
Beauvetetraones A–C, phomaligadione-derived polyketide dimers from the entomopathogenic fungus, Beauveria bassiana†
Christine
Beemelmanns  b
and
Ki Hyun
Kim
*a
b
and
Ki Hyun
Kim
*a
b
and
Ki Hyun
Kim
*a
Abstract
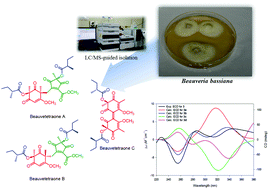
We report the isolation of two novel epimeric phomaligadione-derived polyketides, beauvetetraones A–B (1–2), from the entomopathogenic fungus Beauveria bassiana. Beauvetetraones A and B feature an unprecedented methylene-bridged phloroglucinol skeleton with a highly rearranged scaffold. In addition, a dimer of two phomaligadiones, named beauvetetraone C, was isolated for the first time from a natural source. The structures of compounds 1–3 including their absolute configurations were unambiguously assigned by NMR spectroscopic analyses, phenylglycine methyl ester (PGME) analysis, and quantum chemical ECD calculations. A putative biosynthetic pathway for beauvetetraones A–C is proposed.
 Please wait while we load your content...
Please wait while we load your content...

