
Carbonized polymer dots/TiO2 photonic crystal heterostructures with enhanced light harvesting and charge separation for efficient and stable photocatalysis†
Yue
Zhao‡
,
Qingsen
Zeng‡
,
Tanglue
Feng
,
Chunlei
Xia
,
Chongming
Liu
,
Fan
Yang
,
Kai
Zhang
 and
Bai
Yang
*
and
Bai
Yang
*
State Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, Changchun, 130012, P. R. China. E-mail: byangchem@jlu.edu.cn
First published on 30th September 2019
Abstract
TiO2 photonic crystals (PCs) are widely interesting in photocatalysis due to their slow photon effect for efficient light harvesting. However, their performance is limited by the narrow absorption range and fast charge recombination. In this work, an efficient strategy is developed to enhance the light harvesting and charge lifetime through fabricating TiO2 PC/carbonized polymer dot (CPD) heterostructures. The CPDs with a band gap of 2.08 eV are used to broaden the light absorption range. The energy level of CPDs matches well with TiO2 PCs, forming a type-II heterojunction. The staggered energy levels of CPD/TiO2 PC heterostructures are helpful for charge separation and thus suppressed charge recombination, which is demonstrated by the transient photovoltage (TPV) measurements. Therefore, the CPD/TiO2 PC heterostructures exhibit obviously better photocatalytic performance than individual TiO2 PCs. The combination of CPDs and inorganic semiconductors provides a variety of possibilities for the further development of high-efficiency photocatalysts.
1. Introduction
During the last few decades, environment issues and water pollution have become increasingly serious, which has stimulated scientific research into green, efficient and low-cost photocatalysts, such as metal oxides (TiO2, ZnO, SnO2, ZrO2)1–6 and semiconductor quantum dots (ZnS).7 Among them, TiO2 is one of the most significant photocatalysis materials owing to its excellent stability, low toxicity and low cost.8–12 Many strategies have been adopted to promote the photocatalytic performance, including doping heteroatoms,13 loading co-catalysts,14 and preparing composites with other semiconductors.9,11 However, the utilization efficiency of sunlight of TiO2 is less efficient due to the large bandgap (3.0–3.2 eV).15 Besides, the rapid charge recombination rate primarily limits the photocatalytic performance.16,17 Therefore, strategies for synergetic enhancement of light harvesting and charge lifetime are of great significance to improve photocatalytic efficiency.Photonic crystals (PCs) possess a photonic stop band (PSB) and thus can slow down the photons near the PSB edges, which offers a unique way to promote light harvesting,12,18–20 and have attracted much interest in photocatalysis.21–23 The periodic dielectric structure endows PCs with a unique structure and optical properties, such as large specific surface area with a uniform macroporous structure, and controlling and manipulating incident light with high absorption efficiency. Not only can the PCs be assembled with a conductive substrate to fabricate photoelectrodes for photoelectrocatalysis,24,25 but their powders are able to be prepared on a large scale via substrate-free procedures for photocatalysis.12,22 TiO2 PCs have shown higher photocatalytic activity compared to TiO2 nanocrystals as a result of the slow photon effect, which plays a key role in the enhancement of the photocatalytic performances for TiO2 PCs.26 In spite of the improved photocatalytic properties for TiO2 PCs, the absorption edge still locates at the ultraviolet region and the limited charge lifetime in individual TiO2 PCs restricts the further improvement of photocatalytic performance. Until now, few studies have reported a method to simultaneously achieve enhanced light harvesting and prolonged charge lifetimes in TiO2 PCs (Table S1, ESI†). Therefore, it's urgent to develop new efficient strategies to enhance the light harvesting and charge separation of TiO2 PCs to further improve the photocatalytic efficiency.12
Carbonized polymer dots (CPDs), as a new and significant member of the carbon-based nanomaterials family, have caused increasing interest in recent years,27,28 and are widely applied in bioimaging,29 photoelectronic devices,30 anti-counterfeiting,31 photocatalysis,32 photothermal therapy33,34 and sensing,35 due to their superior light absorption, color-tunable photoluminescence, excellent electron transfer ability, great photo-chemical stability and low toxicity. CPDs are usually prepared through the procedure of hydrothermal condensation crosslinking and carbonization (HCCC) from small molecules and/or polymers, which contain multiple polar carboxyl, amino or hydroxyl groups.36 Very recently, a new strategy of hydrothermal addition polymerization and carbonization (HAPC) was developed by our group to prepare CPDs with ultrahigh yields (ca. 85%).37 Rich surface polar groups of CPDs make them easily anchor with other semiconductor materials. Besides, CPDs have a wide light absorption range and can generate e−/h+ pairs efficiently under light irradiation. Fast charge separation and transfer have been observed in CPD-based hybrid materials.38,39 Accordingly, CPDs have been successfully used in efficient solar cells,30 photocatalysis,40–42 and photoelectrocatalysis43 through coupling with semiconductors. Therefore, it is expected that the light-harvesting ability and charge separation/transfer dynamics of TiO2 PCs can be improved with CPDs to fabricate high-efficiency composite photocatalysts.
In this work, an efficient method that enhances the light harvesting and charge separation of TiO2 PCs is developed through fabricating CPD/TiO2 PC heterostructures. The CPD/TiO2 PC heterostructures exhibit obviously improved photocatalytic performance compared to pure TiO2 PCs. The essential mechanisms of the promoted performance are investigated and discussed in detail. On the one hand, the CPD/TiO2 PC heterostructures have much broader light absorption range than TiO2 PCs because of the narrow bandgap of CPDs. On the other hand, the energy level of CPDs matches well with TiO2 PCs, and type-II heterojunctions are formed between the CPDs and TiO2 PCs. The staggered energy levels of CPD/TiO2 PC heterostructures are helpful for charge separation and thus suppressed charge recombination, which is demonstrated by the transient photovoltage (TPV), transient photocurrent and electrochemical impendence spectra (EIS) measurements.
2. Experimental methods
2.1 Materials
Styrene monomer (99%), methylene blue and anhydrous citric acid (98%) were purchased from Aladdin. p-Aminosalicylic acid (99%) was obtained from J&K Scientific Ltd. Methanol, concentrated hydrochloric acid (HCl, AR) and sodium hydroxide (NaOH, AR) were purchased from Beijing Chemical Works. Tetrabutyl titanate (TBT) was obtained from Guangfu Chemical Reagent. All chemicals were used as received.2.2 Preparation of CPDs
The CPDs with narrow band gaps were synthesized according to the modified method of previous work.27 Citric acid (CA) (1 mmol) and p-aminosalicylic acid (1 mmol) were dissolved in 10 mL of water, and the pH was adjusted by concentrated hydrochloric acid to ∼1. The mixture was heated at 200 °C for 8 h in a poly(tetrafluoroethylene) (Teflon)-lined autoclave. After cooling down, the as-prepared solution was filtered using a 0.22 μm filter to remove the large particles, and then immediately dialyzed in deionized water through a dialysis bag with a molecular weight cut-off of 3500 for 24 h, during which the deionized water was changed every 3–4 h to fully remove the excess precursors and byproducts. Finally, a clear brown solution was achieved, and a red brown CPD powder was obtained after removing the water though freeze-drying.2.3 Preparation of TiO2 PCs
The polystyrene (PS) microspheres were prepared according to the method in previous work.12 The styrene monomer was washed with NaOH solution (0.1 M) and deionized water to remove the 4-tert-butylcatechol stabilizer. The monodispersed PS dispersion with a diameter of 265 nm was synthesized by emulsifier-free emulsion polymerization with potassium peroxydisulfate as an initiator. The PS dispersion was centrifuged at 3000 rpm for 18 h to prepare the PS colloidal crystal templates. The TiO2 precursors (TBT) permeated the voids in the close packed PS colloidal crystal templates through a forced impregnation method.44 In a typical procedure, the PS template was soaked in methanol for 30 min. After pouring off the methanol, the TBT was added into the PS templates, and then the vessel was kept in a vacuum overnight to make the TBT permeate the voids. The excess TBT was poured away, and the solid block was dried at room temperature followed by annealing in the tubular furnace at 450 °C for 12 h to remove the PS microspheres. Finally, the white TiO2 PCs with a well periodic macroporous structure were obtained.2.4 Preparation of CPD/TiO2 PC heterostructures
The CPD/TiO2 PC heterostructures were prepared using a hydrothermal method. In a typical procedure (as described in Scheme 1), 50 mg of TiO2 PCs with different ratios of CPDs were added into the mixed solution of 5 mL of water and 1.5 mL of alcohol. To adequately combine the TiO2 PCs with CPDs, the mixture was transferred into a 25 mL poly(tetrafluoroethylene) (Teflon)-lined autoclave and kept stirring for 4 h before this suspension was heated at 140 °C for 4 h. After cooling down to room temperature naturally, the resulting composites of CPD/TiO2 PC heterostructures were collected through centrifugation and then washed with deionized water three times. Finally, light grey powders of CPDs/TiO2 PCs were obtained after drying.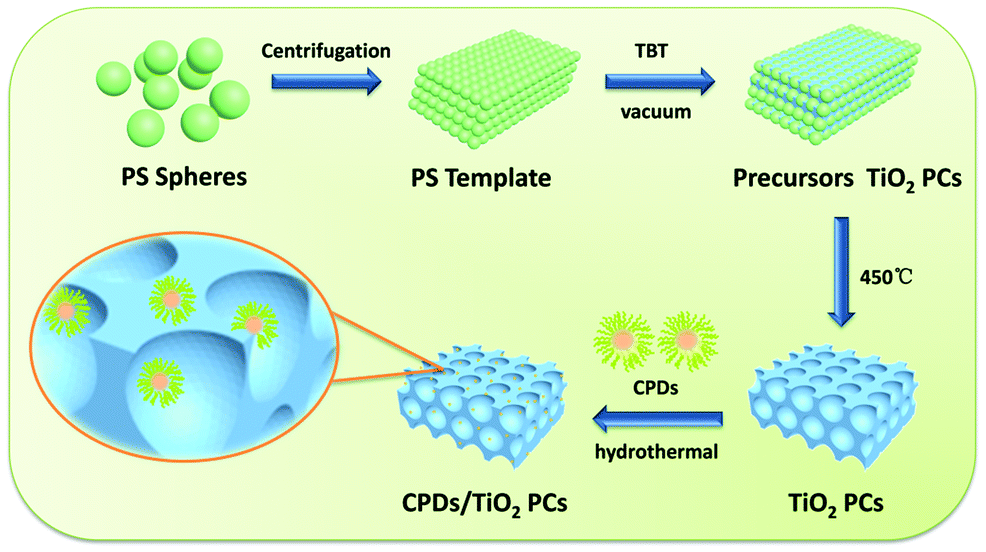 | ||
| Scheme 1 Schematic procedures for the preparation of CPD/TiO2 PC heterostructures. In the magnified scheme of CPD/TiO2 PC heterostructures, the size of the CPDs is enlarged to make them visible. | ||
2.5 Photocatalytic activity and stability measurements
Visible light photocatalytic experiments of Methylene Blue (MB) degradation were carried out to detect the catalytic activity of the CPD/TiO2 PC heterostructures. 20 mg of photocatalyst (CPDs, CPDs/TiO2 PCs heterostructures, TiO2 PCs) was put into 20 mL of MB aqueous solution (20 mg L−1), and the mixture was stirred for 30 min in the dark to reach the adsorption–desorption equilibrium between the dye and catalyst. Next, the solution was cooled with a condensing circulation system and irradiated with a Xe arc lamp fitted with a UV cutoff filter (λ > 420 nm), whose light intensity was set to 100 mW cm−2 (AM1.5 conditions) with the aid of a calibrated Si reference solar cell. During the irradiation, 1 mL of reaction solution was removed at regular intervals, and the change of the MB concentration was analyzed with a ultraviolet-visible spectrophotometer. The stability measurement was performed for 6 repeated cycles under visible (λ > 420 nm, former 3 cycles) and full-spectrum (latter 3 cycles) simulated sunlight radiation. The CPD/TiO2 PC heterostructure photocatalysts were recycled, and carefully collected through centrifugation after each single degradation experiment.2.6 Transient photovoltage (TPV) characterization
The sample chamber consisted of a platinum wire gauze electrode (with the transparency of ca. 50%) as a top electrode, a mica spacer with a thickness of 10 mm as an electron isolator, and a glass substrate covered with indium-tin oxide (ITO) as a bottom electrode. The samples were sandwiched between the electron isolator and bottom electrode, and excited with a laser pulse (365 nm with pulse width of 5 ns) generated by a third harmonic Nd:YAG laser (Polaris, New Wave Research, Inc.). The intensity of the pulse was adjusted with a neutral gray filter and measured by an EM500 single-channel joulemeter (Molectron, Inc.). The TPV data were obtained through a 500 MHz digital phosphor oscilloscope (TDS 5054, Tektronix) with a preamplifier.2.7 Characterization
A shimadzu 3600 UV-visible-NIR spectrophotometer was used to measure the UV-visible absorption spectra. The PL spectra were collected by a Shimadzu RF-5301 PC spectrometer. Fourier transfer infrared (FT-IR) measurements were obtained from a Nicolet AVATAR 360 FT-IR spectrophotometer. Scanning electron microscope (SEM) images were taken using a HITACHI SU800 at 3 kV 10 mA. The morphologies of the PS, TiO2 PCs and CPD/TiO2 PC heterostructures on the super thin carbon film were observed by transmission electron microscopy (TEM), which was performed with a JEM-2100F microscope operated at 200 kV using a CCD camera. XPS spectroscopy was investigated by an ESCA LAB 250. The crystallinity and phase of the TiO2 PCs was studied by XRD using a Empyrean Diffractometer with Cu Kα radiation at 40 kV and 40 mA. The electrochemical impedance (EIS) spectra and transient photocurrent density curves were both collected by a CHI 660E electrochemical workstation in 0.1 M KOH aqueous solution. The transient photocurrent density curves were measured at 1 V (SCE), and the ESI spectra were collected under illumination. Ultraviolet photoelectron spectra (UPS) were collected by a spectrometer (PREVAC sp.z.0.0, R3000).3. Results and discussion
3.1 CPD/TiO2 PC heterostructures
The procedures of preparing CPD/TiO2 PC heterostructures are exhibited in Scheme 1. In this work, the TiO2 PCs are prepared through substrate-free procedures, which can be easily used to enlarge production. First, through centrifuging the polystyrene (PS) solution microspheres, we obtain the close-packed colloidal crystal templates. Subsequently, TiO2 precursors (TBT) permeate the voids in the close packed PS colloidal crystal templates using a forced impregnation method. Third, TiO2 PCs with a three-dimensionally ordered macroporous structure are achieved via a high-temperature annealing process, and the templates are removed completely at the same time. Finally, the CPDs are introduced into TiO2 PCs to form CPD/TiO2 PC heterostructures through a mild hydrothermal process.Herein, PS microspheres with diameters ∼265 nm are used as polymer templates to prepare TiO2 PCs (Fig. S1, ESI†). Fig. 1a shows an SEM image of the close-packed PS microsphere colloidal crystal templates, and they present a highly ordered and face-centered cubic arrangement. Through the processes of precursor infiltration under vacuum, followed by template removal at high temperature, we successfully synthesize the TiO2 PCs with a three-dimensionally ordered macroporous structure. Fig. 1b shows the SEM images of TiO2 PCs. We observe the ordered array of the macropore structure with periodically uniform distribution of macroporous size (∼176 nm). According to the modified Bragg's law:12,45
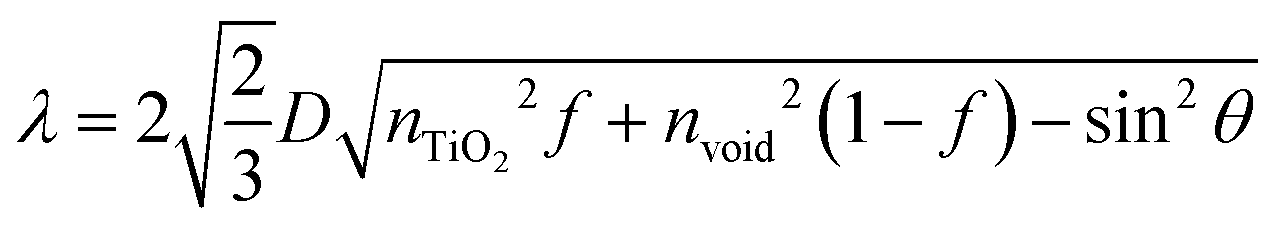
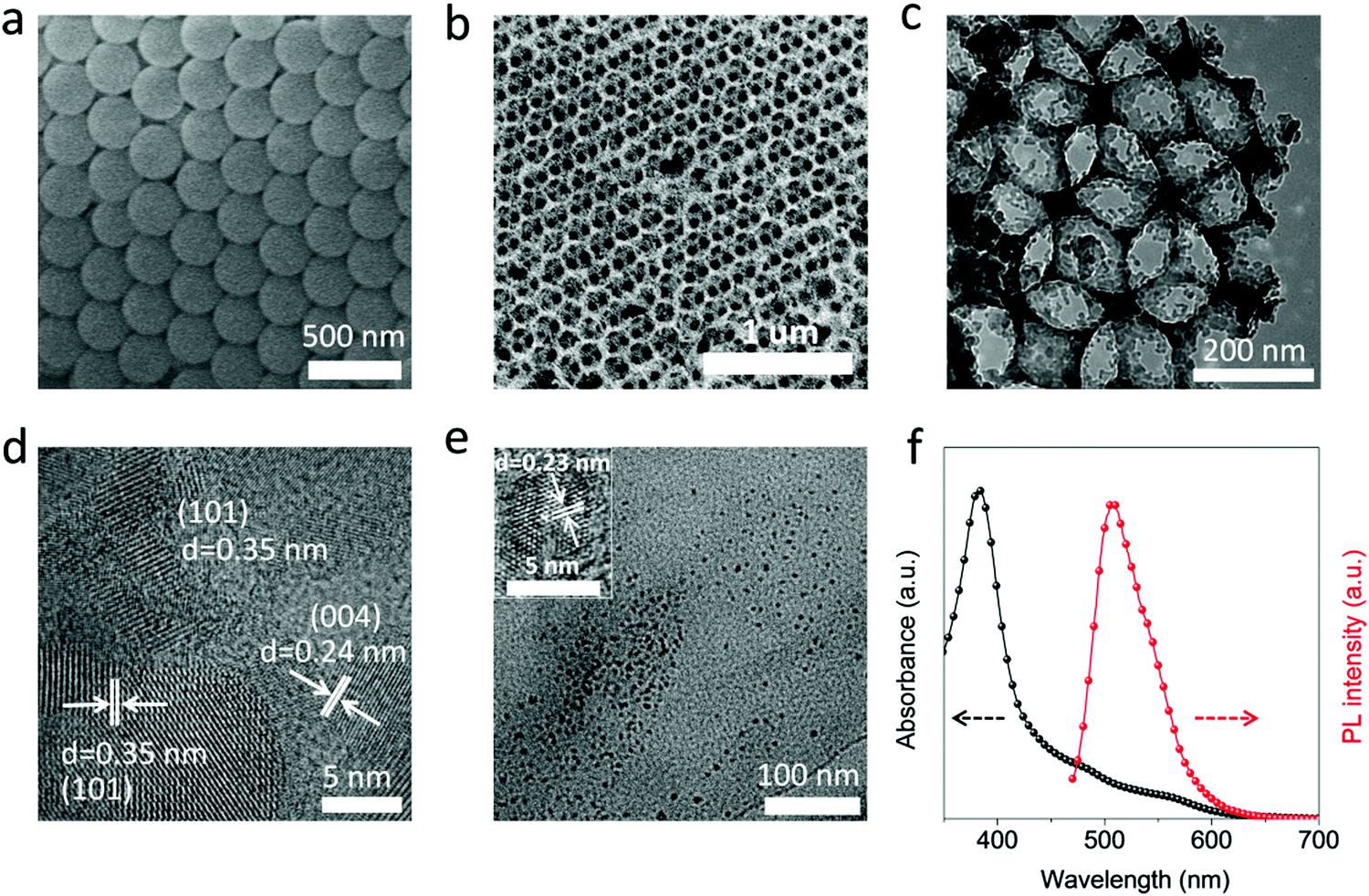 | ||
| Fig. 1 Characterization of TiO2 PCs and CPDs prepared from p-aminosalicylic acid and citric acid precursors. (a) SEM image of closed-packed PS microspheres. (b) SEM, (c) TEM and (d) HR-TEM images of the as-prepared TiO2 PCs. (e) TEM image of the CPDs. Inset shows the HR-TEM image of the CPDs. (f) UV-visible absorption and steady-state photoluminescence spectra of the CPDs. | ||
To further improve the light absorption of anatase TiO2 PCs (3.2 eV), the CPDs with narrow band gap are incorporated into TiO2 PCs to fabricate composite materials. The CPDs with strong visible absorption were synthesized according to previous work.27 Typically, citric acid and p-aminosalicylic acid are selected as the precursors, and the CPDs are prepared under a more acidic hydrothermal environment (pH = 1.0) to increase the carbonization degree, and thus to enhance and broaden the absorption. The TEM image of the CPDs is presented in Fig. 1e, and the CPDs show quasi-spherical shape with the average size of 4.9 nm. The HR-TEM image reveals that the CPDs are crystalline with a lattice spacing of 0.23 nm (inset of Fig. 1e). As shown in Fig. 1f, the UV-vis spectrum of CPDs gives two absorption peaks located at 382 and 507 nm. The CPDs show strong and broad visible absorption, and a band gap of 2.08 eV is calculated from the absorption spectrum (Fig. S4, ESI†). The narrow band gap and strong light absorption of the CPDs are helpful to improve the light harvesting of TiO2 PCs and thus photocatalytic performance. A strong fluorescence-emission peak at 507 nm with negligible excitation-dependent properties is observed in the photoluminescence spectrum (Fig. 1f and Fig. S5, ESI†).27
The CPD/TiO2 PC composite photocatalysts were constructed using a mild hydrothermal method. In the Fourier transform infrared (FT-IR) spectrum of the CPDs (Fig. 2a), the broad peak around 3200 cm−1 belongs to the characteristic absorption of amino, carboxyl and hydroxyl groups, indicating that the CPDs inherit a large amount of polar groups of the precursors (citric acid and p-aminosalicylic acid). These surface polar groups make CPDs easily dissolve in water, and also act as a bridge to link CPDs and TiO2 PCs at the same time. Functional groups of amino and carboxyl can high-effectively bond with TiO2 through coordinating with Ti4+.46 From the FT-IR spectrum of CPD/TiO2 PC composites (Fig. 2a), many characteristic absorption peaks of CPDs appear in the FT-IR spectrum of CPD/TiO2 PC heterostructures. The peaks located at 3425, 1631, 1385 and 1154 cm−1 are attributed to the vibrations of polar groups (amino, carboxyl or hydroxyl), C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N/CC, C–O (phenoxy) and C–O, respectively. Besides, no similar absorption peaks are observed in the FT-IR spectrum of individual TiO2 PCs. Thus, it is suggested that CPDs are successfully introduced into TiO2 PCs.
N/CC, C–O (phenoxy) and C–O, respectively. Besides, no similar absorption peaks are observed in the FT-IR spectrum of individual TiO2 PCs. Thus, it is suggested that CPDs are successfully introduced into TiO2 PCs.
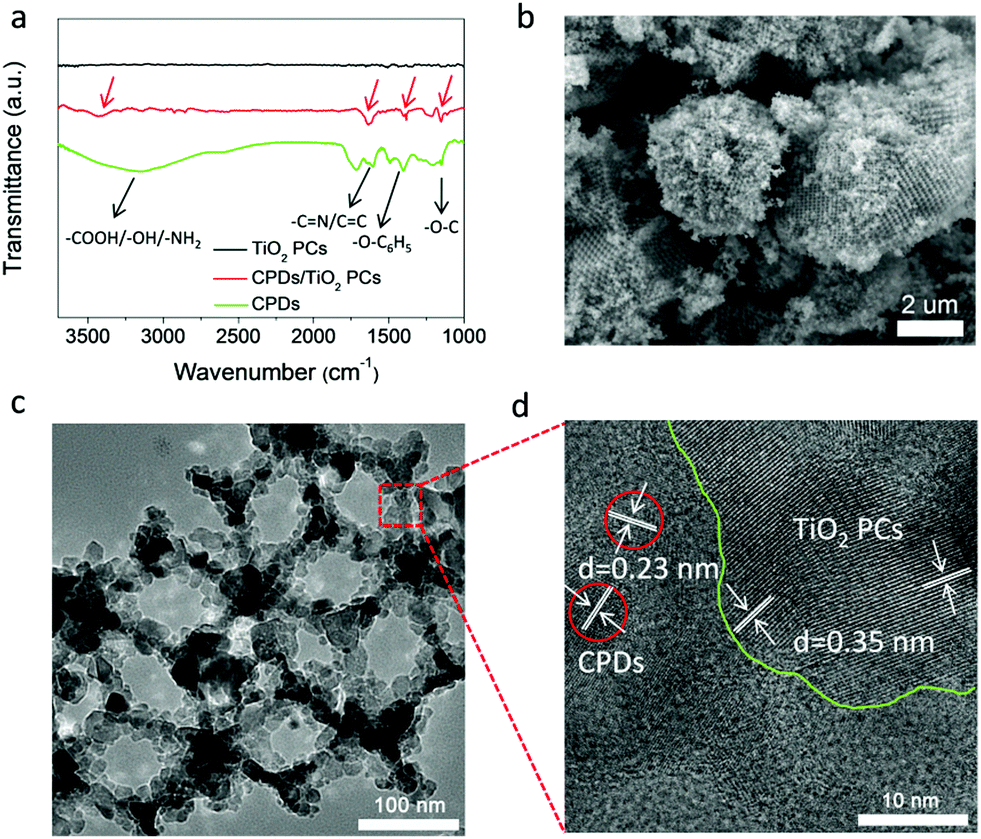 | ||
| Fig. 2 Characterization of a CPD/TiO2 PC heterojunction. (a) FT-IR spectra of CPDs, TiO2 PCs and CPD/TiO2 heterostructures. (b) SEM, (c) TEM and (d) HR-TEM images of CPD/TiO2 PC heterostructures. | ||
SEM and TEM images demonstrate that the CPD/TiO2 PC heterostructures maintain an intact three-dimensionally ordered macroporous structure after hydrothermal and multistep centrifugal purification procedures (Fig. 2b and c). The good structural stability of CPD/TiO2 PC heterostructures is propitious to the durability of photocatalytic performance. HR-TEM measurement was further carried out to study the distinct structure of the CPD/TiO2 PC heterostructures. As shown in Fig. 2d, the crystal fringes of CPDs and TiO2 are both observed, which further indicates that CPDs are anchored on the surface of the TiO2 PCs. Due to the high contrast and crystallinity of TiO2 nanoparticles, their lattice fringes are clearer and more obvious than that of CPDs. Based on the above characterization, it is concluded that CPD/TiO2 PC heterostructures are successfully constructed.
3.2 Photocatalytic performance
In this part, the photocatalytic performance of the CPD/TiO2 PC heterostructures is investigated in detail. Fig. 3a and b present the photocatalytic degradation of methylene blue (MB) and their kinetics in different catalysts under visible light irradiation (λ > 420 nm). The concentration of MB is recorded by UV-visible absorption spectra, and the variation of concentration ratio (final to initial concentration, C/C0) with irradiation time is exhibited in Fig. 3a. MB is slowly degraded and decomposed by about 20% with no catalysts after 120 min irradiation, while sole CPDs show negligible photocatalytic performance of MB degradation (25%). Once TiO2 PC catalysts are adopted, the degradation of MB is promoted to 59%. Furthermore, when the CPDs are introduced into the TiO2 PCs to fabricate a CPD/TiO2 PC heterostructure photocatalyst, the best photocatalytic activity is achieved with enhanced degradation efficiency of up to 85%, indicating that CPDs play a critical role in the improved performance of CPD/TiO2 PC heterostructures. The degradation rate constants of MB for different photocatalysts are studied. It is noted that the kinetics of the degradation process follow a pseudo first order reaction, which can be expressed by the equation: ln(C/C0) = −kappt, where kapp is the apparent reaction rate constant and t is the time.1 The kapp can be obtained from the slope of linear plots. As shown in Fig. 3b, the kapp for CPDs is 0.209 × 10−2 min−1, while that of TiO2 PCs and CPD/TiO2 PC heterostructures is 0.713 × 10−2 min−1 and 1.529 × 10−2 min−1, respectively. The CPD/TiO2 PC heterostructures present an over 2-fold increase of degradation rate compared to the individual TiO2 PCs. The mechanisms of enhanced performance are systematically studied in the next part. Besides, the effects of CPD content on photocatalytic performance are investigated. When the content of CPDs reaches 0.5wt%, the CPD/TiO2 PC heterostructures exhibit the best catalytic activity (Fig. S6, ESI†).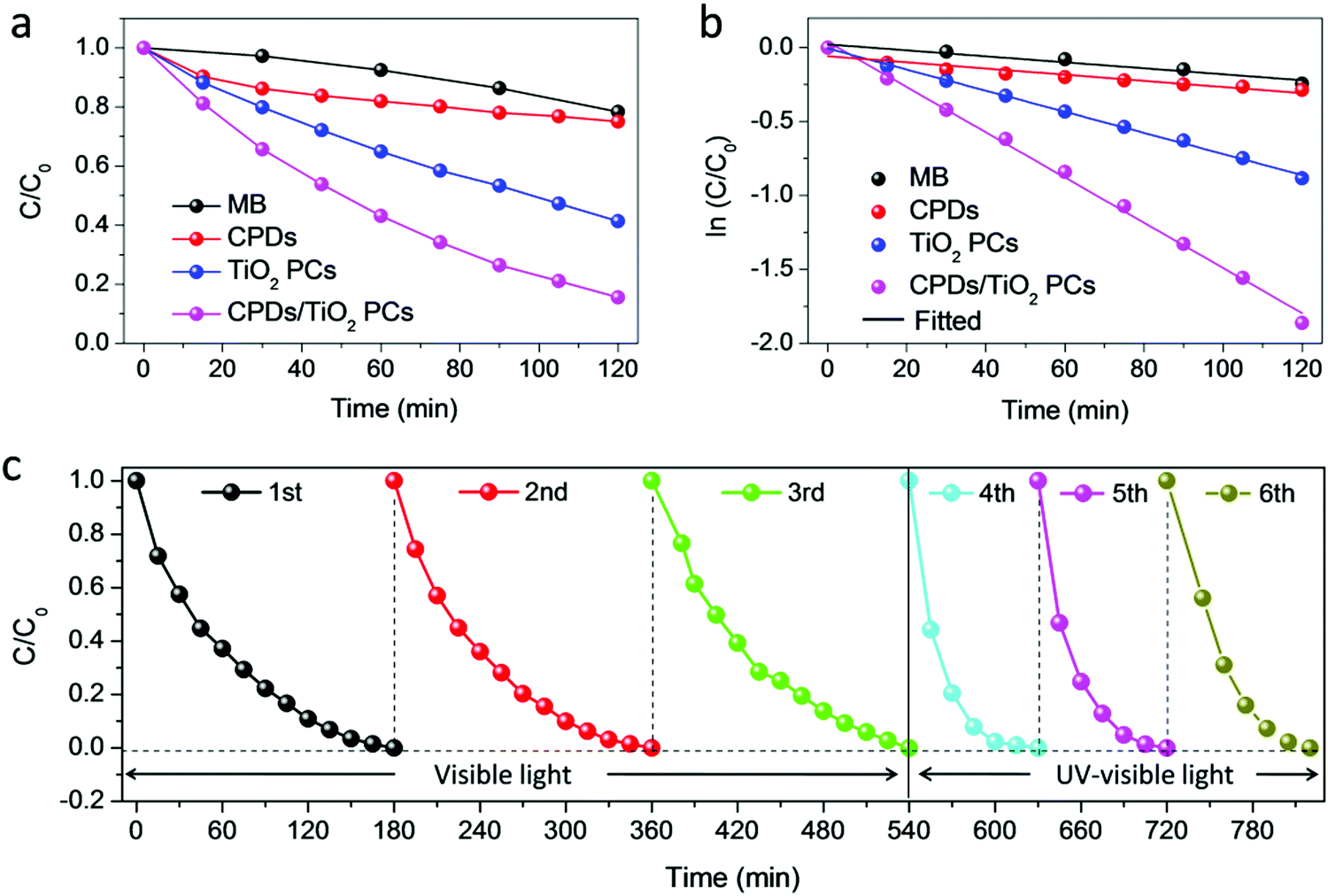 | ||
| Fig. 3 Photocatalytic activity and stability of CPD/TiO2 PC heterostructures for the degradation of MB. (a) Evolution of relative concentration (C/C0) of MB in different photocatalysts under one-sun visible light irradiation (λ > 420 nm). The intensity of the lamp is set to 100 mW cm−2 (AM1.5 conditions) with the aid of a calibrated Si reference solar cell. 20 mg of photocatalyst are used. (b) ln(C/C0) as a function of time. (c) 6 continuous repeated cycles are carried out to test the stability of the CPD/TiO2 PC heterostructure photocatalysts under visible (λ > 420 nm, former 3 cycles) and full-spectrum (latter 3 cycles) simulated sunlight radiation. | ||
The stability and reusability of the CPD/TiO2 PC heterostructures are also studied. As shown in Fig. 3c, 6 continuous repeated cycles are carried out to test the stability of the CPD/TiO2 PC for photocatalytic degradation. The former 3 cycles are performed under visible (λ > 420 nm) simulated sunlight. It is observed that the MB is all degraded within 180 min, and the stability of the CPD/TiO2 PC heterostructures is quite good after 3 recycling photocatalytic experiments. To investigate the stability of the composite photocatalyst in a more real situation, another 3 repeated cycles are conducted under full-spectrum simulated sunlight radiation, where the UV cutoff filter is removed. As presented in Fig. 3c, once the energy of UV-light is introduced, the photocatalytic reaction rate is accelerated obviously and the MB is degraded entirely in less than 90 min. Importantly, the CPD/TiO2 PC heterostructures also maintain excellent stability under complete simulated sunlight radiation.
3.3 Mechanisms of enhanced performance
It is noted that the CPD/TiO2 PC heterostructures exhibit improved photocatalytic efficiency compared with sole TiO2 PCs. The inner synergistic effects between CPDs and TiO2 PCs are explored in detail in this part. Light harvesting is of great importance for a photocatalyst. Fig. 4a presents the absorption spectra of CPDs, TiO2 PCs and CPD/TiO2 heterostructure powders. The TiO2 PCs have strong absorbance in the UV-region (λ < 400 nm), while their absorbance in visible-light-region (λ > 400 nm) is negligible. Nevertheless, once CPDs are introduced, the absorption of both UV and visible light is promoted for CPD/TiO2 heterostructures, which is attributed to the wide absorption range of CPDs. As directly observed in the insets of Fig. 4a, the individual TiO2 PCs are pure white, while their color becomes light grey after introducing CPDs, which indicates enhanced light harvesting for CPD/TiO2 heterostructures. The promoted absorbance of CPD/TiO2 heterostructures is beneficial for a photocatalytic reaction.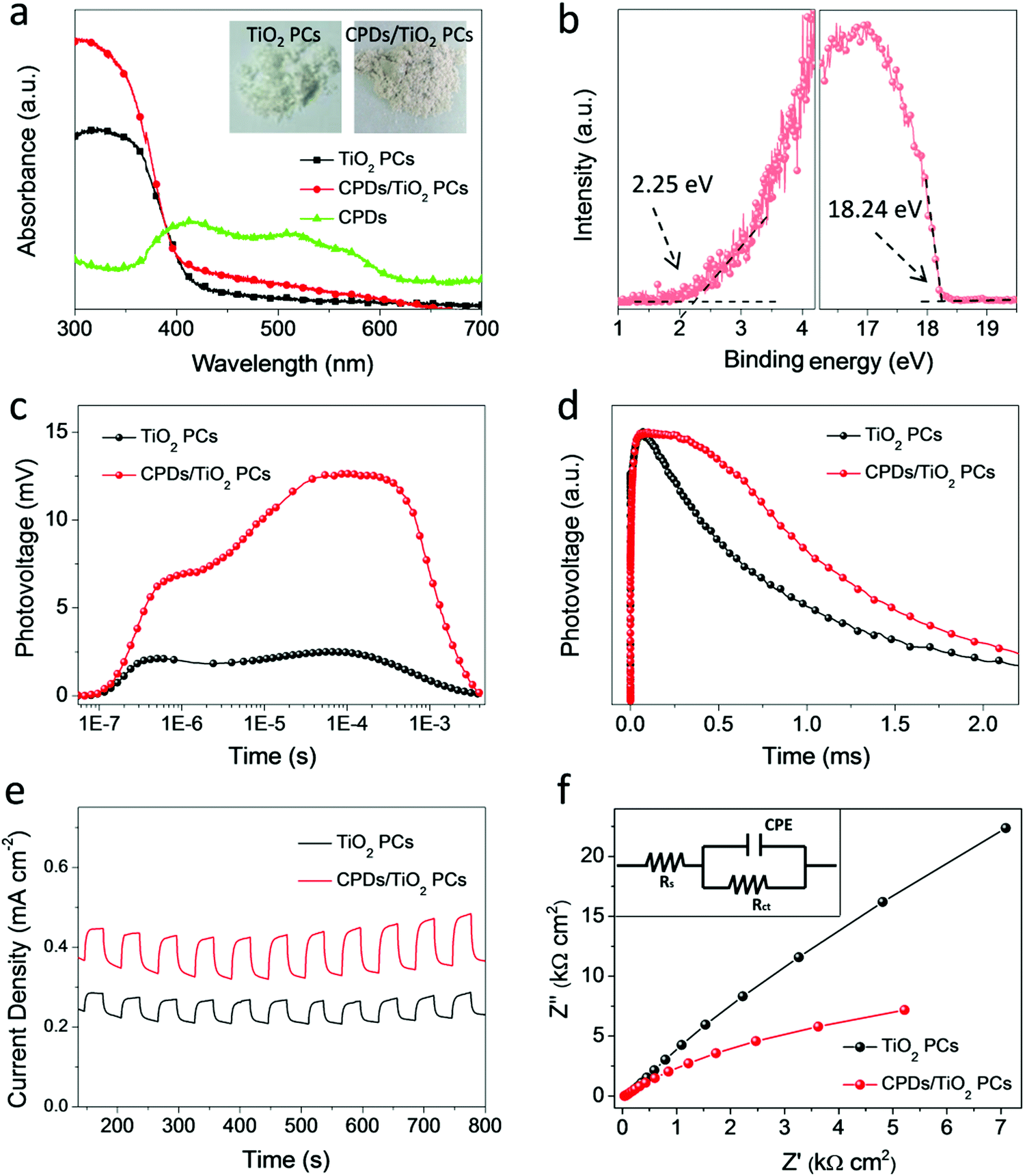 | ||
| Fig. 4 Photocatalytic mechanisms of CPD/TiO2 PC heterostructures. (a) Absorption spectra of CPDs, TiO2 PCs and CPD/TiO2 heterostructure powders. Insets show the optical photographs of TiO2 PCs (left) and CPD/TiO2 heterostructure (right) powders. (b) Ultraviolet photoelectron spectroscopy (UPS) of CPDs: secondary electron cut-off region (left), magnified spectra near the Fermi edge (right). HOMO = −(21.21 − 18.24 + 2.25) eV = −5.32 eV. (c) Transient photovoltage (TPV) curves of TiO2 PCs and CPD/TiO2 PC heterostructures as a function of exponential time. (d) Plots of normalized transient photovoltage versus linear time. (e) Transient photocurrent density, and (f) EIS curves of TiO2 PC and CPD/TiO2 PC heterostructures. The inset in (f) is the equivalent circuit diagram for analyzing charge dynamics. | ||
Besides light harvesting, charge separation is another important factor that affects photocatalytic performance. Charge transfer depends on the energy level alignment between CPDs and TiO2 PCs. Ultraviolet photoelectron spectroscopy (UPS) measurement was used to characterize the energy levels of CPDs. The highest occupied molecular orbital (HOMO) of CPDs is calculated to be −5.32 eV from the UPS spectrum (Fig. 4b). The band gap of CPDs is 2.08 eV, so the lowest unoccupied molecular orbital (LUMO) is determined to be −3.24 eV. The valence band (VB) and conduction band (CB) of anatase TiO2 are −7.40 and −4.20 eV, respectively.15 Therefore, staggered energy levels are established between CPDs and anatase TiO2 PCs. This indicates that CPD/TiO2 PC heterojunctions belong to type-II heterojunctions, which are beneficial for charge separation and thus suppressed charge recombination. Photogenerated electrons in CPDs can transfer into TiO2 PCs, while CPDs can accept the holes of TiO2 PCs. To investigate the charge separation dynamics and carrier lifetime, transient photovoltage (TPV) measurements at 355 nm excitation are performed.47,48 For the TPV response, a positive signal suggests that holes accumulate at the surface of a light-irradiated top electrode. In contrast, a negative signal implies electrons accumulating at the surface of a top electrode.49–51 As shown in Fig. 4c, both of the TiO2 PCs and CPD/TiO2 PC heterostructures present positive signals, which indicates that photogenerated holes are gathered at the top surface. Compared with sole TiO2 PCs, the CPD/TiO2 PC heterostructures exhibit enhanced TPV response, implying promoted charge separation inside the CPD/TiO2 PC heterostructures. This originates from the increased harvesting of incident light and matched energy level alignment of CPD/TiO2 PC heterostructures. The TPV measurement of individual CPDs was also conducted (Fig. S7, ESI†). A very weak and negative TPV response is observed, which demonstrates that, compared with TiO2 PCs and CPD/TiO2 PC heterostructures, charge separation is weak and photogenerated holes and electrons transfer through the opposite direction inside sole CPDs. This further proves that the enhanced TPV response of CPD/TiO2 PC heterostructures is attributed to the synergic actions between TiO2 PCs and CPDs. Plots of normalized TPV intensity versus linear time are presented in Fig. 4d to directly observe the recombination lifetime of photogenerated carriers. In comparison with a half-lifetime of 0.636 ms in TiO2 PCs, the half-lifetime of CPDs/TiO2 PC heterostructures increases to 1.103 ms. The promoted charge lifetime is ascribed to the enhanced charge separation and suppressed charge recombination inside CPD/TiO2 PC heterostructures.
Based on the above discussion, it is concluded that the enhanced light harvesting and charge separation of CPD/TiO2 PC heterostructures accounts for their promoted photocatalytic performance. In order to further investigate the charge generation and separation dynamics in TiO2 PCs and CPD/TiO2 PC heterostructures, the transient photocurrent response and electrochemical impedance spectra (EIS) are measured. As shown in Fig. 4e, the transient photocurrent data are obtained through repeating the on/off illumination cycles at a bias of 1.0 V. The sole TiO2 PCs have a relatively low photocurrent response under each illumination, while CPD/TiO2 PC heterostructures present obviously enhanced reproductive photocurrent response with excellent stability of up to 800 s. This further proves the promoted charge separation in CPD/TiO2 PC heterostructures. Fig. 4f shows the Nyquist plots of TiO2 PCs and CPD/TiO2 PC heterostructures, which are measured under light conditions at a bias of 0 V with a frequency ranging from 0.1 Hz to 200 kHz. Only one circular arc is observed for each sample. In this case, an equivalent circuit containing an electrolyte solution resistance (Rs), interfacial charge-transfer resistance (Rct) and constant phase element (CPE) is used to analyze the charge transfer dynamics of TiO2 PCs and CPD/TiO2 PC heterostructures.7 Through fitting the Nyquist plots according to the above model, the CPD/TiO2 PC heterostructures have much lower Rct of 26.0 kΩ cm2 than the sole TiO2 PCs (301 kΩ cm2), due to the smaller semicircle of CPD/TiO2 PC heterostructures (Table 1). This further proves that the incorporation of CPDs accelerates the charge transport and transfer, which is beneficial for the photocatalytic reaction.
| Samples | R ct (kΩ cm2) | R s (Ω cm2) |
|---|---|---|
| TiO2 PCs | 301 ± 8 | 85.6 ± 11.7 |
| CPDs/TiO2 PCs | 26.0 ± 0.8 | 54.5 ± 5.2 |
According to these control experiments, the photocatalytic MB degradation mechanisms of CPD/TiO2 PC heterojunctions are proposed. As shown in Scheme 2, CPDs are anchored on the TiO2 PCs to form CPD/TiO2 PC type-II heterojunctions with staggered energy level alignment. Therefore, during the photocatalytic process, CPD/TiO2 PC heterostructures absorb incident light and produce photogenerated charge carriers. Driven by the staggered energy level alignment, the photogenerated electrons in CPDs transfer rapidly to TiO2 PCs, while the holes in TiO2 PCs are partly extracted by CPDs as a result of the relatively low content of CPDs in the heterostructures. Compared to individual TiO2 PCs, the charge carriers in CPD/TiO2 PC heterostructures are separated rapidly and have much longer lifetimes, which contributes to a photocatalytic reaction. Based on the reported catalytic mechanisms in TiO2/carbon nanomaterial composites, the separated holes and electrons react with water (H2O) and oxygen (O2), respectively, to produce active ˙OH and ˙O2− radicals,1,52 which are highly energetic to decompose the MB into CO2, water and other inorganic compounds.
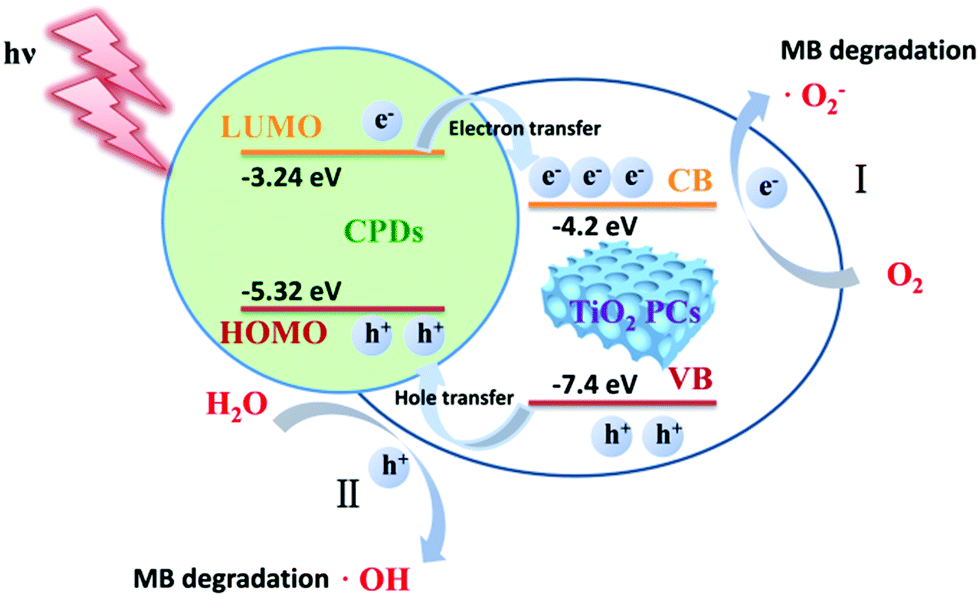 | ||
| Scheme 2 Schematic photocatalytic mechanisms of CPD/TiO2 PC heterostructures. | ||
4. Conclusions
In summary, we develop an efficient method to enhance the light harvesting and charge separation of TiO2 PCs through fabricating CPD/TiO2 PC heterostructures. The charge separation dynamics and photocatalytic performance-improved mechanisms of CPD/TiO2 PC heterostructures are deeply investigated. CPD/TiO2 PC heterostructures have much broader light absorption range than individual TiO2 PCs due to the narrow band gap of CPDs. Besides, the energy level of CPDs matches well with TiO2 PCs, and thus type-II heterojunctions are formed between CPDs and TiO2 PCs. The staggered energy levels of CPD/TiO2 PC heterostructures are beneficial for charge separation, thus leading to enhanced charge lifetime. Therefore, the CPD/TiO2 PC heterostructures exhibit obviously improved photocatalytic performance compared to the individual TiO2 PCs. This work highlights the importance of a heterojunction structure for preparing high-performance photocatalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Science Foundation of China (NSFC) under Grant No. 51433003, and the Fundamental Research Funds for the Central Universities, JLU and JLUSTIRT (2017TD-06). Finally, we thank Kai Zhang and Prof. Tengfeng Xie from College of Chemistry, Jilin University, for the kind help with TPV measurements.References
- G. Rajender, J. Kumar and P. K. Giri, Appl. Catal., B, 2018, 224, 960–972 CrossRef CAS.
- R. Georgekutty, M. K. Seery and S. C. Pillai, J. Phys. Chem. C, 2008, 112, 13563–13570 CrossRef CAS.
- H.-l. Xia, H.-s. Zhuang, T. Zhang and D.-c. Xiao, J. Environ. Sci., 2007, 19, 1141–1145 CrossRef CAS.
- X. Fu, L. A. Clark, Q. Yang and M. A. Anderson, Environ. Sci. Technol., 1996, 30, 647–653 CrossRef CAS.
- X. C. Ma, Y. Dai, L. Yu and B. B. Huang, Light: Sci. Appl., 2016, 5, e16017 CrossRef CAS PubMed.
- F. Pincella, K. Isozaki and K. Miki, Light: Sci. Appl., 2014, 3, e133 CrossRef CAS.
- L. Wang, G. Zhou, Y. Tian, L. Yan, M. Deng, B. Yang, Z. Kang and H. Sun, Appl. Catal., B, 2019, 244, 262–271 CrossRef CAS.
- S. Sakthivel and H. Kisch, Angew. Chem., Int. Ed., 2003, 42, 4908–4911 CrossRef CAS PubMed.
- S. Zhuo, M. Shao and S.-T. Lee, ACS Nano, 2012, 6, 1059–1064 CrossRef CAS PubMed.
- X. Yu, J. Liu, Y. Yu, S. Zuo and B. Li, Carbon, 2014, 68, 718–724 CrossRef CAS.
- D. Pan, J. Jiao, Z. Li, Y. Guo, C. Feng, Y. Liu, L. Wang and M. Wu, ACS Sustainable Chem. Eng., 2015, 3, 2405–2413 CrossRef CAS.
- J. Cai, M. Wu, Y. Wang, H. Zhang, M. Meng, Y. Tian, X. Li, J. Zhang, L. Zheng and J. Gong, Chem, 2017, 2, 877–892 CAS.
- J. C. Yu, J. Yu, W. Ho, Z. Jiang and L. Zhang, Chem. Mater., 2002, 14, 3808–3816 CrossRef CAS.
- J. Yu, L. Qi and M. Jaroniec, J. Phys. Chem. C, 2010, 114, 13118–13125 CrossRef CAS.
- Y. Zhao, Q. Zeng, X. Liu, S. Jiao, G. Pang, X. Du, K. Zhang and B. Yang, J. Mater. Chem. A, 2016, 4, 11738–11746 RSC.
- K. Ozawa, S. Yamamoto, R. Yukawa, R.-Y. Liu, N. Terashima, Y. Natsui, H. Kato, K. Mase and I. Matsuda, J. Phys. Chem. C, 2018, 122, 9562–9569 CrossRef CAS.
- J. C. Calva-Yáñez, M. Solís de la Fuente, M. Ramírez-Vargas and M. E. Rincón, Materials For Renewable and Sustainable Energy, 2018, vol. 7, p. 19 Search PubMed.
- P. Zhou, D. Zhou, L. Tao, Y. Zhu, W. Xu, S. Xu, S. Cui, L. Xu and H. Song, Light: Sci. Appl., 2014, 3, e209 CrossRef CAS.
- X. Chen, C. Chardin, K. Makles, C. Caër, S. Chua, R. Braive, I. Robert-Philip, T. Briant, P.-F. Cohadon and A. Heidmann, Light: Sci. Appl., 2017, 6, e16190 CrossRef CAS PubMed.
- N. V. Konopsky, V. E. Alieva, S. Y. Alyatkin, A. A. Melnikov, V. S. Chekalin and M. V. Agranovich, Light: Sci. Appl., 2016, 5, e16168 CrossRef PubMed.
- J. Du, X. Lai, N. Yang, J. Zhai, D. Kisailus, F. Su, D. Wang and L. Jiang, ACS Nano, 2010, 5, 590–596 CrossRef.
- F. Sordello, C. Duca, V. Maurino and C. Minero, Chem. Commun., 2011, 47, 6147–6149 RSC.
- S. Lee, Y. Lee, D. H. Kim and J. H. Moon, ACS Appl. Mater. Interfaces, 2013, 5, 12526–12532 CrossRef CAS PubMed.
- C. Cheng, S. K. Karuturi, L. Liu, J. Liu, H. Li, L. T. Su, A. I. Tok and H. J. Fan, Small, 2012, 8, 37–42 CrossRef CAS PubMed.
- W. Zhang, X. Zhang, Z. Zhang, W. Wang, A. Xie, C. Xiao, H. Zhang and Y. Shen, J. Electrochem. Soc., 2015, 162, H638–H644 CrossRef CAS.
- J. I. Chen, E. Loso, N. Ebrahim and G. A. Ozin, J. Am. Chem. Soc., 2008, 130, 5420–5421 CrossRef CAS PubMed.
- T. Feng, Q. Zeng, S. Lu, X. Yan, J. Liu, S. Tao, M. Yang and B. Yang, ACS Photonics, 2017, 5, 502–510 CrossRef.
- Y. Song, S. Zhu, J. Shao and B. Yang, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 610–615 CrossRef CAS.
- J. Liu, D. Li, K. Zhang, M. Yang, H. Sun and B. Yang, Small, 2018, 14, e1703919 CrossRef PubMed.
- T. Ji, B. Guo, F. Liu, Q. Zeng, C. Yu, X. Du, G. Jin, T. Feng, S. Zhu, F. Li and B. Yang, Adv. Mater. Interfaces, 2018, 5, 1701519 CrossRef.
- S. Tao, S. Lu, Y. Geng, S. Zhu, S. A. T. Redfern, Y. Song, T. Feng, W. Xu and B. Yang, Angew. Chem., Int. Ed., 2018, 57, 2393–2398 CrossRef CAS PubMed.
- M. Han, S. Zhu, S. Lu, Y. Song, T. Feng, S. Tao, J. Liu and B. Yang, Nano Today, 2018, 19, 201–218 CrossRef CAS.
- X. Bao, Y. Yuan, J. Chen, B. Zhang, D. Li, D. Zhou, P. Jing, G. Xu, Y. Wang, K. Hola, D. Shen, C. Wu, L. Song, C. Liu, R. Zboril and S. Qu, Light: Sci. Appl., 2018, 7, 91 CrossRef PubMed.
- D. Li, D. Han, S. N. Qu, L. Liu, P. T. Jing, D. Zhou, W. Y. Ji, X. Y. Wang, T. F. Zhang and D. Z. Shen, Light: Sci. Appl., 2016, 5, e16120 CrossRef CAS PubMed.
- J. Cui, T. Liu, D. Liang, J. Liu, K. Zhang, B. Yang and Y. Ning, RSC Adv., 2017, 7, 49330–49336 RSC.
- S. Zhu, Q. Meng, L. Wang, J. Zhang, Y. Song, H. Jin, K. Zhang, H. Sun, H. Wang and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 3953–3957 CrossRef CAS PubMed.
- C. Xia, S. Tao, S. Zhu, Y. Song, T. Feng, Q. Zeng, J. Liu and B. Yang, Chem. – Eur. J., 2018, 24, 11303–11308 CrossRef CAS PubMed.
- B. C. M. Martindale, G. A. M. Hutton, C. A. Caputo, S. Prantl, R. Godin, J. R. Durrant and E. Reisner, Angew. Chem., Int. Ed., 2017, 56, 6459–6463 CrossRef CAS PubMed.
- A. Ferrer-Ruiz, T. Scharl, P. Haines, L. Rodriguez-Perez, A. Cadranel, M. A. Herranz, D. M. Guldi and N. Martin, Angew. Chem., Int. Ed., 2018, 57, 1001–1005 CrossRef CAS PubMed.
- H. Yu, R. Shi, Y. Zhao, G. I. Waterhouse, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Mater., 2016, 28, 9454–9477 CrossRef CAS PubMed.
- H. Yu, R. Shi, Y. Zhao, G. I. Waterhouse, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Mater., 2016, 28, 9454–9477 CrossRef CAS PubMed.
- H. Yu, Y. Zhao, C. Zhou, L. Shang, Y. Peng, Y. Cao, L.-Z. Wu, C.-H. Tung and T. Zhang, J. Mater. Chem. A, 2014, 2, 3344–3351 RSC.
- K.-H. Ye, Z. Wang, J. Gu, S. Xiao, Y. Yuan, Y. Zhu, Y. Zhang, W. Mai and S. Yang, Energy Environ. Sci., 2017, 10, 772–779 RSC.
- X. Chen, Z. Li, J. Ye and Z. Zou, Chem. Mater., 2010, 22, 3583–3585 CrossRef CAS.
- J. Xu, B. Yang, M. Wu, Z. Fu, Y. Lv and Y. Zhao, J. Phys. Chem. C, 2010, 114, 15251–15259 CrossRef CAS.
- J. Wu, Z. Lan, J. Lin, M. Huang, Y. Huang, L. Fan and G. Luo, Chem. Rev., 2015, 115, 2136–2173 CrossRef CAS PubMed.
- Q. Bu, S. Li, K. Zhang, Y. Lin, D. Wang, X. Zou and T. Xie, ACS Sustainable Chem. Eng., 2019, 7, 10971–10978 CrossRef CAS.
- L. Bi, X. Gao, L. Zhang, D. Wang, X. Zou and T. Xie, ChemSusChem, 2018, 11, 276–284 CrossRef CAS PubMed.
- Q. Bu, S. Li, Q. Wu, L. Bi, Y. Lin, D. Wang, X. Zou and T. Xie, ChemSusChem, 2018, 11, 3486–3494 CrossRef CAS.
- L. Bi, X. Gao, Z. Ma, L. Zhang, D. Wang and T. Xie, ChemCatChem, 2017, 9, 3779–3785 CrossRef CAS.
- Q. Zeng, Z. Chen, F. Liu, G. Jin, X. Du, T. Ji, Y. Zhao, Y. Yue, H. Wang, D. Meng, T. Xie, H. Zhang and B. Yang, Sol. RRL, 2017, 1, 1600020 CrossRef.
- A. Qu, H. Xie, X. Xu, Y. Zhang, S. Wen and Y. Cui, Appl. Surf. Sci., 2016, 375, 230–241 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qm00556k |
| ‡ Yue Zhao and Qingsen Zeng contributed equally to this work. |
| This journal is © the Partner Organisations 2019 |