
G-quadruplex-based antiviral hydrogels by direct gelation of clinical drugs†
Jingjing
Hu
a,
Hui
Wang
b,
Qianyu
Hu
a and
Yiyun
Cheng
 *ab
*ab
aShanghai Key Laboratory of Regulatory Biology, School of Life Sciences, East China Normal University, Shanghai, 200241, P. R. China
bSouth China Advanced Institute for Soft Matter Science and Technology, South China University of Technology, Guangzhou 510640, China. E-mail: yycheng@mail.ustc.edu.cn
First published on 25th April 2019
Abstract
Supramolecular hydrogels made from guanine derivatives have attracted great interest for various applications. The gelation of guanine analogues usually involves the formation of guanine/K+ quartets. Functional cargoes such as fluorophores and drugs are usually decorated onto the guanine quartet gels via non-covalent π–π interactions or dynamic covalent linkages to render the gel with new functionalities. Here, a class of antiviral guanine quartet hydrogels have been developed via direct gelation of clinically available antiviral drugs such as entecavir, penciclovir, and ganciclovir in the presence of K+ ions. The prepared gels were stable at ambient temperature, non-toxic to normal cell lines, and maintained their inherent antiviral activities against hepatitis B virus and Herpes simplex virus, respectively. This study expands the function of guanine-based supramolecular gels and permits the development of a new class of antiviral gels.
Introduction
Supramolecular assembly of biomolecule gelators such as peptides, nucleobases, lipids and their analogues to construct supramolecular hydrogels has received great attention in drug delivery, immunotherapy, cell culture, and tissue regeneration.1 These biomolecule-derived supramolecular gels show unique properties, i.e. excellent biocompatibility, facile and reproducible preparation without involving complicated synthesis, predictable degradation and bio-responsiveness.2 Guanosine analogues such as guanosine monophosphate have been known for a long period to form hydrogels via the π–π stacking of the guanosine quartet, a hydrogen bonded quadruplex coordinated with metal ions such as K+ in the center (G4/K+).1e,3 The stacked G-quartets generate long fibers, and further connect with each other to form crosslinking networks at ambient temperature. To stabilize the G-quartet hydrogels, various guanosine derivatives were synthesized by increasing the gelator hydrophobicity.4 In an alternative strategy, borate anions were introduced to stabilize G-quartet gels via the formation of dynamic covalent borate/diol linkages.5 Interestingly, the G-quartet structures were widely recognized in telomeric DNA with repeated G-rich sequences, and were relevant to specific biological functions as well as various pathological processes.6 G-rich DNA motifs were widely used to fabricate shape memory and ion-responsive hydrogels.7 The G-quartet gels were generally decorated with fluorophores, dyes, hemin, and drugs via π–π stacking interactions or dynamic covalent bonds to render the gels with specific functionalities for various applications.5c,8 Though there have been promising advances in this field, the direct gelation of bioactive components via the G-quartet supramolecular chemistry is yet to be investigated. There is a family of clinically available antiviral drugs that contain a guanosine unit. We hypothesize that these guanosine-containing drugs may gelate with the K+ to form supramolecular gels with inherent antiviral activity.Experimental section
Materials
Entecavir, penciclovir, ganciclovir, lithium chloride (LiCl), rubidium chloride (RuCl), and cesium chloride (CsCl) were purchased from Dalian Meilun Biotechnology Co., Ltd (Dalian, China). Sodium chloride (NaCl) and potassium chloride (KCl) were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Sangon Biotech (Shanghai, China). Deuterium oxide was purchased from Sigma-Aldrich (St. Louis, USA).Preparation of antiviral gels
Entecavir was dissolved in 80 mM KCl solution at a concentration of 40 mM. The solution was heated to 90 °C until entecavir was dissolved completely, then the hot solution was cooled down to room temperature, and a transparent gel was obtained. The ganciclovir gel and penciclovir gel were prepared in a similar manner at ganciclovir and penciclovir concentrations of 60 mM and 80 mM, respectively. The concentration of KCl was twice that of the antiviral drugs in the gels. To give an insight into entecavir gelation in the presence of K+, the concentration of entecavir ranged from 30 to 80 mM, and the KCl concentration ranged from 10 mM to 80 mM. The mixture was heated and cooled down to room temperature as described above, and then the status of the entecavir/K+ mixture was observed to plot the gelation phase diagram.Characterization
For TEM, the gels were diluted to a final drug concentration of 50 μM, and 1.5 μL of the samples were dropped onto the copper grid and dried naturally for TEM characterization. The TEM experiments were performed on a Hitachi microscope (HT7700, Hitachi, Japan) with an acceleration voltage of 100 kV. For AFM characterization, 30 μL samples were dropped onto the silicon chip and automatically dried before characterization by AFM (Veeco Dimension 3100, Bruker). The experiments were carried out in air tapping mode with an antimony (n) doped Si tip (spring constant cantilever: 40 N m−1). For CD characterization, 0.34 mM entecavir solution, or 0.39 mM ganciclovir/penciclovir solution was characterized by CD spectroscopy (CD, J-815, Jasco International). The experiments were carried out in a quartz cell with a 0.1 mm path length, and the scans were recorded from 200 to 500 nm. For ESI-MS, the samples were diluted to a drug concentration of 100 μM and characterized by ESI-MS (ABI 4800 plus, USA). For NMR studies, entecavir (5.545 mg), D2O (295 μL) and KCl solution (200 μL, 200 mM) were mixed together and added with 5 μL of dioxane (300 mM in D2O) as an inner standard, and then the solutions were added into the NMR tubes. The mixture was gently heated to 90 °C to dissolve the entecavir and then cooled down to room temperature to form the gel. The NMR spectra were recorded at 10 °C, 15 °C, 25 °C, 30 °C, 35 °C, 45 °C, 50 °C, 60 °C, and 70 °C, respectively by 500 MHz NMR spectroscopy (Bruker Advance, Germany). The percentage of free entecavir in the gel was calculated by the integrated area of the H1 peak on the guanine group in entecavir according to that of dioxane.Rheology study
The melting temperature of the gel was determined by temperature-dependent rheology measurement. Generally, 500 μL of the gel was placed on the steel plate of the rheometer (Discovery HR-3, TA Instrument, USA). The angular frequency and strain were set at 10 rad s−1 and 1%, respectively, and the entecavir concentration in the gels was 30 mM, 35 mM, 40 mM, 45 mM, and 50 mM, respectively. A temperature-dependent rheology study ranging from 25 °C to 60 °C was performed. For the shear-shining experiment, the strain was set at a constant of 1%, and the angular frequency was swept from 0.1 to 628 rad s−1, and the viscosity of the gel (40 mM) was recorded. 200 μL of the gel was injected through a syringe (0.45 × 16 mm) to support the property. The thixotropic property was investigated through a continuous step strain measurement, and the angular frequency was set at a constant of 10 rad s−1 with an alternative strain of 0.1% and 300% for three times.Gel degradation and drug release
400 μL entecavir gels (40 mM) were prepared in vials and added with 3 mL of deionized water, then the vials were incubated at 20 °C, 37 °C and 45 °C, respectively. At scheduled time intervals, the supernatant was collected to analyze the released entecavir from the gel, and the remaining gels in the vial were weighed simultaneously for the degradation assay. The entecavir concentration was analyzed by injecting 10 μL of the sample into a high-performance liquid chromatography (HPLC, Agilent 1200) instrument equipped with a C18 column (4.6 × 150 mm, 5 μm diameter). The mobile phase consisted of 95% water and 5% acetonitrile, and pumped at a flow rate of 1.0 mL min−1. The samples were detected at a wavelength of 254 nm and the obtained standard curve is y = 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 606x + 267.44 (x is the concentration of entecavir, mg mL−1, and y is the peak area, R2 = 0.9998).
606x + 267.44 (x is the concentration of entecavir, mg mL−1, and y is the peak area, R2 = 0.9998).
Antiviral activity and cytotoxicity assay
The anti-HBV activity and cytotoxicity of the entecavir gels were investigated by HD Biosciences (Shanghai, China). Generally, the entecavir solution was prepared starting from 160 μM with a serial 4-fold, 8-point dilution for the HBV-assay. 1 μL of the serially diluted compounds was added to the assay plates with a final top concentration of 800 nM for the test. Dulbecco's phosphate buffered saline (DPBS, Ca2+ and Mg2+ free), DMSO and tenofovir were used as the negative control, solvent control, and positive control, respectively. The virus-infected HepG2.2.15 cells were incubated with the samples in plates for 7 days, and the medium was refreshed with the serially diluted compounds every other day. 150 μL of supernatant in each well was collected on day 7 for qPCR assay. For the cytotoxicity assay, the entecavir solution was diluted 3-fold and 11-point with the top test concentration of 1 mM. 5 μL of the serially diluted solutions was added to 384-well plates for the cytotoxicity assay. Then, 45 μL of the HepG2.2.15 cells (4000 cells per well in 10% FBS) were added to each well and incubated for 4 days in 5% CO2 atmosphere at 37 °C. After incubation, the cytotoxicity of the compounds was measured by chemiluminescence detection using a Lumi-Glow reagent (Suzhou J&C Laboratories Co., Ltd).For the anti-HSV assay, the ganciclovir and penciclovir solutions were prepared starting from 1 mM with a serial 3-fold, 11-point dilution for the HSV-1 assay. The serially diluted compounds were added to assay plates by Echo 550:2.5 μL per well to 384-well antiviral and cytotoxicity plates. Acyclovir (ACV) was used as an antiviral reference compound, and it was prepared starting from 10 mM in a serial 3-fold, 11-point dilution. The Vero cells are cultured in completed DMEM culture medium supplemented with 10% FBS, 1% penicillin and 1% streptomycin, and the cells should be maintained in high viability (>95%) and diluted to 8 × 104 cell mL−1 before the assay. The HSV-1 virus was thawed and added to the Vero cells in an optimal density of 100 TCID50 per well, and mixed intensively. Then, 47.5 μL of the cells and virus mixture per well were added into the 384-well assay plates containing 2.5 μL of the sample for the antiviral test. The final concentration of the tested samples was 20-fold diluted, and the top concentration is 50 μM. The cells were incubated under a 90% humidified 5% CO2 atmosphere at 37 °C for four days, and the infection CPE was measured by a Promega CellTiter Glo kit. For the cytotoxicity assay, 47.5 μL of the cells without virus were added into each well of the 384-well assay plates containing 2.5 μL of the sample. The cytotoxicity of the compounds was measured by a Lumi-Glow reagent. The antiviral activity (% inhibition) of the sample is calculated as:
| % inhibition = (test value − AVE ZPE)/(AVE HPE − AVE ZPE) × 100 |
The cytotoxicity is calculated as % cytotoxicity = 100 − (test value)/(AVE CELL only wells) × 100. Then, a four-parameter model was used to calculate the EC50 and CC50. In addition, a standard MTT assay was used to evaluate the cytotoxicity of antiviral gels on the NIH 3T3 cells.
The hemolytic activity of the antiviral gels was evaluated using red blood cell (RBC) suspensions from 8 week old Balb/c mice. Briefly, 1 mL of the RBC suspension (2%) was added into the centrifuge tube with 20 μL of the hydrogel (40 mM) and treated for 1 h at 37 °C. The RBC suspensions treated with Triton X-100 (0.5%) and PBS (pH 7.4) were tested as a positive control and negative control, respectively. Then, the mixture was centrifuged at 2000 rpm for 5 min, and the supernatant was collected and transferred into a 96-well plate to measure the absorption of the solution at 540 nm. The RBC suspension treated with Triton X-100 was set as 100% hemolysis.
Results and discussion
Entecavir is a potent antiviral drug with high selectivity and activity against hepatitis B virus (HBV).9 The drug has a guanosine unit in the structure and may form G-quartet fibers with the help of K+ (Fig. 1a). As expected, the drug formed a transparent and stable hydrogel at 40 mM in the presence of K+ at ambient temperature. Other alkali cations such as the Li+, Na+, Rb+ and Cs+ failed to gelate entecavir under the same conditions (Fig. 1b). The gelation is influenced by both components, and the concentrations of entecavir and K+ should be above 30 mM (Fig. 1c). The transmission electron microscopy (TEM) and atomic force microscopy (AFM) images revealed a uniform fibrous network for the entecavir gel (Fig. 2a and b), and the fibers are several micrometers in length (Fig. S1, ESI†). These fibers are cross-linked with each other to build up the gel network. Circular dichroism (CD) was widely used to characterize the conformation of the guanosine quartet fibers. As shown in Fig. 2c, CD spectroscopy of the entecavir gel showed opposite signed bands in the range of 240 to 260 nm, which is a head-to-tail signature of the guanosine quartet. The transition at 250 nm explained the inversion of polarity, from the same polarity of a head-to-tail stacking to a head-to-head or tail-to-tail one. In addition, a positive band at 280 nm was observed, indicating the stacking of quartets with alternating polarities.10 The CD result provided molecular-level evidence for the assembly of entecavir, and the conformation of the assembled nanofibers is distinct from the guanosine quartets. We further used an electrospray ionization mass spectrometer (ESI-MS) to characterize the assembled entecavir/K+ quartets. The MS results showed peaks at m/z 316, 593, and 1147, which represent the entecavir monomer (E/K+), dimer (E2/K+) and tetramer (E4/K+), respectively. These results together proved the assembly of entecavir into quartets in the presence of K+.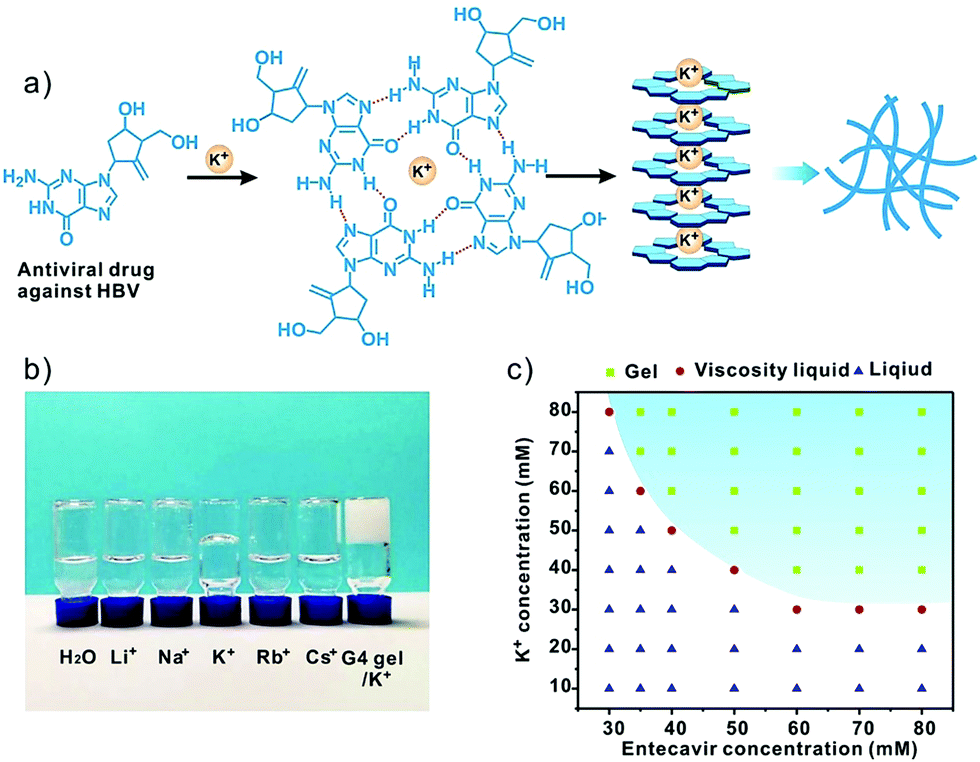 | ||
| Fig. 1 (a) Preparation of the entecavir gel via G-quadruplex formation in the presence of K+. (b) The gelation behavior of entecavir in the presence of various alkali metal ions. The entecavir and ion concentrations were 40 and 80 mM, respectively. The guanosine/K+ gel (G4 gel) is shown as a control. (c) Phase diagram of entecavir/K+ mixtures at different entecavir and K+ concentrations. | ||
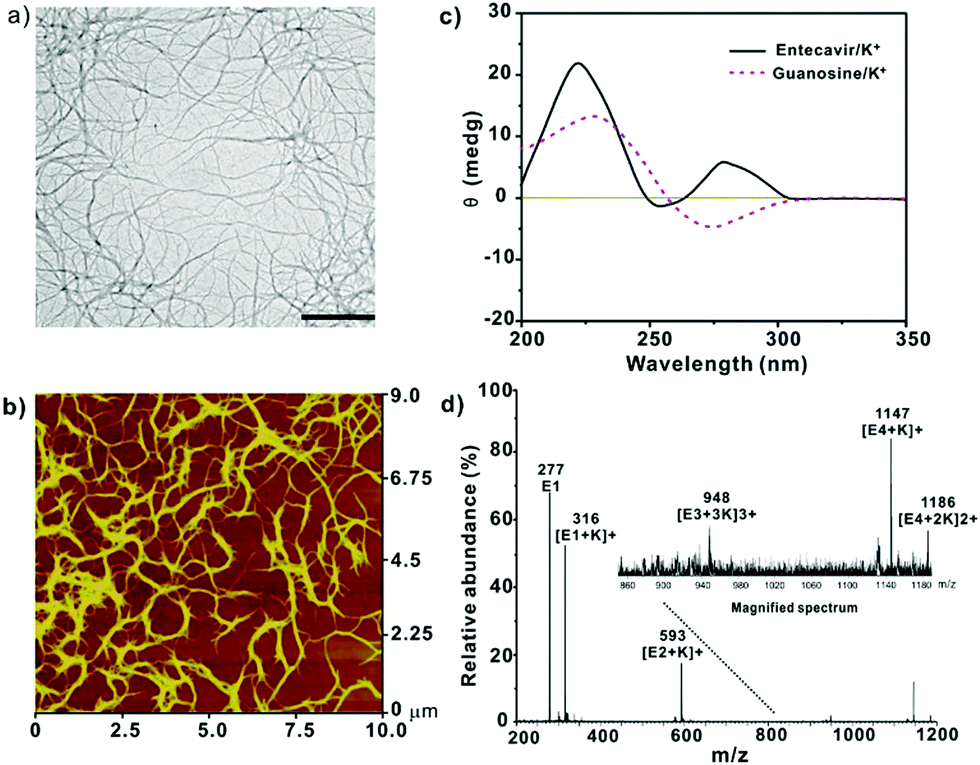 | ||
| Fig. 2 (a) TEM image of the entecavir gel (50 μM entecavir). Scale bar 500 nm. (b) AFM image of the entecavir gel (50 μM entecavir). (c) CD spectra of the entecavir gel prepared at 0.34 mM. The G4/K+ gel was measured as a control. (d) ESI-MS spectra of the entecavir/K+ solution. The entecavir concentration was 2 mM. The molar ratio of entecavir/K+ was 1:2 for all samples. | ||
Since the assembly of guanosine into quartets is mainly driven by hydrogen bonding interactions between the guanosine units, we investigated the temperature-responsiveness of the entecavir gel. We could directly observe the transition of the gel state into a liquid state when the environmental temperature increased from 25 °C to 55 °C (Fig. 3a). Since the proton signal for entecavir in the gel state cannot be detected by solution 1H NMR, the ratio of free entecavir can be determined by the integrated area of the entecavir peaks according to that of the dioxane, which was used as an internal standard.3c The free fraction of entecavir versus temperature showed a sigmoidal shaped curve, and the transition temperature was analyzed to be around 35 °C (Fig. 3a). In addition, a down-field shift of the H1 signal for entecavir was observed with increasing temperature (Fig. S2, ESI†), indicating the disassembly of the entecavir quartets at higher temperatures. A temperature-dependent rheology measurement was further performed to measure the melting temperature (Tm) of the entecavir gel. As shown in Fig. 3b, the storage modulus (G′) decreased below the loss modulus (G′′) when the temperature was above 39 °C. Besides, the melting temperature of the entecavir gel increased with the entecavir concentration (Fig. 3c). This is because higher entecavir concentration may contribute to higher cross-linking degree and higher modulus of the gel, and thus, higher temperature is required to destroy the hydrogen bonding interactions in the gels. The temperature responsiveness indicates the temperature-responsive drug release and gel degradation behaviors. As shown in Fig. 3d, about 20% entecavir molecules were released after incubation at 20 °C for 8 h, while nearly all the drugs were released at 37 °C during the same period. When the temperature was 45 °C, which is higher than the melting temperature, 87.31% of the drugs were released within an hour. Similar results were obtained for the gel weights when incubated at different temperatures (Fig. S3, ESI†). The entecavir gel also showed a shear-thinning property (Fig. 3e) and could be injected through a syringe (Fig. S4, ESI†). The injected liquid could recover to a gel state and be fabricated into different shapes. Moreover, a continuous step strain measurement was performed to evaluate the thixotropic property. The result showed that the gel collapsed at 300% strain, but recovered at 0.1% strain (Fig. S5, ESI†), and the step strain measurement was repeated three times with a high recovery efficiency, confirming a good thixotropic property of the entecavir gel. The in vitro antiviral effect of the entecavir gel against HBV was further evaluated. As shown in Fig. 3f, the EC50 of the entecavir gel against HBV is 0.33 μM, which is similar to that of the free entecavir (0.3 μM). The presence of K+ in the entecavir gel did not significantly influence the bioactivity of entecavir. In addition, both the G4 gel and KCl showed poor antiviral activity at equal molar concentrations to those in the entecavir gel. The entecavir gel also showed low toxicity to normal cells, such as HepG2.2.15 and NIH 3T3 cells, and exhibited no hemolytic activity (Fig. S6, ESI†).
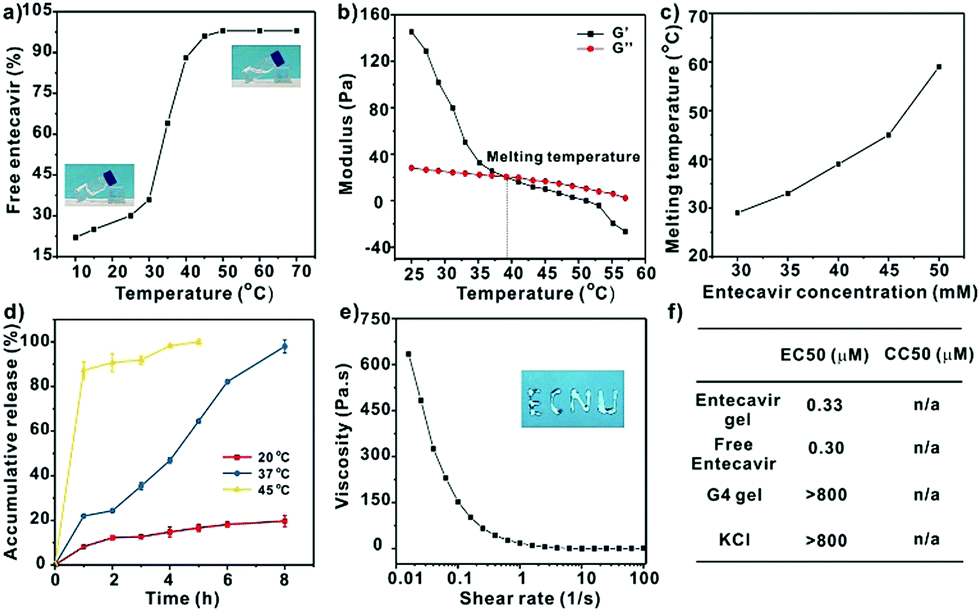 | ||
| Fig. 3 (a) The ratio of free entecavir in the entecavir gel as a function of temperature. The entecavir concentration in the gel was 40 mM. The inserts are the images of the gel at 25 °C and 55 °C, respectively. (b) Temperature-dependent modulus measurement of the entecavir gel (40 mM). (c) Melting temperature of the entecavir gel prepared at different entecavir concentrations. (d) Accumulative release of entecavir from the gel at different temperatures. (e) The shear-thinning property of the entecavir gel (40 mM), and the insert is the image of the entecavir gel (40 mM) injected from a syringe. (f) Antiviral activities (EC50) of the entecavir gel, free entecavir, G4 gel, and KCl against HBV, and their cytotoxicity (CC50) to HepG2.2.15 cells. Molar ratio of the entecavir/K+ was 1:2 for all samples. | ||
Except for entecavir, ganciclovir and penciclovir can also form hydrogels in the presence of K+ (Fig. 4a). These molecules with a guanosine unit in the chemical structure were highly active against herpes simplex virus (HSV).11 The TEM images showed that both drugs form uniform nanofibers in the presence of K+. The molecules were arranged in head-to-tail and head-to-head orientations, which is similar to that observed for the entecavir gel. Similarly, both the ganciclovir and penciclovir gels maintained the high antiviral activity of the free antiviral drugs against HSV-1 and showed low toxicity to normal cells.
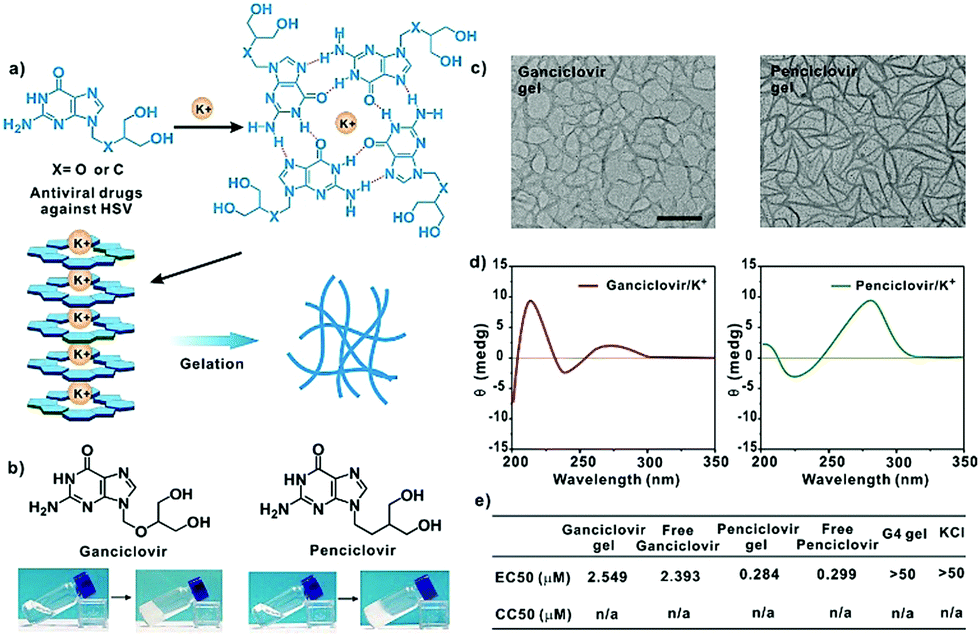 | ||
| Fig. 4 (a) The gelation mechanism of the ganciclovir and penciclovir gels. (b) Photographs of the ganciclovir (60 mM) and penciclovir gels (80 mM) in the presence of K+. (c) The TEM images and (d) CD spectra of the ganciclovir gel (left) and penciclovir gel (right), respectively. 100 μM for TEM, and 0.39 mM for CD, respectively. Scale bar, 500 nm. (e) Antiviral activities (EC50) of the ganciclovir gel, free ganciclovir, penciclovir gel, free penciclovir, G4 gel, and KCl against HSV-1, and their cytotoxicity (CC50) to Vero cells. The molar ratio of the ganciclovir or penciclovir/K+ was 1:2 for all samples. | ||
Conclusions
In summary, we developed a class of guanosine quartet hydrogels by direct gelation of clinically available antiviral drugs such as entecavir, penciclovir, and ganciclovir in the presence of K+. The developed gels showed temperature responsiveness and were capable of releasing the antiviral components in a temperature-dependent manner. Moreover, the prepared gels were injectable, biocompatible and showed similar antiviral activities in comparison to the free antiviral drugs. The developed antiviral hydrogels, especially those which are active against HSV, are promising for the local treatment of viral infections in clinical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (21725402) and the Shanghai Municipal Science and Technology Commission (17XD1401600).References
- (a) Y. Cai, H. Shen, J. Zhan, M. Lin, L. Dai, C. Ren, Y. Shi, J. Liu, J. Gao and Z. Yang, J. Am. Chem. Soc., 2017, 139, 2876 CrossRef CAS PubMed; (b) H. Wang, Z. Feng, Y. Wang, R. Zhou, Z. Yang and B. Xu, J. Am. Chem. Soc., 2016, 138, 16046 CrossRef CAS PubMed; (c) J. Li, X. Du, S. Hashim, A. Shy and B. Xu, J. Am. Chem. Soc., 2017, 139, 71 CrossRef CAS PubMed; (d) J. K. Sahoo, C. G. Pappas, I. R. Sasselli, Y. M. Abul-Haija and R. V. Ulijn, Angew. Chem., Int. Ed., 2017, 56, 6828 CrossRef CAS PubMed; (e) G. M. Peters and J. T. Davis, Chem. Soc. Rev., 2016, 45, 3188 RSC; (f) A. Rotaru, G. Pricope, T. N. Plank, L. Clima, E. L. Ursu, M. Pinteala, J. T. Davis and M. Barboiu, Chem. Commun., 2017, 53, 12668 RSC.
- (a) T. Gajanayake, R. Olariu, F. M. Leclère, A. Dhayani, Z. Yang, A. K. Bongoni, Y. Banz, M. A. Constantinescu, J. M. Karp, P. K. Vemula, R. Rieben and E. Vögelin, Sci. Transl. Med., 2014, 6, 249ra110 CrossRef PubMed; (b) X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165 CrossRef CAS PubMed; (c) Y. Shao, H. Jia, T. Cao and D. Liu, Acc. Chem. Res., 2017, 50, 659 CrossRef CAS PubMed.
- (a) J. T. Davis, Angew. Chem., Int. Ed., 2004, 43, 668 CrossRef CAS PubMed; (b) J. T. Davis and G. P. Spada, Chem. Soc. Rev., 2007, 36, 296 RSC; (c) N. Sreenivasachary and J.-M. Lehn, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5938 CrossRef CAS PubMed; (d) E. Buhler, N. Sreenivasachary, S.-J. Candau and J.-M. Lehn, J. Am. Chem. Soc., 2007, 129, 10058 CrossRef CAS PubMed.
- (a) J. L. Sessler, M. Sathiosatham, K. Doerr, V. Lynch and K. A. Abboud, Angew. Chem., Int. Ed., 2000, 39, 1300 CrossRef CAS; (b) K. B. Sutyak, P. Y. Zavalij, M. L. Robinson and J. T. Davis, Chem. Commun., 2016, 52, 11112 RSC; (c) L. Ma, M. Melegari, M. Colombini and J. T. Davis, J. Am. Chem. Soc., 2008, 130, 2938 CrossRef CAS PubMed; (d) Y.-L. Wu, N. E. Horwitz, K.-S. Chen, D. A. Gomez-Gualdron, N. S. Luu, L. Ma, T. C. Wang, M. C. Hersam, J. T. Hupp, O. K. Farha, R. Q. Snurr and M. R. Wasielewski, Nat. Chem., 2017, 9, 466 CrossRef CAS PubMed.
- (a) G. M. Peters, L. P. Skala, T. N. Plank, B. J. Hyman, G. N. Manjunatha Reddy, A. Marsh, S. P. Brown and J. T. Davis, J. Am. Chem. Soc., 2014, 136, 12596 CrossRef CAS PubMed; (b) G. M. Peters, L. P. Skala, T. N. Plank, H. Oh, G. N. Manjunatha Reddy, A. Marsh, S. P. Brown, S. R. Raghavan and J. T. Davis, J. Am. Chem. Soc., 2015, 137, 5819 CrossRef CAS PubMed; (c) G. M. Peters, L. P. Skala and J. T. Davis, J. Am. Chem. Soc., 2015, 138, 134 CrossRef PubMed.
- (a) C. Kang, X. Zhang, R. Ratliff, R. Moyzis and A. Rich, Nature, 1992, 356, 126 CrossRef CAS PubMed; (b) J. U. Guo and D. P. Bartel, Science, 2016, 353, aaf5371 CrossRef PubMed; (c) G. Guilbaud, P. Murat, B. Recolin, B. C. Campbell, A. Maiter, J. E. Sale and S. Balasubramanian, Nat. Chem., 2017, 9, 1110 CrossRef CAS PubMed; (d) N. Shioda, Y. Yabuki, K. Yamaguchi, M. Onozato, Y. Li, K. Kurosawa, H. Tanabe, N. Okamoto, T. Era, H. Sugiyama, T. Wada and K. Fukunaga, Nat. Med., 2018, 24, 802 CrossRef CAS PubMed.
- (a) C.-H. Lu, X.-J. Qi, R. Orbach, H.-H. Yang, I. Mironi-Harpaz, D. Seliktar and I. Willner, Nano Lett., 2013, 13, 1298 CrossRef CAS PubMed; (b) J. S. Kahn, A. Trifonov, A. Cecconello, W. Guo, C. Fan and I. Willner, Nano Lett., 2015, 15, 7773 CrossRef CAS PubMed; (c) C.-H. Lu, W. Guo, Y. Hu, X.-J. Qi and I. Willner, J. Am. Chem. Soc., 2015, 137, 15723 CrossRef CAS PubMed; (d) C. Wang, M. Fadeev, J. Zhang, M. Vázquez-González, G. Davidson-Rozenfeld, H. Tian and I. Willner, Chem. Sci., 2018, 9, 7145 RSC; (e) C.-H. Lu, W. Guo, X.-J. Qi, A. Neubauer, Y. Paltiel and I. Willner, Chem. Sci., 2015, 6, 6659 RSC; (f) C. Wang, M. Fadeev, M. Vázquez-González and I. Willner, Adv. Funct. Mater., 2018, 28, 1803111 CrossRef.
- (a) V. Venkatesh, N. K. Mishra, I. Romero-Canelón, R. R. Vernooij, H. Shi, J. P. C. Coverdale, A. Habtemariam, S. Verma and P. J. Sadler, J. Am. Chem. Soc., 2017, 139, 5656 CrossRef CAS PubMed; (b) R. Zhong, Q. Tang, S. Wang, H. Zhang, F. Zhang, M. Xiao, T. Man, X. Qu, L. Li, W. Zhang and H. Pei, Adv. Mater., 2018, 30, 1706887 CrossRef PubMed; (c) T. N. Plank and J. T. Davis, Chem. Commun., 2016, 52, 5037 RSC; (d) D. M. Harraz and J. T. Davis, Chem. Commun., 2018, 54, 1587 RSC; (e) N. Sreenivasachary and J.-M. Lehn, Chem. – Asian J., 2008, 3, 134 CrossRef CAS PubMed.
- L. S. Y. Tang, E. Covert, E. Wilson and S. Kottilil, JAMA, J. Am. Med. Assoc., 2018, 319, 1802 CrossRef CAS PubMed.
- S. Masiero, R. Trotta, S. Pieraccini, S. D. Tito, R. Perone, A. Randazzo and G. P. Spada, Org. Biomol. Chem., 2010, 8, 2683 RSC.
- D. J. Maggs and H. E. Clarke, Am. J. Vet. Res., 2004, 65, 399 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9qm00209j |
| This journal is © the Partner Organisations 2019 |