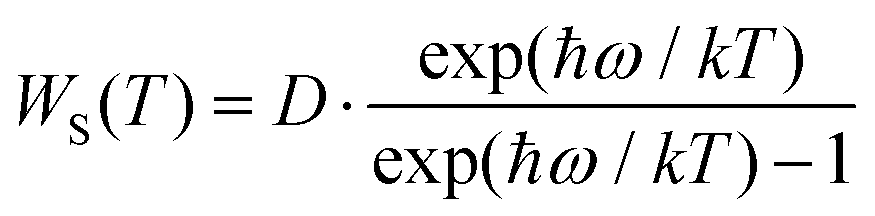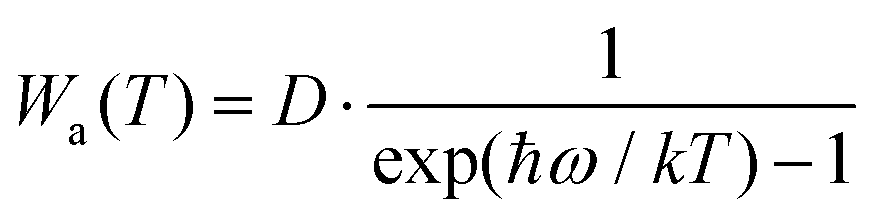DOI:
10.1039/C8QM00603B
(Research Article)
Mater. Chem. Front., 2019,
3, 403-413
Ca3La2Te2O12:Mn4+,Nd3+,Yb3+: an efficient thermally-stable UV/visible–far red/NIR broadband spectral converter for c-Si solar cells and plant-growth LEDs†
Received
24th November 2018
, Accepted 19th December 2018
First published on 20th December 2018
Abstract
A series of novel Mn4+,Nd3+,Yb3+-doped Ca3La2Te2O12 (CLTO) phosphors were prepared by substituting Te6+ for W6+ in the Ca3La2W2O12 compound using a high-temperature solid-state reaction method. A Mn4+ singly-doped CLTO phosphor (CLTO:0.004Mn4+) showed a bright deep red-light emission corresponding to a narrow band around 707 nm (14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 144 cm−1) due to a Mn4+ 2Eg → 4A2g spin-forbidden transition upon 365 nm UV excitation, which largely overlapped the absorption spectrum of plant phytochrome Pfr. The excitation spectrum (λem = 707 nm) presented a broad band ranging from 250 nm (40000 cm−1) to 600 nm (16667 cm−1), which can be decomposed into four Gaussian bands peaking at 318 nm (31431 cm−1), 355 nm (28148 cm−1), 410 nm (24385 cm−1), and 476 nm (20992 cm−1), corresponding to a Mn4+–O2− charge transfer (CT) transition, and Mn4+ transitions 4T1g ← 4A2g, 2T2g ← 4A2g and 4T2g ← 4A2g, respectively. Moreover, three kinds of Mn4+ emission sites were analyzed with the assistance of time-resolved spectra. With the single incorporation of Nd3+/Yb3+ into CLTO:Mn4+, energy transfer phenomena from Mn4+ to Nd3+/Yb3+ ions can be observed, which proceeded via non-radiative resonant and phonon-assisted mechanisms, respectively, resulting in the broadband spectral conversion of the UV/blue to NIR light. When Nd3+ and Yb3+ were co-doped into CLTO:Mn4+ to form the tri-doped CLTO phosphors, a successive energy transfer process, Mn4+ → Nd3+ → Yb3+, was determined based on the simultaneous energy transfer processes Mn4+ → Nd3+ and Nd3+ → Yb3+ in the co-doped samples, further enhancing the broadband spectral conversion process of the UV/blue to NIR region, which can be absorbed by photosynthetic bacteria and show high response when applied to c-Si solar cells. More attractively, the luminescence thermal stabilities of both CLTO:0.004Mn4+ and CLTO:0.004Mn4+,0.04Nd3+,0.20Yb3+ showed excellent performance, and the temperature-dependent luminescence properties of the Mn4+,Nd3+,Yb3+ tri-doped materials have been investigated for the first time. These results indicate that this kind of phosphor can be potentially applied to improving spectral conversion efficiency for c-Si solar cells and plant-growth far-red/NIR LEDs. In addition, this report provides a strategy wherein hosts for Mn4+ doping can be well enriched by substituting Te6+ for W6+ in certain tungstate compounds, which is highly desired in searching for novel red-emitting phosphors.
144 cm−1) due to a Mn4+ 2Eg → 4A2g spin-forbidden transition upon 365 nm UV excitation, which largely overlapped the absorption spectrum of plant phytochrome Pfr. The excitation spectrum (λem = 707 nm) presented a broad band ranging from 250 nm (40000 cm−1) to 600 nm (16667 cm−1), which can be decomposed into four Gaussian bands peaking at 318 nm (31431 cm−1), 355 nm (28148 cm−1), 410 nm (24385 cm−1), and 476 nm (20992 cm−1), corresponding to a Mn4+–O2− charge transfer (CT) transition, and Mn4+ transitions 4T1g ← 4A2g, 2T2g ← 4A2g and 4T2g ← 4A2g, respectively. Moreover, three kinds of Mn4+ emission sites were analyzed with the assistance of time-resolved spectra. With the single incorporation of Nd3+/Yb3+ into CLTO:Mn4+, energy transfer phenomena from Mn4+ to Nd3+/Yb3+ ions can be observed, which proceeded via non-radiative resonant and phonon-assisted mechanisms, respectively, resulting in the broadband spectral conversion of the UV/blue to NIR light. When Nd3+ and Yb3+ were co-doped into CLTO:Mn4+ to form the tri-doped CLTO phosphors, a successive energy transfer process, Mn4+ → Nd3+ → Yb3+, was determined based on the simultaneous energy transfer processes Mn4+ → Nd3+ and Nd3+ → Yb3+ in the co-doped samples, further enhancing the broadband spectral conversion process of the UV/blue to NIR region, which can be absorbed by photosynthetic bacteria and show high response when applied to c-Si solar cells. More attractively, the luminescence thermal stabilities of both CLTO:0.004Mn4+ and CLTO:0.004Mn4+,0.04Nd3+,0.20Yb3+ showed excellent performance, and the temperature-dependent luminescence properties of the Mn4+,Nd3+,Yb3+ tri-doped materials have been investigated for the first time. These results indicate that this kind of phosphor can be potentially applied to improving spectral conversion efficiency for c-Si solar cells and plant-growth far-red/NIR LEDs. In addition, this report provides a strategy wherein hosts for Mn4+ doping can be well enriched by substituting Te6+ for W6+ in certain tungstate compounds, which is highly desired in searching for novel red-emitting phosphors.
1. Introduction
Solving the energy crisis is a global issue mankind is currently facing. As one of the most clean and sustainable energies, solar energy is always harnessed by utilizing solar cell devices to convert it into electricity. At present, there are several kinds of solar cells and technologies that are well developed such as crystalline silicon (c-Si) (mono/polycrystalline silicon), non-crystalline silicon, GaAs, CdTe, perovskite, organic, and dye-sensitized solar cells.1–8 So far, c-Si type solar cells, first-generation solar energy-conversion devices, have occupied the major market on account of their relatively mature fabrication technology and low price compared to others. However, their low energy conversion efficiencies of around 22% and 25% for industrial application and lab research, respectively, which are under the Shockley–Queisser limit of about 33.7%,9 still need to be improved. The main issue that limits their solar energy conversion efficiency is the spectral mismatch between the phonon energy of the incident sunlight and the band gap of c-Si semiconductor materials. This indicates that only a small portion of the solar spectrum lying at 900–1100 nm can be utilized and converted into electricity; in addition, low-energy photons cannot be absorbed and the excess energy of high-energy photons will be released in the form of heat via charge carrier thermalization or electron–hole recombination effects.10–12 Therefore, spectral converters showing giant potential for integration with solar cells to reduce the non-absorption and thermal losses are developed. They are often used as photo-luminescent layers to absorb the photons that cannot be effectively captured by solar cells and convert them to suitable wavelengths for specific type solar cells.13 This strategy offers the advantages of unnecessarily modifying the solar cells’ standard structure and flexible optimization toward a specific type solar cell.
Rare earth ion doped luminescent materials converting UV/n-UV/visible light into near infrared emission around 1000 nm have attracted lots of attention because of their potential application in improving the photovoltaic efficiency in c-Si solar cells, among which Yb3+ is considered to be the most promising candidate for this purpose since it possesses only one excited state level, 2F5/2, and approaches the excellent spectral response of c-Si solar cells.14,15 As reported before, diverse ions such as Tm3+, Pr3+, Tb3+, Er3+, and Ho3+ were first selected as the sensitizers for Yb3+ ions to realize spectral conversion.16–22 It is unsatisfactory that only a small portion of the sunlight can be converted as a result of their natural f–f transition. In order to overcome this disadvantage and obtain better conversion results, several other kinds of ions such as Ce3+, Eu2+, Yb2+, Bi3+, and Cr3+23–28 with broadband absorptions were adopted as the effective sensitizers to realize broadband spectral conversion for Yb3+, mainly through the quantum cutting method. Apart from Yb3+, Nd3+ can also be considered as an alternative activated luminescent ion owing to its emission bands around 900 (4F3/2 → 4I9/2) and 1064 nm (4F3/2 → 4I11/2), which are also within the high spectral response range of 900–1100 nm for c-Si semiconductor materials.29
Mn4+, a transition metal ion, is popular as an activator in a large number of compounds,30–35 typically in oxides and fluorides, to produce red or far-red emissions around wavelengths ranging from 620 nm to 750 nm when it is located in octahedra, and often acts as a red-component supplement for white light-emitting diodes (w-LEDs) to improve their device performance. It often shows broadband absorption originating from Mn4+–O2− charge transfer transition and Mn4+ d–d inner transitions 4T1 ← 4A2, 4T1 ← 4A2 and 4T2 ← 4A2 in the UV and visible regions, which indicates its potential for broadband spectral conversion of UV/visible to red light. It was reported before that an effective energy transfer from Mn4+ to Yb3+ ions took place in certain phosphors, indicating their potential applications in improving the conversion efficiency of c-Si solar cells.36–39 Moreover, the energy transfer process from Mn4+ to Yb3+ ions would be a probable phonon-assisted non-resonant energy transfer process, which often needs nearly five phonons since the energy gap between the Mn4+ 2Eg and Yb3+ 2F5/2 levels is quite large. In spite of the visible spectral overlap between common singly doped Nd3+ excitation and Mn4+ emission bands in certain systems, few reports were focused on Mn4+ → Nd3+ resonant energy transfer properties. In addition, rarely has attention been paid to the energy transfer from Nd3+ to Yb3+ ions via a down-shifting process, which is different from previously reported down-conversion processes40 in some phosphor systems. Recently, the luminescence properties of Ca3La2W2O12:Mn4+ as a red phosphor have been investigated.41 On the basis of the similar ionic radii of Te6+ and W6+, we search for a suitable host (Ca3La2Te2O12) through the reasonable substitution of Te6+ for W6+ in Ca3La2W2O12 to be doped with Mn4+, Nd3+ and Yb3+ ions, which show improved luminescence thermal stability in Mn4+ doped systems and clear Mn4+ → Yb3+, Mn4+ → Nd3+ and Nd3+ → Yb3+ energy transfer processes in co-doped systems, in addition to a successive energy transfer process, Mn4+ → Nd3+ → Yb3+, which has rarely been investigated in Mn4+,Nd3+,Yb3+ tri-doped systems. All these energy transfer processes would effectively realize the spectral conversion from UV/visible to NIR emission in phosphors. Furthermore, blue (400–480 nm), red (600–680 nm) and far-red (FR, 680–780 nm) lights play important roles in photosynthetic, phototropic and photomorphogenetic processes,42,43 respectively, for plant cultivation, and the near infrared (NIR, 715–1050 nm) light absorbed by photosynthetic bacteria can accelerate the synthesis of nutrients for assisting biological nitrogen fixation,44 which can indirectly prompt plant growth. Currently, light-emitting diodes (LEDs) with the merits of high-efficiency, energy saving, non-polluting nature, and long lifetimes are considered to replace traditional incandescent and fluorescent lamps as next generation light source candidates. In particular, the spectral composition of LEDs can be flexibly controlled via the phosphor-converted technology to match well with the target emission wavelengths that different plants and photosynthetic bacteria require. Therefore, it is attractive to achieve a single-phase red to NIR emission to meet the requirements of plant growth and photosynthetic bacteria based on the Mn4+ → Yb3+, Mn4+ → Nd3+ and Mn4+ → Nd3+ → Yb3+ energy transfer processes in this kind of Ca3La2Te2O12:Mn4+,Nd3+,Yb3+ phosphor, which can avert the complicated combination of single color-emitting phosphors, spectral re-absorption and color instability arising from the different aging speeds of phosphors. In fact, spectral conversion materials should exhibit good spectral conversion, and physical, chemical and thermal stability properties for their real applications. Herein, the luminescence thermal stability of the as-prepared Mn4+,Nd3+,Yb3+ tri-doped CLTO phosphors, which exhibit excellent properties, has been investigated for the first time. Accordingly, this kind of phosphor would be a possible component candidate for c-Si solar cells and plant-growth LEDs. In addition, this report provides a good example to enrich hosts for Mn4+ doping with red emission by substituting Te6+ for W6+ in certain compounds.
2. Experimental
2.1 Material synthesis
Polycrystalline powders with the chemical composition formulae Ca3La2Te2−xO12:xMn4+ (CLTO:xMn4+, x = 0–0.02), Ca3La2−a/bTe1.996O12:0.004Mn4+,aNd3+/bYb3+ (CLTO:0.004Mn4+,aNd3+/bYb3+, a = 0–0.06, b = 0–0.40), Ca3La1.96−yTe2O12:0.04Nd3+,yYb3+ (CLTO:0.04Nd3+,yYb3+, y = 0–0.40), Ca3La1.8−cTe1.996O12:0.004Mn4+,0.20Yb3+,cNd3+ (CLTO:0.004Mn4+,0.20Yb3+,cYb3+, c = 0–0.0.06), Ca3La1.96−dTe1.996O12:0.004Mn4+,0.04Nd3+,dYb3+ (CLTO:0.004Mn4+,0.04Nd3+,dYb3+, d = 0–0.40), Ca3La1.8T2O12:0.20Yb3+ (CLTO:0.20Yb3+), and Ca3La1.96Te2O12:0.04Nd3+ (CLTO:0.04Nd3+) were prepared via a high-temperature solid-state reaction route. Raw materials CaCO3 (A.R.), TeO2 (99.9%), MnCO3 (99.9%), and high purity La2O3 (99.99%), Yb2O3 (99.99%), and Nd2O3 (99.99%) were first weighed according to the chemical stoichiometric ratios and then thoroughly mixed and ground in an agate mortar for about 15 min with the addition of proper amount of ethanol. After that, the homogenous mixtures were dried in an oven followed by grinding for another 1 min and were transferred to ceramic crucibles one by one. They were sintered in an oven at 1100 °C for 10 h with the increasing rate of 5 °C min−1. Eventually, the as-prepared naturally cooled products were ground again for the following measurements.
2.2 Characterization
The crystal structure and phase purity were investigated using powder X-ray diffraction (PXRD) performed using a Thermo Scientific ARLX’TRA diffractometer equipped with a Cu Kα (λ = 1.5405 Å) source, maintaining the scanning rate of 5° min−1 with the scattering angle range (2θ) from 15° to 65°. The Fourier transform infrared (FT-IR) and Raman spectra were recorded on a FT-IR-RAMAN-DRIFT NICOLET 6700 setup. Photoluminescence (PL), quantum yield (QY) and luminescence lifetimes in the visible and infrared regions were recorded on an Edinburgh Instruments FLSP 920 UV-vis-NIR spectrofluorimeter, equipped with a 450 W continuous xenon lamp and a 60 W pulsed μF 920 xenon lamp and an integrated sphere. The setup has a Hamamatsu R928P red-sensitive photomultiplier tube (PMT) to detect the luminescence located in the 200–870 nm wavelength range, together with a liquid-nitrogen cooled (−80 °C) Hamamatsu R5509-72 PMT to record near-infrared luminescence up to 1700 nm. All the measurements above were performed at room temperature. In addition, the measurements of temperature-dependent PL spectra of the selected samples were performed on an FLS 920 spectrofluorimeter with the assistance of a temperature controller Model 336 from Lakeshore Company.
3. Results and discussion
3.1 Phase, structure, FT-IR and Raman spectra
Fig. 1a shows the PXRD patterns of the as-prepared CLTO host and Mn4+, Nd3+, Yb3+ doped CLTO samples. It is observed that all the patterns are assigned to the reported Ca3La2W2O12 compound (JCPDS no. 49-0965),45 indicating that a pure phase has been obtained with the rational substitution of Te6+ for W6+ atoms in phosphors due to their similar ionic radii and valence states; moreover, the introduction of Mn4+, Nd3+ or Yb3+ dopants into the CLTO host does not induce any significant structural change. Although the crystal structures of neither the CLTO nor Ca3La2W2O12 compounds have been obtained up to now, it is reported that they have similar structures to the Ca5Re2O12 compound.46 As approximately depicted in Fig. 1b, it crystallizes in a 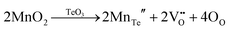 rhombohedral system with the space group R
rhombohedral system with the space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m. There are three kinds of Te6+ sites [coordination number (CN) = 6] for Mn4+ occupancy and four kinds of La3+ sites for Nd3+/Yb3+ (CN = 6, 8, 9) substitution based on their similar ionic radii [Mn4+ ions (CN = 6, radius (r) = 0.53 Å), Te6+ (CN = 6, r = 0.56 Å); Yb3+/Nd3+ (CN = 6, r = 0.87/0.98 Å; CN = 8, r = 0.98/1.12 Å; CN = 9, r = 1.05/1.19 Å), La3+ (CN = 6, r = 1.05 Å; CN = 8, r = 1.18 Å; CN = 9, r = 1.20 Å)]. As proposed, a difference in charge between Mn4+ and Te6+ can be found, which would result in oxygen vacancies according to the balanced formula, and simultaneously induce a change in the TeO6 octahedra; this may prompt the luminescence of Mn4+. The FT-IR spectra of the representative CLTO host and Mn4+, Nd3+, Yb3+ doped CLTO samples in the range of 500–3000 cm−1, depicted in Fig. 1c, show two main peaks at 673 and 704 cm−1, which can be assigned to the stretching Te–O vibrations in [TeO6]6− groups.47 Another observed peak at 1463 cm−1 commonly originates from the vibration of the OH− bonds in absorbed water from air on the surfaces of the samples. It can be found that the profiles of the FT-IR spectra for the three representatives are similar to each other, indicating little change in the structure after the incorporation of dopants Mn4+, Nd3+, and Yb3+ into the CLTO host, which is consistent with the XRD analysis result above. The Raman spectrum for the CLTO host in Fig. 1d presents several main peaks at 314, 438, 566 and 715 cm−1, due to the valence vibrations of O–Te–O and Te–O bonds,48 in which the maximum phonon energy in the crystal lattice is around 715 cm−1. This may be beneficial for the luminescence thermal stability of the as-prepared Mn4+,Yb3+,Nd3+ doped materials.
m. There are three kinds of Te6+ sites [coordination number (CN) = 6] for Mn4+ occupancy and four kinds of La3+ sites for Nd3+/Yb3+ (CN = 6, 8, 9) substitution based on their similar ionic radii [Mn4+ ions (CN = 6, radius (r) = 0.53 Å), Te6+ (CN = 6, r = 0.56 Å); Yb3+/Nd3+ (CN = 6, r = 0.87/0.98 Å; CN = 8, r = 0.98/1.12 Å; CN = 9, r = 1.05/1.19 Å), La3+ (CN = 6, r = 1.05 Å; CN = 8, r = 1.18 Å; CN = 9, r = 1.20 Å)]. As proposed, a difference in charge between Mn4+ and Te6+ can be found, which would result in oxygen vacancies according to the balanced formula, and simultaneously induce a change in the TeO6 octahedra; this may prompt the luminescence of Mn4+. The FT-IR spectra of the representative CLTO host and Mn4+, Nd3+, Yb3+ doped CLTO samples in the range of 500–3000 cm−1, depicted in Fig. 1c, show two main peaks at 673 and 704 cm−1, which can be assigned to the stretching Te–O vibrations in [TeO6]6− groups.47 Another observed peak at 1463 cm−1 commonly originates from the vibration of the OH− bonds in absorbed water from air on the surfaces of the samples. It can be found that the profiles of the FT-IR spectra for the three representatives are similar to each other, indicating little change in the structure after the incorporation of dopants Mn4+, Nd3+, and Yb3+ into the CLTO host, which is consistent with the XRD analysis result above. The Raman spectrum for the CLTO host in Fig. 1d presents several main peaks at 314, 438, 566 and 715 cm−1, due to the valence vibrations of O–Te–O and Te–O bonds,48 in which the maximum phonon energy in the crystal lattice is around 715 cm−1. This may be beneficial for the luminescence thermal stability of the as-prepared Mn4+,Yb3+,Nd3+ doped materials.
 |
| | Fig. 1 (a) PXRD patterns for the as-prepared representative samples of the Ca3La2Te2O12 host and Mn4+,Nd3+,Yb3+ doped CLTO samples, in addition to the standard reference of the Ca3La2W2O12 compound (JCPDS card no. 49-0965). (b) Approximate crystal structure of the Ca3La2Te2O12 compound and the coordination environments of the cations. (c) FT-IR spectra of the representative CLTO host and Mn4+,Nd3+,Yb3+ doped CLTO samples. (d) The Raman spectrum for the CLTO host. | |
3.2 PL properties of Mn4+ doped CLTO phosphors
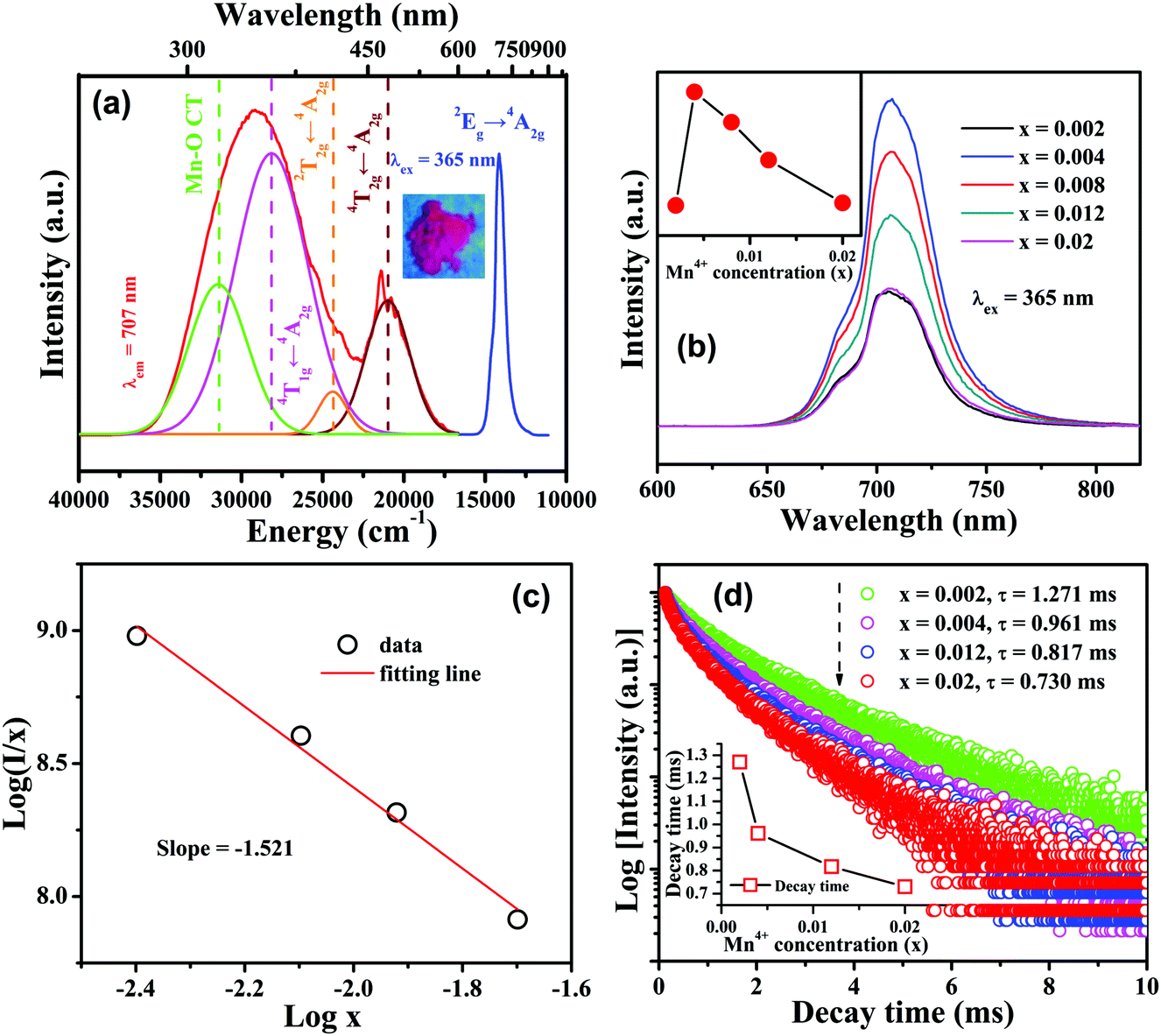
Fig. 2a exhibits the PL excitation and emission spectra of the CLTO:0.004Mn4+ sample. It can be observed that the emission spectrum consists of a narrow band ranging from 640 to 800 nm around 707 nm (14144 cm−1) upon 365 nm excitation, corresponding to the Mn4+ 2Eg → 4A2g transition. The bright deep-red color under 365 nm UV lamp excitation (the inset in Fig. 2a) shows its intensive emission. The quantum yield of this representative sample is determined to be 53.6%, a relatively high one, as shown in Fig. S1 (ESI†). The excitation spectrum detected at 707 nm comprises a broad band from 250 nm (40000 cm−1) to 600 nm (16667 cm−1), which can be decomposed into four Gaussian bands peaking at 318 nm (31431 cm−1), 355 nm (28148 cm−1), 410 nm (24385 cm−1), and 476 nm (20992 cm−1), corresponding to the Mn4+–O2− charge transfer transition, and Mn4+ transitions 4T1g ← 4A2g, 2T2g ← 4A2g and 4T2g ← 4A2g, respectively, as presented in Fig. 2a. With increasing doped Mn4+ content into CLTO, the intensity of the emission spectrum first increases due to more luminescent centers and then decreases owing to the concentration quenching effect in spite of their similar profiles. The optimal Mn4+ concentration is determined to be x = 0.004, as shown in the inset of Fig. 2b. The critical distance (Rc) between Mn4+ in this kind of phosphor can be approximately determined via the following formula:49,50| | 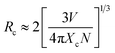 | (1) |
where Xc is the critical content of the dopant Mn4+, N is the number of available Te6+ sites in a unit cell, and V is the unit cell volume. As the ionic radii of W6+ and Te6+ are the same (0.56 Å), we propose that the substitution of Te6+ for W6+ seldom causes a change in crystal lattice parameters. As clarified above, herein, Xc = 0.004, N = 2 × 18 = 36, and V = 4569.7 Å3. Therefore, the value of Rc is calculated to be 38.28 Å, indicating a little possibility of exchange interaction (Rc ≤ 5 Å) for the non-radiative energy transfer process here. Consequently, the electric multi-pole interactions would take place in this kind of phosphor, which can be determined using the following formula derived by Blasse and Van Uitert:51,52| | 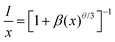 | (2) |
where x, I and β refer to the activator (Mn4+) concentration beyond the critical content, integrated emission intensity and a constant for a given matrix, respectively. θ = 6, 8 and 10 refer to the electric dipole–dipole, dipole–quadrupole and quadrupole–quadrupole interaction mechanisms, respectively. In order to determine the specific θ value, the relationship between log(x) and log(I/x) is plotted and displayed in Fig. 2c, which can be fitted with a straight line, showing a slope of −1.521 = −θ/3; thus θ = 4.563 is close to 6, implying that an electric dipole–dipole interaction predominates the energy transfer process between Mn4+ ions in the CLTO:Mn4+ phosphors. The decay curves (λex = 365 nm, λem = 707nm) of the selected samples CLTO:xMn4+ (x = 0.002, 0.004, 0.012 and 0.020) are used to further illustrate the concentration quenching in this kind of phosphor in detail, as presented in Fig. 2d, which can be fitted with a double-exponential function:53| | | I(t) = I0 + A1exp(−t/τ1) + A2exp(−t/τ2) | (3) |
where I(t) and I0 are the emission intensities at times t and 0, respectively, both A1 and A2 are the constants, and τ1 and τ2 refer to the luminescence lifetimes for the quick and slow decays, respectively. As a result, the average lifetimes (τ) are determined to be 1.271, 0.961, 0.817 and 0.730 ms corresponding to x = 0.002, 0.004, 0.012 and 0.02, respectively, by calculation using the following equation:| | | τ1 = (A1τ12 + A2τ22)/(A1τ1 + A2τ2) | (4) |
A decreasing trend (the inset in Fig. 2d) is clearly seen for the lifetime upon increasing the Mn4+ concentration from x = 0.002 to 0.02, which is also related to the total relaxation rate expressed using the following equation:54| | 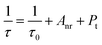 | (5) |
where τ0 is the radiative lifetime, Anr is the non-radiative rate attributed to multiphonon relaxation, and Pt is the energy transfer rate between Mn4+ ions. As the Mn4+ concentration is increased, the distance between Mn4+ ions becomes shorter and shorter, which implies that both the probability of energy transfer to luminescent killer sites and the energy transfer rate between Mn4+–Mn4+ ion groups increase. As a consequence, the lifetime decreases monotonically with the increase of Mn4+ content in CLTO.
 |
| | Fig. 2 (a) PL excitation and emission spectra of CLTO:0.004Mn4+ and Gaussian deconvolution for the emission spectrum; the inset shows the luminescence image under 365 nm UV lamp excitation for this sample. (b) Variation of emission spectra for CLTO:xMn4+ (x = 0–0.02) samples as a function of Mn4+ concentration x; the inset shows the variation of the corresponding emission intensity. (c) Linear fitting for log(I/x) versus logx beyond the critical concentration (x ≥ 0.004). (d) Decay curves for CLTO:xMn4+ (x = 0.002–0.02) samples; the inset shows the dependence of the decay lifetime on Mn4+ concentration. | |
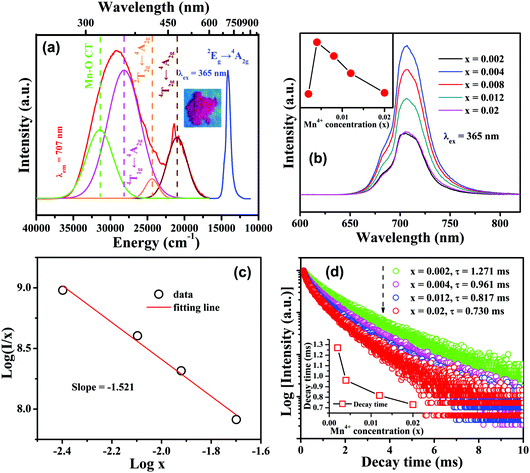
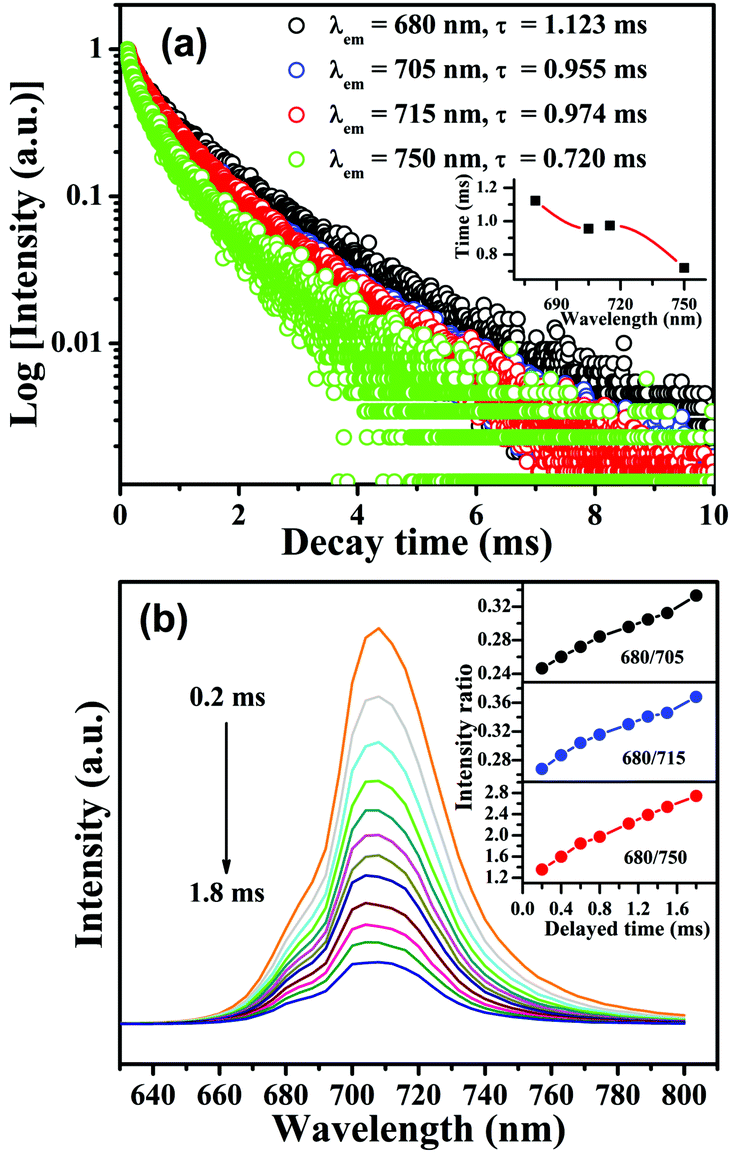
In order to distinguish three possible kinds of Mn4+ luminescent centers, the decay curves for CLTO:0.004Mn4+ (λex = 365 nm) monitored at different wavelengths of 680, 705, 715 and 750 nm are shown in Fig. 3a, all of which can be fitted with a bi-exponential function mentioned above; therefore, the decay lifetimes are determined to be 1.123, 0.955, 0.974 and 0.720 ms, respectively. It can be found that the lifetime decreases first followed by a slight increase and it subsequently decreases again upon increasing the wavelength from 680 to 750 nm. This phenomenon illustrates that three kinds of emission bands tend to be located between 680 and 705 nm, 705 and 715 nm and 715 and 750 nm, respectively. Moreover, the time-resolved spectra, shown in Fig. 3b, were recorded with a delayed time from 0.2 to 1.8 ms to further show the different kinds of sites for Mn4+ in the CLTO:0.004Mn4+ phosphor. It can be seen that the profile of the emission spectrum changes a little; that is to say, it varies and the emission peak gradually shifts to a shorter wavelength with increasing delayed time. These phenomena can be reflected by the homogeneous increments of the emission intensity ratios for 680/705 nm, 680/715 nm and 680/750 nm in the inset of Fig. 3b.
 |
| | Fig. 3 (a) Decay curves monitored at different emission wavelengths (λex = 365 nm, λem = 680, 705, 715 and 750 nm) for CLTO:0.004Mn4+; the inset shows the variation of the corresponding decay lifetime. (b) Time-resolved emission spectra for CLTO:0.004Mn4+ excited at 365 nm; the inset shows the variations of the intensity ratios for 680/705 nm, 680/715 nm and 680/750 nm as a function of delayed time. | |
3.3 Energy transfer properties in Mn4+,Nd3+,Yb3+ doped CLTO phosphors
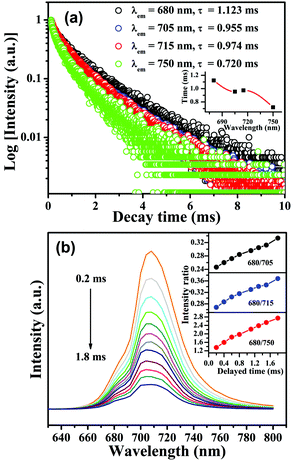
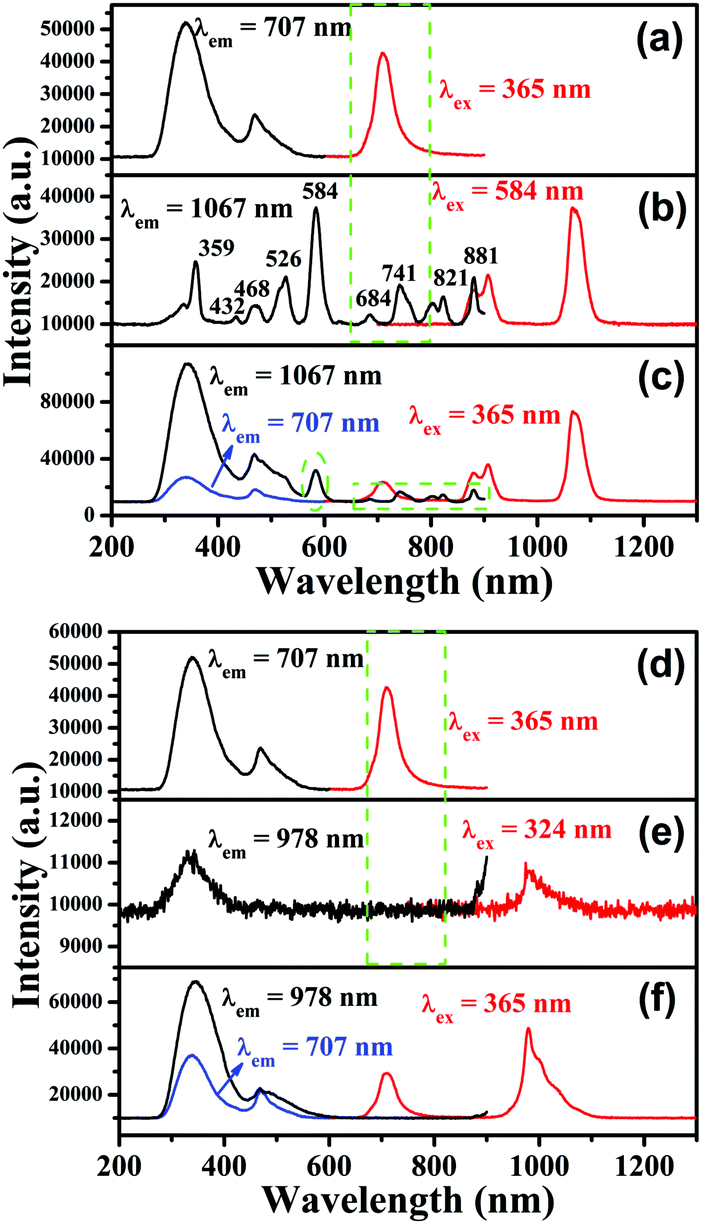
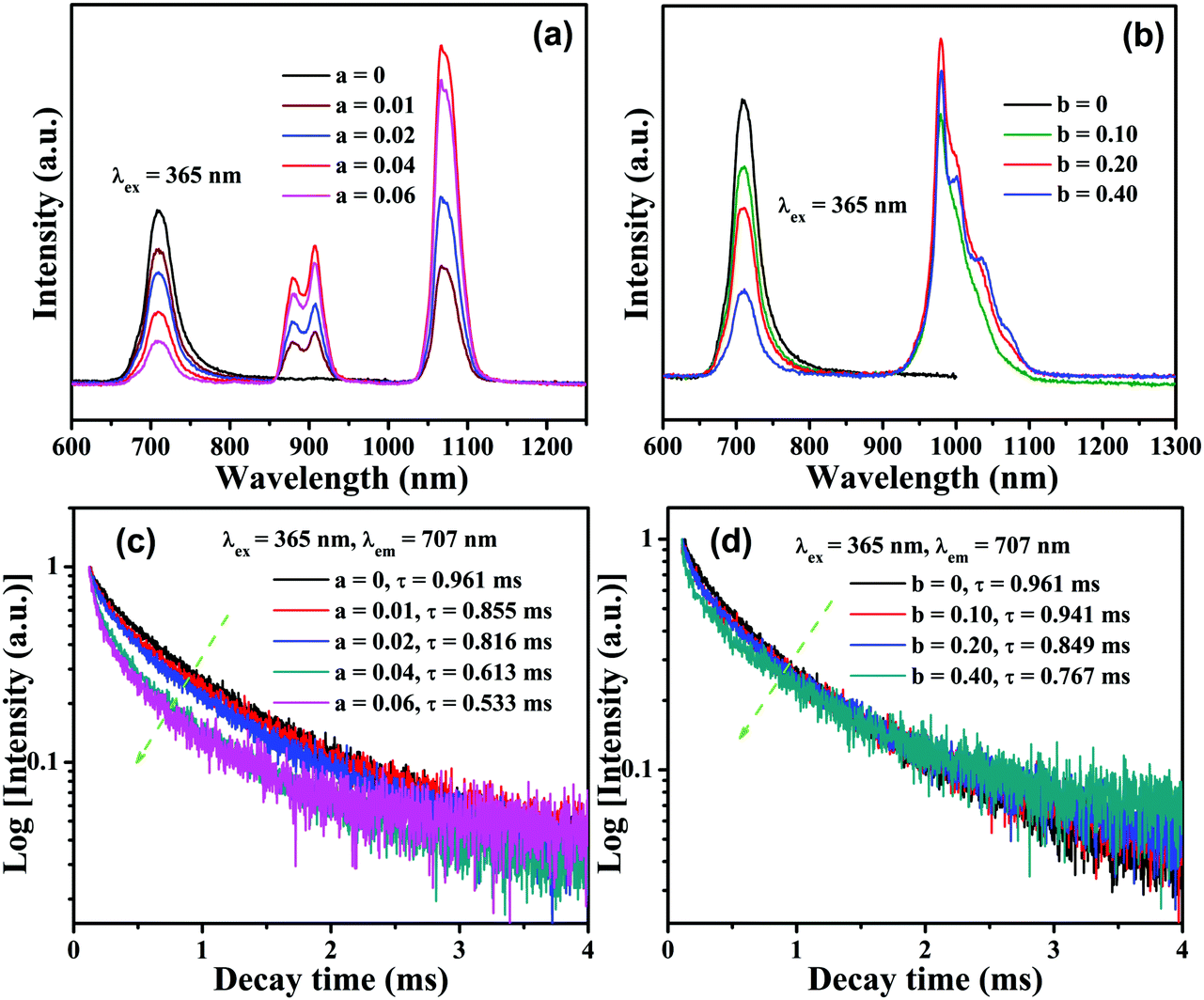
PL excitation and emission spectra for CLTO:0.004Mn4+ are shown in Fig. 4a and d, and have been described above in Section 3.2. Fig. 4b shows the PL excitation and emission spectra for CLTO:0.04Nd3+, in which two primary narrow bands around 907 nm (4F3/2 → 4I9/2) and 1067 nm (4F3/2 → 4I11/2) due to Nd3+ upon maximum excitation at 584 nm are observed. Monitored at 1067 nm, several sharp excitation lines peaking at 359 nm (4D1/2 ← 4I9/2), 432 nm (2P1/2 ← 4I9/2), 468 nm (4G9/2 ← 4I9/2), 526 nm (4G7/2 ← 4I9/2), 584 nm (4G5/2 ← 4I9/2), 684 nm (4F9/2 ← 4I9/2), 741 nm (4S3/2 ← 4I9/2), 821 nm (4F5/2 ← 4I9/2) and 881 nm (4F3/2 ← 4I9/2) can be observed.55,56 By comparing the emission spectrum of Mn4+ in Fig. 4a and the Nd3+ excitation spectrum in Fig. 4b, a significant spectral overlap can be found (marked with a green dashed line frame), indicating that a possible resonant energy transfer from Mn4+ to Nd3+ ions would take place in Mn4+,Nd3+ co-doped CLTO samples. In order to elucidate this energy transfer possibility, the excitation and emission spectra for Mn4+,Nd3+ co-doped CLTO:0.004Mn4+,0.04Nd3+ are recorded and plotted in Fig. 4c, in which the emission spectrum shows both Mn4+ and Nd3+ emission bands upon 365 nm excitation, and the excitation spectra monitored at 707 nm (Mn4+ emission) and 1067 nm (Nd3+ emission) exhibit similar broadband profiles to Mn4+ singly doped samples, besides the characteristic excitation band around 584 nm (marked with a green dashed line oval) and other bands (marked with a green dashed line frame) due to Nd3+. These phenomena imply that the excitation energy of Nd3+ mainly originates from Mn4+; that is, the energy transfer from Mn4+ to Nd3+ ions occurs in this kind of co-doped sample. As to CLTO:0.20Yb3+, its weak emission shows a narrow band with a peak at 978 nm upon 324 nm excitation originating from the Yb3+ 2F5/2 → 2F7/2 transition, see Fig. 4e. In addition, an excitation spectral band centered at 324 nm appears when monitored at an emission wavelength of 978 nm, which may have originated from the Yb3+–O2− charge transfer transition.57 In Fig. 4f, the emission spectrum for CLTO:0.004Mn4+,0.20Yb3+ presents both Mn4+ and Yb3+ bands upon 365 nm excitation, in which the emission intensity of Yb3+ is enhanced by over 50 times compared to that of CLTO:0.20Yb3+ upon 324 nm excitation. Moreover, two excitation spectra monitored at 709 and 978 nm exhibit analogous profiles to Mn4+ singly doped samples. These phenomena confirm that efficient energy transfer from Mn4+ to Yb3+ ions in CLTO:Mn4+,Yb3+ can also take place despite the scarcely observed spectral overlap between the Mn4+ emission and Yb3+ excitation spectra in the green dashed line frame in Fig. 4d and e. It is proposed that the energy transfer process can proceed via a phonon-assisted process similar to a Cr3+–Yb3+ group,27 which generally requires five phonons. As mentioned in Fig. 1d, the phonon energy is about 715 cm−1; upon multiplying by five, the value is about 3575 cm−1, which is close to the energy gap of 3919 cm−1 between the Yb3+ 2F5/2 and 2Eg levels. This suggests its possible phonon-assistant energy transfer process from Mn4+ to Yb3+ ions in this system. From the emission spectrum of CLTO:0.04Nd3+ in Fig. S2a (ESI†) and the excitation spectrum of CLTO:0.20Yb3+ in Fig. S2b (ESI†), a rough overlap between them (marked with a green dashed frame) can be found, which indicates energy transfer from Nd3+ to Yb3+ ions in Nd3+,Yb3+ co-doped systems. This can be validated by the similar excitation spectra monitored at around 1067 nm (Nd3+ emission band) and around 978 nm (Yb3+ emission band) for CLTO:0.04Nd3+,0.20Yb3+, as illustrated in Fig. S2c (ESI†). In addition, the emission band upon 584 nm excitation including both Nd3+ and Yb3+ emissions further confirms the energy transfer process from Nd3+ to Yb3+ ions here. In order to show the influence of luminescence on the concentration of dopants, the content-dependent emission spectra (λex = 365 nm) for CLTO:0.004Mn4+,aNd3+ (a = 0–0.06) and CLTO:0.004Mn4+,bYb3+ (b = 0–0.40) are depicted in Fig. 5a and b, respectively. It can be found that the emission intensity of Mn4+ monotonically decreases with increasing Nd3+/Yb3+ concentration, while that of Nd3+/Yb3+ increases to a maximum a = 0.04/b = 0.20 first and then decreases with further dopants owing to their self-concentration quenching effect. Analogous results are obtained for CLTO:0.04Nd4+,yYb3+ (y = 0–0.40), as illustrated in Fig. S3 (ESI†); that is, the Nd3+ emission intensity (λex = 584 nm) monotonically decreases with increasing Yb3+ content y and the maximum intensity of Yb3+ occurs when y = 0.20. These phenomena elucidate that the energy transfers from Mn4+ to Nd3+, Mn4+ to Yb3+, and Nd3+ to Yb3+ ions can proceed in the respective co-doped CLTO systems. Decay lifetimes, an efficient supplement for the confirmation of the energy transfer phenomenon, of Mn4+ for CLTO:0.004Mn4+,aNd3+ (a = 0–0.06) and CLTO:0.004Mn4+,bYb3+ (b = 0–0.40) (λex = 365 nm, λem = 707 nm) are determined to be 0.961, 0.855, 0.816, 0.613 and 0.533 ms corresponding to a = 0, 0.01, 0.02, 0.04 and 0.06, and 0.961, 0.941, 0.849 and 0.767 ms corresponding to b = 0, 0.10, 0.20 and 0.40, respectively, by fitting the decay curves for these samples in Fig. 5c and d with a bi-exponential function mentioned above in eqn (3). Similarly, the Nd3+ decay lifetimes (λex = 584 nm, λem = 907 nm) are determined to be 117, 79.9, 47.2 and 37.5 μs corresponding to y = 0, 0.10, 0.20 and 0.40, respectively, for CLTO:0.04Nd3+,yYb3+ samples (y = 0–0.40) in Fig. S4 (ESI†). We can observe that the decay lifetimes of Mn4+ and Nd3+ monotonically decline with the corresponding increasing Nd3+/Yb3+ and Yb3+ concentrations in the respective co-doped samples, further indicating the energy transfer processes of Mn4+ → Yb3+, Mn4+ → Nd3+ and Nd3+ → Yb3+.
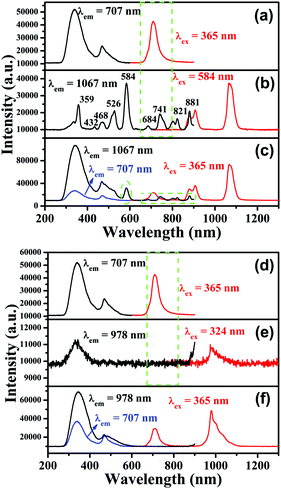 |
| | Fig. 4 PL excitation and emission spectra for CLTO:0.004Mn4+ (a and d), CLTO:0.04Nd3+ (b), CLTO:0.004Mn4+,0.04Nd3+ (c), CLTO:0.20Yb3+ (e) and CLTO:0.004Yb3+,0.20Yb3+ (f). | |
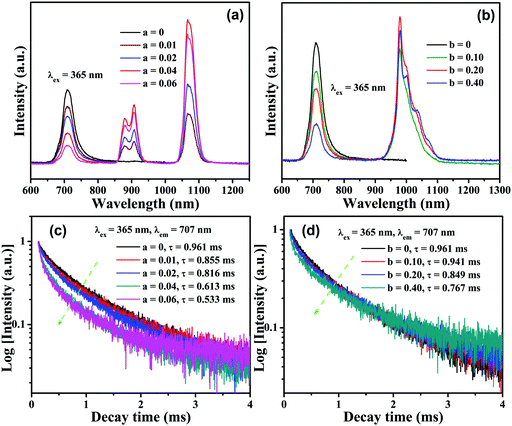 |
| | Fig. 5 PL emission spectra of CLTO:0.004Mn4+,aNd3+ (a = 0–0.06) (a) and CLTO:0.004Mn4+,bNd3+ (b = 0–0.40) (b) excited at 365 nm. Decay curves of CLTO:0.004Mn4+,aNd3+ (a = 0–0.06) (c) and CLTO:0.004Mn4+,bNd3+ (b = 0–0.40) (d) (λex = 365 nm, λem = 707 nm). | |
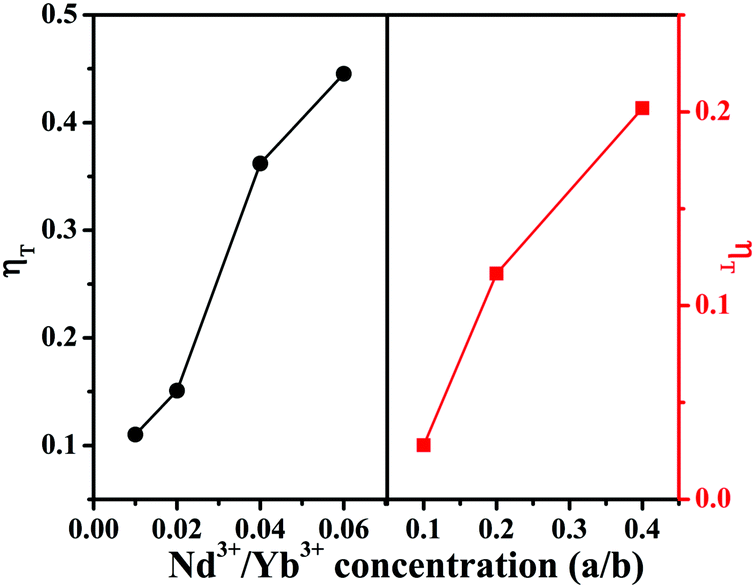
The energy transfer efficiencies (ηT) from Mn4+ to Nd3+/Yb3+ ions can be approximately calculated using the formula below:58
where
τ0 and
τ correspond to the decay lifetimes of Mn
4+ in the absence and presence of Nd
3+/Yb
3+ in the CLTO:0.004Mn
4+,
aNd
3+/
bYb
3+ samples. Accordingly, the values of
ηT (listed in
Table 1) increase from 11.03%/2.8% to 44.54%/20.89%, as illustrated in
Fig. 6 left and right, respectively, for
a/
b = 0/0 to 0.06/0.40. The increasing trends of
ηT indicate more and more efficient energy transfer processes from Mn
4+ to Nd
3+/Yb
3+ ions in the CLTO:0.004Mn
4+,Nd
3+/Yb
3+ samples with increasing dopant Nd
3+/Yb
3+ concentration.
Table 1 Decay lifetimes and energy transfer efficiencies (ηT) (λex = 365 nm, λem = 707 nm) for CLTO:0.004Mn4+,aNd3+ (a = 0–0.06) and CLTO:0.004Mn4+,bYb3+ (b = 0–0.40) samples
| Concentration (a/b) |
Decay time (ms) |
Energy transfer efficiency (ηT) |
|
a = 0 |
0.961 |
— |
|
a = 0.01 |
0.855 |
0.1103 |
|
a = 0.02 |
0.816 |
0.1509 |
|
a = 0.04 |
0.613 |
0.3621 |
|
a = 0.06 |
0.533 |
0.4454 |
|
|
|
b = 0 |
0.961 |
— |
|
b = 0.10 |
0.941 |
0.028 |
|
b = 0.20 |
0.849 |
0.1165 |
|
b = 0.40 |
0.767 |
0.2019 |
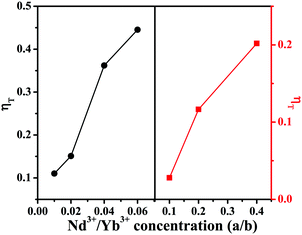 |
| | Fig. 6 Energy transfer efficiencies (ηT) for CLTO:0.004Mn4+,aNd3+ (a = 0.01–0.06) (left) and CLTO:0.004Mn4+,bYb3+ (b = 0.10–0.40) (right). | |
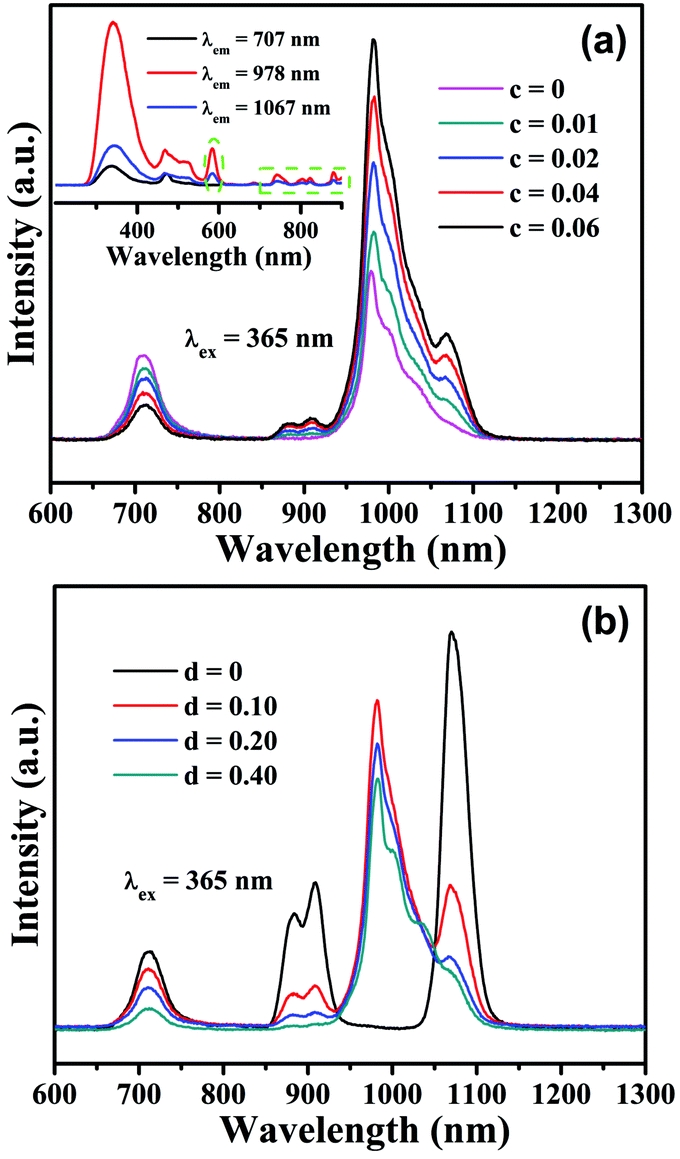
In the subsequent section, we will investigate the PL properties of Mn4+,Nd3+,Yb3+ tri-doped CLTO phosphors based on their validated Mn4+ → Yb3+, Mn4+ → Nd3+ and Nd3+ → Yb3+ energy transfer processes in this system. As shown in Fig. 7a, upon 365 nm excitation, with the increase of Nd3+ concentration, the emission intensities of Mn4+ in the spectra for CLTO:0.004Mn4+,0.20Yb3+,cNd3+ (c = 0–0.06) decrease monotonically, and the emission intensities of Nd3+ and Yb3+ gradually increase, indicating that the addition of Nd3+ in CLTO:0.004Mn4+,0.20Yb3+ will result in energy transfer from Mn4+ to Nd3+ ions, and simultaneously, some energy from Nd3+ will be transferred to Yb3+ to enhance the Yb3+ emission. The excitation spectra monitored at 707 (Mn4+), 978 (Yb3+) and 1067 nm (Nd3+), as shown in the inset in Fig. 7a, exhibit similar profiles, among which the excitation spectra for 978 and 1067 nm display an exceptional excitation band around 584 nm(the green dashed oval) and several peaks over 700 nm (the green dashed frame) attributed to Nd3+. These phenomena imply the existence of the successive energy transfer process from Mn4+ to Nd3+ and then to Yb3+ ions. The emission intensities of the Mn4+ and Nd3+ bands in CLTO:0.004Mn4+,0.04Nd3+,dYb3+ (d = 0–0.40) in Fig. 7b show a monotonic decline, with increasing Yb3+ concentration, while the Yb3+ emission intensity increases to the maximum when d = 0.10, beyond which it decreases as a result of its self-concentration quenching. The decay times (λex = 584 nm, λem = 907 nm) for CLTO:0.004Mn4+,0.04Nd3+,dYb3+ (d = 0–0.40) samples also decrease upon increasing the Yb3+ concentration, as shown in Fig. S5 (ESI†), from 187.8 μs to 44.7 μs. These results likewise indicate the Mn4+ → Nd3+ → Yb3+ successive energy transfer process in the Mn4+,Nd3+,Yb3+ tri-doped CLTO phosphor.
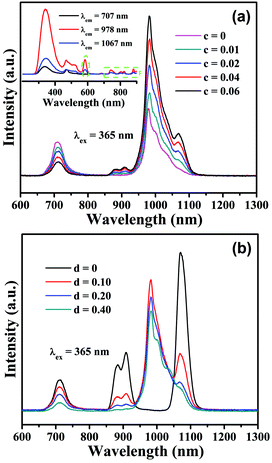 |
| | Fig. 7 PL emission spectra (λex = 365 nm) of CLTO:0.004Mn4+,0.20Yb3+,cNd3+ (c = 0–0.06) (a) and CLTO:0.004Mn4+,0.04Nd3+,dYb3+ (d = 0–0.40) (b). The inset in (a) shows the comparison of excitation spectra monitored at 707, 978 and 1067 nm for CLTO:0.004Mn4+,0.20Yb3+,0.04Nd3+. | |
The emission and excitation spectra of the representative sample CLTO:0.004Mn4+,0.20Yb3+,0.04Nd3+ and the general solar spectrum are exhibited in Fig. 8a, showing that the broadband UV/visible spectrum can be converted to the NIR region to better match the high response region of c-Si semiconductor materials, which would be possible to promote the utilization of rare solar spectral response parts (300–600 nm) in the solar spectrum. In addition, the absorption spectrum of plant far-red absorption type phytochrome Pfr,37 a color protein promoting plant cultivation, which is compared to the emission spectra of the CLTO:0.04Mn4+ and CLTO:0.004Mn4+,0.04Nd3+,0.10Yb3+ samples shown in Fig. 8b. It is manifested that there is a significant overlap between the absorption spectrum of Pfr and the Mn4+ emission band, which indicates the possibility of using CLTO:0.04Mn4+ in plant cultivation far-red LEDs. An extra NIR emission appears for CLTO:0.004Mn4+,0.04Nd3+,0.10Yb3+, besides the Mn4+ far-red emission, which can be absorbed by photosynthetic bacteria (extensively distributed in oil, water and floral tissue) to accelerate the synthesis of nutrients for assisting biological nitrogen fixation to indirectly boost plant cultivation. This indicates that far-red light and NIR emissions from the CLTO:0.004Mn4+,0.04Nd3+,0.10Yb3+ phosphor can cooperatively promote plant growth.
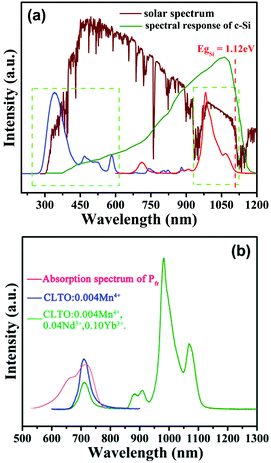 |
| | Fig. 8 (a) The general solar spectrum, spectral response of c-Si solar cells, and PL emission (λex = 365 nm) and excitation (λem = 978 nm) spectra for the CLTO:0.004Mn4+,0.20Yb3+,0.04Nd3+ sample. (b) The comparison of the absorption spectrum of plant phytochrome Pfr and the emission spectra (λex = 365 nm) of the CLTO:0.04Mn4+ and CLTO:0.004Mn4+,0.04Nd3+,0.10Yb3+ samples. | |
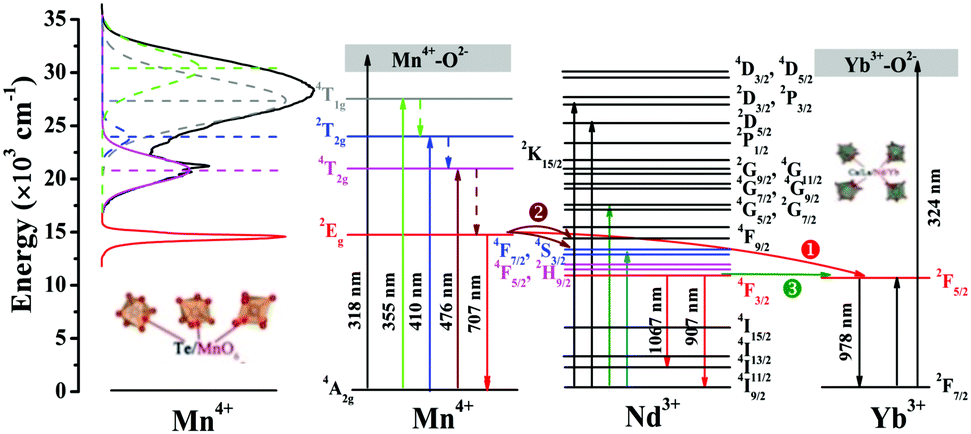
A simple schematic with respect to the electronic energy level transitions and energy transfer processes in Mn4+,Nd3+,Yb3+ doped CLTO systems is depicted in Fig. 9. Mn4+ singly doped CLTO would occupy three kinds of Te6+ sites to generate an approximate overall far-red emission around 707 nm (14144 cm−1) attributed to the Mn4+ 2Eg → 4A2g transition under UV/visible light excitation with wavelengths from about 250 nm (40000 cm−1) to 600 nm (16667 cm−1). Four excitation bands peaking at 318 nm (31431 cm−1), 355 nm (28148 cm−1), 410 nm (24385 cm−1), and 476 nm (20992 cm−1) correspond to the Mn4+–O2− charge transfer transition and Mn4+ transitions 4T1g ← 4A2g, 2T2g ← 4A2g and 4T2g ← 4A2g, respectively. For Nd3+ and Yb3+ ions, substituting four kinds of La3+ sites in the CLTO host produces their characteristic excitations and emissions. In the Mn4+/Nd3+/Yb3+ double-doped CLTO systems, the energy at the Mn4+ 2Eg level can be transferred to Nd3+ 4F7/2,4S3/2 or 4F5/2,2H9/2 levels (process  ) to enhance the emission bands around 907 nm (11025 cm−1) (4F3/2 → 4I9/2) and 1067 nm (9372 cm−1) (4F3/2 → 4I11/2) followed by relaxation to the 4F3/2 level via a resonant process due to the spectral overlap between the Mn4+ emission and Nd3+ excitation bands, and the Yb3+ 2F5/2 level to reinforce the emission band around 978 nm (10225 cm−1) (process
) to enhance the emission bands around 907 nm (11025 cm−1) (4F3/2 → 4I9/2) and 1067 nm (9372 cm−1) (4F3/2 → 4I11/2) followed by relaxation to the 4F3/2 level via a resonant process due to the spectral overlap between the Mn4+ emission and Nd3+ excitation bands, and the Yb3+ 2F5/2 level to reinforce the emission band around 978 nm (10225 cm−1) (process  ) with the assistance of approximately five phonons mentioned above due to the large energy gap (3919 cm−1) between the Mn4+ 2Eg and Yb3+ 2F5/2 levels. Besides, the energy at the Nd3+ 4F3/2 level can also be transferred to the 2F5/2 level to enhance the Yb3+ 978 nm emission (process
) with the assistance of approximately five phonons mentioned above due to the large energy gap (3919 cm−1) between the Mn4+ 2Eg and Yb3+ 2F5/2 levels. Besides, the energy at the Nd3+ 4F3/2 level can also be transferred to the 2F5/2 level to enhance the Yb3+ 978 nm emission (process  ) ascribed to their close energy levels. When Mn4+,Nd3+ and Yb3+ are tri-doped into CLTO, the Mn4+ → Nd3+ → Yb3+ successive energy transfer process can be implied to further enhance the Yb3+ emission based on the energy transfer processes mentioned above to better match the spectral response of c-Si solar cells and the absorption spectra of photosynthetic bacteria.
) ascribed to their close energy levels. When Mn4+,Nd3+ and Yb3+ are tri-doped into CLTO, the Mn4+ → Nd3+ → Yb3+ successive energy transfer process can be implied to further enhance the Yb3+ emission based on the energy transfer processes mentioned above to better match the spectral response of c-Si solar cells and the absorption spectra of photosynthetic bacteria.
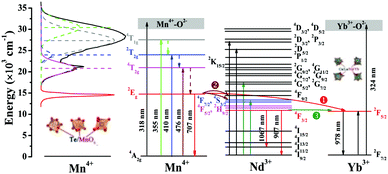 |
| | Fig. 9 A schematic energy level diagram illustrating the energy level transitions and energy transfer in CLTO:Mn4+,Nd3+,Yb3+ phosphors. | |
3.4 Luminescence thermal stability
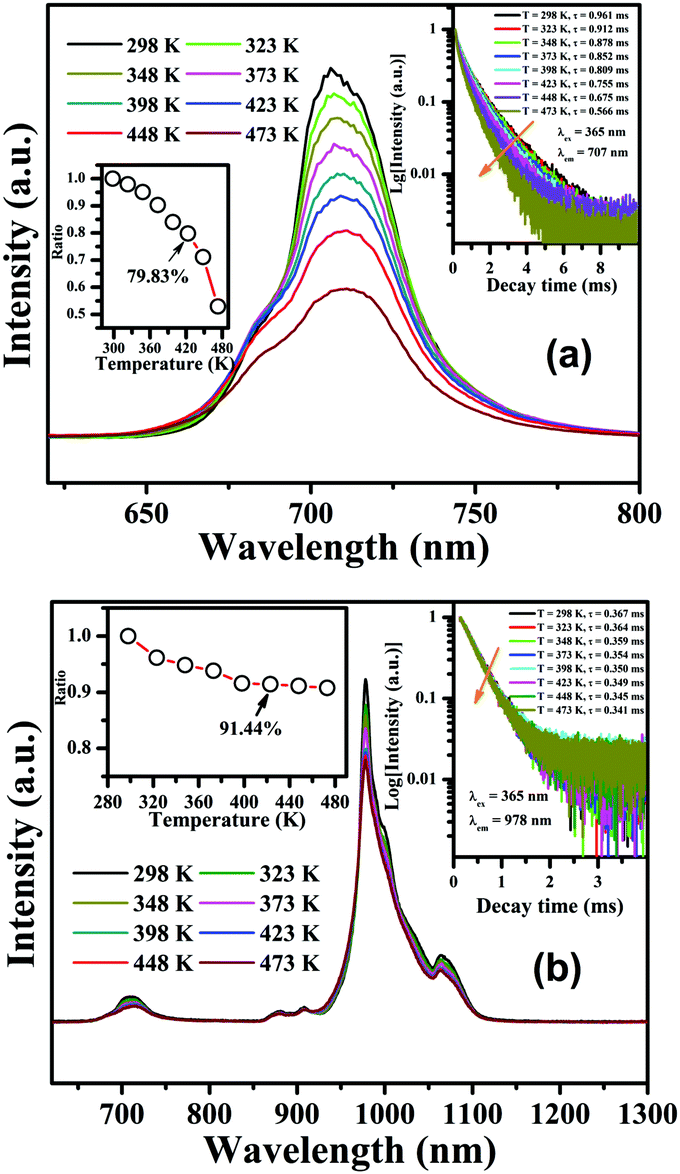
The temperature-dependent luminescent properties are some of the most important parameters for phosphors used for LEDs or c-Si solar cells because they often need to work at a relatively high temperature. Herein, the temperature-dependent luminescence emission spectra for CLTO:0.004Mn4+ and CLTO:0.004Mn4+,0.04Nd3+,0.20Yb3+, upon 365 nm excitation, are depicted in Fig. 10a and b, respectively. Generally, with increasing operating temperature, in both of them, the higher the temperature, the lower the emission intensities of the phosphors, which is attributed to the increasing non-radiative transition process. The insets on the left show that the integrated emission intensities of CLTO:0.004Mn4+ in Fig. 10a and CLTO:0.004Mn4+,0.04Nd3+,0.20Yb3+ in Fig. 10b are maintained at 79.83% (much higher than that of Ca3La2W2O12:Mn4+41) and 91.44% (150 °C) compared to those at room temperature, respectively, which indicate their excellent luminescence thermal stabilities. Moreover, the Mn4+ emission peaks present a slight red-shift and become wider and wider with increasing temperature in Fig. 10a and b, which can be attributed to the expansion of the cell unit and the reinforcement of the vibration mode under higher and higher temperature treatments.59
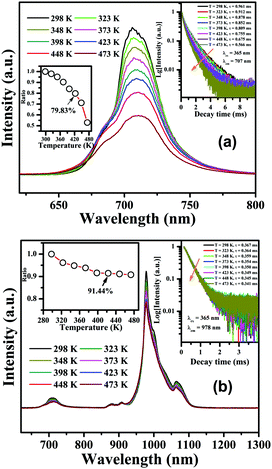 |
| | Fig. 10 Temperature-dependent PL emission spectra (λex = 365 nm) for CLTO:0.004Mn4+ (a) and CLTO:0.004Mn4+,0.04Nd3+,0.20Yb3+ (b) samples. The insets in (a) and (b) show variations of emission integral intensity on temperature and decay curves for CLTO:0.004Mn4+ (λex = 365 nm, λem = 707 nm) and CLTO:0.004Mn4+,0.04Nd3+,0.20Yb3+ (λex = 365 nm, λem = 978 nm) samples, respectively. | |
To further characterize the thermal stabilities of the as-prepared CLTO:0.004Mn4+ and CLTO:0.004Mn4+,0.04Nd3+,0.20Yb3+ phosphors, the temperature-dependent decay curves of Mn4+ (λex = 365 nm, λem = 707 nm) in the CLTO:0.004Mn4+ phosphor and Yb3+ (λex = 365 nm, λem = 978 nm) in the CLTO:0.004Mn4+,0.04Nd3+,0.20Yb3+ phosphor are shown in the right insets of Fig. 10a and b, respectively. It is found that the decay lifetime of Mn4+ decreases monotonically upon increasing the temperature from 298 K to 473 K. The lifetime of Mn4+ can be calculated by fitting the curves with a bi-exponential function well mentioned in eqn (4), which is determined to be 0.961 ms at room temperature (298 K) and decreases to 0.566 ms at 473 K. In order to clarify the dynamic process of Mn4+ in Mn4+ doped CLTO with increasing temperature, a simple model based on the temperature-dependent Stokes and anti-Stokes transition possibilities of vibronic emission can be expressed as:60
| | 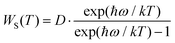 | (8) |
| | 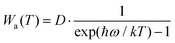 | (9) |
where
D is the offset coefficient,
ħω refers to the energy of the coupled vibronic mode,
T is the temperature using the K mode, and
k is the Boltzmann constant. By considering
eqn (8) and (9), we can deduce that both Stokes and anti-Stokes transition possibilities will increase with increasing temperature. Moreover, it is reported that the decay lifetime is inversely proportional to the sum of the radiative transition possibilities [
Wa(
T) +
WS(
T)] and non-radiative transition probabilities, which therefore decrease monotonically with increasing temperature. As for Yb
3+ in the CLTO:0.004Mn
4+,0.04Nd
3+,0.20Yb
3+ sample, the decay curves can be fitted well with a single exponential equation:
61| | | I = I0exp(−t/τ) | (10) |
where
I and
I0 are the luminescence intensities at times
t and 0, and
τ is the decay lifetime for the fitting result. Accordingly, the
τ value is determined to be 0.367 ms at room temperature (298 K) and slightly decreases to 0.341 ms at 473 K, which is ascribed to the declined lifetimes of the excited states of Yb
3+ ions arising from the increase of the non-radiative relaxation rate.
62 The results for decay lifetimes are consistent with the variation of emission intensities above, further indicating that the Mn
4+,Nd
3+,Yb
3+ doped CLTO phosphors have excellent thermal stability due to the luminescence quenching effect, suggesting that this kind of phosphor system can be a possible component candidate for plant growth far-red and NIR LEDs and c-Si solar cells.
4. Conclusions
In conclusion, a series of novel Mn4+,Nd3+,Yb3+ doped CLTO phosphors were synthesized by substituting Te6+ for W6+ in the Ca3La2W2O12 compound via a high-temperature solid-state reaction route. A Mn4+ singly-doped CLTO phosphor showed broadband excitation (λem = 707 nm, 14144 cm−1) ranging from 250 nm (40000 cm−1) to 600 nm (16667 cm−1), which could be decomposed into four Gaussian bands peaking at 318 nm (31431 cm−1), 355 nm (28148 cm−1), 410 nm (24385 cm−1), and 476 nm (20992 cm−1), corresponding to Mn4+–O2− charge transfer transition and Mn4+ transitions 4T1g ← 4A2g, 2T2g ← 4A2g and 4T2g ← 4A2g, respectively. A narrow far-red emission arising from Mn4+ 2Eg → 4A2g spin-forbidden transition appeared upon 365 nm UV excitation. Moreover, three kinds of Mn4+ sites were analyzed using the time-resolved spectral technology. After co-doping Nd3+/Yb3+ into CLTO:Mn4+, energy transfer from Mn4+ to Nd3+/Yb3+ was observed, which realized the effective spectral conversion of UV/visible to NIR light for possible utilization in c-Si solar cells. In addition, the Mn4+,Nd3+,Yb3+ tri-doped CLTO further enhanced this kind of spectral conversion based on the confirmed successive energy transfer process Mn4+ → Nd3+ → Yb3+ in phosphors. More importantly, the Mn4+,Nd3+,Yb3+ phosphors show excellent luminescence thermal stabilities. Results indicate that the as-prepared materials can be possible component candidates for use in c-Si solar cells and plant-growth far-red and NIR LEDs. In addition, this work provides a strategy wherein hosts for Mn4+ doping can be well enriched by substituting Te6+ for W6+ in certain tungstate compounds, which is highly desired in the search for novel red-emitting phosphors.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This project was financially supported by Ghent University's Special Research Fund (BOF; postdoctoral fellowship BOF16/PDO/159).
References
- L. Wondraczek, M. Batentschuk, M. A. Schmidt, R. Borchardt, S. Scheiner, B. Seemann, P. Schweizer and C. J. Brabec, Nat. Commun., 2013, 4, 2047 CrossRef PubMed.
- B. McKenna and R. C. Evans, Adv. Mater., 2017, 29, 1606491 CrossRef PubMed.
- M. A. Green, K. Emery, Y. Hishikawa, W. Warta and E. D. Dunlop, Prog. Photovoltaics Res. Appl., 2016, 24, 905 CrossRef.
- D. Deng, Y. Zhang, J. Zhang, Z. Wang, L. Zhu, J. Fang, B. Xia, Z. Wang, K. Lu, W. Ma and Z. Wei, Nat. Commun., 2016, 7, 13740 CrossRef CAS PubMed.
- K. Yoshikawa, H. Kawasaki, W. Yoshida, T. Irie, K. Konishi, K. Nakano, T. Uto, D. Adachi, M. Kanematsu, H. Uzu and K. Yamamoto, Nat. Energy, 2017, 2, 17032 CrossRef CAS.
-
B. M. Kayes, H. Nie, R. Twist, S. G. Spruytte and F. Reinhardt, Proceedings of the 37th IEEE Photovoltaic Specialists Conference, 2011.
- Y. Jiao, L. Mao, S. Liu, T. Tan, D. Wang, D. Cao, B. Mi, Z. Gao and W. Huang, Dyes Pigm., 2018, 158, 165 CrossRef CAS.
- A. Mashreghi and K. Maleki, Mater. Sci. Semicond. Process., 2018, 87, 92 CrossRef CAS.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510 CrossRef CAS.
- P. Kumar, S. Kanika, S. Singh, R. Lahon, A. Gundimed, G. Gupt and B. K. Gupta, J. Lumin., 2018, 196, 207 CrossRef CAS.
- P. Tumram, P. D. Sahare and S. V. Moharil, J. Lumin., 2018, 199, 78 CrossRef CAS.
- B. M. V. D. Ende, L. Aarts and A. Meijerink, Adv. Mater., 2009, 21, 3073 CrossRef.
- X. Huang, S. Han, W. Huang and X. Liu, Chem. Soc. Rev., 2013, 42, 173 RSC.
- X. Luo, X. Yang and S. Xiao, Mater. Res. Bull., 2018, 101, 73 CrossRef CAS.
- P. Vergeer, T. J. H. Vlugt, M. H. F. Kox, M. I. den Hertog, J. van der Eerden and A. Meijerink, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 014119 CrossRef.
- L. Li, Y. Pan, W. Chang, Z. Feng, P. Chen, C. Li, Z. Zeng and X. Zhou, Mater. Res. Bull., 2017, 93, 144 CrossRef CAS.
- G. Gao and L. Wondraczek, Opt. Mater. Express, 2013, 5, 633 CrossRef.
- L. Fu, H. Xia, Y. Dong, S. Li, H. Jiang and B. Chen, IEEE Photonics, 2014, 6, 2600209 Search PubMed.
- A. Verma and S. K. Sharma, Ceram. Int., 2017, 43, 8879 CrossRef CAS.
- G. Xiang, Y. Ma, X. Zhou, S. Jiang, L. Li, X. Luo, Z. Hao, X. Zhang, G.-H. Pan, Y. Luo and J. Zhang, Inorg. Chem., 2017, 56, 1498 CrossRef CAS PubMed.
- Y. Yan, Z. Chen, X. Jia and S. Li, Opt. Mater., 2018, 75, 465 CrossRef CAS.
- L. J. Borrero-Gonzalez, L. A. O. Nunes, G. S. Bianchi, F. B. G. Astrath and M. L. Baesso, J. Appl. Phys., 2013, 114, 013103 CrossRef.
- Y. Hao, Y. Wang, X. Hu, X. Liu, E. Liu, J. Fa, H. Miao and Q. Sun, Ceram. Int., 2016, 42, 9396 CrossRef CAS.
- L. Lin, J. Chen, Z. Wang, Z. Feng, F. Huang, B. Zheng, L. Huang, Z. Yu and Z. Zheng, Mater. Res. Bull., 2017, 93, 35 CrossRef CAS.
- Y. Teng, J. Zhou, X. Liu, S. Ye and J. Qiu, Opt. Express, 2010, 18, 9671 CrossRef CAS PubMed.
- L.-T. Lin, X.-M. Wu, T. Wang, D.-J. Chen, C. Deng, J.-X. Meng and L.-W. Cao, J. Alloys Compd., 2016, 673, 411 CrossRef CAS.
- M. Rai, K. Mishra, S. B. Rai and P. Morthekai, Mater. Res. Bull., 2018, 105, 192 CrossRef CAS.
- L. Wang, Y.-F. Li, Z. Wang, R. Yang, Y. Tong and L. Sun, J. Lumin., 2018, 203, 121 CrossRef CAS.
- A. Vyas, C. P. Joshi, P. D. Sahare and S. V. Moharil, J. Alloys Compd., 2018, 743, 789 CrossRef CAS.
- B. Wang, H. Lin, F. Huang, J. Xu, H. Chen, Z. Lin and Y. Wang, Chem. Mater., 2016, 28, 3515 CrossRef CAS.
- P. Cai, X. Wang and H. J. Seo, Phys. Chem. Chem. Phys., 2018, 20, 2028 RSC.
- H. Lin, T. Hu, Q. Huang, Y. Cheng, B. Wang, J. Xu, J. Wang and Y. Wang, Laser Photonics Rev., 2017, 11, 1700148 CrossRef.
- Q. Zhou, L. Dolgov, A. M. Srivastava, L. Zhou, Z. Wang, J. Shi, M. D. Dramićanin, M. G. Brik and M. Wu, J. Mater. Chem. C, 2018, 6, 2652 RSC.
- H. Lin, T. Hu, Y. Cheng, M. Chen and Y. Wang, Laser Photonics Rev., 2018, 12, 1700344 CrossRef.
- W. Chen, Y. Cheng, L. Shen, C. Shen, X. Liang and W. Xiang, J. Alloys Compd., 2018, 762, 688 CrossRef CAS.
- W. Lü, M. Jiao, B. Shao, L. Zhao, Y. Feng and H. You, Dalton Trans., 2016, 45, 466 RSC.
- J. Xiang, J. Chen, N. Zhang, H. Yao and C. Guo, Dyes Pigm., 2018, 154, 257 CrossRef CAS.
- W. Li, T. Chen, W. Xia, X. Yang and S. Xiao, J. Lumin., 2018, 194, 547 CrossRef CAS.
- J. Liao, L. Kong, Q. Wang, J. Li and H. Wen, Opt. Mater., 2018, 84, 82 CrossRef CAS.
- Y. Tai, X. Li and B. Pan, J. Lumin., 2018, 195, 102 CrossRef CAS.
- X. Huang and H. Guo, Dyes Pigm., 2018, 152, 36 CrossRef CAS.
- Z. Zhou, M. Xia, Y. Zhong, S. Gai, S. Huang, Y. Tian, X. Lu and N. Zhou, J. Mater. Chem. C, 2017, 5(32), 8201 RSC.
- L. Li, Y. Pan, Y. Huang, S. Huang and M. Wu, J. Alloys Compd., 2017, 724, 735 CrossRef CAS.
- S. W. Lee, J. M. Seo, M. K. Lee, J. H. Chun, P. Antonisamy, M. V. Arasu, T. Suzuki, N. A. Al-Dhabi and S.-J. Kim, Ind. Crops Prod., 2014, 54, 320 CrossRef CAS.
- Q. Zeng, H. Liang, M. Gong and Q. Su, J. Electrochem. Soc., 2008, 155, H730 CrossRef CAS.
- H. A. Mons, M. S. Schriewer and W. Jeitschko, J. Solid State Chem., 1992, 9, 149 CrossRef.
- J. Llanosa, R. Castillo, D. Barrionuevo, D. Espinoza and S. Conejeros, J. Alloys Compd., 2009, 485, 565 CrossRef.
- L. Vanek, I. Nemec, I. Cisarova, Z. Micka and V. Fajnor, J. Solid State Chem., 2000, 150, 410 CrossRef CAS.
- G. Blasse, Phys. Lett., 1968, 28(6), 444 CrossRef CAS.
- G. Blasse, J. Solid State Chem., 1986, 62, 207 CrossRef CAS.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836 CrossRef CAS.
- L. G. Van Uitert, J. Electrochem. Soc., 1967, 114, 1048 CrossRef CAS.
- C. Li, H. Zheng, H. Wei, S. Qiu, L. Xu, X. Wang and H. Jiao, Dalton Trans., 2018, 47, 6860 RSC.
- D. Y. Wang, C. H. Huang, Y. C. Wu and T. M. Chen, J. Mater. Chem., 2011, 21, 10818 RSC.
- Y. Yu, Y. Huang, L. Zhang, Z. Lin and G. Wang, J. Alloys Compd., 2015, 651, 164 CrossRef CAS.
- Ł. Marciniak, A. Bednarkiewicz, D. Hreniak and W. Strek, J. Mater. Chem. C, 2016, 4, 11284 RSC.
- Z. Yang, J. Yang, J. Qiu and Z. Song, Phys. Chem. Chem. Phys., 2017, 19, 31997 RSC.
- D. Böhnisch, F. Baur and T. Jüstel, Dalton Trans., 2018, 47, 1520 RSC.
- L. L. Wei, C. C. Lin, M. H. Fang, M. G. Brik, S. Hu, H. Jiao and R. S. Liu, J. Mater. Chem. C, 2015, 3, 1655 RSC.
- E. H. Song, J. Q. Wang, S. Ye, X. F. Jiang, M. Y. Peng and Q. Y. Zhang, J. Mater. Chem. C, 2016, 4, 2480 RSC.
- K. Li and R. Van Deun, Dalton Trans., 2018, 47, 6995 RSC.
- A. Fu, L. Zhou, S. Wang and Y. Li, Dyes Pigm., 2018, 148, 9 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00603b |
|
| This journal is © the Partner Organisations 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 * and
Rik
Van Deun
* and
Rik
Van Deun
 rhombohedral system with the space group R
rhombohedral system with the space group R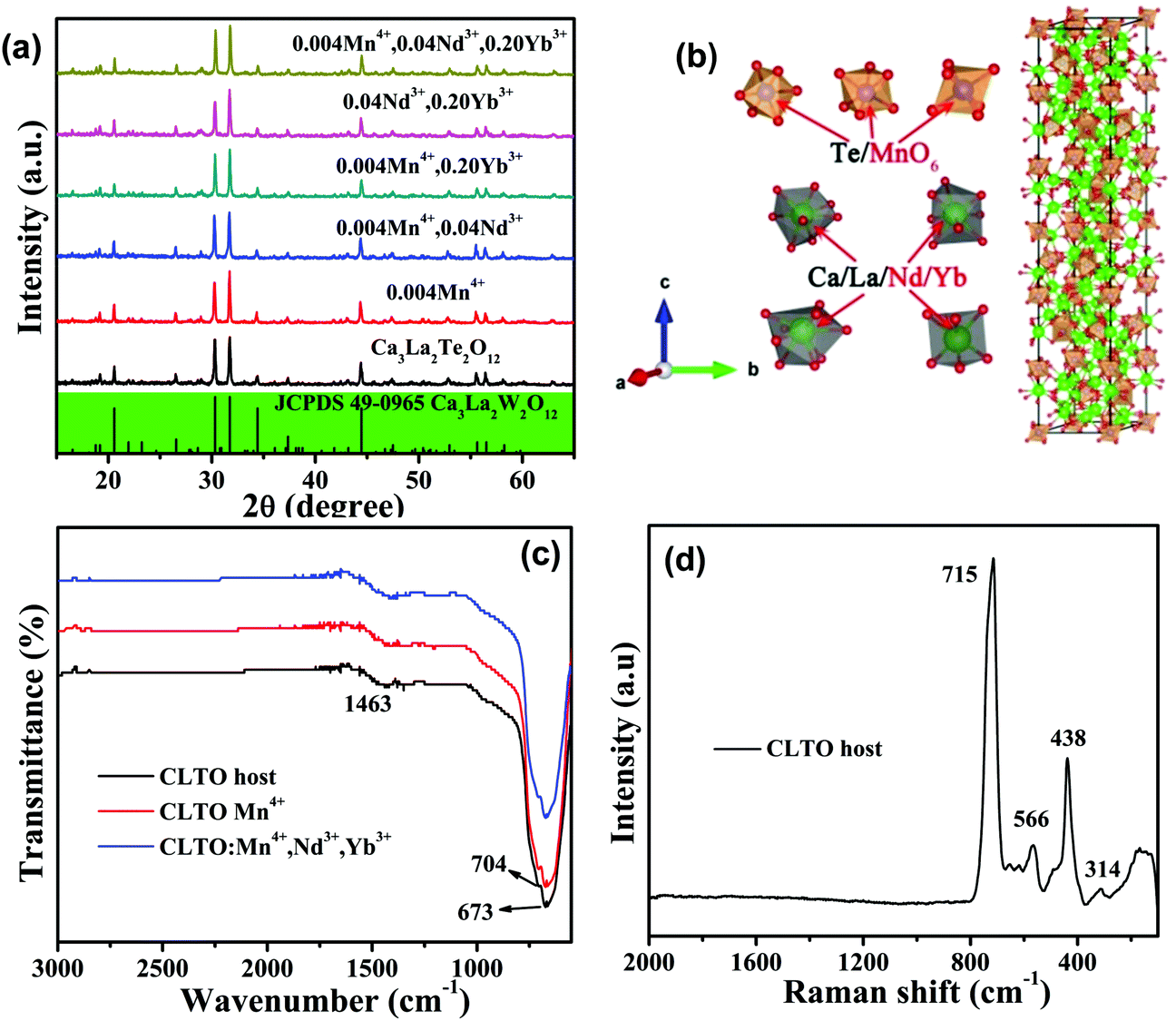
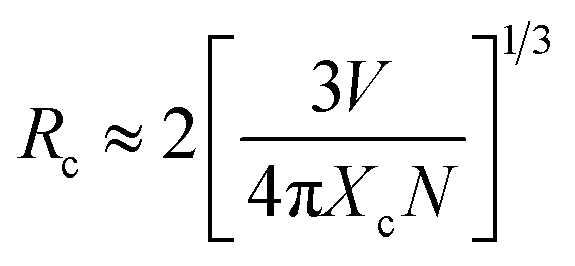
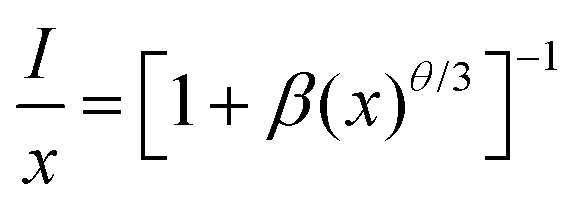
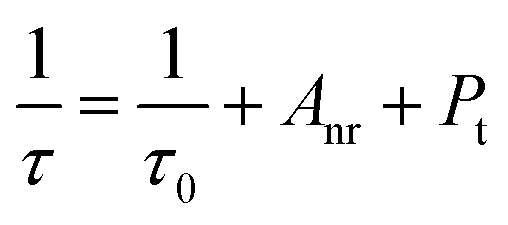

 ) to enhance the emission bands around 907 nm (11
) to enhance the emission bands around 907 nm (11 ) with the assistance of approximately five phonons mentioned above due to the large energy gap (3919 cm−1) between the Mn4+ 2Eg and Yb3+ 2F5/2 levels. Besides, the energy at the Nd3+ 4F3/2 level can also be transferred to the 2F5/2 level to enhance the Yb3+ 978 nm emission (process
) with the assistance of approximately five phonons mentioned above due to the large energy gap (3919 cm−1) between the Mn4+ 2Eg and Yb3+ 2F5/2 levels. Besides, the energy at the Nd3+ 4F3/2 level can also be transferred to the 2F5/2 level to enhance the Yb3+ 978 nm emission (process  ) ascribed to their close energy levels. When Mn4+,Nd3+ and Yb3+ are tri-doped into CLTO, the Mn4+ → Nd3+ → Yb3+ successive energy transfer process can be implied to further enhance the Yb3+ emission based on the energy transfer processes mentioned above to better match the spectral response of c-Si solar cells and the absorption spectra of photosynthetic bacteria.
) ascribed to their close energy levels. When Mn4+,Nd3+ and Yb3+ are tri-doped into CLTO, the Mn4+ → Nd3+ → Yb3+ successive energy transfer process can be implied to further enhance the Yb3+ emission based on the energy transfer processes mentioned above to better match the spectral response of c-Si solar cells and the absorption spectra of photosynthetic bacteria.