
Trifluoromethylation of α-diazoesters and α-diazoketones with fluoroform-derived CuCF3: synergistic effects of co-solvent and pyridine as a promoter†
Qiao
Ma
and
Gavin Chit
Tsui
 *
*
Department of Chemistry, The Chinese University of Hong Kong, Shatin, New Territories, Hong Kong SAR. E-mail: gctsui@cuhk.edu.hk
First published on 6th November 2018
Abstract
We herein describe a new protocol for copper-mediated trifluoromethylation of α-diazoesters and α-diazoketones, both using readily available fluoroform-derived CuCF3. The synergistic effect of using 1,4-dioxane as a co-solvent and pyridine as a promoter is revealed. Reaction conditions are mild and easy to handle, allowing the synthesis of a range of α-trifluoromethyl esters and ketones in one-step from α-diazo carbonyl compounds. The ultimate CF3 source is the inexpensive, nontoxic, industrial waste fluoroform.
Introduction
Organofluorine compounds play a central role in pharmaceutical, agrochemical, material and PET imaging applications.1 Introduction of fluorine-containing functional groups, particularly the trifluoromethyl (CF3) group, into organic molecules has been the subject of intense research.2 In this context, α-trifluoromethyl carbonyl compounds are highly sought-after building blocks for synthesizing a wide range of valuable fluorinated molecules.3 Traditional methods for the preparation of α-trifluoromethyl carbonyl compounds mainly relied on electrophilic or radical trifluoromethylation of enolates, silyl enol ethers and enamines,4 generated in situ or in advance from the corresponding carbonyl compounds. More recent advancement in the oxidative trifluoromethylation of alkenes via photoredox catalysis and radical processes allowed the use of abundant olefin feedstocks as starting materials.5 Other unique approaches have also been described in isolated reports.6 Majority of the above methods used electrophilic CF3 sources and catalysts (organo-/photo-/precious metals) that are commercially available at high prices or need to be prepared, therefore rendering them difficult for industrial-scale production. The search for novel substrate classes and readily available trifluoromethylating reagents towards the direct synthesis of α-trifluoromethyl carbonyl compounds is still much needed.Cross-coupling of diazo compounds in the presence of copper is a versatile method for C–C bond formation.7 Rare examples of copper-mediated trifluoromethylation of α-diazoesters 1 exist,8 which offer a unique advantage for the direct installation of a C(sp3)–CF3 bond at the α-position of an aliphatic ester. Hu and co-workers were the first to demonstrate such an approach by using a combination of CuI/TMSCF3/CsF to pre-generate “CuCF3” (Scheme 1).8a The key to their success was adding water to activate the otherwise inactive [Cu(CF3)I]−A by promoting ligand exchange on Cu (scavenging I− by the formation of a hydrated iodide ion), thus affording the reactive complex [CuCF3]·1. Extrusion of N2 leads to the trifluoromethylcopper-carbene species C. Subsequent CF3-migratory insertion followed by protonolysis of D delivers the α-trifluoromethyl ester 2. In continuation of our interest in developing new trifluoromethylation methods using the fluoroform (CF3H)-derived CuCF3,9 we herein report that [CuCF3] B, generated from CuCl/CF3H/t-BuOK,10 was also effective in the trifluoromethylation of α-diazoesters 1. This reagent is a stable, pre-formed CuCF3 solution in DMF without any added ligand. It does not require extra proton sources due to the presence of t-BuOH and Et3N·3HF (stabilizer) from the preparation, which facilitates the protonolysis of D. Most attractively, the CF3 source is fluoroform, a nontoxic, non-flammable, inexpensive (commercially available at <$0.1 mol−1)11 gas byproduct from Teflon manufacturing.10c For the first time, the synergistic effect of using a co-solvent and pyridine as a promoter to enhance the reactivity of fluoroform-derived CuCF3 is disclosed.
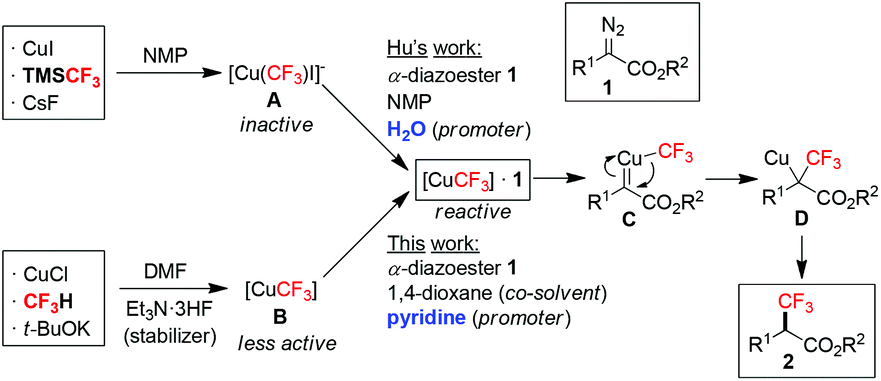 | ||
| Scheme 1 Copper-mediated trifluoromethylation of α-diazoesters. | ||
Results and discussion
The optimization studies began with α-diazoester 1a (Table 1, see the ESI† for full details). The CuCF3 reagent was prepared from CuCl, t-BuOK and fluoroform in DMF, stabilized with Et3N·3HF, according to the standard procedure.9a,10a When substrate 1a was subjected to 2.5 equivalents of CuCF3 (0.6 mL in DMF) at room temperature and stirred overnight under argon, we observed only 21% (19F NMR yield) of the desired α-trifluoromethyl ester 2a due to incomplete conversion (entry 1).|
|
|||
|---|---|---|---|
| Entry | Co-solvent (mL) | Additive (equiv.) | Yieldb (%) |
| a Unless specified otherwise, reactions were carried out using 1a (0.1 mmol), CuCF3 (0.42 M in DMF, 0.6 mL, 2.5 equiv., prepared from CuCl/t-BuOK/CF3H and stabilized with Et3N·3HF), under argon. b Yield was determined by 19F NMR analysis using benzotrifluoride as the internal standard. c Isolated yield. d 0.6 mL DMF was added. e 1.5 mL DMF was added. | |||
| 1 | None | None | 21 |
| 2 | THF (0.6) | None | 22 |
| 3 | Et2O (0.6) | None | 22 |
| 4 | Toluene (0.6) | None | 32 |
| 5 | CH2Cl2 (0.6) | None | 32 |
| 6 | CH3CN (0.6) | None | 52 |
| 7d | None | None | 12 |
| 8 | CH3CN (1.5) | None | 66 |
| 9 | CH3CN (1.5) | Pyridine (2.5) | 81 |
| 10 | THF (1.5) | Pyridine (2.5) | 76 |
| 11 | Et2O (1.5) | Pyridine (2.5) | 78 |
| 12 | Toluene (1.5) | Pyridine (2.5) | 80 |
| 13 | CH2Cl2 (1.5) | Pyridine (2.5) | 69 |
| 14 | 1,4-Dioxane (1.5) | Pyridine (2.5) | 86, 83 |
| 15e | None | Pyridine (2.5) | 24 |
| 16 | 1,4-Dioxane (1.5) | None | 37 |
Screening of co-solvents (equal volume to that of the CuCF3 solution) revealed a significant improvement in product yield by adding acetonitrile (entries 2–6).12 The co-solvent effect was not simply due to the change in concentration because we observed no yield increase by diluting the reaction with DMF alone (entry 7). Using more acetonitrile further increased the yield to 66% (entry 8). We have previously found the use of amine additives such as TMEDA, presumably acting as a ligand, was important for the reactivity of fluoroform-derived CuCF3 in the trifluoromethylation of terminal alkynes.9a Therefore, a variety of nitrogen-containing additives (2.5 equiv.) including Et3N, TMEDA, piperidine, 2,2,6,6-tetramethylpiperidine, imidazole, 1,10-phenanthroline and DMAP were screened but showed no beneficial effects on yields (see the ESI†). However, adding pyridine dramatically enhanced the yield to 81% (entry 9). Furthermore, previously tested co-solvents also gave significantly higher yields now with pyridine (entries 10–13, cf. entries 2–5). Other additives such as phosphine PPh3 and inorganic bases K2CO3 and Na2CO3 gave inferior yields than pyridine (see the ESI†). Finally, 1,4-dioxane was identified as the optimal co-solvent, the desired product 1a was obtained in 86% (83% isolated) yield (entry 14). This was a synergistic effect since reactions run without 1,4-dioxane (entry 15) or pyridine (entry 16) gave much poorer yields. The amount of pyridine was controlled at 2.5 equivalents, a larger amount (5.0 equiv.) did not lead to a further increase in yield. Other important information gained from the optimization studies included (see the ESI†): higher reaction temperature (50 °C) decreased the yield; 2.5 equivalents of CuCF3 were optimal (1.5 or 3.5 equiv. gave lower yields); using HF-pyridine as the stabilizer9d for CuCF3 was not effective; no conversion was observed when the reaction was open to air; using pyridine as the co-solvent (1.5 mL) or pyrazine as the additive gave traces of product.
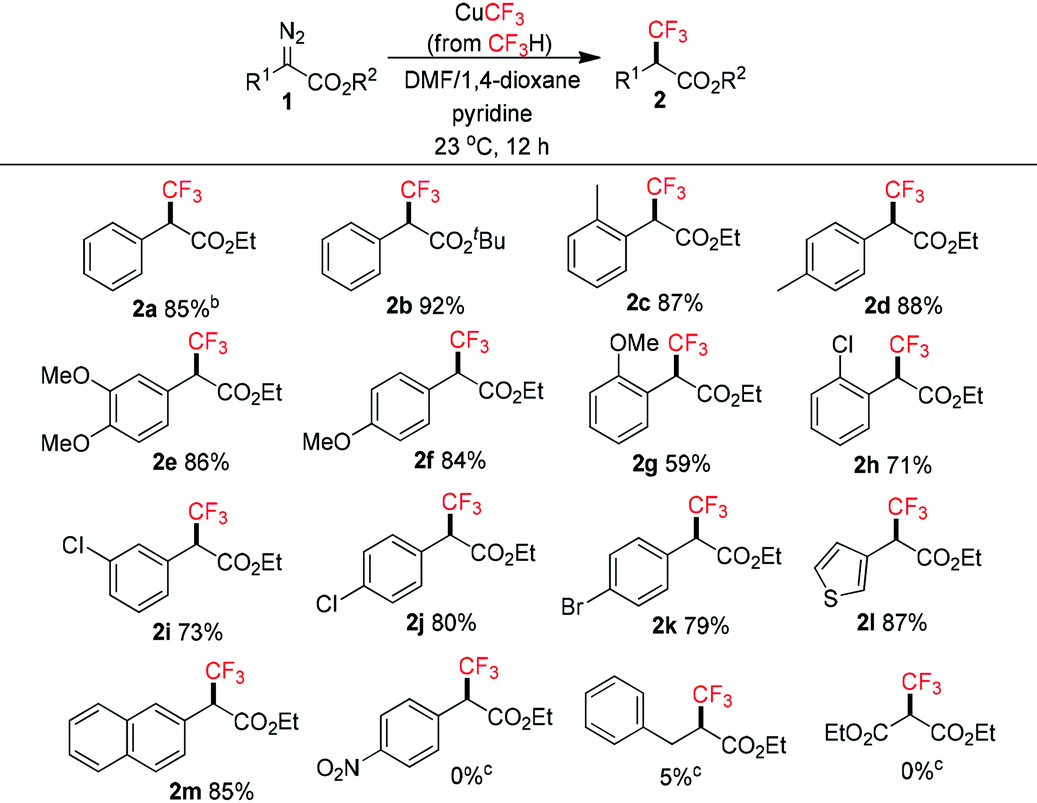
The scope of α-trifluoromethyl esters 2 was subsequently investigated under the optimized conditions (Table 2). α-Diazoesters 1 were easily prepared by diazo-transfer reaction of the corresponding esters.13 In general, aryl-substituted α-diazoesters reacted smoothly to give the products in moderate to good yields. Steric bulk in the ester group did not affect the reactivity (2b). Substituents at the ortho, meta and para positions of the benzene ring were tolerated, although in some cases the ortho groups gave lower yields (2g, 2h). Halogens such as chloro and bromo groups (2h–k) remained intact even though trifluoromethylation of aryl halides with fluoroform-derived CuCF3 is known.10b Heteroaryl such as the thienyl group (2l) was also tolerated. These yields were comparable to Hu's method using CuI/TMSCF3.8a On the other hand, the strong electron-withdrawing nitro group and alkyl-substituted α-diazoesters were not compatible.8b
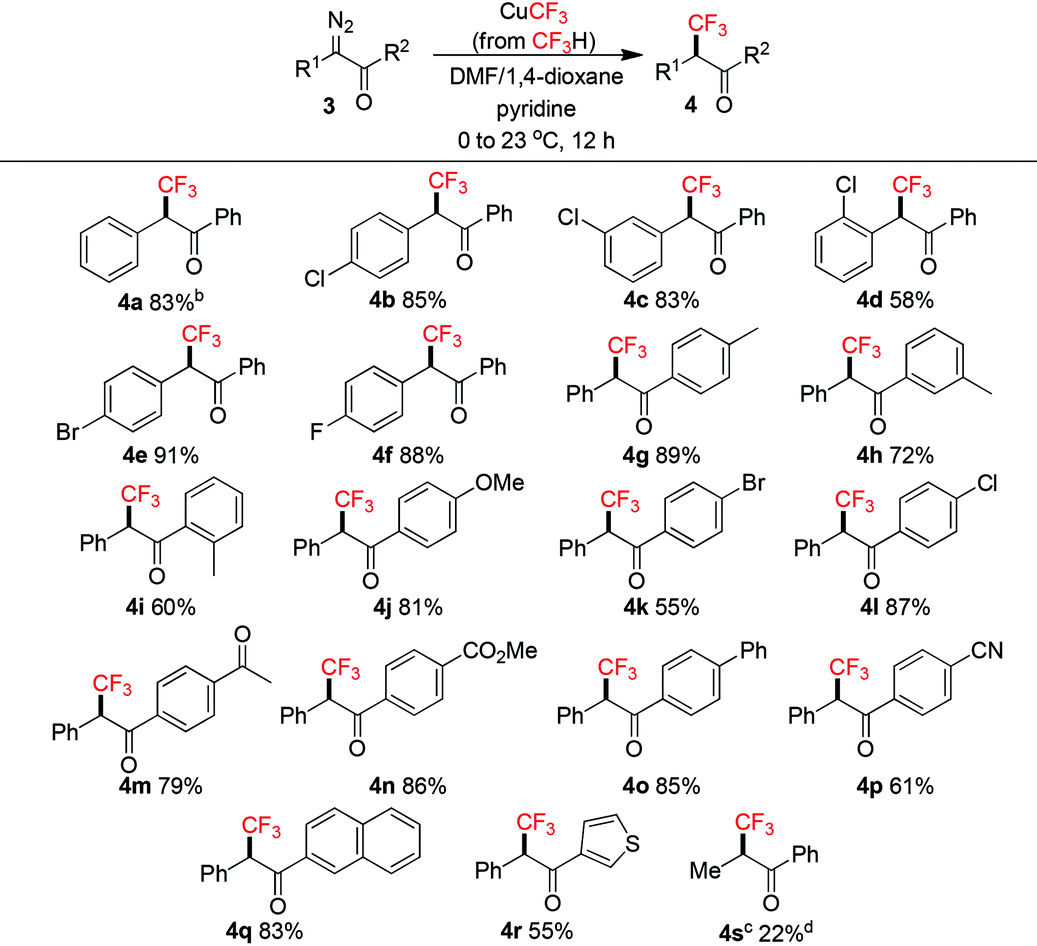
Trifluoromethylation of α-diazoketones 3 with CuCF3 has not been reported and would be challenging using the CuI/TMSCF3/CsF protocol8a due to the facile addition of CF3− to ketones.2g We decided to apply our conditions to tackle this transformation (Table 3). Slight modifications of the reaction conditions, i.e. adding CuCF3 at 0 °C to the substrate mixture and slowly warmed to room temperature, and decreasing the amounts of CuCF3 and pyridine, were necessary to ensure good yields (see the ESI†). We found that these procedures helped in diminishing the formation of unidentified side products. As a result, a series of unique α-CF3-α-aryl ketones 4a–r were successfully synthesized. α-Aryl halides were tolerated (4b–f). Functional groups on the benzene ring at the carbonyl position were also well-tolerated, including alkyl (4g–i, para/meta/ortho positions), electron-donating methoxy (4j), halogens (4k–l), and electron-withdrawing acetyl (4m)/ester (4n)/nitrile (4p) groups. In general, ortho substituents led to lower yields (4d, 4i). Naphthyl (4q) and heteroaryl (4r) groups were also compatible. Disappointingly, the α-alkyl group (4s) was not suitable for this reaction and even with larger amounts of CuCF3, only a low yield was obtained due to incomplete conversion. It is worth mentioning that Grushin and co-workers have used fluoroform-derived CuCF3 in the trifluoromethylation of α-haloketones,14 however, the reaction only tolerated substrates with primary α-carbons.
We initially suspected that pyridine acted as a ligand; however, it could also act as an external base affecting the protonolysis step. To distinguish these roles, we studied the effects of a series of 2- and 2,6-disubstituted pyridine derivatives in the trifluoromethylation of α-diazoester 1a and compared the results with that of using pyridine and without pyridine (Table 4).15a Toluene was chosen as a non-coordinating solvent to single out the effects of pyridine derivatives. As the steric environment around the pyridine nitrogen atom increased, the yield decreased. For example, moving from unsubstituted pyridine to 2-picoline and 2,6-lutidine (coordinating ligands)15b caused a significant drop in yield (Table 4, entries 1–3). Highly sterically encumbered 2,6-di-tert-butylpyridine (non-coordinating) gave a similar yield to that without pyridine (Table 4, entries 4–5). These results suggested the role of pyridine as a ligand. 19F NMR experiments were also conducted to monitor the yield of 2a and the consumption of CuCF3 over time (as a molar ratio of CuCF3 at t vs. initial CuCF3 at 5 min) (Fig. 1). For the product formation (y-axis, left-hand side), the rate-enhancing effect (as approximated by the yield within the first 60 min) of pyridine was very clear. Reaction without pyridine was much more sluggish (e.g. 13% vs. 40% at t = 37 min, see the ESI†). For the consumption of CuCF3 (y-axis, right-hand side), adding pyridine significantly increased the rate of disappearance of CuCF3. Intriguingly, using a sub-stoichiometric amount of pyridine gave the yield comparable to that using excess pyridine (50 mol% pyridine, 71% yield vs. 250 mol% pyridine, 80% yield, cf.Table 1, entry 12), indicating a catalytic turnover of pyridine as a labile ligand (Scheme 2).
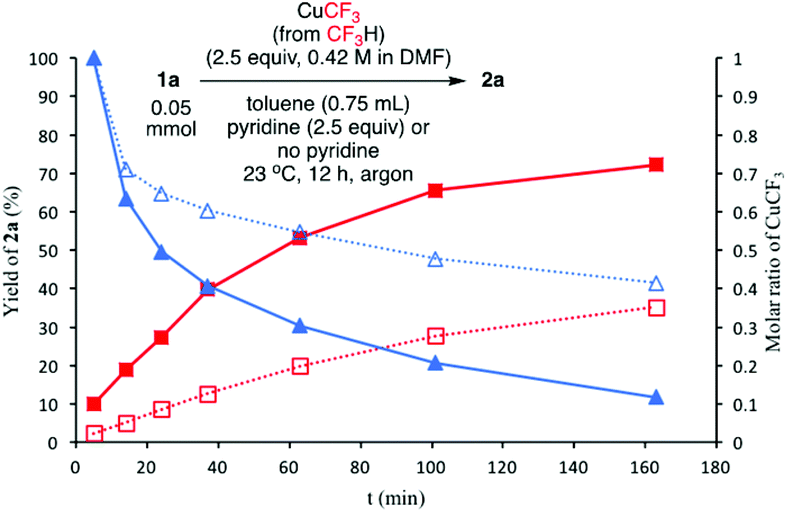 | ||
Fig. 1
19F NMR yield (%) of 2a and the molar ratio of CuCF3 (at t vs. t = 5 min) against time t (min). Internal standard = PhCF3; yield of 2a with pyridine ( ); yield of 2a without pyridine ( ); yield of 2a without pyridine ( ); molar ratio of CuCF3 with pyridine ( ); molar ratio of CuCF3 with pyridine ( ); molar ratio of CuCF3 without pyridine ( ); molar ratio of CuCF3 without pyridine ( ). ). | ||
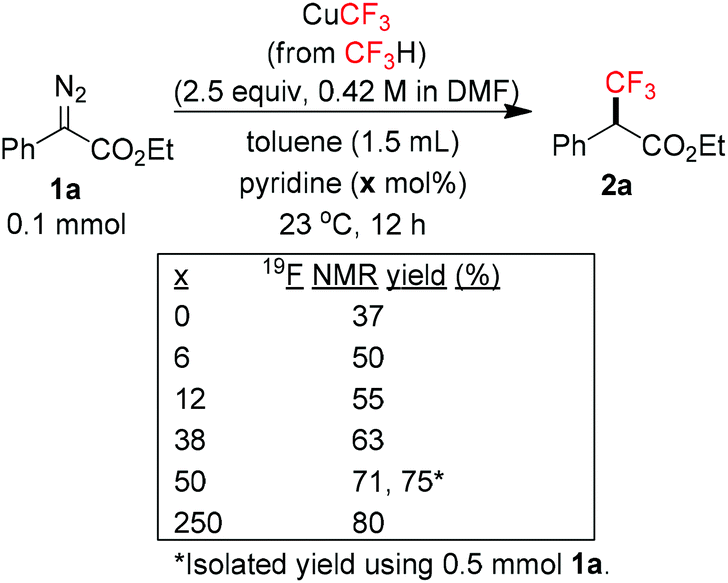 | ||
| Scheme 2 Sub-stoichiometric amounts of pyridine. | ||
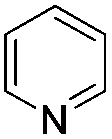
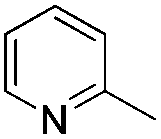
| Entry | Pyridine or pyridine derivative | Yieldb (%) |
|---|---|---|
| a General conditions: 1a (0.1 mmol), fluoroform-derived CuCF3 (2.5 equiv., 0.42 M in DMF), toluene (1.5 mL), pyridine or pyridine derivative (2.5 equiv.), 23 °C, 12 h, under argon. b Yield was determined by 19F NMR analysis using benzotrifluoride as the internal standard. c cf. Table 1. | ||
| 1c |
|
80 |
| 2 |
|
61 |
| 3 |
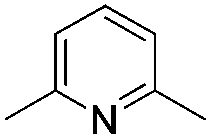
|
33 |
| 4 |
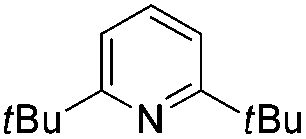
|
38 |
| 5 | None | 37 |
Combining these data, we concluded that pyridine acts as a ligand on copper to activate the [CuCF3] species B for the generation of a pyridine-ligated [CuCF3]·1 complex, although the exact nature is unclear at the moment, leading to subsequent product formation steps (cf.Scheme 1). The action of pyridine should be in the upstream of the reaction mechanism, i.e. not the protonolysis of D to 2, based on the observation of increased consumption of CuCF3 reagent when adding pyridine (cf.Fig. 1). It is noteworthy that the initially formed [CuCF3] species B should be solvent-stabilized, in which the solvent molecules are effectively “ligands”.8a,10a Therefore, the synergistic effects of co-solvent and pyridine are to promote the formation of reactive complex [CuCF3]·1 through ligand exchange between solvent molecules and substrate 1, possibly by displacing the weakly coordinating solvent molecules from the Cu centre.12b
Conclusions
In summary, we have developed a protocol using low-cost fluoroform-derived CuCF3 reagent for the trifluoromethylation of α-diazoesters and α-diazoketones. Various α-trifluoromethyl esters and ketones can be prepared by this method in one-step from α-diazo carbonyl compounds, which is highly desirable for medicinal chemistry research. Furthermore, the synergistic effects of co-solvent and pyridine in promoting the reaction were disclosed, which could have a significant impact on copper-catalyzed/-mediated cross-couplings of diazo compounds, and further investigation is ongoing in our laboratory.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Research Grants Council of Hong Kong (CUHK 24301217) and the Chinese University of Hong Kong (the Faculty Strategic Fund for Research from the Faculty of Science and the Direct Grant for Research 4053276).References
- (a) P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, Wiley-VCH, Weinheim, 2004 CrossRef; (b) I. Ojima, Fluorine in Medicinal Chemistry and Chemical Biology, Wiley-Blackwell, Chichester, 2009 CrossRef; (c) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef PubMed; (d) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (e) J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS PubMed; (f) T. Fujiwara and D. O'Hagan, J. Fluorine Chem., 2014, 167, 16 CrossRef CAS; (g) S. Preshlock, M. Tredwell and V. Gouverneur, Chem. Rev., 2016, 116, 719 CrossRef CAS; (h) N. A. Meanwell, J. Med. Chem., 2018, 61, 5822 CrossRef CAS.
- (a) O. A. Tomashenko and V. V. Grushin, Chem. Rev., 2011, 111, 4475 CrossRef CAS; (b) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214 CrossRef CAS; (c) L. Chu and F.-L. Qing, Acc. Chem. Res., 2014, 47, 1513 CrossRef CAS PubMed; (d) H. Egami and M. Sodeoka, Angew. Chem., Int. Ed., 2014, 53, 8294 CrossRef CAS; (e) J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef CAS PubMed; (f) X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826 CrossRef CAS PubMed; (g) X. Liu, C. Xu, M. Wang and Q. Liu, Chem. Rev., 2015, 115, 683 CrossRef CAS PubMed; (h) C. Alonso, E. Martínez de Marigorta, G. Rubiales and F. Palacios, Chem. Rev., 2015, 115, 1847 CrossRef CAS; (i) T. Chatterjee, N. Iqbal, Y. You and E. J. Cho, Acc. Chem. Res., 2016, 49, 2284 CrossRef CAS; (j) T. Koike and M. Akita, Acc. Chem. Res., 2016, 49, 1937 CrossRef CAS.
- (a) G. Alvernhe, B. Langlois, A. Laurent, I. Le Drean, A. Selmi and M. Weissenfels, Tetrahedron Lett., 1991, 32, 643 CrossRef CAS; (b) N. A. Petasis and M. Myslinska, US Patent Appl20090247766, 2009 Search PubMed; (c) P. F. Wang, N. Li, W. M. Liu, S. T. Lee and C. S. Lee, US Patent Appl20090102356, 2009 Search PubMed; (d) H. A. Schenck, P. W. Lenkowski, I. Choudhury-Mukhejee, S. H. Ko, J. P. Stables, M. K. Patel and M. L. Brown, Bioorg. Med. Chem., 2004, 12, 979 CrossRef CAS.
- For representative examples, see: (a) T. Umemoto and S. Ishihara, J. Am. Chem. Soc., 1993, 115, 2156 CrossRef CAS; (b) Y. Itoh and K. Mikami, Org. Lett., 2005, 7, 649 CrossRef CAS; (c) I. Kieltsch, P. Eisenberger and A. Togni, Angew. Chem., Int. Ed., 2007, 46, 754 CrossRef CAS; (d) V. Matoušek, A. Togni, V. Bizet and D. Cahard, Org. Lett., 2011, 13, 5762 CrossRef; (e) A. T. Herrmann, L. L. Smith and A. Zakarian, J. Am. Chem. Soc., 2012, 134, 6976 CrossRef CAS; (f) Q.-H. Deng, H. Wadepohl and L. H. Gade, J. Am. Chem. Soc., 2012, 134, 10769 CrossRef CAS PubMed; (g) K. Miura, M. Taniguchi, K. Nozaki, K. Oshima and K. Utimoto, Tetrahedron Lett., 1990, 31, 6391 CrossRef CAS; (h) K. Mikami, Y. Tomita, Y. Ichikawa, K. Amikura and Y. Itoh, Org. Lett., 2006, 8, 4671 CrossRef CAS; (i) K. Sato, T. Yuki, R. Yamaguchi, T. Hamano, A. Tarui, M. Omote, I. Kumadaki and A. Ando, J. Org. Chem., 2009, 74, 3815 CrossRef CAS; (j) P. V. Pham, D. A. Nagib and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2011, 50, 6119 CrossRef CAS PubMed; (k) D. A. Nagib, M. E. Scott and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 10875 CrossRef CAS PubMed; (l) A. E. Allen and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 4986 CrossRef CAS.
- (a) C. P. Zhang, Z. L. Wang, Q. Y. Chen, C. T. Zhang, Y. C. Gu and J. C. Xiao, Chem. Commun., 2011, 47, 6632 RSC; (b) A. Deb, S. Manna, A. Modak, T. Patra, S. Maity and D. Maiti, Angew. Chem., Int. Ed., 2013, 52, 9747 CrossRef CAS PubMed; (c) R. Tomita, Y. Yasu, T. Koike and M. Akita, Angew. Chem., Int. Ed., 2014, 53, 7144 CrossRef CAS; (d) Y. B. Wu, G. P. Lu, T. Yuan, Z. B. Xu, L. Wan and C. Cai, Chem. Commun., 2016, 52, 13668 RSC; (e) P. Panday, P. Garg and A. Singh, Asian J. Org. Chem., 2018, 7, 111 CrossRef CAS.
- (a) B. Morandi and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 123, 9251 CrossRef; (b) Z. He, R. Zhang, M. Hu, L. Li, C. Ni and J. Hu, Chem. Sci., 2013, 4, 3478 RSC; (c) T. Kawamoto, R. Sasaki and A. Kamimura, Angew. Chem., Int. Ed., 2017, 56, 1342 CrossRef CAS; (d) H. T. Qin, S. W. Wu, J. L. Liu and F. Liu, Chem. Commun., 2017, 53, 1696 RSC.
- (a) X. Zhao, Y. Zhang and J. Wang, Chem. Commun., 2012, 48, 10162 RSC; (b) A. Ford, H. Miel, A. Ring, C. N. Slattery, A. R. Maguire and M. A. McKervey, Chem. Rev., 2015, 115, 9981 CrossRef CAS; (c) Y. Xia, D. Qiu and J. Wang, Chem. Rev., 2017, 117, 13810 CrossRef CAS.
- (a) M. Hu, C. Ni and J. Hu, J. Am. Chem. Soc., 2012, 134, 15257 CrossRef CAS PubMed. For a related report, see: (b) X.-Q. Hu, J.-B. Han and C.-P. Zhang, Eur. J. Org. Chem., 2016, 324 Search PubMed.
- (a) L. He and G. C. Tsui, Org. Lett., 2016, 18, 2800 CrossRef CAS; (b) L. He, X. Yang and G. C. Tsui, J. Org. Chem., 2017, 82, 6192 CrossRef CAS; (c) X. Yang, L. He and G. C. Tsui, Org. Lett., 2017, 19, 2446 CrossRef CAS; (d) X. Yang and G. C. Tsui, Org. Lett., 2018, 20, 1179 CrossRef CAS; (e) Y. Ye, K. P. S. Cheung, L. He and G. C. Tsui, Org. Lett., 2018, 20, 1676 CrossRef CAS; (f) Y. Ye, K. P. S. Cheung, L. He and G. C. Tsui, Org. Chem. Front., 2018, 5, 1511 RSC; (g) X. Yang and G. C. Tsui, Chem. Sci., 2018 10.1039/c8sc03754j.
- (a) A. Zanardi, M. A. Novikov, E. Martin, J. Benet-Buchholz and V. V. Grushin, J. Am. Chem. Soc., 2011, 133, 20901 CrossRef CAS; (b) A. Lishchynskyi, M. A. Novikov, E. Martin, E. C. Escudero-Adán, P. Novák and V. V. Grushin, J. Org. Chem., 2013, 78, 11126 CrossRef CAS. For a review, see: (c) V. V. Grushin, Chem. Today, 2014, 32, 81 CAS.
- J. B. Geri and N. K. Szymczak, J. Am. Chem. Soc., 2017, 139, 9811 CrossRef CAS.
- For the importance of co-solvent with fluoroform-derived CuCF3, see: (a) A. Lishchynskyi, G. Berthon and V. V. Grushin, Chem. Commun., 2014, 50, 10237 RSC; (b) A. I. Konovalov, A. Lishchynskyi and V. V. Grushin, J. Am. Chem. Soc., 2014, 136, 13410 CrossRef CAS.
- M. P. Doyle, M. A. Mckervey and T. Ye, Modern Catalytic Methods for Organic Synthesis with Diazo Compounds, John Wiley & Sons, New York, 1998 Search PubMed.
- P. Novák, A. Lishchynskyi and V. V. Grushin, J. Am. Chem. Soc., 2012, 134, 16167 CrossRef.
- (a) For analogous studies of the pyridine ligand effect on palladium centres, see: M. H. Emmert, A. K. Cook, Y. J. Xie and M. S. Sanford, Angew. Chem., Int. Ed., 2011, 123, 9581 CrossRef; (b) L. M. Engelhardt, C. Pakawatchai and A. H. White, J. Chem. Soc., Dalton Trans., 1985, 117 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectral data for all new compounds. See DOI: 10.1039/c8qo00834e |
| This journal is © the Partner Organisations 2019 |