. These protocols afforded alkoxy derivatives, e.g.178a–c, in low to moderate yields (Scheme 56).118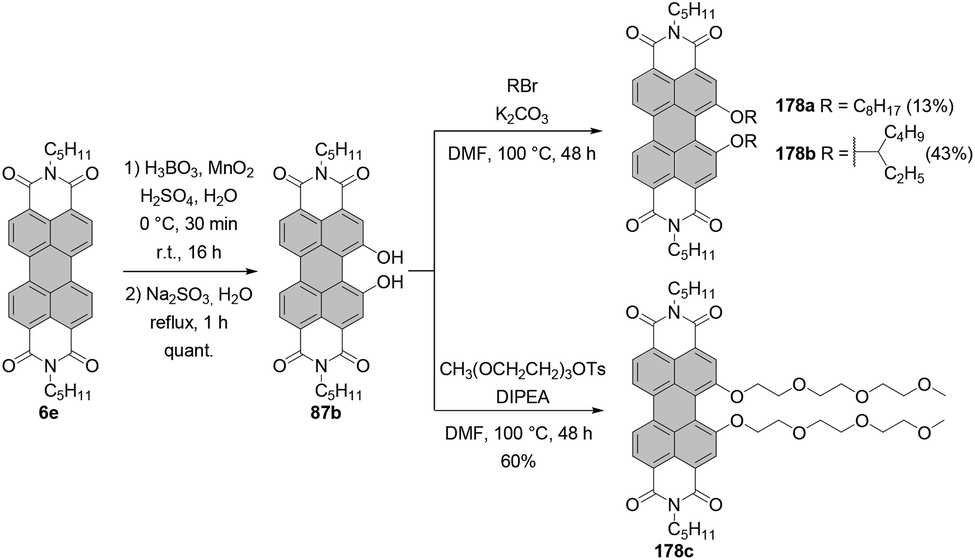 | ||
| Scheme 56 Synthesis of 1,12-dialkoxy-PBIs via direct hydroxylation of core-unsubstituted PBI. | ||
The literature survey may create some confusion, as it was reported that similar conditions led to 1,7-dihydroxy-PBI.52 However, in the latter case the assignment was based exclusively on the 1H NMR spectrum. To verify the 1,12-substitution pattern, Schenning and Brunsveld not only carried out 2D NMR studies, but also synthesized 1,7-dialkoxy-PBI via alkoxylation of the 1,7-dibromo precursor. Comparison of the spectra of the di-n-octyl-substituted compounds obtained by these two methods revealed that they differed, hence precluding the formation of 1,7-dihydroxy-PBI in the reaction with MnO2.
Another means to access bay-alkoxylated PBIs from core-unsubstituted PBIs is by fluoride-mediated alkoxylation as proposed by Fernández-Lázaro and co-workers.173 This reaction, as well as the analogous thiolation,174 has, however, some limitations. Firstly, the scope of this novel reaction is still limited and the introduction of alkoxy or thio substituents occurs preferably for alkyl starting materials rather than aryl counterparts. Accordingly, the reaction with phenols did not take place for core-unsubstituted PBIs.173 Likewise, introduction of two tert-butylthio substituents into bay areas was low-yielding, while the reaction of core-unsubstituted PBI with ethanedithiol, which was otherwise successful for halogenated precursors (see Scheme 30), proved ineffective.174 It is also interesting to note that even for halogenated PBIs, fluoride-assisted reactions with thiophenols are not well-suited as the yields are mediocre, whereas direct functionalization of the PBI core with thiophenols did not proceed at all.174 Secondly, the reactions are not regioselective and twofold functionalization typically provides inseparable mixtures of 1,6 and 1,7-regioisomers.
Other direct C–H functionalization reactions at ortho-positions
C–H functionalization at ortho-positions of a PBI core has been rarely reported. In addition to the commonly applied Rh, Ru and Ir-catalyzed reactions for the synthesis of 2,5,8,11-substituted PBIs, ortho-substitution was reported for the reactions of ortho-unfunctionalized PBIs with a lithiated organophosphorus reagent175 and N-heterocyclic carbene (Scheme 57).176 | ||
| Scheme 57 C–H functionalization of ortho-position with a lithiated organophosphorus reagent and N-heterocyclic carbene. | ||
The diphenylphosphide anion (PPh2P−) is known to substitute halogen atoms on the aromatic scaffolds. Therefore, it was surprising that lithium diphenylphosphide, when reacted with 1,7-dibromo-PBI 4a, yielded diphenylphosphinoyl-PBI 179 rather than a product of bromide displacement. The reaction was also tested for tetrachloro- and core-unsubstituted PBIs 22u and 6a and in both cases, it produced 2-diphenylphosphinoyl products 180 and 181. Presumably, the reaction proceeded via intermediate P(III) compounds, which were readily oxidized into more stable P(V) derivatives during purification. The isolated yields varied between 21 and 42%. The diphenylphosphinoyl-PBIs exhibited slightly bathochromically shifted absorption spectra and most importantly, retained high fluorescence quantum yields of their precursors in the range of 83–100%.175
Likewise, the attack of 1,3-di-isopropyl-imidazolin-2-ylidene on the ortho-position of tetrachloro-PBI 22d was unexpected. The reaction produced an ambient stable zwitterionic PBI-centered radical 182 whose structure and radical characteristics were unambiguously confirmed by single crystal structural analysis and EPR spectroscopy in addition to other techniques.176
Direct functionalization of core-unsubstituted perylene scaffolds provides an attractive atom-economical alternative to the standard substitution or cross-coupling reaction employing pre-functionalized building blocks. The benefits include the reduction of the synthetic steps and byproducts, including organometallic byproducts. Indeed, in many cases, C–H activation of perylene dyes still gives inferior results in comparison with the halogenated or borylated starting materials. However, as was shown in this section, unsubstituted building blocks offer very often unusual reactivity, high selectivity towards products of different locations of substituents and straightforward access to otherwise difficult to synthesize products.
Core-extended and annulated PBIs
Organic dyes consisting of two or more PBI units were particularly intensively investigated during the last decade as novel n-type semiconductors for utilization in polymer-based organic solar cells (OSCs). These compounds differ in the character of linkage, such that two or more PBI cores may be directly linked to each other by one, two or three C–C bonds or they may be attached to a central unit. Annulation of the latter affords fully conjugated aromatic systems, which are particularly attractive for application in organic solar cells. A dynamic progress in the field of OSCs resulted in the appearance of a rich variety of PBI-based scaffolds, some of them featuring sophisticated non-planar structures. This section aims at summarizing the advances in the synthesis of PBIs whose π-scaffold was annulated with other π-systems or PBIs themselves. Since the progress in the field of OSCs, including those derived from PBIs, has been thoroughly reviewed,11,15,177 only the structures of general importance along with the selected remarkable PBI derivatives are discussed. The molecules are presented with a significant emphasis on their synthesis rather than their photovoltaic properties. In addition, we discuss other important annulation methods leading to single-core PBIs or multichromophoric PBI scaffolds that are investigated in other fields.Core-extended PBIs by bridging of bay positions with heteroatoms
The simplest examples of core-extended PBIs are molecules with the bay area bridged with sulfur, nitrogen, oxygen, selenium and silicon heteroatoms. Methods for the inclusion of a nitrogen atom are depicted in Scheme 58. The reaction of 45c and 45e with triethyl phosphite (method A), known as a Cadogan cyclization, produced N-annulated PBIs 183a and 183b in 56% and 63% yields, respectively, along with diethyl phosphonates 184a and 184b as minor products. P(OEt)3 acts here either as a reducing agent via a nitrene insertion mechanism or as a nucleophile replacing the nitro group. Pyrrole nitrogen of the first compounds can be readily deprotonated with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in THF or KOH in EtOH to form a strong nucleophile and subjected to alkylation or acylation reactions with methyl iodide, benzyl bromide, bromoacetic acid ethyl ester and acetyl or benzoyl chloride to produce the corresponding compounds in yields of ca. 70–90%.79 Welch and co-workers observed the formation of an N-ethyl side-product for 1-nitro PBI bearing 1-ethylpropyl imide substituents under these conditions, decreasing the yield of the desired product. To avoid the unwanted alkylation of the five-membered ring, they exchanged the reducing agent for triphenylphosphine,159 which previously proved an effective solution in the synthesis of carbazoles.178 This modification afforded a cyclic NH-product in yields above 60%. N-Annulated PBIs were also prepared by the Buchwald–Hartwig amination (method B). The reaction of tetrachloro-PBI with aniline in the presence of Pd(OAc)2, PCy3 and KOt-Bu produced only a singly annulated product with an additional amine substituent in the other bay area, whereas the fourth chlorine atom was cleaved. This reaction outcome is most probably due to the high strain imparted by the N-bridged first bay area. The realization of twofold N-bridging was, however, possible in the triply fused dimeric PBI scaffold depicted in Scheme 12.179 By analogy to the successful halogenation with bulky iodine of fused bis-PBI, the second inclusion of a N atom was aided by the contorted structure of the precursor. As suggested by the authors, the contorted π-system accordingly reduced the energy cost of the formation of the second 5-membered ring. Li proposed substitution with azide catalyzed by Cu(I)/N,N′-dimethylethane-1,2-diamine, followed by the ring closure in one synthetic step to access these compounds (method C).180 The method produced singly N-annulated products bearing one or two amino groups in the second bay area in 81% and 59% yields when 1,6,7-tribromo- or 1,6,7,12-tetrabromo-PBIs were used as starting materials, respectively. | ||
| Scheme 58 N-Annulation of PBIs with a concomitant formation of 5-membered heteroaromatic rings. | ||
Mononitro-PBIs are versatile starting materials for the preparation of monobay S-heterocyclic annulated derivatives. Langhals reported that treatment of 45c,e,f with sulfur in DMF afforded compounds 186a–c bearing swallow-tails or 2,5-di-tert-butylphenyl groups at imide positions in 30–47% yields along with 1,2-dithiin molecules 187a–c in yields of 40–55% (Scheme 59). The ratio of both products could be enhanced in favor of the five-membered compound when the reaction was carried out in NMP at a similar temperature or in refluxing DMF.79 The reaction of sulfur with pyran-annulated PBI derivative 18880 bearing a nitro group in the second bay area gave comparable results, that is, S-annulated product 189 and a 1,2-dithiin derivative 190 in 51% and 28%, respectively (Scheme 59).181 While the monosulfur-bridged compound exhibited fluorescence quantum yields above 60% in DCM, emission of the 1,2-dithiin product was quenched.
 | ||
| Scheme 59 S- and Si-annulation of PBIs with a concomitant formation of 5-membered heteroaromatic rings. | ||
Li reported a substantially more efficient incorporation of a sulfur atom in the bay area of PBI starting from mononitro-PBI. The reaction thereof with the sulfur powder in NMP afforded the S-annulated product in 85% yield when the temperature was increased to 190 °C.182 Zhu noted that the approach employing nitro-PBI as a starting material is not suitable for the integration of intramolecular sulfur bridges into both bay regions as cyclization of 1,7-dinitro-PBIs yields a complex mixture without the target twofold S-annulated PBIs.183 These compounds were accessed via Stille-type coupling between bay-chlorinated PBIs and bis(tributyltin)sulfide. Palladium-catalyzed reactions of 22w and 22l with Bu3SnSSnBu3 yielded twofold S-bridged products 164a and 164b in very good yields (Scheme 59).167,183 This convenient method was also used for S-annulation of C–C single bond-linked bis-PBIs.158 In 2017, Jiang, Wang and co-workers reported the first examples of sila-annulated PBIs. These compounds were formed by Pd-catalyzed reactions between tetrachloro-PBIs and diethylsilane. The reactions tolerated both polyfluoroalkyl and aryl imide substituents and produced in each case a mixture of di- and monosila-products 191a,b and 192a,b in ca. 55% and 30% yields, respectively. The absorption spectra, in contrast to S-annulated compounds, were shifted bathochromically relative to the parent PBIs and the shift increased with the number of sila 5-membered rings. One-fold and twofold annulated PBIs exhibited strong emission with fluorescence quantum yields of around 90% and small Stokes shifts due to the rigid structures.184
Inclusion of selenium was carried out by reacting nitro-PBIs with selenium powder at 190 °C. The reaction was straightforward and high-yielding for both a single-core PBI81 and a complex multi-PBI array 193 (Scheme 60). Not only did the incorporation of selenium modulate the FMO levels of a parent scaffold, but also it resulted in a closer packing owing to additional chalcogen bonding interactions.185
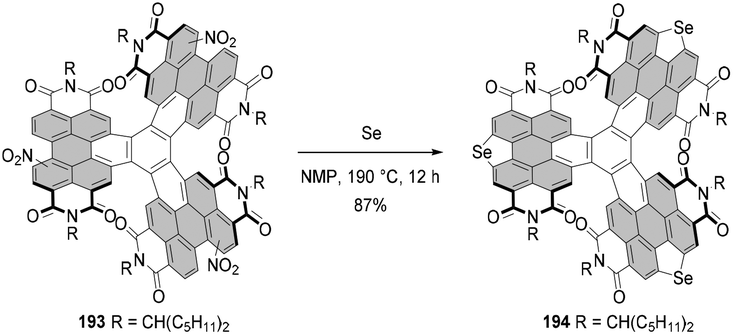 | ||
| Scheme 60 Synthesis of Se-annulated PBI dyes. | ||
As revealed by single crystal X-ray analysis the short nitrogen–carbon bonds in N-annulated PBIs induce a noticeable distortion of the PBI framework from planarity.159 Accordingly, N-annulated triply fused PBI dimers displayed doubly bowl-shaped structures due to the combined effect of the repulsion between ortho hydrogens and the imide oxygen atoms of the adjacent PBI and the strain imposed by the N-heterocyclic ring.179 Therefore, the distortion of the nitrogen derivative is markedly more pronounced than that of its sulfur congener which benefits from the significantly longer sulfur–carbon bonds.49 Thus, single crystals of single-core S-167,183 and Se-annulated PBIs and of the Se-annulated three-bladed three-dimensional compound185 revealed essentially planar PBI units. Likewise, crystallographic analysis showed that the sila-annulated PBIs possess nearly planar aromatic scaffolds. Sila-annulated PBIs bearing polyfluoroalkyl imide groups were tested in single-crystal OFET devices. Monosila-product 192a showed electron mobilities of up to 0.3 cm2 V−1 s−1 under an inert atmosphere.184 It was recognized that bay-annulation to form a five-membered ring has a beneficial impact on the geometry, packing arrangement and photovoltaic performance of polymer solar cells containing these PBI dyes as an acceptor component. While S and Se PBI derivatives feature high planarity, nitrogen in N-annulated PBIs offers an additional site for a solubilizing chain. Introduction thereof allows a reduction of the size of the imide alkyl chains required to solubilize the compounds, hence affecting the blend morphology. Essentially in each case integration of a heteroatom enhanced the key parameters of OSCs, including PCEs, in comparison with bay-unsubstituted counterparts.185–187 Thus, heterocyclic 5-membered rings are, meanwhile, common motifs in novel compounds composed of two or more PBI units for application as acceptor materials in OSCs. For instance, simple bay-linked bis-PBI with N, S or Se bridges, when paired with appropriate donor materials, produced OSC devices with PCEs of 7.2–8.4%.81,158,159 An even higher PCE value of 9.3% was achieved for the structurally more sophisticated highly twisted propeller dye 194 with selenophene rings (Scheme 60).185
Core-extended PBIs by annulation at the bay area
Nitro-PBI was also an intermediate in the synthesis of ruthenium(II) and iridium(III) PBI complexes. The corresponding amine 48d was subjected to acid-catalyzed condensation with 2-pyridinecarboxaldehyde to give the intermediate imine and to consecutive Pictet–Spengler ring closure providing azabenzannulated PBI 195.188 The conditions employing triflic acid as a catalyst provided 195 in 37% yield. Replacement of TfOH with trifluoroacetic acid increased the yield to 52% (Scheme 61).188 Bipyridine-PBI ligand 195 was then reacted with a ruthenium salt, followed by anion exchange to give the remarkably intensely phosphorescent PBI complex 196 (Scheme 61).188 It should be noted that the phosphorescence emission has been seldom reported for PBIs,188 which are otherwise well known for their excellent fluorescent properties. Azabenzannulation gave also positive results for variously substituted 2-pyridinecarboxaldehydes and benzaldehydes producing the corresponding cyclized products in moderate yields (∼20–60%).78,189 Moreover, the conditions proved effective for the preparation of twofold azabenzannulated PBIs. Thus, a 3:2 mixture of 1,7- and 1,6-dinitro-PBIs obtained from the corresponding dinitro precursors afforded the corresponding mixture of the anti and syn azabenzannulated isomers anti-197 and syn-197 (Scheme 61). Multiple column chromatography afforded the pure compounds in 23% and 14% yields, respectively.189
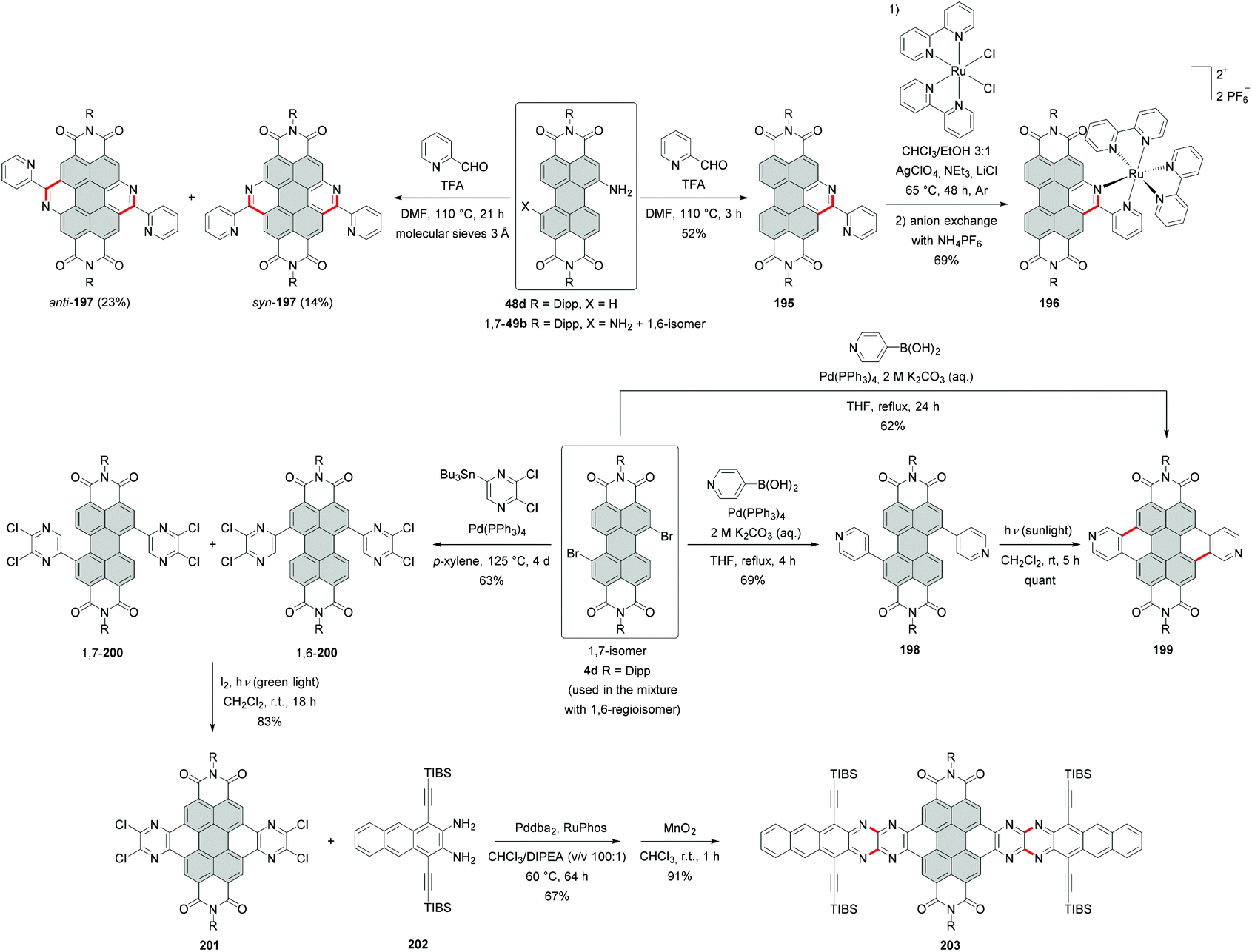 | ||
| Scheme 61 Synthesis of pyridine- and pyrazine-annulated PBIs. TIBS = triisobutylsilyl. | ||
Exposing a solution of pyridyl-substituted PBI 198 in methylene chloride to sunlight induced regiospecific annulation thereof to the pyridyl-bay-fused 199. Interestingly, compound 199 can be synthesized in one pot directly from 4d without isolation of the intermediate coupling product upon extension of the Suzuki reaction time from 4 h to 24 h. A two-step one-pot process afforded 199 in 62% yield, which is only slightly lower compared to the overall efficiency of the stepwise synthesis (Scheme 61).190 Bunz reported laterally extended PBIs with up to 13 annulated rings along the shorter PBI axis.191 The rational synthesis of these molecules utilized a regioisomeric mixture of 1,6- and 1,7-dibromo PBIs. Accordingly, a Stille coupling of the mixture afforded the corresponding pyrazine-substituted PBIs 1,7-200 and 1,6-200 whose subsequent annulation led to the same compound 201 (Scheme 61). As opposed to pyridine derivatives, fusion of the pyrazine ring to the perylene core proved ineffective under irradiation by sunlight. Yet, compound 201 could be obtained in as high as 83% yield when the chloroform solution of isomeric pyrazinyl-PBIs was irradiated with green light at room temperature in the presence of a catalytic amount of iodine. The subsequent annulation was accomplished by the Buchwald–Hartwig reaction between tetrachloro derivative 201 and various diamines, e.g.202, on increasing the number of fused benzene rings in the presence of Pddba2 and RuPhos. Subsequent oxidation with MnO2 provided the largest conjugated π-scaffold 203 (Scheme 61). Single crystal X-ray analysis of 203 confirmed the full planarity of this azaacene-functionalized PBI. Even though the compound is highly electron-deficient, as indicated by the first reduction potential at −0.37 V vs. Fe+/Fe, the OTFT devices (bottom-contact, top-gate architecture) employing 203 as an active layer showed only moderate electron mobility due to unfavorable packing arrangement in the solid state.
As depicted already in Scheme 37, exposure of monoarylated PBI 127a to sunlight promoted spontaneous cyclization of this compound with the formation of the benzene-fused PBI 128a.63 Photocyclization of phenyl-substituted molecule 127b was more sluggish. The process was monitored by thin-layer chromatography and terminated after three hours to give 128b in 92% yield (Scheme 62).192 Remarkably, when diaryl-substituted PBI 1,12-126a was treated with sunlight or irradiated using a halogen lamp, photocyclization reaction proceeded towards 128b through elimination of one benzene from intermediate 204, though at a much lower reaction rate. Accordingly, 128b was isolated in 83% yield after two days of keeping the solution in sunlight (Scheme 62). Importantly, this reaction was not observed for tetraaryl derivatives.63
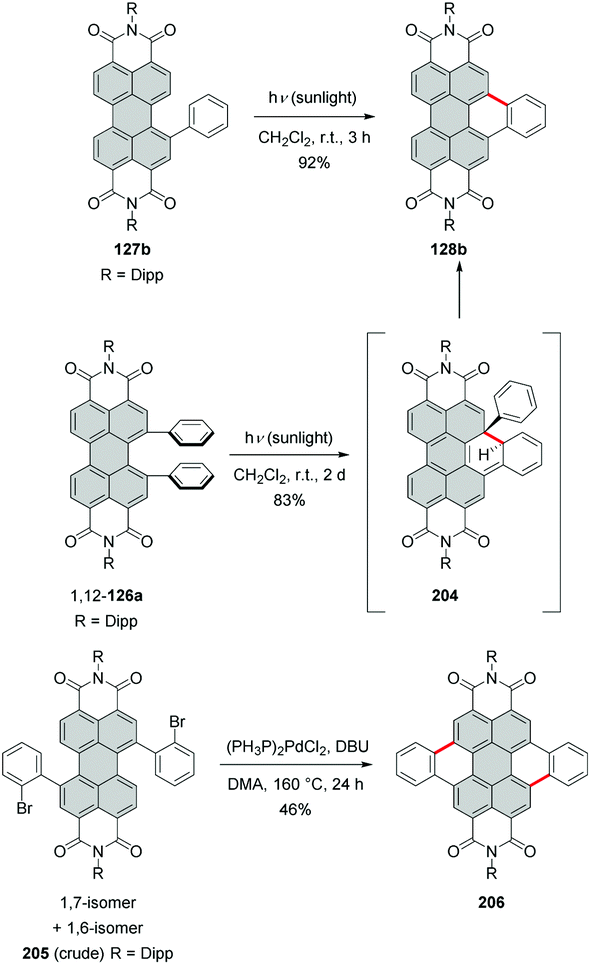 | ||
| Scheme 62 Annulation of arylated PBIs. DMA = dimethylacetamide. | ||
Müllen presented another synthetic approach towards benzene-fused PBIs. Dibenzocoronene 206 was synthesized by a Pd-catalyzed reaction of bis(bromophenyl)-substituted precursor 205, which was used in a mixture with its 1,6-isomer. The reaction was carried out in the presence of DBU at 160 °C and provided 206 in 46% yield (Scheme 62).193 However, the efficiency of this transformation can be somewhat underestimated, since dehydrohalogenation was carried out for the crude starting material containing around 10% of dehalogenated molecules. The product features markedly higher photostability than the coronene derivative 207a. As pointed out, this situation can be most likely associated with the partial double bond character of the C–C bonds of the newly formed six-membered rings of molecule 207a (Scheme 63).193
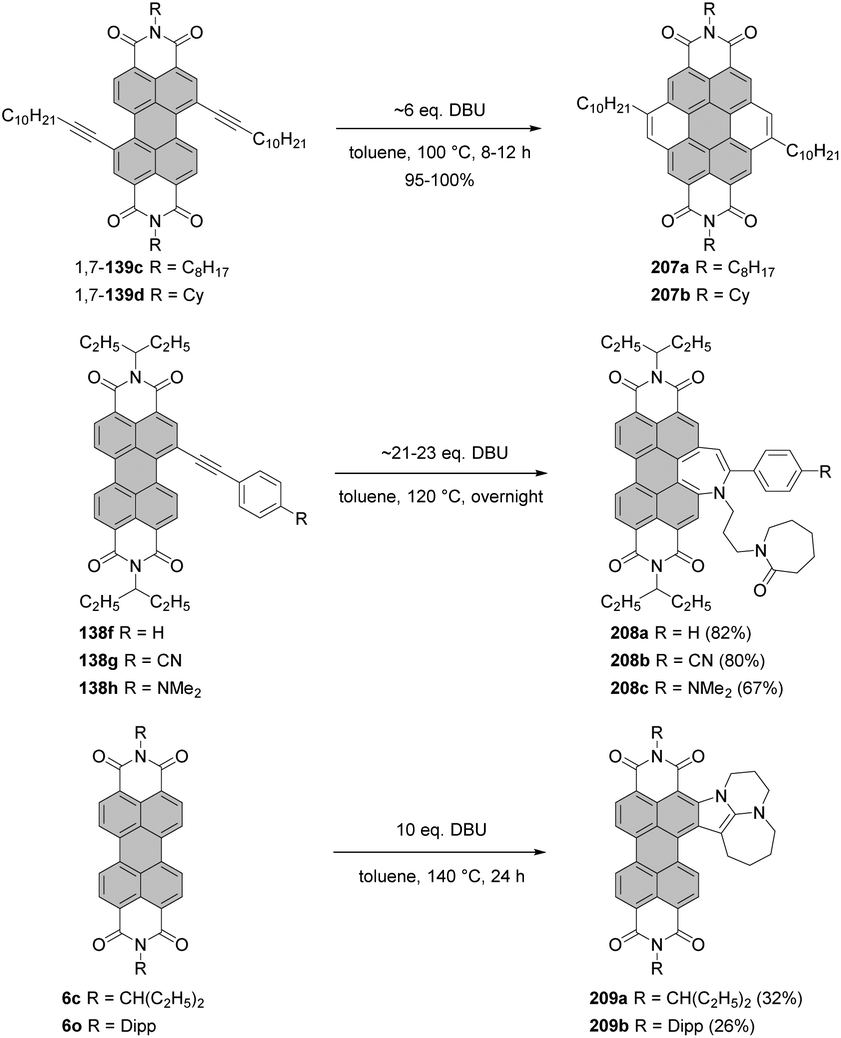 | ||
| Scheme 63 Cyclization of ethynyl-PBIs in the presence of DBU and reaction of core-unsubstituted PBIs with DBU. | ||
Coronene bis(dicarboximide)s, such as 207a and 207b, have already been described for the first time in 1998 by the same group. The synthesis of these molecules involved cyclization of the corresponding dialkynyl PBIs 1,7-139c,d triggered by DBU. This highly efficient process gave rise to liquid-crystalline materials, e.g.207b (Scheme 63), when appropriate imide groups and alkyl substituents on the coronene core were selected.194 Interestingly, when a significantly higher excess of DBU was used and the concentration of starting materials was increased by 6–7 times, the reaction of phenylethynyl 138f–h yielded azepine derivatives 208a–c with the appended caprolactam moiety in good yields, independent of the end substituent of the phenyl ring (Scheme 63).195 DBU, which is commonly used as nonnucleophilic base, participated as a substrate in the construction of a new molecular framework here. This finding, though surprising, is more common than expected and was observed for other compounds leading to the formation of e.g. DBU-annulated naphthalene bisimide196 or 4,7-diazaindoles.197 The plausible mechanism involves an attack of DBU nitrogen on the carbon alkyne atom, addition of water and concomitant ring opening of DBU, followed by the second formation of the C–N bond. Examination of the impact of other nonnucleophilic bases on the reaction outcome revealed that DABCO did not trigger this reaction, while 1,5-diazabicyclo[4.3.0]non-5-ene (DBN) provided only a trace amount of the desired seven-membered ring annulated product, being a component of a complex mixture.
A further increase in the concentration of the starting material and an increase in the temperature to 140 °C allowed the synthesis of ortho- and bay-annulated derivatives 209a,b from core-unsubstituted PBIs 6c/6o and 10 eq. of DBU, although the efficiencies of these transformations were rather low. The products were formed in 32% and 26% yields, respectively (Scheme 63). DBU acted here as N- and C-nucleophiles forming a new five-membered N-heterocyclic ring. Compound 209a exhibited panchromatic absorption spanning up to 950 nm. The lowest absorption energy band was of charge transfer origin and could be attributed to the presence of an electron-donating dialkylamino group in the annulated ring. CT is also partly responsible for fluorescence quenching. Yet, fluorescence could be recovered upon protonation with TFA,198 as in the case of azepine derivatives 208a.195
Twofold nucleophilic aromatic substitution of 1,7-dibromo PBI 4d with imidazole required only direct heating of the reaction mixture at 100 °C, whereas substitution reactions with the less reactive pyrazole and triazole were carried out in the presence of K2CO3 as a base and crown ether (Scheme 64). The ring closure was executed by photocyclization. The corresponding imidazole and 1,2,4-triazole derivatives 210 and 211 underwent twofold annulation, although the rate of the latter reaction was much slower. While exposure of 210 to sunlight over 3 h furnished an imidazole-fused product in 75% yield, compound 214 was formed in barely 40% yield after 6 days. On the other hand, irradiation of dipyrazolyl-PBI 175b gave rise to singly cyclized 212 in 75% yield, whereas the formation of a doubly cyclized derivative was not observed.199
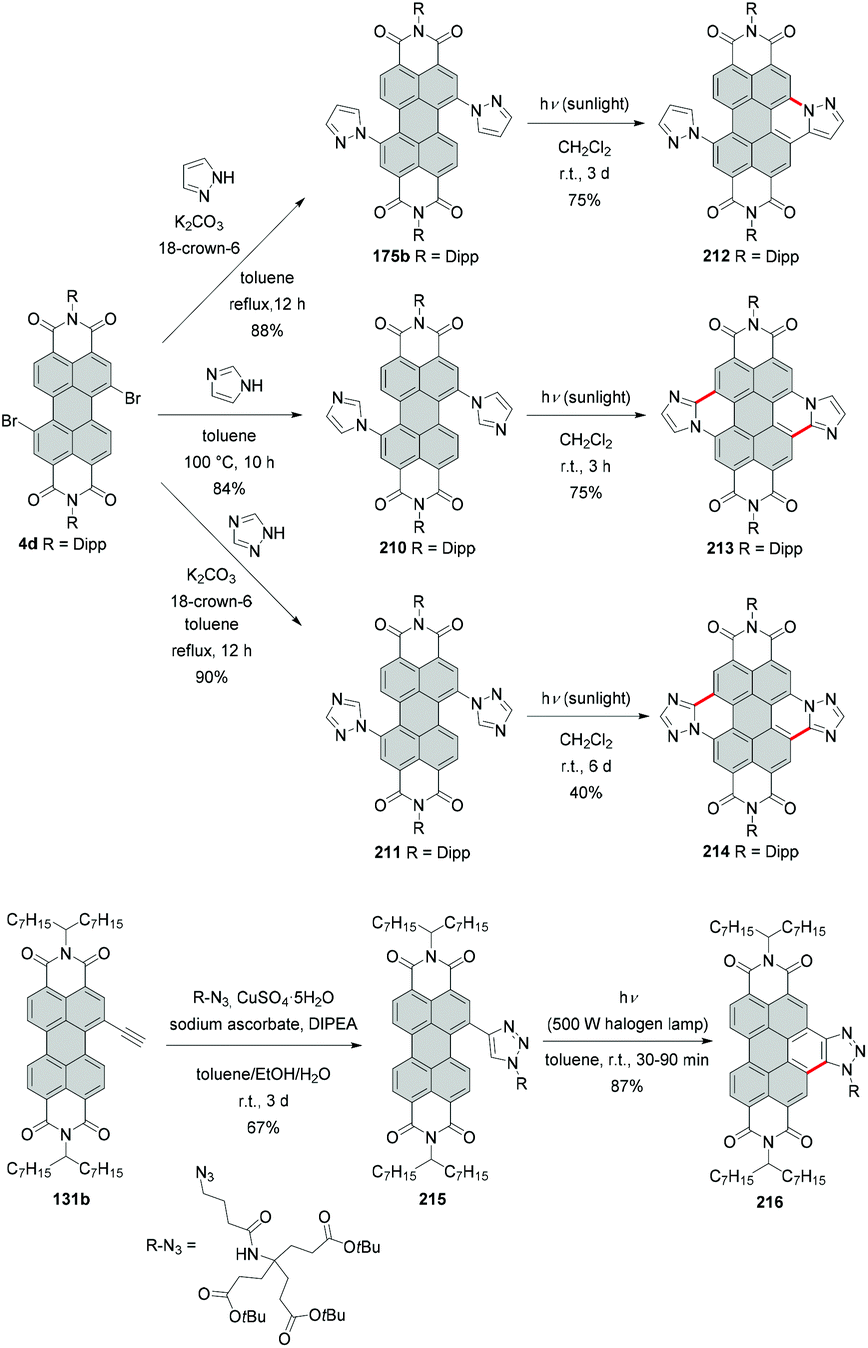 | ||
| Scheme 64 Synthesis of N-heterocycle annulated PBIs. | ||
The synthesis of triazole-functionalized PBIs differing in connectivity such that the triazole ring is attached to the PBI core via C–C single bonds required a different approach. An obvious choice for the preparation of 1,2,3-triazole-PBIs is copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) leading to the formation of a 1,4-substituted triazole derivative. Accordingly, ethynyl-PBI 131b was reacted with a dendritic azide to produce triazolyl-PBI 215 in 67% yield. The reaction was catalyzed by a copper salt in combination with sodium ascorbate in the absence of light. The triazolyl ring can be readily cyclized onto the perylene core by a photochemical electrocyclic reaction. For instance, mono-functionalized 215 was irradiated using a 500 W halogen lamp for 30–90 min to give triazole-fused 216 in an excellent yield. The preparation of a coronene derivative from a 1,7-ditriazolyl-PBI used as a mixture with its 1,6-regioisomer required a longer irradiation time of ca. 8–10 h.200
Thiophene-annulation for high-performance organic solar cells
Thiophene-annulation has evolved as the most useful strategy towards multichromophoric PBI systems that have proved to be very successful alternatives to fullerenes in bulk heterojunction solar cells in combination with polymer-based p-type semiconductors.22 Fusion of thiophene in the PBI bay area is typically accomplished either by treatment with FeCl3 or photocyclization. Selected examples are depicted in Scheme 65. Although the fusion site is sometimes of importance for the success of the reaction, the thiophene derivatives were efficiently fused to the PBI core either via the α- or β-position. For instance, compound 217 was obtained by coupling of thiophene at the bay position of the perylene core via the β-position. In the next step, photocyclization of 217 in the presence of a catalytic amount of iodine induced the formation of a C–C bond between the more reactive α-position of thiophene and a neighboring carbon atom of a perylene bay area. Thiophene-annulated compound 218 was readily functionalized by bromination of α-positions of thiophene followed by Suzuki coupling with pinacol boronate ester to yield 219, which was used as a donor component in bulk heterojunction (BHJ) organic solar cells.201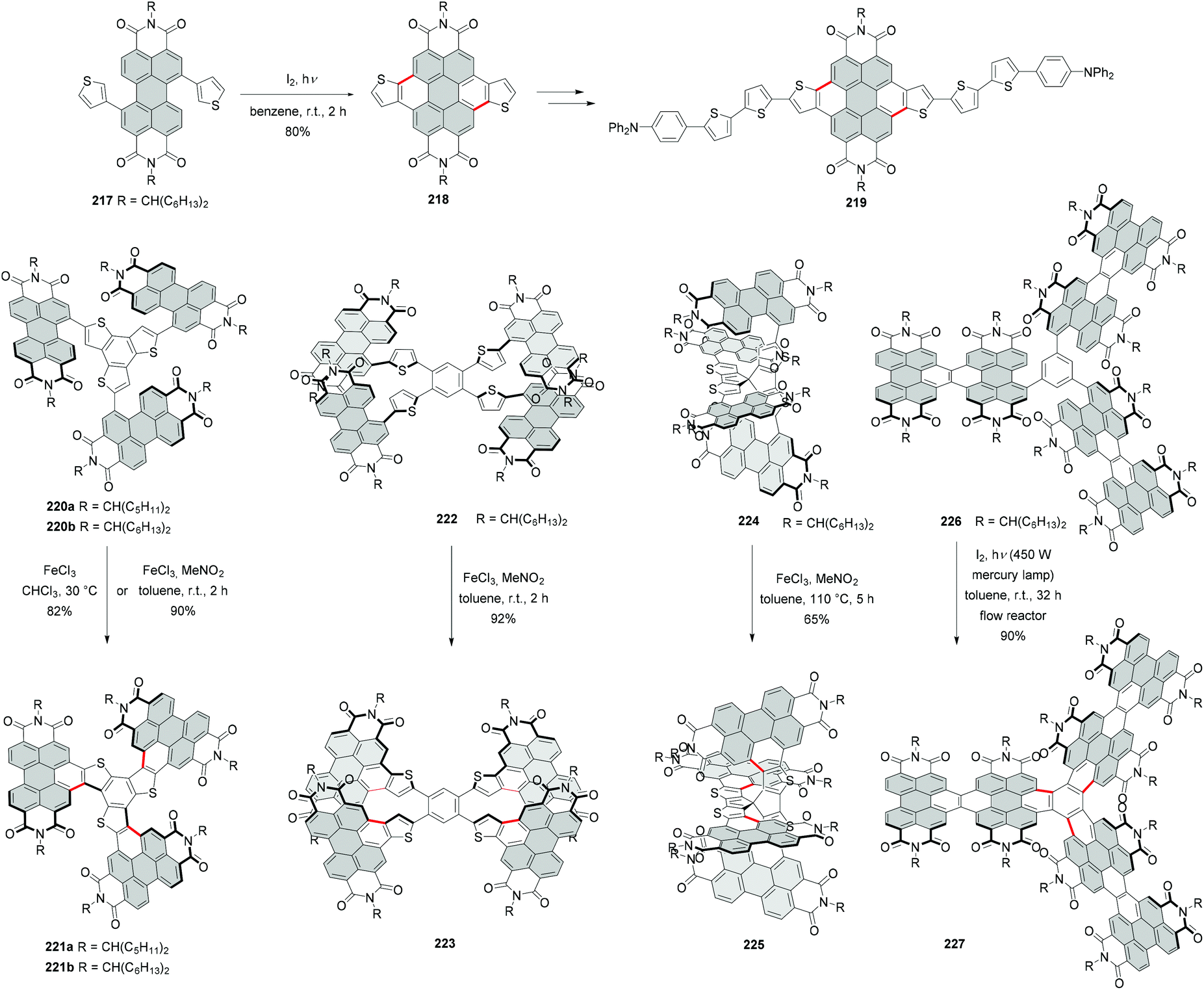 | ||
| Scheme 65 Synthesis of 3D multichromophoric PBIs by fusion of thiophene or carbocyclic rings with PBI cores. | ||
From a synthetic point of view, the preparation of the majority of planar or even more common highly twisted 3D (fully-)fused multi-PBIs involving thiophene or any other carbocyclic or heterocyclic ring follows the same scenario: arylation by Suzuki or Stille coupling between bromo-PBI and suitably functionalized coupling partners, followed by oxidative aromatic coupling with FeCl3 or photocyclization in the presence of iodine. Using these synthetic tools, it was possible to gather an impressive collection of complex contorted PBI structures of this prospering field. In this section, only selected examples that evolved as game changers in bulk heterojunction solar cells with p-type semiconducting polymers are discussed. A set of 3D fused molecules, including 221a,b and 223, were derived from multichromophoric arrays in which PBI units were appended to central thiophene-based aryl units. The precursors of 221b and 223 were prepared by microwave-assisted Stille and Suzuki couplings, respectively, between 1-bromo PBI and stannylated benzotrithiophene (BTT) or aryl boronic pinacol ester in 62–78% yields.16 Compound 220a was synthesized by the conventional Stille coupling under similar conditions in 65% yield.202 Fusion of these molecules was carried out by oxidative aromatic coupling using FeCl3 as an oxidant. Coupling of BTT derivatives 220a,b with FeCl3 provided three-dimensional trimeric PBIs 221a,b by fusion of β-carbon atoms of thiophene rings with bay carbon atoms of PBI units (Scheme 65).202 Compound 221a showed a higher electron mobility than that of non-fused tris-PBI 220a due to the favorable packing arrangement and afforded polymer OSCs with a PCE of >6%, which is three times higher than that of 220a.202 The related synthesis of 223 is also shown in Scheme 65 and the additional C–C bonds are highlighted in red.16 The consistently observed improvement of the OPV performance upon fusion in these molecules was attributed to the increased rigidity of the molecular frameworks. This in turn reduced the intermixing with the donor polymer, hence improving domain purity in the blend films of the OSCs. Another important effect of fusion was enhancement of the molar absorption coefficients of the systems in comparison with the more flexible precursors. Consequently, a record PCE value of above 10% was achieved for devices based on 223 of a double-decker geometry.15,16
An important positive effect of ring-fusion on packing patterns, blend film morphology and photovoltaic properties was also recognized for other twisted PBI-based molecules.203,204 A spiro compound 225 with perpendicularly arranged two pairs of PBI moieties was obtained by Stille coupling of stannylated 4,4′-spirobi[cyclopenta[2,1-b:3,4-b′]dithiophene] with 1-bromo PBI followed by fusing PBI units with the spiro core via oxidative coupling using FeCl3 as an oxidant. Cyclization of 224 was carried out at elevated temperature to give 225 in 65% yield. The compound was used as an acceptor in polymer solar cells reaching a PCE of around 9%.205
In contrast to ubiquitous thiophene derivatives, reports on furan-annulated PBIs are scarce in the literature. Single-core PBIs fused with one or two furan rings were obtained in moderate yields by exposing the reaction mixtures to sunlight.206 The addition of iodine was indispensable for the synthesis of the doubly cyclized product.
PBI arrays by fusion of carbocyclic and heterocyclic rings
Three-bladed rylene propellers, such as molecule 194 (see Scheme 60), were synthesized by a Suzuki coupling between 1-bromo PBI and 1,3,5-benzenetriboronic acid tris(pinacol) ester and a subsequent fusion. Cyclization of the intermediate was executed by irradiation in the presence of iodine. The yields varied between 83 and 93% depending on the imide substituents, that is, perfluoroalkyl or branched alkyl chains of different lengths.185,207 Due to the strong steric repulsion between PBI units attached to the small central core, the molecules are significantly twisted with torsion angles between neighboring PBI moieties of ca. 25 and 42°.185In their endeavor to achieve large three-dimensional graphene nanostructures, Xia and co-workers carried out the photocyclization of 226 in the presence of iodine in a flow reactor to give a propeller-shaped PBI array 227 (Scheme 65). This compound was used as an acceptor material in polymer solar cells showing a PCE of around 8%.208 Similarly, related 3D nanostructures with a central triptycene core and molecular weights up to 6500 Da could be synthesized by Nuckolls and co-workers by light-induced fusion of PBI nanoribbons at the triptycene unit in the presence of iodine. These novel materials were used as electron-extracting layers in perovskite solar cells.209
Another elegant approach towards non-planar PBI-based scaffolds was reported by Steigerwald and Nuckolls.210 Target molecules 229 and 231 were synthesized in two steps from bromo-PBI. Suzuki coupling thereof with 2,7-diborylated naphthalene or anthracene was followed by ultraviolet light-induced photocyclization in the presence of iodine as an oxidant. The corresponding double fusion of an acene unit with two PBI units gave rise to racemic helical PBIs 229 or 231 in quantitative yields (Scheme 66). In accord with the intrinsic reactivity of the acenes, the reaction took place at the peri-positions of both naphthalene and anthracene. Interestingly, compound 228 is prone to gradual oxidative photocyclization under ambient conditions to yield monocyclized bis-PBI. Separation of (P)- and (M)-enantiomers was executed by chiral HPLC. The studies of thermal stability against racemization revealed that compound 229 consisting of a [6]helicene subunit features an exceptionally high racemization barrier, whereas enantiomers of the anthracene analog interconvert already at room temperature in solution. Using this approach, also three PBI scaffolds could be fused to give a chiral superstructure with a large Cotton effect.211
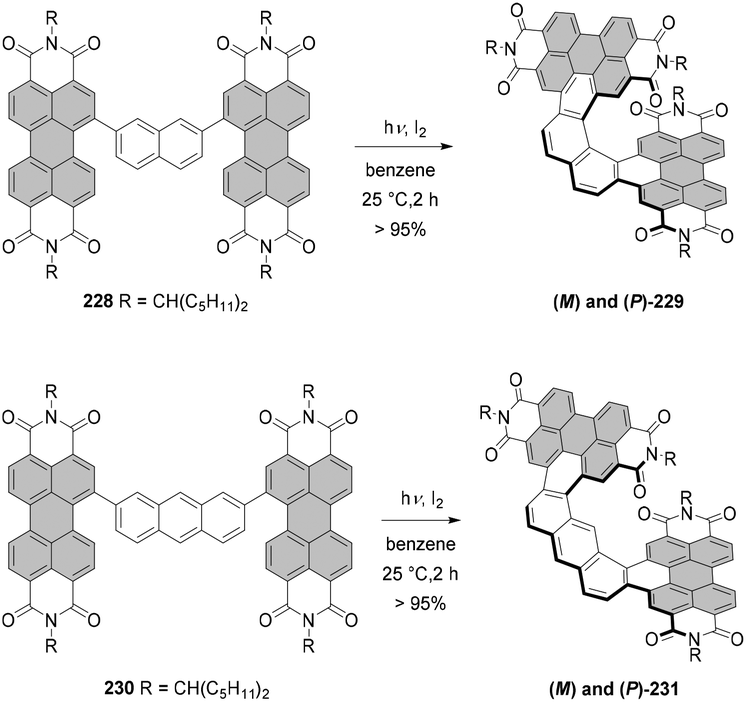 | ||
| Scheme 66 Synthesis of helical bis-PBI systems. | ||
Vinylene-bridged, spiro and directly linked PBI systems
The first synthesis of PBI molecules fused by ethylene bridges was accomplished by the cyclization of alkynyl-linked perylene tetracarboxylic acid esters and their subsequent conversion into the ethylene-fused dimeric PBI by Wang in moderate yield.152 A more efficient approach which proved to be also useful for the further lateral extension of the PBI core was accomplished by Nuckolls via the Mallory oxidation of ethylene-linked bis-PBI 134 with iodine as an oxidant.149 The reaction led to the efficient fusion of PBI units with vinylene bridges. This effective approach allowed the synthesis of fully π-conjugated dimer 232 and nanoribbons composed of up to four PBI units such as 233 (Scheme 67). Due to the steric repulsion between the C–H bonds of the two neighboring PBI subunits, the molecules are nonplanar and may consist of waggling and/or helical conformations. According to the DFT calculations, all the considered conformations are isoenergetic.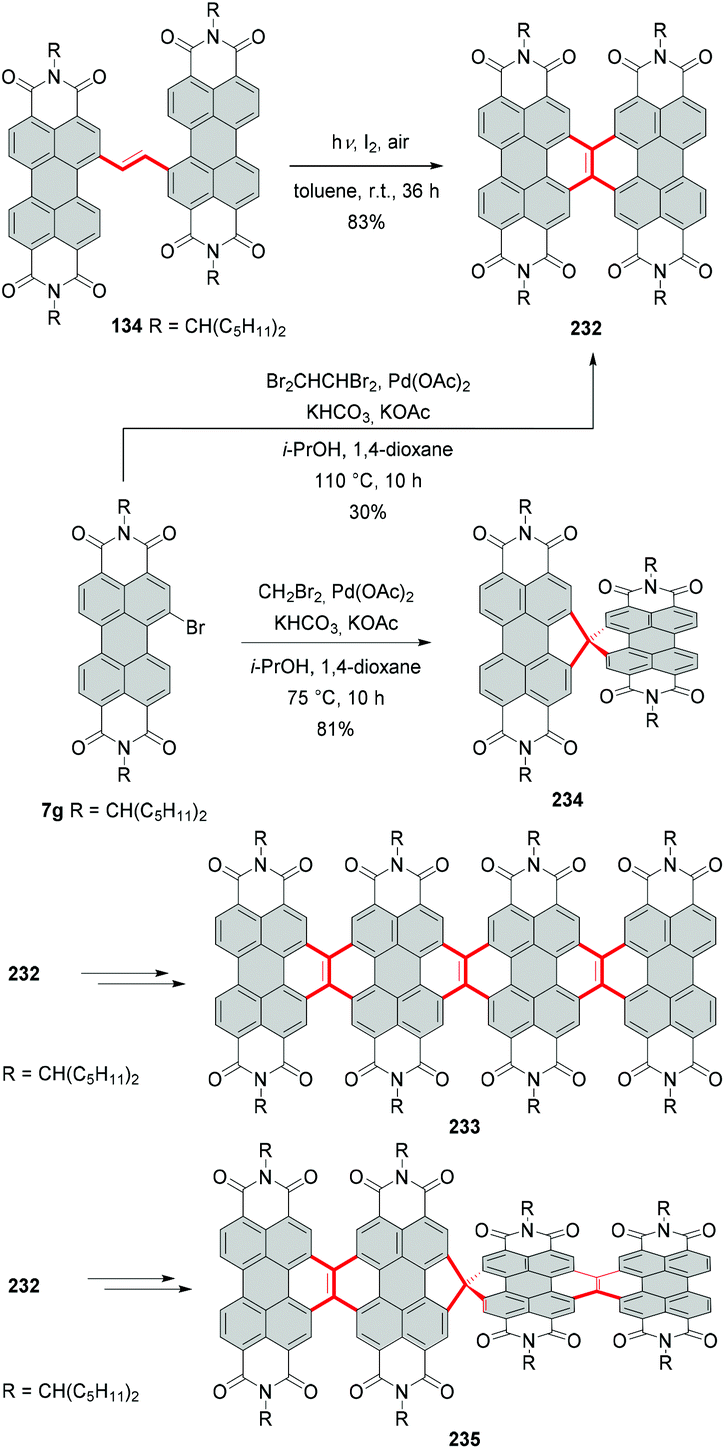 | ||
| Scheme 67 Synthesis of ethylene-fused and spiro multichromophoric PBI arrays. | ||
In 2017, Wang proposed the synthesis of ethylene-fused dimers directly from bromo-PBI. Thus, palladium-catalyzed reaction of 7g with tetrabromoethane in the presence of a base and i-PrOH as a reductant afforded product 232 in 30% yield.212 The unquestionable advantage of this method is its simplicity. Nevertheless, based on yields Nuckolls’ approach seems to be the preferable synthetic method for these compounds. Accordingly, the protocol was adapted to the synthesis of polymers with ethylene-fused dimeric PBI repeat units150,151 and as building blocks for large arrays composed of fused PBI units.208 The latter compound as well as Nuckolls’ tetrameric ribbon 233 and naphthoperylenediimide-vinylene polymer150,213 showed high potential for application in polymer OSCs as acceptor materials leading to PCEs above 8%.
A method introduced by Wang for the synthesis of the ethylene-fused PBI was also applied for the synthesis of spiro-fused PBI 234 of a cruciform configuration. Formation of the spirocyclic skeleton was realized by palladium-catalyzed reaction of 7g with dibromomethane. This facile and straightforward method afforded 234 in 81% yield and was used to prepare spiro-fused tetrameric system 235 (Scheme 67). The compound features a broad and intense absorption band (εmax = 143400) in the range of 450–600 nm and was used as an acceptor material in polymer solar cells with a PCE of 7.2%.212
Wang also explored the synthesis of directly linked multichromophoric PBIs via a combination of Ullmann coupling and C–H transformation (Scheme 68). Due to the steric repulsion between imide oxygen and core hydrogen of the neighboring PBI unit a considerable out-of-plane twisting of the molecule is observed.
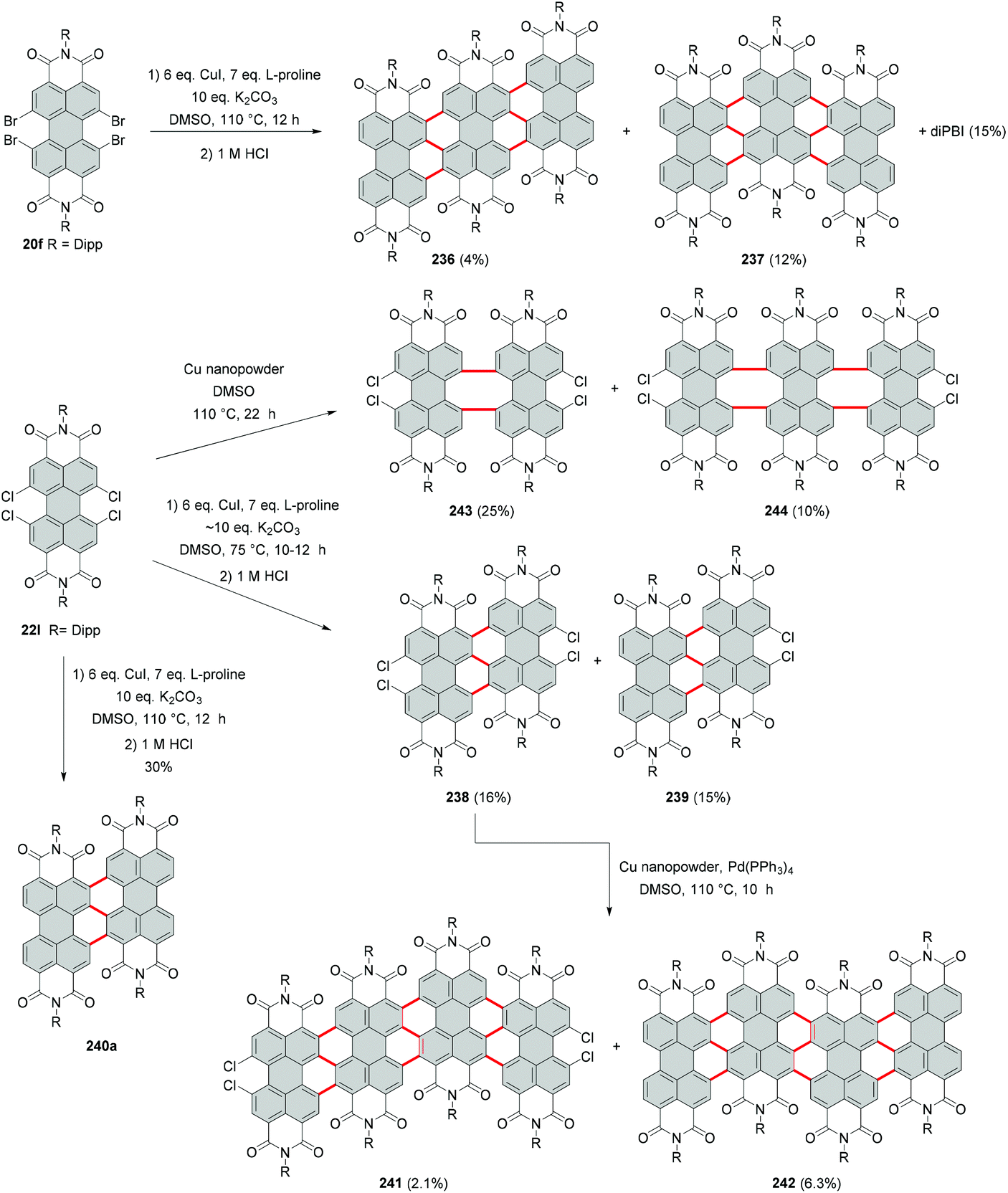 | ||
| Scheme 68 Synthesis of fused multichromophoric PBI arrays by copper-mediated coupling reactions. | ||
Likewise, fusion of three PBI chromophores leads to a significant distortion of the aromatic scaffold. Two low-energy conformations computed for both 236 and 237 differ in the geometry. In the first conformation, each next PBI unit is twisted in the same direction giving rise to a helical structure ((P)- and (M)-enantiomers), whereas the second conformation represents a meso form. A comparison of the experimental and computed NMR spectra suggests the preferable formation of meso-237.214 A similar transformation utilizing chlorinated precursor 22l as a starting material afforded preferentially dimeric PBI 238, presumably due to lower reactivity of chloride in copper-mediated coupling. Another key parameter affecting the reaction is the temperature. Accordingly, heating tetrachloro-PBI 22l with CuI, L-proline and K2CO3 at 75 °C produced bay-chlorinated fused bis-PBI 238 and 239 in 16% and 15% yields, respectively (Scheme 68).62,215 When the temperature was elevated to 110 °C, fully dehalogenated congener 240a was isolated in 30% yield.215 Importantly, when the reaction is conducted in the absence of a base, the cleavage of two halogen atoms in one bay area can be executed rather than the coupling of PBI chromophores.62
To obtain PBI nanoribbons consisting of four PBI units from chlorinated precursor, addition of a palladium catalyst was indispensable. Thus, Pd(PPh3)4-promoted, copper-mediated coupling of tetrachloro-diPBI 238 produced 241 and 242 in low yields.216 Utilization of nanosized copper powder promoted the formation of doubly linked PBIs 243 and 244 bearing chlorine atoms in bay areas.217 As opposed to almost planar triply linked multi-PBIs, subunits of 243 and 244 are oriented nearly perpendicularly versus each other, which reduces electronic communication between the chromophores. The presence of the twisted eight-membered ring between particular perylene cores restricts the interconversion process of (P)- to (M)-enantiomers. Dimeric compound 245a was successfully synthesized from 1,12-dibromo-PBI in 69% yield along with a small amount of triply linked 240a under similar conditions (Scheme 69).64 Analogous reaction of the dichloro precursor failed to point out lower reactivity of the chlorinated starting material. However, addition of Pd catalyst promoted the formation of the desired dimer 245b in 33% yield from dichloro-PBI 31b. On the other hand, the reaction thereof with CuI, L-proline and K2CO3 led preferentially to triply linked product 240b (Scheme 69).157
 | ||
| Scheme 69 Synthesis of doubly and triply linked PBIs from monobay dihalo-PBIs by copper-mediated coupling reactions. | ||
Fusion at the bay- and ortho-positions of a PBI scaffold
Apart from bay-annulated PBI dyes, there are few examples of PBI derivatives with aromatic units fused to the ortho- and bay-positions of a PBI scaffold. In this case a substituent is initially introduced at the ortho-position and later fused to the PBI core via the formation of a C–C bond with a bay carbon atom. For instance, extension of the π-conjugated system was achieved by the intramolecular ring fusion of tetrachloro-bay-substituted PBIs bearing aniline moieties at ortho positions. Palladium-catalyzed fourfold annulation of 149 provided propeller-like molecule 151 in 45% (Scheme 43).69 Treatment of unsubstituted PBI with sodium amide in benzonitrile gave rise to a derivative with an imidazole ring fused to 1,2-positions.218 Recently, Jiang and co-workers reported the formation of fourfold thiophene-annulated PBIs 247 by cyclization of tetraethynyl-PBIs 246 with elemental sulfur (Scheme 70). The reaction provided the annulated helical products in only ∼10% yield. Subsequent removal of TIPS groups afforded target molecules 247. Single crystal transistors based on these compounds showed high electron mobility reaching 0.90 cm2 V−1 s−1 for the compound bearing polyfluoroalkyl imide substituents.163 | ||
| Scheme 70 Synthesis of thiophene-annulated PBIs with thiophene rings fused to bay and ortho-positions. | ||
Conclusions
Hundred years after its discovery, the parent π-skeleton of perylene bisimide has replicated into a huge family of colorants with an enormous number of derivatives. Driven by new functional demands and supported by the availability of novel synthetic methodologies, a structural evolution became possible giving access to structures which twenty years ago were elusive. Indeed, this progress becomes obvious if we compare the contents of the previous two main reviews dedicated to the synthesis of perylene bisimides published in 1995 and 2011 with the current one. The first one by Langhals primarily focused on the variation of the imide substituents and absorption and emission properties of these dyes,2 and the second one by Marder on imide and bay-functionalization for organic electronic applications.12 But only from the structures and synthetic methods discussed in the given review, we see the proficiency to tailor PBI molecules fully according to the chemists’ will. This includes control of optical, redox and conformational properties of PBIs and their functionalization by synthetic methods beyond the formerly favored halogenations and the reactions of halogenated PBIs with nucleophiles. Thus, efficient synthetic protocols became available for the preparation of perylene bisimide dyes with almost any desired substitution pattern. Outstanding highlights of this research are provided by annulation reactions towards linear and star-shaped PBI oligomers, prompted by the development of synthetic methodologies and analytical tools to characterize such compounds with masses of 6000 Da and beyond.Nevertheless, while such large-sized π-scaffolds are intriguing examples of academic research, the complexity of their synthesis may hamper their prospects for commercialization as functional dyes in new high technology products such as organic solar cells. Thus, straightforward synthetic routes as well as efficient reactions under mild conditions with a broad substrate scope and excellent functional group tolerance would not only boost the progress of PBI research, but also would make possible the commercialization of PBI dyes. From the perspective of commercial application, high selectivity and efficiency are particularly important, as they give a chance to easily purify material by crystallization rather than costly chromatography, and decrease material consumption in the form of side-products. In this context, methods employing inexpensive metal catalysts would also be highly appreciated. For instance, they could replace cost-intensive Rh, Ru or Ir catalysts commonly used for ortho-functionalization of PBIs. Accordingly, for the continuation of the success story of the highly (photo-)stable perylene bisimides as market products, the development of new synthetic methods such as direct CH-functionalizations is of equal importance as the exploration of new functional applications for PBIs. The establishment of new synthetic protocols is therefore invaluable to the dynamically growing field of perylene bisimide dyes and the search for more efficient synthetic methods for the preparation of perylene dyes is warranted. Such endeavors will focus on reducing the number of synthetic steps and limiting the production of by-products.
An important role will also be played by smart strategies for harnessing the intrinsic reactivity of particular perylene derivatives that would allow the installation of mixed substituents in a given substitution pattern and with high precision. Likewise, fourfold functionalization of bay-positions remains an open challenge. Despite some interesting reports in the last few years, the number of functional groups that can be introduced into bay areas is still very limited. Moreover, these reactions are typically not high-yielding and, as such, are very often a bottleneck to the synthesis. These factors also explain the dearth of configurationally stable chiral PBI compounds, in which the chirality stems from the twisting of the PBI core. Such materials may be attractive for application in new technologies requiring excellent optical properties and chirality, e.g. circularly polarized light emitting diodes (CP-LEDs) and other optoelectronic devices. Therefore, the synthesis of 1,6,7,12-tetra-substituted PBIs, including configurationally stable molecules, certainly deserves some attention.
The synthesis of materials with tailored properties, scalability of the technique, and development of new methods that would be more attractive to the industrial sector and would enable large-scale production are all important issues that should be addressed in the future to make PBIs competitive in the field of functional dyes. As we experience a steadily growing interest in perylene bisimide scaffolds for a continuously increasing number of applications, we expect that this trend will not change during the coming years. With the areas of organic electronics and photovoltaics entering a more mature stage, new applications in photonics and sensing already appear on the horizon. These may again take advantage of the superb optical and photostability properties of perylene dyes, in particular their outstanding photoluminescence, as opposed to organic electronics and photovoltaics which primarily relied on the electron acceptor character of perylene bisimides. Any kind of new application can now profit from the substantial progress that has already been achieved for the synthesis of perylene bisimides covered herein. Accordingly, we anticipate that this review can give the reader the overview of available synthetic transformations to successfully master sighted goals in the field of PBIs. We also hope that the compilation of reactions and structures presented in this review will encourage chemists to further explore the reactivity of perylene dyes as well as their larger congeners. The limitations or ambiguity related to some reactions may serve as impetus for further investigations and the development of new synthetic methods for interesting new perylene bisimide based architectures.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful to all our co-workers and collaboration partners who contributed to our research in the field of perylene dyes during the last two decades. ANK thanks the Alexander-von-Humboldt Foundation for a postdoctoral fellowship and the University of Würzburg for supporting her entry into an independent career by the Emil-Hilb Program.Notes and references
- W. Herbst and K. Hunger, Industrial Organic Pigments, Wiley-VCH, Weinheim, 2004 Search PubMed.
- H. Langhals, Heterocycles, 1995, 40, 477 CrossRef CAS.
- R. Iden and G. Seybold, (BASF AG), Ger. Pat. Appl, DE3434059A1, 1985 Search PubMed.
- G. Seybold and G. Wagenblast, Dyes Pigm., 1989, 11, 303 CrossRef.
- F. Würthner, Chem. Commun., 2004, 1564 RSC.
- R. Gronheid, A. Stefan, M. Cotlet, J. Hofkens, J. Qu, K. Müllen, M. Van der Auweraer, J. W. Verhoeven and F. C. De Schryver, Angew. Chem., Int. Ed., 2003, 42, 4209 CrossRef CAS PubMed.
- P. Tinnefeld, J. Hofkens, D.-P. Herten, S. Masuo, T. Vosch, M. Cotlet, S. Habuchi, K. Müllen, F. C. De Schryver and M. Sauer, ChemPhysChem, 2004, 5, 1786 CrossRef CAS PubMed.
- T. Weil, T. Vosch, J. Hofkens, K. Peneva and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 9068 CrossRef CAS PubMed.
- M. Sun, K. Müllen and M. Yin, Chem. Soc. Rev., 2016, 45, 1513 RSC.
- S. Betzold, S. Herbst, A. A. P. Trichet, J. M. Smith, F. Würthner, S. Höfling and C. P. Dietrich, ACS Photonics, 2018, 5, 90 CrossRef CAS.
- A. Nowak-Król, K. Shoyama, M. Stolte and F. Würthner, Chem. Commun., 2018, 54, 13763 RSC.
- C. Huang, S. Barlow and S. R. Marder, J. Org. Chem., 2011, 76, 2386 CrossRef CAS PubMed.
- S. Rajaram, R. Shivanna, S. K. Kandappa and K. S. Narayan, J. Phys. Chem. Lett., 2012, 3, 2405 CrossRef CAS PubMed.
- R. Shivanna, S. Shoaee, S. Dimitrov, S. K. Kandappa, S. Rajaram, J. R. Durrant and K. S. Narayan, Energy Environ. Sci., 2014, 7, 435 RSC.
- J. Zhang, Y. Li, J. Huang, H. Hu, G. Zhang, T. Ma, P. C. Y. Chow, H. Ade, D. Pan and H. Yan, J. Am. Chem. Soc., 2017, 139, 16092 CrossRef CAS PubMed.
- H. Hu, Y. Li, J. Zhang, Z. Peng, L.-K. Ma, J. Xin, J. Huang, T. Ma, K. Jiang, G. Zhang, W. Ma, H. Ade and H. Yan, Adv. Energy Mater., 2018, 1800234 CrossRef.
- J. Dostál, F. Fennel, F. Koch, S. Herbst, F. Würthner and T. Brixner, Nat. Commun., 2018, 9, 2466 CrossRef PubMed.
- C. Kaufmann, W. Kim, A. Nowak-Król, Y. Hong, D. Kim and F. Würthner, J. Am. Chem. Soc., 2018, 140, 4253 CrossRef CAS PubMed.
- P. Spenst, R. M. Young, M. R. Wasielewski and F. Würthner, Chem. Sci., 2016, 7, 5428 RSC.
- S. W. Eaton, L. E. Shoer, S. D. Karlen, S. M. Dyar, E. A. Margulies, B. S. Veldkamp, C. Ramanan, D. A. Hartzler, S. Savikhin, T. J. Marks and M. R. Wasielewski, J. Am. Chem. Soc., 2013, 135, 14701 CrossRef CAS PubMed.
- G. Zhang, J. Zhao, P. C. Y. Chow, K. Jiang, J. Zhang, Z. Zhu, J. Zhang, F. Huang and H. Yan, Chem. Rev., 2018, 118, 3447 CrossRef CAS PubMed.
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K.-Y. Jen, S. R. Marder and X. Zhan, Nat. Rev. Mater., 2018, 3, 18003 CrossRef CAS.
- Z. Chen, B. Fimmel and F. Würthner, Org. Biomol. Chem., 2012, 10, 5845 RSC.
- S. Demmig and H. Langhals, Chem. Ber., 1988, 121, 225 CrossRef CAS.
- R. Schmidt, J. H. Oh, Y.-S. Sun, M. Deppisch, A.-M. Krause, K. Radacki, H. Braunschweig, M. Könemann, P. Erk, Z. Bao and F. Würthner, J. Am. Chem. Soc., 2009, 131, 6215 CrossRef CAS PubMed.
- N. E. Aksakal, Y. Chumakov and F. Yuksel, J. Chem. Crystallogr., 2018 DOI:10.1007/s10870-018-0735-1.
- F. Würthner, C. Thalacker, A. Sautter, W. Schärtl, W. Ibach and O. Hollricher, Chem. – Eur. J., 2000, 6, 3871 CrossRef.
- T. E. Kaiser, V. Stepanenko and F. Würthner, J. Am. Chem. Soc., 2009, 131, 6719 CrossRef CAS PubMed.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962 CrossRef PubMed.
- R. K. Dubey, N. Westerveld, E. J. R. Sudholter, F. C. Grozema and W. F. Jager, Org. Chem. Front., 2016, 3, 1481 RSC.
- S. Sengupta, R. K. Dubey, R. W. M. Hoek, S. P. P. van Eeden, D. D. Gunbaş, F. C. Grozema, E. J. R. Sudhölter and W. F. Jager, J. Org. Chem., 2014, 79, 6655 CrossRef CAS PubMed.
- M. Takahashi, K. Asaba, T. T. Lua, T. Inuzuka, N. Uemura, M. Sakamoto, T. Sengoku and H. Yoda, J. Org. Chem., 2018, 83, 624 CrossRef CAS PubMed.
- R. Wang, Z. Shi, C. Zhang, A. Zhang, J. Chen, W. Guo and Z. Sun, Dyes Pigm., 2013, 98, 450 CrossRef CAS.
- S. Alibert-Fouet, I. Seguy, J.-F. Bobo, P. Destruel and H. Bock, Chem. – Eur. J., 2007, 13, 1746 CrossRef CAS.
- Á. J. Jiménez, M.-J. Lin, C. Burschka, J. Becker, V. Settels, B. Engels and F. Würthner, Chem. Sci., 2014, 5, 608 RSC.
- P. Osswald and F. Würthner, J. Am. Chem. Soc., 2007, 129, 14319 CrossRef CAS PubMed.
- A. Böhm, H. Arms, G. Henning and P. Blaschka, (BASF AG), Ger. Pat. Appl, DE19547209A1, 1997 Search PubMed.
- F. Würthner, V. Stepanenko, Z. Chen, C. R. Saha-Möller, N. Kocher and D. Stalke, J. Org. Chem., 2004, 69, 7933 CrossRef PubMed.
- P. Rajasingh, R. Cohen, E. Shirman, L. J. W. Shimon and B. Rybtchinski, J. Org. Chem., 2007, 72, 5973 CrossRef CAS PubMed.
- Y. Yang, Y. Wang, Y. Xie, T. Xiong, Z. Yuan, Y. Zhang, S. Qian and Y. Xiao, Chem. Commun., 2011, 47, 10749 RSC.
- R. K. Gupta and A. S. Achalkumar, J. Org. Chem., 2018, 83, 6290 CrossRef CAS PubMed.
- W. Qiu, S. Chen, X. Sun, Y. Liu and D. Zhu, Org. Lett., 2006, 8, 867 CrossRef CAS PubMed.
- F. Rigodanza, E. Tenori, A. Bonasera, Z. Syrgiannis and M. Prato, Eur. J. Org. Chem., 2015, 5060 CrossRef CAS.
- M. Könemann and G. Mattern, (BASF SE), PCT Int. Appl, WO2009/000831A1, 2008 Search PubMed.
- Y. Kumar, S. Kumar, S. K. Keshri, J. Shukla, S. S. Singh, T. S. Thakur, M. Denti, A. Facchetti and P. Mukhopadhyay, Org. Lett., 2016, 18, 472 CrossRef CAS PubMed.
- A. Jozeliūnaitė, R. Striela, L. Labanauskas and E. Orentas, Synthesis, 2017, 49, 5176 CrossRef.
- K. Tambara, N. Ponnuswamy, G. Hennrich and G. D. Pantoş, J. Org. Chem., 2011, 76, 3338 CrossRef CAS PubMed.
- X. Gao, W. Qiu, X. Yang, Y. Liu, Y. Wang, H. Zhang, T. Qi, Y. Liu, K. Lu, C. Du, Z. Shuai, G. Yu and D. Zhu, Org. Lett., 2007, 9, 3917 CrossRef CAS PubMed.
- C. Röger, S. Ahmed and F. Würthner, Synthesis, 2007, 1872 Search PubMed.
- C. Röger and F. Würthner, J. Org. Chem., 2007, 72, 8070 CrossRef PubMed.
- J. H. Hurenkamp, W. R. Browne, R. Augulis, A. Pugžlys, P. H. M. van Loosdrecht, J. H. van Esch and B. L. Feringa, Org. Biomol. Chem., 2007, 5, 3354 RSC.
- L. Perrin and P. Hudhomme, Eur. J. Org. Chem., 2011, 5427 CrossRef CAS.
- M. Sadrai, L. Hadel, R. R. Sauers, S. Husain, K. Krogh-Jespersen, J. D. Westbrook and G. R. Bird, J. Phys. Chem., 1992, 96, 7988 CrossRef CAS.
- W. Wang, A. D. Shaller and A. D. Q. Li, J. Am. Chem. Soc., 2008, 130, 8271 CrossRef CAS PubMed.
- M. Queste, C. Cadiou, B. Pagoaga, L. Giraudet and N. Hoffmann, New J. Chem., 2010, 34, 2537 RSC.
- R. K. Dubey, N. Westerveld, F. C. Grozema, E. J. R. Sudhölter and W. F. Jager, Org. Lett., 2015, 17, 1882 CrossRef CAS PubMed.
- J. Baggerman, D. C. Jagesar, R. A. L. Vallée, J. Hofkens, F. C. De Schryver, F. Schelhase, F. Vögtle and A. M. Brouwer, Chem. – Eur. J., 2007, 13, 1291 CrossRef CAS PubMed.
- A. Shaygan Nia, C. Enders and W. H. Binder, Tetrahedron, 2012, 68, 722 CrossRef.
- Z. Chen, M. G. Debije, T. Debaerdemaeker, P. Osswald and F. Würthner, ChemPhysChem, 2004, 5, 137 CrossRef CAS PubMed.
- F. Würthner, A. Sautter and J. Schilling, J. Org. Chem., 2002, 67, 3037 CrossRef.
- E. Nuin, V. Lloret, K. Amsharov, F. Hauke, G. Abellán and A. Hirsch, Chem. – Eur. J., 2018, 24, 4671 CrossRef CAS PubMed.
- Y. Zhen, H. Qian, J. Xiang, J. Qu and Z. Wang, Org. Lett., 2009, 11, 3084 CrossRef CAS PubMed.
- B. Pagoaga, L. Giraudet and N. Hoffmann, Eur. J. Org. Chem., 2014, 5178 CrossRef CAS.
- W. Jiang, C. Xiao, L. Hao, Z. Wang, H. Ceymann, C. Lambert, S. Di Motta and F. Negri, Chem. – Eur. J., 2012, 18, 6764 CrossRef CAS PubMed.
- M. Gsänger, J. H. Oh, M. Könemann, H. W. Höffken, A.-M. Krause, Z. Bao and F. Würthner, Angew. Chem., Int. Ed., 2010, 49, 740 CrossRef PubMed.
- M.-M. Ling, P. Erk, M. Gomez, M. Koenemann, J. Locklin and Z. Bao, Adv. Mater., 2007, 19, 1123 CrossRef CAS.
- M. Sadrai, G. R. Bird, J. A. Potenza and H. J. Schugar, Acta Crystallogr., Sect. C: Struct. Chem., 1990, 46, 637 CrossRef.
- M. Koenemann, G. Mattern and G. Weber, (BASF SE), PCT Int. Appl, WO2009/024512A1, 2009 Search PubMed.
- W. Yue, W. Jiang, M. Böckmann, N. L. Doltsinis and Z. Wang, Chem. – Eur. J., 2014, 20, 5209 CrossRef CAS PubMed.
- F. Würthner, P. Osswald, R. Schmidt, T. E. Kaiser, H. Mansikkamäki and M. Könemann, Org. Lett., 2006, 8, 3765 CrossRef PubMed.
- S. Seifert, D. Schmidt and F. Würthner, Chem. Sci., 2015, 6, 1663 RSC.
- R. Schmidt, P. Osswald, M. Könemann and F. Würthner, Z. Naturforsch., B: Chem. Sci., 2009, 64, 735 CAS.
- R. Schmidt, M. M. Ling, J. H. Oh, M. Winkler, M. Könemann, Z. Bao and F. Würthner, Adv. Mater., 2007, 19, 3692 CrossRef CAS.
- Y. Shi, H. Qian, Y. Li, W. Yue and Z. Wang, Org. Lett., 2008, 10, 2337 CrossRef CAS PubMed.
- D. Ma, Y. Zhang, J. Yao, S. Wu and F. Tao, J. Am. Chem. Soc., 1998, 120, 12459 CrossRef CAS.
- A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 14844 CrossRef CAS PubMed.
- K.-Y. Chen and T. J. Chow, Tetrahedron Lett., 2010, 51, 5959 CrossRef CAS.
- L. Hao, W. Jiang and Z. Wang, Tetrahedron, 2012, 68, 9234 CrossRef CAS.
- H. Langhals and S. Kirner, Eur. J. Org. Chem., 2000, 365 CrossRef CAS.
- R. Wang, G. Li, A. Zhang, W. Wang, G. Cui, J. Zhao, Z. Shi and B. Tang, Chem. Commun., 2017, 53, 6918 RSC.
- D. Meng, D. Sun, C. Zhong, T. Liu, B. Fan, L. Huo, Y. Li, W. Jiang, H. Choi, T. Kim, J. Y. Kim, Y. Sun, Z. Wang and A. J. Heeger, J. Am. Chem. Soc., 2016, 138, 375 CrossRef CAS PubMed.
- H.-Y. Tsai, C.-W. Chang and K.-Y. Chen, Tetrahedron Lett., 2014, 55, 884 CrossRef CAS.
- H.-Y. Tsai and K.-Y. Chen, Dyes Pigm., 2013, 96, 319 CrossRef CAS.
- Y. Zhang, Z. Zhao, X. Huang, Y. Xie, C. Liu, J. Li, X. Guan, K. Zhang, C. Cheng and Y. Xiao, RSC Adv., 2012, 2, 12644 RSC.
- R. K. Gupta, D. S. Shankar Rao, S. K. Prasad and A. S. Achalkumar, Chem. – Eur. J., 2018, 24, 3566 CrossRef CAS PubMed.
- R. K. Gupta, S. K. Pathak, B. Pradhan, M. Gupta, S. K. Pal and A. A. Sudhakar, ChemPhysChem, 2016, 17, 859 CrossRef CAS PubMed.
- G. Li, Y. Zhao, J. Li, J. Cao, J. Zhu, X. W. Sun and Q. Zhang, J. Org. Chem., 2015, 80, 196 CrossRef CAS PubMed.
- K.-Y. Chen, T.-C. Fang and M.-J. Chang, Dyes Pigm., 2012, 92, 517 CrossRef CAS.
- H.-Y. Tsai, C.-W. Chang and K.-Y. Chen, Molecules, 2014, 19, 327 CrossRef PubMed.
- X. Zhang, C. Zhan, X. Zhang and J. Yao, Tetrahedron, 2013, 69, 8155 CrossRef CAS.
- K. Khokhlov, N. J. Schuster, F. Ng and C. Nuckolls, Org. Lett., 2018, 20, 1991 CrossRef CAS PubMed.
- T. Teraoka, S. Hiroto and H. Shinokubo, Org. Lett., 2011, 13, 2532 CrossRef CAS PubMed.
- G. Battagliarin, C. Li, V. Enkelmann and K. Müllen, Org. Lett., 2011, 13, 3012 CrossRef CAS PubMed.
- S. Nakazono, Y. Imazaki, H. Yoo, J. Yang, T. Sasamori, N. Tokitoh, T. Cédric, H. Kageyama, D. Kim, H. Shinokubo and A. Osuka, Chem. – Eur. J., 2009, 15, 7530 CrossRef CAS PubMed.
- S. Nakazono, S. Easwaramoorthi, D. Kim, H. Shinokubo and A. Osuka, Org. Lett., 2009, 11, 5426 CrossRef CAS PubMed.
- F. Kakiuchi, Y. Matsuura, S. Kan and N. Chatani, J. Am. Chem. Soc., 2005, 127, 5936 CrossRef CAS PubMed.
- J. Wu, D. He, L. Zhang, Y. Liu, X. Mo, J. Lin and H.-J. Zhang, Org. Lett., 2017, 19, 5438 CrossRef CAS PubMed.
- D. T. Racys, S. A. I. Sharif, S. L. Pimlott and A. Sutherland, J. Org. Chem., 2016, 81, 772 CrossRef CAS PubMed.
- N. Schröder, F. Lied and F. Glorius, J. Am. Chem. Soc., 2015, 137, 1448 CrossRef PubMed.
- G. Battagliarin, Y. Zhao, C. Li and K. Müllen, Org. Lett., 2011, 13, 3399 CrossRef CAS PubMed.
- J. Zhang, S. Singh, D. K. Hwang, S. Barlow, B. Kippelen and S. R. Marder, J. Mater. Chem. C, 2013, 1, 5093 RSC.
- S. Ito, S. Hiroto and H. Shinokubo, Org. Lett., 2013, 15, 3110 CrossRef CAS PubMed.
- Y. Zhao and M. R. Wasielewski, Tetrahedron Lett., 1999, 40, 7047 CrossRef CAS.
- L. Fan, Y. Xu and H. Tian, Tetrahedron Lett., 2005, 46, 4443 CrossRef CAS.
- R. K. Dubey, A. Efimov and H. Lemmetyinen, Chem. Mater., 2011, 23, 778 CrossRef CAS.
- L. George, Z. Ahmed, H. Lemmetyinen and A. Efimov, Eur. J. Org. Chem., 2015, 584 CrossRef CAS.
- G. Rauch and S. Höger, Chem. Commun., 2014, 50, 5659 RSC.
- Y. Shibano, H. Imahori and C. Adachi, J. Phys. Chem. C, 2009, 113, 15454 CrossRef CAS.
- H. Wang, T. E. Kaiser, S. Uemura and F. Würthner, Chem. Commun., 2008, 1181 RSC.
- C. Zhao, Y. Zhang, R. Li, X. Li and J. Jiang, J. Org. Chem., 2007, 72, 2402 CrossRef CAS PubMed.
- M. J. Ahrens, M. J. Tauber and M. R. Wasielewski, J. Org. Chem., 2006, 71, 2107 CrossRef CAS PubMed.
- C. Domínguez, M. J. Baena, S. Coco and P. Espinet, Dyes Pigm., 2017, 140, 375 CrossRef.
- M.-J. Lin, Á. J. Jiménez, C. Burschka and F. Würthner, Chem. Commun., 2012, 48, 12050 RSC.
- M.-J. Lin, B. Fimmel, K. Radacki and F. Würthner, Angew. Chem., Int. Ed., 2011, 50, 10847 CrossRef CAS PubMed.
- D. Schmidt, M. Son, J. M. Lim, M.-J. Lin, I. Krummenacher, H. Braunschweig, D. Kim and F. Würthner, Angew. Chem., Int. Ed., 2015, 54, 13980 CrossRef CAS PubMed.
- C. Kohl, T. Weil, J. Qu and K. Müllen, Chem. – Eur. J., 2004, 10, 5297 CrossRef CAS PubMed.
- Y. Li, Z. Qing, Y. Yu, T. Liu, R. Jiang and Y. Li, Chem. – Asian J., 2012, 7, 1934 CrossRef CAS PubMed.
- J. Schill, S. van Dun, M. J. Pouderoijen, H. M. Janssen, L.-G. Milroy, A. P. H. J. Schenning and L. Brunsveld, Chem. – Eur. J., 2018, 24, 7734 CrossRef CAS PubMed.
- A. Fin, I. Petkova, D. A. Doval, N. Sakai, E. Vauthey and S. Matile, Org. Biomol. Chem., 2011, 9, 8246 RSC.
- N. Zink-Lorre, E. Font-Sanchis, Á. Sastre-Santos and F. Fernández-Lázaro, Org. Chem. Front., 2017, 4, 2016 RSC.
- Z. Xie, V. Stepanenko, K. Radacki and F. Würthner, Chem. – Eur. J., 2012, 18, 7060 CrossRef CAS.
- Z. Xie and F. Würthner, Org. Lett., 2010, 12, 3204 CrossRef CAS PubMed.
- P. Leowanawat, A. Nowak-Król and F. Würthner, Org. Chem. Front., 2016, 3, 537 RSC.
- W. Zhang, X. Zhou, Z. Xie, B. Yang, L. Liu and Y. Ma, Chem. Commun., 2013, 49, 11560 RSC.
- A. Nowak-Król, M. I. S. Röhr, D. Schmidt and F. Würthner, Angew. Chem., Int. Ed., 2017, 56, 11774 CrossRef PubMed.
- C. Hippius, I. H. M. van Stokkum, E. Zangrando, R. M. Williams, M. Wykes, D. Beljonne and F. Würthner, J. Phys. Chem. C, 2008, 112, 14626 CrossRef CAS.
- M. Weiser and H.-A. Wagenknecht, Chem. Commun., 2015, 51, 16530 RSC.
- M.-J. Lin, M. Schulze, K. Radacki and F. Würthner, Chem. Commun., 2013, 49, 9107 RSC.
- G. Bélanger-Chabot, A. Ali and F. P. Gabbaï, Angew. Chem., Int. Ed., 2017, 56, 9958 CrossRef PubMed.
- X. Kong, J. Gao, T. Ma, M. Wang, A. Zhang, Z. Shi and Y. Wei, Dyes Pigm., 2012, 95, 450 CrossRef CAS.
- M. Zhou, L. Zhu, Z. Sun, Z. Yang, D. Cao and Q. Li, Synlett, 2017, 28, 2121 CrossRef CAS.
- B. A. Jones, M. J. Ahrens, M.-H. Yoon, A. Facchetti, T. J. Marks and M. R. Wasielewski, Angew. Chem., Int. Ed., 2004, 43, 6363 CrossRef CAS PubMed.
- M. J. Ahrens, M. J. Fuller and M. R. Wasielewski, Chem. Mater., 2003, 15, 2684 CrossRef CAS.
- H. E. Katz, A. J. Lovinger, J. Johnson, C. Kloc, T. Siegrist, W. Li, Y. Y. Lin and A. Dodabalapur, Nature, 2000, 404, 478 CrossRef CAS PubMed.
- Y. Li, L. Tan, Z. H. Wang, H. L. Qian, Y. B. Shi and W. P. Hu, Org. Lett., 2008, 10, 529 CrossRef CAS PubMed.
- V. V. Roznyatovskiy, D. M. Gardner, S. W. Eaton and M. R. Wasielewski, Org. Lett., 2014, 16, 696 CrossRef CAS PubMed.
- H. Morimoto, T. Tsubogo, N. D. Litvinas and J. F. Hartwig, Angew. Chem., Int. Ed., 2011, 50, 3793 CrossRef CAS PubMed.
- Z. Yuan, Y. Xiao, Z. Li and X. Qian, Org. Lett., 2009, 11, 2808 CrossRef CAS PubMed.
- T. T. Clikeman, E. V. Bukovsky, X.-B. Wang, Y.-S. Chen, G. Rumbles, S. H. Strauss and O. V. Boltalina, Eur. J. Org. Chem., 2015, 6641 CrossRef CAS.
- T. Maeda and F. Würthner, Chem. Commun., 2015, 51, 7661 RSC.
- Q. Yan, Y. Zhou, Y.-Q. Zheng, J. Pei and D. Zhao, Chem. Sci., 2013, 4, 4389 RSC.
- S. Li, W. Liu, C.-Z. Li, T.-K. Lau, X. Lu, M. Shi and H. Chen, J. Mater. Chem. A, 2016, 4, 14983 RSC.
- H. Sun, P. Sun, C. Zhang, Y. Yang, X. Gao, F. Chen, Z. Xu, Z.-K. Chen and W. Huang, Chem. – Asian J., 2017, 12, 721 CrossRef CAS PubMed.
- Q. Yan and D. Zhao, Org. Lett., 2009, 11, 3426 CrossRef CAS PubMed.
- A. Takai and M. Takeuchi, Bull. Chem. Soc. Jpn., 2018, 91, 44 CrossRef CAS.
- S. Xie, J. Zhang, L. Wu, J. Zhang, C. Li, X. Chen, Z. Wei and Z. Bo, Dyes Pigm., 2017, 146, 143 CrossRef CAS.
- J. Baram, E. Shirman, N. Ben-Shitrit, A. Ustinov, H. Weissman, I. Pinkas, S. G. Wolf and B. Rybtchinski, J. Am. Chem. Soc., 2008, 130, 14966 CrossRef CAS PubMed.
- C. Shahar, J. Baram, Y. Tidhar, H. Weissman, S. R. Cohen, I. Pinkas and B. Rybtchinski, ACS Nano, 2013, 7, 3547 CrossRef CAS PubMed.
- Y. Zhong, B. Kumar, S. Oh, M. T. Trinh, Y. Wu, K. Elbert, P. Li, X. Zhu, S. Xiao, F. Ng, M. L. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2014, 136, 8122 CrossRef CAS PubMed.
- Y. Guo, Y. Li, O. Awartani, H. Han, J. Zhao, H. Ade, H. Yan and D. Zhao, Adv. Mater., 2017, 29, 1700309 CrossRef PubMed.
- M. Liu, J. Yang, C. Lang, Y. Zhang, E. Zhou, Z. Liu, F. Guo and L. Zhao, Macromolecules, 2017, 50, 7559 CrossRef CAS.
- Y. Li, C. Wang, C. Li, S. Di Motta, F. Negri and Z. Wang, Org. Lett., 2012, 14, 5278 CrossRef CAS PubMed.
- Q. Yan, K. Cai and D. Zhao, Phys. Chem. Chem. Phys., 2016, 18, 1905 RSC.
- R. Mishra, J. M. Lim, M. Son, P. Panini, D. Kim and J. Sankar, Chem. – Eur. J., 2014, 20, 5776 CrossRef CAS PubMed.
- R. Mishra, Z. Mushtaq, R. Regar, B. Mallik, V. Kumar and J. Sankar, ChemBioChem, 2018, 19, 1386 CrossRef CAS PubMed.
- N. V. Handa, K. D. Mendoza and L. D. Shirtcliff, Org. Lett., 2011, 13, 4724 CrossRef CAS PubMed.
- W. Jiang, L. Ye, X. Li, C. Xiao, F. Tan, W. Zhao, J. Hou and Z. Wang, Chem. Commun., 2014, 50, 1024 RSC.
- D. Sun, D. Meng, Y. Cai, B. Fan, Y. Li, W. Jiang, L. Huo, Y. Sun and Z. Wang, J. Am. Chem. Soc., 2015, 137, 11156 CrossRef CAS PubMed.
- A. D. Hendsbee, J.-P. Sun, W. K. Law, H. Yan, I. G. Hill, D. M. Spasyuk and G. C. Welch, Chem. Mater., 2016, 28, 7098 CrossRef CAS.
- F. Würthner, S. Ahmed, C. Thalacker and T. Debaerdemaeker, Chem. – Eur. J., 2002, 8, 4742 CrossRef.
- I. Pugliesi, U. Megerle, S.-L. Suraru, F. Würthner, E. Riedle and S. Lochbrunner, Chem. Phys. Lett., 2011, 504, 24 CrossRef CAS.
- C. W. Liskey, X. Liao and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 11389 CrossRef CAS PubMed.
- C. Zeng, C. Xiao, X. Feng, L. Zhang, W. Jiang and Z. Wang, Angew. Chem., Int. Ed., 2018, 57, 10933 CrossRef CAS PubMed.
- Q. Wu, D. Zhao, A. M. Schneider, W. Chen and L. Yu, J. Am. Chem. Soc., 2016, 138, 7248 CrossRef CAS PubMed.
- L. Zhang, D. He, Y. Liu, K. Wang, Z. Guo, J. Lin and H.-J. Zhang, Org. Lett., 2016, 18, 5908 CrossRef CAS PubMed.
- K. Padala and M. Jeganmohan, Org. Lett., 2011, 13, 6144 CrossRef CAS PubMed.
- X. Li, H. Wang, J. A. Schneider, Z. Wei, W.-Y. Lai, W. Huang, F. Wudl and Y. Zheng, J. Mater. Chem. C, 2017, 5, 2781 RSC.
- W. Yue, Y. Li, W. Jiang, Y. Zhen and Z. Wang, Org. Lett., 2009, 11, 5430 CrossRef CAS PubMed.
- Y. Li, C. Li, W. Yue, W. Jiang, R. Kopecek, J. Qu and Z. Wang, Org. Lett., 2010, 12, 2374 CrossRef CAS PubMed.
- Z. Yuan, J. Li, Y. Xiao, Z. Li and X. Qian, J. Org. Chem., 2010, 75, 3007 CrossRef CAS PubMed.
- M. Kremer, M. Kersten and S. Höger, Org. Chem. Front., 2018, 5, 1825 RSC.
- D. Gutiérrez-Moreno, Á. Sastre-Santos and F. Fernández-Lázaro, Org. Chem. Front., 2018, 5, 1830 RSC.
- N. Zink-Lorre, E. Font-Sanchis, Á. Sastre-Santos and F. Fernández-Lázaro, Dyes Pigm., 2016, 127, 9 CrossRef CAS.
- N. Zink-Lorre, E. Font-Sanchis, Á. Sastre-Santos and F. Fernández-Lázaro, Org. Biomol. Chem., 2016, 14, 9375 RSC.
- X. Wu, C. Yin, Z. Shi, M. Xu, J. Zhang and J. Sun, New J. Chem., 2010, 34, 61 RSC.
- D. Schmidt, D. Bialas and F. Würthner, Angew. Chem., Int. Ed., 2015, 54, 3611 CrossRef CAS PubMed.
- N. Liang, W. Jiang, J. Hou and Z. Wang, Mater. Chem. Front., 2017, 1, 1291 RSC.
- A. W. Freeman, M. Urvoy and M. E. Criswell, J. Org. Chem., 2005, 70, 5014 CrossRef CAS PubMed.
- H. Qian, W. Yue, Y. Zhen, S. Di Motta, E. Di Donato, F. Negri, J. Qu, W. Xu, D. Zhu and Z. Wang, J. Org. Chem., 2009, 74, 6275 CrossRef CAS PubMed.
- Y. Li, L. Xu, R. Jiang, H. Liu and Y. Li, Eur. J. Org. Chem., 2013, 7076 CrossRef CAS.
- R. Wang, G. Li, Y. Zhou, P. Hao, Q. Shang, S. Wang, Y. Zhang, D. Li, S. Yang, Q. Zhang, Z. Shi and B. Tang, Asian J. Org. Chem., 2018, 7, 702 CrossRef CAS.
- W. Fan, N. Liang, D. Meng, J. Feng, Y. Li, J. Hou and Z. Wang, Chem. Commun., 2016, 52, 11500 RSC.
- H. Qian, C. Liu, Z. Wang and D. Zhu, Chem. Commun., 2006, 4587 RSC.
- Z. Ma, C. Xiao, C. Liu, D. Meng, W. Jiang and Z. Wang, Org. Lett., 2017, 19, 4331 CrossRef CAS PubMed.
- D. Meng, H. Fu, C. Xiao, X. Meng, T. Winands, W. Ma, W. Wei, B. Fan, L. Huo, N. L. Doltsinis, Y. Li, Y. Sun and Z. Wang, J. Am. Chem. Soc., 2016, 138, 10184 CrossRef CAS PubMed.
- Z. Luo, T. Liu, W. Cheng, K. Wu, D. Xie, L. Huo, Y. Sun and C. Yang, J. Mater. Chem. C, 2018, 6, 1136 RSC.
- Z. Luo, W. Xiong, T. Liu, W. Cheng, K. Wu, Y. Sun and C. Yang, Org. Electron., 2017, 41, 166 CrossRef CAS.
- M. Schulze, A. Steffen and F. Würthner, Angew. Chem., Int. Ed., 2015, 54, 1570 CrossRef CAS PubMed.
- M. Schulze, M. Philipp, W. Waigel, D. Schmidt and F. Würthner, J. Org. Chem., 2016, 81, 8394 CrossRef CAS PubMed.
- W. Jiang, Y. Li, W. Yue, Y. Zhen, J. Qu and Z. Wang, Org. Lett., 2010, 12, 228 CrossRef CAS PubMed.
- A. H. Endres, M. Schaffroth, F. Paulus, H. Reiss, H. Wadepohl, F. Rominger, R. Krämer and U. H. F. Bunz, J. Am. Chem. Soc., 2016, 138, 1792 CrossRef CAS PubMed.
- Y. Li, H. Zheng, Y. Li, S. Wang, Z. Wu, P. Liu, Z. Gao, H. Liu and D. Zhu, J. Org. Chem., 2007, 72, 2878 CrossRef CAS PubMed.
- S. Müller and K. Müllen, Chem. Commun., 2005, 4045 RSC.
- U. Rohr, P. Schlichting, A. Böhm, M. Gross, K. Meerholz, C. Bräuchle and K. Müllen, Angew. Chem., Int. Ed., 1998, 37, 1434 CrossRef CAS PubMed.
- R. Mishra, P. Panini and J. Sankar, Org. Lett., 2014, 16, 3994 CrossRef CAS PubMed.
- C. Zhou, Y. Li, Y. Zhao, J. Zhang, W. Yang and Y. Li, Org. Lett., 2011, 13, 292 CrossRef CAS PubMed.
- D. T. Gryko, J. Piechowska, M. Tasior, J. Waluk and G. Orzanowska, Org. Lett., 2006, 8, 4747 CrossRef CAS PubMed.
- R. Regar, R. Mishra, P. K. Mondal and J. Sankar, J. Org. Chem., 2018, 83, 9547 CrossRef CAS PubMed.
- Y. Li, Y. Li, J. Li, C. Li, X. Liu, M. Yuan, H. Liu and S. Wang, Chem. – Eur. J., 2006, 12, 8378 CrossRef CAS PubMed.
- C. D. Schmidt, N. Lang, N. Jux and A. Hirsch, Chem. – Eur. J., 2011, 17, 5289 CrossRef CAS PubMed.
- H. Choi, S. Paek, J. Song, C. Kim, N. Cho and J. Ko, Chem. Commun., 2011, 47, 5509 RSC.
- B. Wang, W. Liu, H. Li, J. Mai, S. Liu, X. Lu, H. Li, M. Shi, C.-Z. Li and H. Chen, J. Mater. Chem. A, 2017, 5, 9396 RSC.
- Q. Wu, D. Zhao, J. Yang, V. Sharapov, Z. Cai, L. Li, N. Zhang, A. Neshchadin, W. Chen and L. Yu, Chem. Mater., 2017, 29, 1127 CrossRef CAS.
- P. E. Hartnett, H. S. S. R. Matte, N. D. Eastham, N. E. Jackson, Y. Wu, L. X. Chen, M. A. Ratner, R. P. H. Chang, M. C. Hersam, M. R. Wasielewski and T. J. Marks, Chem. Sci., 2016, 7, 3543 RSC.
- H. Sun, X. Song, J. Xie, P. Sun, P. Gu, C. Liu, F. Chen, Q. Zhang, Z.-K. Chen and W. Huang, ACS Appl. Mater. Interfaces, 2017, 9, 29924 CrossRef CAS PubMed.
- Y. Yu, Y. Li, Z. Qin, R. Jiang, H. Liu and Y. Li, J. Colloid Interface Sci., 2013, 399, 13 CrossRef CAS PubMed.
- H. Fu, D. Meng, X. Meng, X. Sun, L. Huo, Y. Fan, Y. Li, W. Ma, Y. Sun and Z. Wang, J. Mater. Chem. A, 2017, 5, 3475 RSC.
- M. Wu, J.-P. Yi, L. Chen, G. He, F. Chen, M. Y. Sfeir and J. Xia, ACS Appl. Mater. Interfaces, 2018, 10, 27894 CrossRef CAS PubMed.
- S. R. Peurifoy, E. Castro, F. Liu, X. Y. Zhu, F. Ng, S. Jockusch, M. L. Steigerwald, L. Echegoyen, C. Nuckolls and T. J. Sisto, J. Am. Chem. Soc., 2018, 140, 9341 CrossRef CAS PubMed.
- N. J. Schuster, D. W. Paley, S. Jockusch, F. Ng, M. L. Steigerwald and C. Nuckolls, Angew. Chem., Int. Ed., 2016, 55, 13519 CrossRef CAS PubMed.
- N. J. Schuster, R. Hernández Sánchez, D. Bukharina, N. A. Kotov, N. Berova, F. Ng, M. L. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2018, 140, 6235 CrossRef CAS PubMed.
- G. Gao, N. Liang, H. Geng, W. Jiang, H. Fu, J. Feng, J. Hou, X. Feng and Z. Wang, J. Am. Chem. Soc., 2017, 139, 15914 CrossRef CAS PubMed.
- Y. Zhong, M. T. Trinh, R. Chen, G. E. Purdum, P. P. Khlyabich, M. Sezen, S. Oh, H. Zhu, B. Fowler, B. Zhang, W. Wang, C.-Y. Nam, M. Y. Sfeir, C. T. Black, M. L. Steigerwald, Y.-L. Loo, F. Ng, X. Y. Zhu and C. Nuckolls, Nat. Commun., 2015, 6, 8242 CrossRef CAS PubMed.
- H. Qian, F. Negri, C. Wang and Z. Wang, J. Am. Chem. Soc., 2008, 130, 17970 CrossRef CAS PubMed.
- H. Qian, Z. Wang, W. Yue and D. Zhu, J. Am. Chem. Soc., 2007, 129, 10664 CrossRef CAS PubMed.
- Y. Zhen, C. Wang and Z. Wang, Chem. Commun., 2010, 46, 1926 RSC.
- Y. Zhen, W. Yue, Y. Li, W. Jiang, S. Di Motta, E. Di Donato, F. Negri, S. Ye and Z. Wang, Chem. Commun., 2010, 46, 6078 RSC.
- H. Langhals and A. Hofer, J. Org. Chem., 2012, 77, 9585 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2019 |