DOI:
10.1039/C8QM00599K
(Research Article)
Mater. Chem. Front., 2019,
3, 505-512
A general concept for white light emission formation from two complementary colored luminescent dyes†
Received
22nd November 2018
, Accepted 22nd January 2019
First published on 25th January 2019
Abstract
White-light emitting materials have attracted extensive research interest in recent years, which is largely ascribed to their potential applications in lighting devices and display media. Scientists have reported that organic white-light emitters mostly depend on the integration of several components that simultaneously emit different colors of light. However, it is a challenge to rationally tune the ratios of colored luminescent dyes to produce white-light emitting materials since we still cannot find the general principles to guide their preparation. In this paper, we demonstrate that “equal is important”, which means that once the emission intensities from two complementary colored luminescent dyes are equally maintained, white light emission can be generated in a range of states, including in solids, solutions, and gels. During the research process, we also found that cationic fluorescent cores are highly sensitive to the polarity of solvents. Therefore, our results could inform the design of visual indicators for the polarity and electrical conductivity of solvents using fluorescence signals.
1. Introduction
White-light emissive materials have gained significant attention in the fields of information display, fluorescent sensors, and optical-recording systems, etc.1 Researchers have developed white-light emitting materials using fluorescence energy transfer from donor to acceptor in solution, gel, and solid states.2 The common method for obtaining white-light emitting materials has focused on the integration of three primary colored (red-green-blue) or two complementary colored luminescent dyes covering the entire visible range.3 Based on the idea of integration, white-light emitting materials have been obtained by the methods of polymerization,4 nanotechnology,5 supramolecular chemistry,6 gelation of multicomponent emitters7 and fine tuning of the structures of small organic molecules etc.8 However, several obstacles have been encountered. First of all, although the design of single white-light emitting molecules has been a research focus in recent years, the fine tuning of the structures of small organic molecules requires tedious organic synthesis, which largely limits their high-throughput testing and totally prevents facile access to large libraries for light harvesting.9 Meanwhile, the other procedures also inevitably suffer from drawbacks, such as phase segregation, color aging, poor reproducibility, and small scale preparation. Not only that, for these integration procedures, it is unknown how best to finely tune the ratios of the primary colored or complementary colored luminescent dyes to produce the white-light emitting materials. Obviously, we still cannot find the general principles to guide the preparation of white-light emitting materials. The aforementioned strategies have proven effective in some cases.10 However, the resultant white-light emitting materials are usually formed in either solution or gel, or in the solid state.11 It is a real challenge to generate white-light emitting materials that can emit pure white light in both water and organic solvents, and also in the gel as well as in the solid state.12 In this context, we explored the white-light emitting materials by copolymerizing two complementary colored fluorescent monomers with N-isopropyl acrylamide. After carefully judging the ratios of the two colored monomers, the resultant polymers (P-Red&Blue) exhibited white light emission in powders, with the Commission Internationale de l’Eclairage (CIE) coordinates of 0.34, 0.33.13 From an analysis of fluorescence in different solvents, we observed white light emission in water only (CIE coordinates: 0.29, 0.30). The corresponding fluorescence spectrum clearly displayed that when the emission intensity of two colored cores is almost equally maintained, white light could be emitted. Beyond the white light emission in water, the emission in organic solvents was of particular interest for applications. Consequently, we synthesized blue emitting polymers of P-Blue and red emitting polymers of P-Red, and then rationally mixed them with different ratios until we found almost equal emission intensities for the two colored cores. Finally, we successfully achieved white-light emitting solutions in organic solvents. Therefore, the obtained general principles are applicable and convenient and can guide the preparation of white-light emitting materials in a very reasonable manner. The most popular strategies for white light emission involve single molecular systems in powders or in solution, supramolecular assembly systems in diluted solutions, and hydrogels obtained by physically mixing three primary colored small molecules in a polymer matrix. In comparison, our explored polymer systems could generate white light emissions in a versatile way in powders, hydrogels, organic solvents, and water, leading to a wide range of potential uses for our white-light emitting materials. In our research program, we also found that cationic fluorescent cores are highly sensitive to the polarity of solvents. Using this property, our polymers could be employed as polarity indicators for different solvents, and as electrical conductivity indicators in the aqueous phase.
2. Results and discussion
2.1 Synthesis of monomers of TPA-Py-CVB, TPA-VPy-CVB, and TFBP-VB
From our previous reports,14 we were conscious that triphenylamine (TPA) could serve as a very strong electron-donating core, and that the connection of TPA with pyridinium was able to produce long-wavelength emitting materials, partially due to the formation of a strong electron D–A system. Therefore, we synthesized the orange color emissive monomer of 4-(4-(diphenylamino)phenyl)-1-(4-vinylbenzyl)pyridin-1-ium chloride (TPA-Py-CVB) from the starting material of TPA-Br. After consecutive steps of Suzuki and Menshutkin reactions, TPA-Py-CVB could be produced with a high yield of 85%. The corresponding maximum emission wavelength and the fluorescence quantum yield were determined as 560 nm and 5.13% respectively. The maximum emission wavelength of the dyes has an important influence on white light formation. Thus we next synthesized the more red-shifted compound of 4-(4-(diphenylamino)styryl)-1-(4-vinylbenzyl)pyridin-1-ium chloride (TPA-VPy-CVB),15 which is slightly different from TPA-Py-CVB due to the presence of a small bridge between the TPA core and pyridinium. Surprisingly, this slight change led to a red shift of almost 65 nm when compared to TPA-Py-CVB. However, a low fluorescence quantum yield of only 0.16% was observed. Finally, we designed and synthesized the bluest emissive partner, namely 4′-(trifluoromethyl)-N-(4′-(trifluoromethyl)-[1,1′-biphenyl]-4-yl)-N-(4′-vinyl-[1,1′-biphenyl]-4-yl)-[1,1′-biphenyl]-4-amine (TFBP-VB) by two steps of Suzuki coupling reactions. During these synthetic processes, we achieved a high fluorescence quantum yield of 12.02% which was attributed to the presence of the strong electron accepting trifluoromethyl groups. Overall, we successfully obtained the monomers of TPA-Py-CVB, TPA-VPy-CVB, and TFBP-VB (Fig. 1). All three of them were modified by the same ethylene group, which was required as this is the reactive group for carrying out free radical polymerization with N-isopropyl acrylamide (NIPAM). The corresponding structural characterizations are detailed in the ESI.†
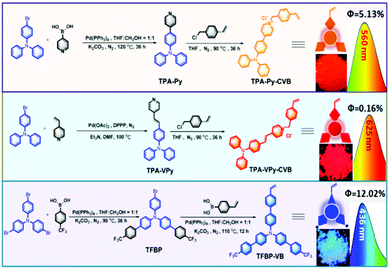 |
| | Fig. 1 The synthetic processes for the three monomers and the corresponding maximum emission wavelengths, fluorescence quantum yields, and images of the powders when illuminated by a 365 nm UV lamp. | |
2.2 Synthesizing polymers of P-Orange&Blue from monomers of TPA-Py-CVB and TFBP-VB with N-isopropyl acrylamide (NIPAM), and the corresponding fluorescence performances
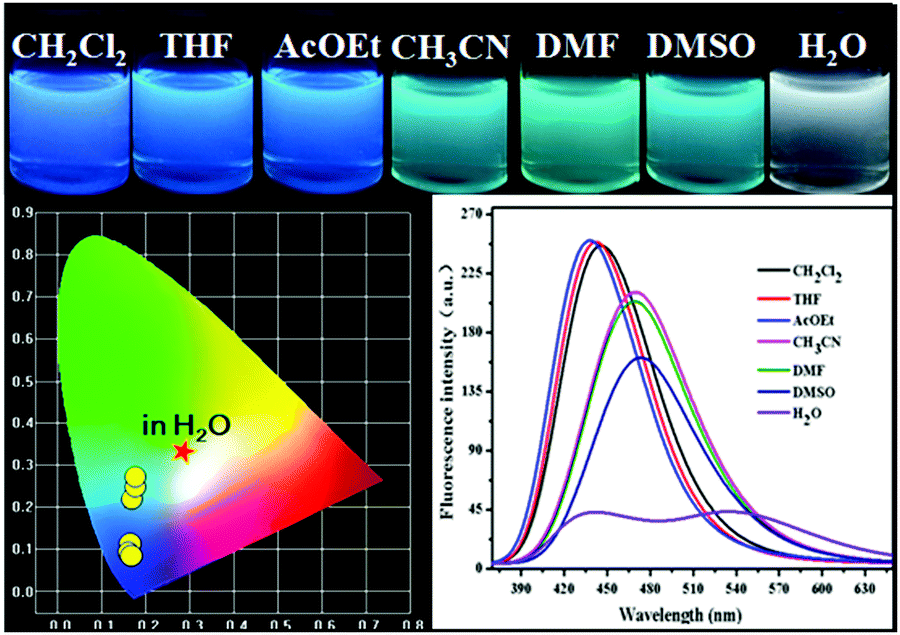
Referring to the classical free radical polymerization method,16 we employed AIBN as an initiator, and then copolymerized TPA-Py-CVB and TFBP-VB with NIPAM in THF at 90 °C for 9 h. We used a constant quantity of NIPAM, and carefully tuned the ratio of TPA-Py-CVB and TFBP-VB. In all, eight samples (abbreviated as P-Orange&Blue) were produced at the macroscale. Clearly, from the CIE coordinate diagrams, we could see that the emissions of the P-Orange&Blue powders were totally outside the white light region. This phenomenon possibly demonstrated the miss-match of blue TFBP-VB and orange TPA-Py-CVB for the generation of white light emission (Fig. S14, ESI†). However, the CIE coordinate plot from Fig. S14 (ESI†) showed that the sixth sample (abbreviated as P-Orange&Blue-6) was the nearest to the boundary of the white light region. Then, we dissolved all samples into different solvents, and further studies of fluorescence performance were performed. Surprisingly, P-Orange&Blue-3 was particularly sensitive to the solvation media. For instance, in organic solvents with moderate polarity (CH2Cl2, THF, and AcOEt), the orange monomers' emission was totally suppressed, leading to solutions with blue emission. In the most polar solvents (CH3CN, DMF, and DMSO), the emission of the blue monomers was red-shifted by almost 25 nm and they had a high intensity, which also shielded the orange emission color. Notably, it was only in water that both of the emission colors could be detected with almost the same intensity; the CIE coordinate was cross-located at 0.22, 0.30. The corresponding water solution also exhibited a near white emission color. The very different emission behaviors induced us to conclude that by maintaining almost the same emission intensities from the blue and orange dyes, there might be a general possibility of producing the fascinating white colors (Fig. 2). In the case of P-Orange&Blue, the pure white emission color was not achieved, but this result served as the foundation for further work on white-light emitting materials.
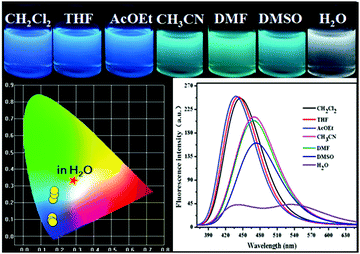 |
| | Fig. 2 The photographs of P-Orange&Blue-3 in different solvents under UV light (365 nm); the fluorescence spectra of P-Orange&Blue-3 in different solvents; the CIE chromaticity diagram of P-Orange&Blue-3 in different solvents. | |
2.3 Synthesizing polymers of P-Red&Blue from monomers of TPA-VPy-CVB and TFBP-VB with N-isopropyl acrylamide (NIPAM), and the corresponding fluorescence performances
As mentioned above, we were unable to obtain pure white emission polymers in the case of P-Orange&Blue, which is possibly due to the short emission wavelength (560 nm) of orange colored TPA-Py-CVB. From a survey of the previous literature,7c,8a we were aware that the more red-shifted colored cores might be beneficial to the formation of white emission. Therefore, we synthesized the compound TPA-VPy-CVB which has a longer emission wavelength (625 nm) than TPA-Py-CVB because of the presence of a small bridge. By a similar copolymerization method, we produced eight serial polymers, all termed P-Red&Blue (Fig. 3).
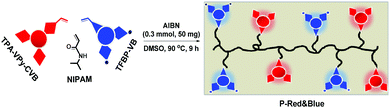 |
| | Fig. 3 Synthetic routes for P-Red&Blue. | |
First of all, we measured the fluorescence performances of all of the powders, and the CIE coordinates are listed in Fig. 4. After serially tuning the ratio of TPA-VPy-CVB to TFBP-VB, pure white emission could be generated in the case of P-Red&Blue-3 (CIE coordinates of 0.34, 0.33). These coordinates are almost the same as those for real white emission (CIE coordinates of 0.33, 0.33). As well as the white emission, there were two notable findings: (1) the rational, rather than random adjustment of the two cores led to an almost linear relationship between the ratios and the CIE coordinate locations; (2) the resulting polymers demonstrated enhanced fluorescence quantum yields that were larger than those of the monomers, which was probably due to the restricted intramolecular rotations after polymerization that led to the occurrence of aggregation induced emission (AIE). Both of these advantages are technologically very beneficial for the design of white-light emitting materials and also for the application of AIE in a wide range of fields.
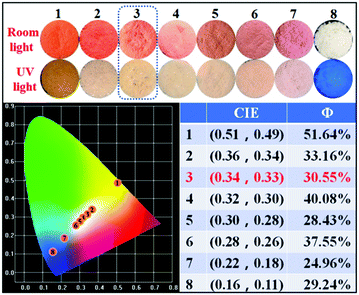 |
| | Fig. 4 The photographs of P-Red&Blue in the solid state under room light and UV light (365 nm); the fluorescence spectra of P-Red&Blue in the solid state; the CIE chromaticity diagram of P-Red&Blue in the solid state with the fluorescence quantum yields (%). | |
After dissolving all of the samples in water at the same concentrations, the CIE coordinates and the fluorescence quantum yields were recorded (Fig. 5). Similar to the result for the solid state, P-Red&Blue-3 gave the nearest CIE coordinates (0.29, 0.30) to those of real white light emission and the highest fluorescence quantum yield of 30.14%. Again, once almost the same emission intensity from the two partners was achieved, emission closest to white light could be generated. This conclusion firmly supported our strategy for white-light emission formation from two complementary colored luminescent dyes.
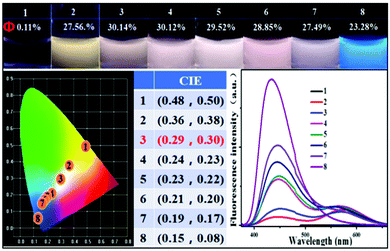 |
| | Fig. 5 The photographs of P-Red&Blue in aqueous solutions under UV light (365 nm) with the fluorescence quantum yields (%); the fluorescence spectra of P-Red&Blue in aqueous solutions; the CIE chromaticity diagram of P-Red&Blue in aqueous solutions. | |
2.4 White-light emissive hydrogel formation by P-Red&Blue-3
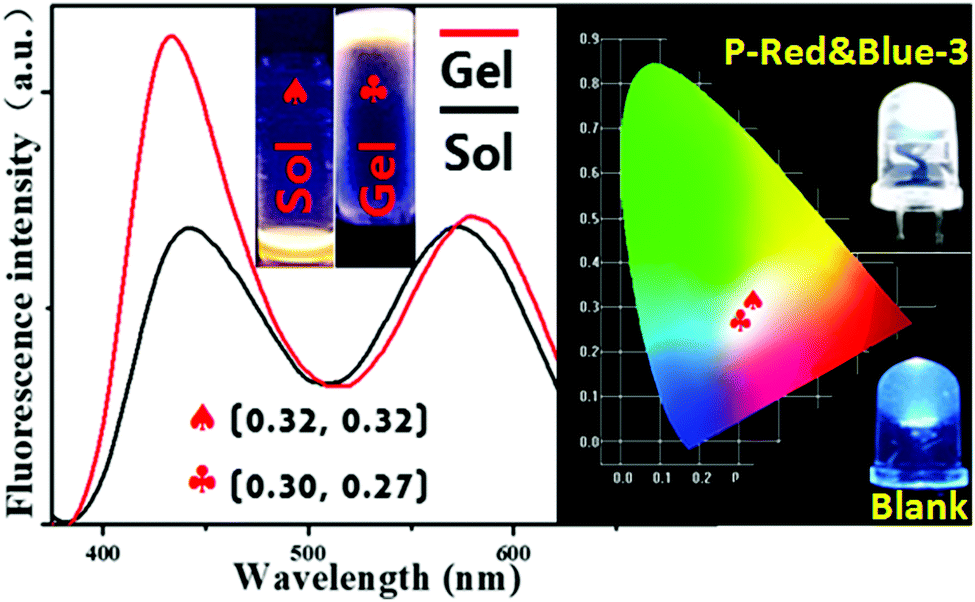
As we mentioned above, the typical method for forming white-light emitting hydrogels involves mechanically dissolving colored small molecules in a water soluble polymer matrix, but this method suffers from the disadvantages of phase segregation, and color aging etc. Covalent bond formation between colored segments and the polymer matrix is expected to readily overcome these problems. Herein, we heated the hydrogels with P-Red&Blue-3 at the concentration of 50 mg/0.5 mL, leading to a very stable soft matter at room temperature (30 °C) which was highly emissive. The corresponding CIE coordinates slightly moved from white emission (0.32, 0.32) to the position 0.30, 0.27 (Fig. 6). This result demonstrated the minor effect on the color due to the sol–gel reversible transformations. Needless to say, the obtained white-light emitting materials will have a wide range of applications in the different states. Also, due to the temperature sensitive property of poly-N-isopropylacrylamide (PNIPAM), the multi-functional sensing behavior of P-Red&Blue could be explored. This part of the work is underway in our laboratory, and will be reported in the near future.
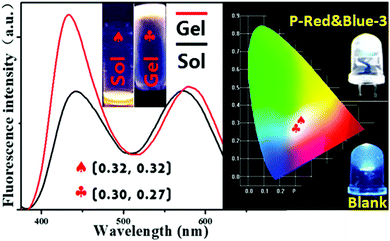 |
| | Fig. 6 The fluorescence spectra, photographs (under UV light (365 nm)) and CIE chromaticity diagram of P-Red&Blue-3 in aqueous solution, and in the sol and gel states; an image of a commercially available UV-LED (λem = 365 nm, 3.2–3.4 V, 20 mA) coated with hydrogel, when turned on. | |
2.5 The polarity indicator of P-Red&Blue in organic solvents and in water
We next screened the emission behaviors of P-Red&Blue-3 in different solvents. The changes were similar to those seen for P-Orange&Blue-3. That is to say, both polymers were very sensitive to the polarity of the solvents. P-Red&Blue-3 exhibited white light emission only in the aqueous phase. These similarities of solvation effects on the P-Orange&Blue-3 and P-Red&Blue-3 systems made us aware that neutral fluorescent cores (for instance TFBP-VB) are more emissive in the less polar solvents, but less emissive in the more polar solvents such as water. In contrast, the ionic fluorescent cores (for instance TPA-Py-CVB or TPA-VPy-CVB) are especially emissive in the polar solvents, but not in the less polar solvents (Fig. 7).
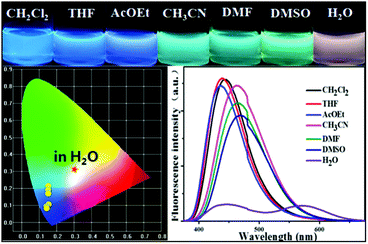 |
| | Fig. 7 The photographs of P-Red&Blue-3 in different solvents under UV light (365 nm); the fluorescence spectra and the CIE chromaticity diagram of P-Red&Blue-3 in different solvents. | |
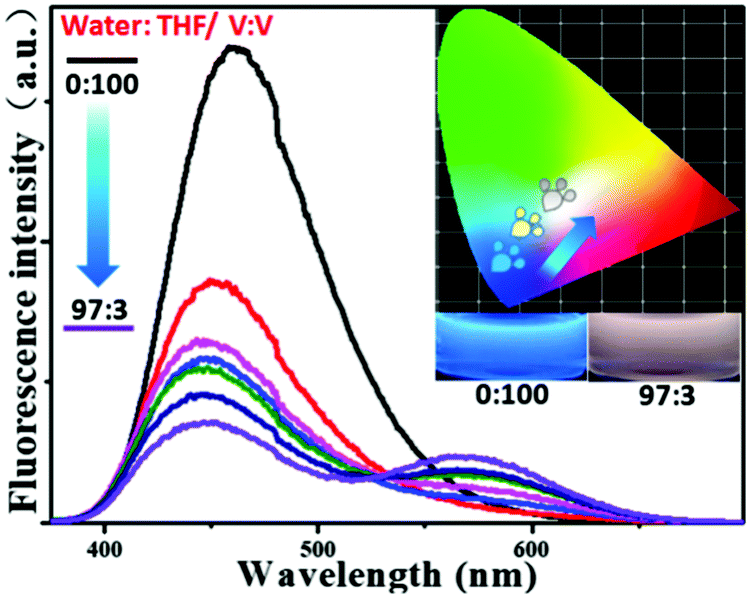
From this research, we postulated that an enhancement of the solvent polarity, going beyond simply organic solvents or the aqueous phase, could be used to tune the emission intensity ratios between the neutral and ionic fluorescent cores. This result could be used to design polarity indicators on the basis of the fluorescence signals. Therefore, we gradually added water into the solution of P-Red&Blue-3 in THF to change the polarity of the solvent. Initially, blue emission (λmax = 428 nm) could be observed in the less polar and water miscible THF. Obviously, after adding water into the solution, the blue emission was suppressed, and the red emission was progressively enhanced.
Finally, in a mixture of H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :THF with a ratio of 97:3 by volume, the CIE coordinates went into the white emission region from the blue light region (Fig. 8). This result perfectly agreed with our original expectations that P-Red&Blue has the potential to be used as a polarity indicator.
:THF with a ratio of 97:3 by volume, the CIE coordinates went into the white emission region from the blue light region (Fig. 8). This result perfectly agreed with our original expectations that P-Red&Blue has the potential to be used as a polarity indicator.
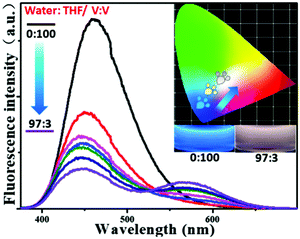 |
| | Fig. 8 The fluorescence spectra and the CIE chromaticity diagram of P-Red&Blue-3 in mixed solvents of THF and H2O. | |
2.6 The electrical conductivity indicator of P-Red&Blue in NaCl aqueous solutions
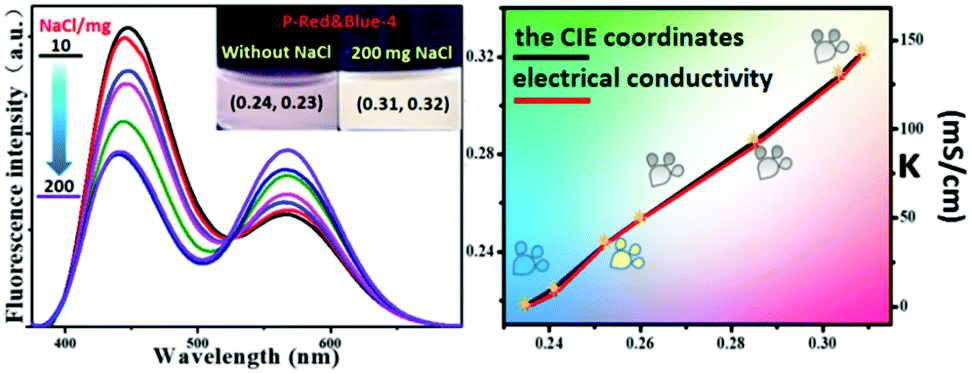
Aside from its use as a polarity indicator in organic solvents, it was of interest to see whether our system could be used to determine the polarity or electrical conductivity of an aqueous phase. Thus, for the purpose of elevating the polarity and electrical conductivity of the aqueous phase, different quantities of NaCl were dissolved in solutions of P-Red&Blue-4 in water, and the emission performances were screened systematically. It should be noted that the CIE coordinates of P-Red&Blue-4 were 0.24, 0.23 in the pure aqueous phase, which are far from those of real white light (Fig. 9). Experimentally, we added 10 to 200 mg NaCl solid into P-Red&Blue-4 aqueous solution (0.5 mg mL−1, 3 mL) and the corresponding CIE coordinates gradually shifted into the white light region showing an almost linear relationship with the quantity of added NaCl (Fig. 9). Then, we detected the electrical conductivities of the aqueous solutions containing the different amounts of NaCl. Reasonably, the CIE coordinates depended on the electrical conductivity in a perfect linear relationship. Generally speaking, because of the different emission properties from the neutral fluorescent cores and the ionic fluorescent cores, both the polarity of the aqueous phase and the electrical conductivity could be determined by the fluorescence signal in a very convenient manner. This rational design will be of definite general interest to future studies of photoelectro-chemistry.
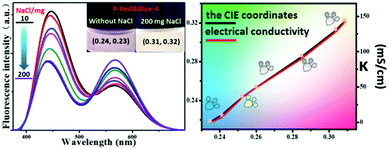 |
| | Fig. 9 The fluorescence spectra and the CIE chromaticity diagram of P-Red&Blue-4 in NaCl solution. | |
2.7 The rational design of white-light emitting materials from P-red and P-blue in organic solvents and organic gels
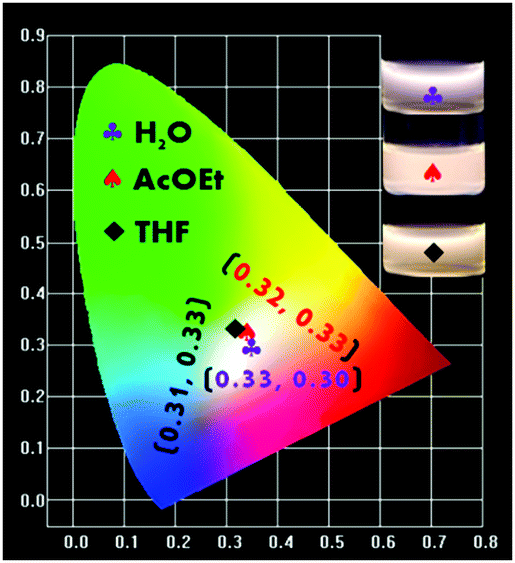
During the studies of the emission performances of the P-Red&Blue polymers in different conditions, we achieved a clear understanding that the almost contradictory emission properties from two colored cores are largely dependent on the polarity of the solvents. We found that in the less polar organic solvents, the neutral blue core was mostly emissive, and totally shielded the emissions from the ionic red cores. Thus, up to now, we have not obtained white-light emitting polymers in the organic solvents. By successfully dealing with these issues we might be able to extend the applications of the polymers. We thought that by greatly increasing the ratio of the ionic red cores (TPA-VPy-CVB) during the copolymerization, we might elevate the emission intensity of the ionic red cores, leading to an almost equal emission intensity from the two types of core. However, obstacles existed, in particular the poor solubility of TPA-VPy-CVB in THF and its low fluorescence quantum yield of 5.13%. Thus, we independently synthesized the P-Red polymer from TPA-VPy-CVB with NIPAM, and the P-Blue polymer from TFBP-VB with NIPAM under the same conditions. After tuning the ratios of P-Red and P-Blue in the organic solvents of THF and AcOEt, white emission light was rationally generated. The corresponding CIE coordinates were located at 0.32, 0.33 in AcOEt and 0.31, 0.33 in THF (Fig. 10). As expected, the white light emissions could also be obtained in water solutions. It is important to point out that the mixing of P-Blue and P-Red in one pot is totally different from the previous methods in which small molecule dyes were dispersed in the polymer matrix, aggregation of the organic molecule dyes was inevitable, and so phase segregation was hard to avoid. In our method, the two colored cores harmoniously resided in PNIPAM networks at a molecular level, and so phase segregation and color aging should be excluded completely.
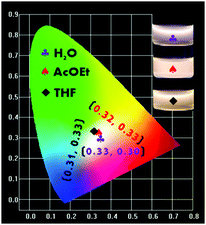 |
| | Fig. 10 The CIE chromaticity diagram obtained from different mass ratios of P-Red and P-Blue in AcOEt, THF and water. | |
3. Materials and methods
3.1 Materials
N-Isopropylacrylamide (NIPAM, 98%), tetrakis(triphenylphosphine)-palladium(0) (Pd(PPh3)4, 99%), palladium acetate (Pd(OAc)2, 99%), 1-(chloromethyl)-4-ethenylbenzene (99%) and 4-vinylpyridine (96%) were purchased from Aladdin. Azobis(isobutyronitrile) (AIBN, 98%) was purchased from Aladdin and purified by recrystallization using ethanol. 4-Trifluoromethylphenylboronic acid (98%), 4-vinylphenylboronic acid (98%), pyridine-4-boronic acid, 4-bromotriphenylamine (98%) and tris(4-bromophenyl)amine (98%) were purchased from Suzhou Sukai Road Chemical Technology Company Limited. 1,3-Bis(diphenylphosphino) propane (DPPP, 98%) was purchased from Beijing Greenland Keemer Technology Company Limited. Nitrogen (N2) with a purity of 99.99% was provided from a commercial source. Other reagents, such as triethylamine (Et3N), N,N-dimethylformamide (DMF), dimethyl sulfoxide (DMSO), methanol, ethanol, tetrahydrofuran (THF) and chloroform were of analytical grade and were purchased from Energy Chemical Company. 1H NMR and 13C NMR spectra were recorded on a MERCURY spectrometer at 25 °C, and all NMR spectra were referenced to the solvent. Mass spectrometry (MS) was carried out using a Thermoscientific Q Exactive instrument. UV-visible absorption spectra (UV) were recorded on a TU-1901 spectrometer. Fluorescence spectra and fluorescence quantum yields were measured using a FluoroSENS 9003 fluorescence spectrophotometer. All the samples were prepared according to the standard methods. Conductivity measurements were obtained using a NDCH-1 ethyl acetate saponification reactor. The CIE chromaticity diagrams were obtained using the CIE1931xy.V.1.6.0.2 program.
3.2 Synthesis of N,N-diphenyl-4-(pyridin-4-yl)aniline (TPA-Py)14
4-Bromotriphenylamine (8 mmol, 324.21 g mol−1, 2.594 g), pyridine-4-boronic acid (12 mmol, 122.92 g mol−1, 1.475 g), tetrakis(triphenylphosphine)-palladium(0) (5% equiv., 1155 g mol−1, 0.462 g) and potassium carbonate (8 mmol, 138.21 g mol−1, 1.105 g) were weighed out, and a mixture of THF (50 mL) and MeOH (50 mL) was added. The mixture was refluxed at 120 °C for 36 h under a N2 atmosphere and then concentrated by rotary evaporation. TPA-Py was purified by column chromatography on silica gel (200–300 mesh) with a mixture of petroleum ether and ethyl acetate as the eluent (20:1 by volume) to obtain a white solid (1.9 g, 76% yield); known compound.141H NMR (600 MHz, CDCl3) δ (TMS, ppm): 8.61 (d, J = 6.0 Hz, 2H), 7.52 (d, J = 8.7 Hz, 2H), 7.47 (d, J = 6.1 Hz, 2H), 7.29 (dd, J = 8.2, 7.7 Hz, 4H), 7.14 (dd, J = 8.1, 3.9 Hz, 6H), 7.08 (t, J = 7.4 Hz, 2H). 13C NMR (151 MHz, CDCl3) δ (TMS, ppm): 150.19, 148.91, 147.59, 147.22, 130.94, 129.40, 127.60, 124.95, 123.57, 122.89, 120.83. HRMS (ESI): m/z calculated for C23H19N2 322.15482; found 323.15259 [M + H+].
3.3 Synthesis of 4-(4-(diphenylamino)phenyl)-1-(4-vinylbenzyl)pyridin-1-ium chloride (TPA-Py-CVB)14
A mixture of TPA-Py (1 mmol, 322.14 g mol−1, 0.332 g), 1-(chloromethyl)-4-ethenylbenzene (1.5 mmol, 152.62 g mol−1, 0.229 g) and THF (20 mL) was stirred at 90 °C for 36 h. After that, the reaction mixture was precipitated from THF (dry), and washed several times using THF and n-hexane. An orange solid powder (TPA-Py-CVB) was obtained by filtration, and dried under vacuum at 30 °C overnight (0.404 g, 85% yield); unknown compound. 1H NMR (600 MHz, CD3OD) δ (TMS, ppm): 8.82 (d, J = 7.1 Hz, 2H), 8.28 (d, J = 7.0 Hz, 2H), 7.90 (d, J = 9.0 Hz, 2H), 7.55–7.46 (m, 4H), 7.41–7.37 (m, 4H), 7.20 (dd, J = 20.4, 7.6 Hz, 6H), 7.06 (d, J = 9.0 Hz, 2H), 6.76 (d, J = 4.4 Hz, 1H), 5.83 (s, 1H), 5.71 (s, 2H), 5.31 (d, J = 10.9 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ (TMS, ppm): 163.65, 152.31, 145.70, 135.71, 129.82, 129.74, 128.94, 127.31, 126.37, 126.32, 126.30, 125.45, 122.56, 120.16, 115.66, 105.90, 93.97. HRMS (ESI): m/z calculated for C32H27N2ClNa 497.17605; found 497.23428 [M + Na].
3.4 Synthesis of N-(4-bromophenyl)-4′-(trifluoromethyl)-N-(4′-(trifluoromethyl)-[1,1′-biphenyl]-4-yl)-[1,1′-biphenyl]-4-amine (TFBP)
4-Bromotriphenylamine (4 mmol, 324.21 g mol−1, 1.928 g), 4-trifluoromethylphenylboronic acid (8 mmol, 189.93 g mol−1, 1.519 g), tetrakis(triphenylphosphine)-palladium(0) (5% equiv., 1155 g mol−1, 0.231 g) and potassium carbonate (4 mmol, 138.21 g mol−1, 0.553 g) were weighed out, and a mixture of THF (50 mL) and MeOH (50 mL) was added. The mixture was refluxed at 90 °C for 9 h under a N2 atmosphere and then concentrated by rotary evaporation. TFBP was purified by column chromatography on silica gel (200–300 mesh) with petroleum ether as the eluent, to obtain a white solid (1.7 g, 70% yield); unknown compound. 1H NMR (600 MHz, CDCl3) δ (TMS, ppm): 7.68 (d, J = 9.4 Hz, 8H), 7.52 (dd, J = 6.7, 2.0 Hz, 4H), 7.41 (d, J = 8.9 Hz, 2H), 7.20 (d, J = 8.7 Hz, 4H), 7.06 (d, J = 8.8 Hz, 2H). 13C NMR (151 MHz, CDCl3) δ (TMS, ppm): 147.17, 146.25, 143.85, 134.38, 132.50, 129.12, 128.91, 128.20, 126.85, 126.16, 125.72, 124.30, 116.15. HRMS (ESI): m/z calculated for C32H20NBrF6 611.06833; found 611.06616 [M+].
3.5 Synthesis of 4′-(trifluoromethyl)-N-(4′-(trifluoromethyl)-[1,1′-biphenyl]-4-yl)-N-(4′-vinyl-[1,1′-biphenyl]-4-yl)-[1,1′-biphenyl]-4-amine (TFBP-VB)
TFBP (2 mmol, 611.06 g mol−1, 1.225 g), 4-vinylphenylboronic acid (3 mmol, 147.94 g mol−1, 1.475 g), potassium carbonate (2 mmol, 138.21 g mol−1, 0.276 g) and tetrakis(triphenylphosphine)-palladium(0) (5% equiv., 1155 g mol−1, 0.115 g) were weighed out, and a mixture of THF (50 mL) and MeOH (50 mL) was added. The mixture was refluxed at 110 °C for 12 h under a N2 atmosphere and then concentrated by rotary evaporation. TFBP-VB was purified by column chromatography on silica gel (200–300 mesh) with a mixture of petroleum ether and ethyl acetate as the eluent (500:1 by volume) to obtain a yellow solid (1.1 g, 86% yield); unknown compound. 1H NMR (400 MHz, CDCl3) δ (TMS, ppm): 7.69 (s, 8H), 7.62–7.41 (m, 10H), 7.27 (d, J = 4.6 Hz, 5H), 7.25 (d, J = 3.9 Hz, 2H), 6.76 (dd, J = 17.5, 11.0 Hz, 1H), 5.80 (d, J = 17.7 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ (TMS, ppm): 147.46, 146.40, 143.96, 139.73, 136.40, 136.35, 135.84, 134.06, 128.11, 127.86, 126.74, 126.68 125.73, 125.71, 124.97, 124.29, 113.80. HRMS (ESI): m/z calculated for C40H27NF6 635.20477; found 635.20251 [M+].
3.6 Synthesis of N,N-diphenyl-4-(2-(pyridin-4-yl)vinyl)aniline (TPA-VPy)15
4-Bromotriphenylamine (4 mmol, 324.21 g mol−1, 1.928 g), 4-vinylpyridine (4 mmol, 105.14 g mol−1, 0.621 g), 1,3-bis(diphenylphosphino)propane (5% equiv., 1155 g mol−1, 0.083 g), Pd(OAc)2 (2.5% equiv., 224.51 g mol−1, 0.022 g), and Et3N (8 mmol, 101.19 g mol−1, 0.808 g) were added into 20 mL of dry DMF. After stirring for 12 h at 100 °C under a N2 atmosphere, 20 mL of water was added, and the solution was extracted with CH2Cl2 (3 × 20 mL). The organic phases were then dried with anhydrous magnesium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel (200–300 mesh) with a mixture of petroleum ether and ethyl acetate as the eluent (50:1 by volume) to obtain a yellow solid (0.7 g, 50% yield); unknown compound.151H NMR (600 MHz, CDCl3) δ (TMS, ppm): 8.54 (d, J = 5.6 Hz, 2H), 7.40 (d, J = 8.6 Hz, 2H), 7.33 (d, J = 5.8 Hz, 2H), 7.28 (d, J = 7.7 Hz, 3H), 7.26–7.22 (m, 2H), 7.12 (d, J = 7.8 Hz, 4H), 7.08–7.04 (m, 4H), 6.88 (d, J = 16.3 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ (TMS, ppm): 150.08, 148.42, 147.24, 144.95, 132.63, 129.78, 129.34, 127.91, 124.86, 123.87, 123.45, 122.79, 120.59. HRMS (ESI): m/z calculated for C40H27NF6 348.16265; found 348.16199 [M+].
3.7 Synthesis of 4-(4-(diphenylamino)styryl)-1-(4-vinylbenzyl)pyridin-1-ium chloride (TPA-VPy-CVB)15
A mixture of TPA-VPy (1 mmol, 348.16 g mol−1, 0.348 g), 1-(chloromethyl)-4-ethenylbenzene (1.5 mmol, 152.62 g mol−1, 0.229 g) and THF (20 mL) was stirred at 90 °C for 36 h. After that, the reaction mixture was precipitated from THF (dry), and washed several times using THF and n-hexane. A red solid powder (TPA-Py-CVB) was obtained by filtration, and dried under vacuum at 30 °C overnight (0.4435 g, 87% yield); unknown compound. 1H NMR (600 MHz, CDCl3) δ (TMS, ppm): 9.24 (d, J = 6.5 Hz, 2H), 7.80 (d, J = 6.6 Hz, 2H), 7.56–7.52 (m, 3H), 7.45–7.42 (m, 4H), 7.36–7.29 (m, 4H), 7.18–7.08 (m, 6H), 7.01 (d, J = 8.7 Hz, 2H), 6.89 (d, J = 15.9 Hz, 1H), 6.68 (dd, J = 17.6, 10.9 Hz, 1H), 6.08 (s, 2H), 5.77 (d, J = 17.6 Hz, 1H), 5.31 (d, J = 10.9 Hz, 1H). 13C NMR (151 MHz, CDCl3) δ (TMS, ppm): 183.80, 153.92, 146.33, 143.80, 142.15, 139.09, 135.71, 135.48, 129.83, 129.65, 129.61, 129.57, 127.30, 126.82, 125.82, 124.68, 123.16, 120.84, 115.64, 109.99. HRMS m/z calculated for C34H29N2 465.23307; found 465.23243 [M*+].
3.8 Synthesis of P-Orange&Blue polymers16
TPA-Py-CVB (0.02 mmol, 0.01 mmol, 0.005 mmol, 0.0045 mmol, 0.004 mmol, 0.003 mmol, 0.002 mmol, 0 mmol) and TFBP-VB (0 mmol, 0.03 mmol, 0.035 mmol, 0.036 mmol, 0.036 mmol, 0.036 mmol, 0.038 mmol, 0.02 mmol, respectively) were added to eight flasks. N-Isopropylacrylamide (1.0 g, 8.85 mmol), AIBN (50 mg) and THF (20 mL) were added and the reaction mixtures were stirred at 90 °C under N2 for 9 h. Then the THF was evaporated and the resulting precipitates were washed several times with n-hexane to obtain solid powders. 1H NMR (600 MHz, CDCl3) δ (TMS, ppm): 8.82–8.80 (m), 7.68 (m), 7.54 (m), 7.46 (m), 6.59–5.96 (m), 5.74–5.27 (m), 4.40–4.31 (m), 4.21–4.14 (m), 4.10–3.90 (m), 3.89–3.78 (m), 3.76–3.59 (m), 3.37 (m), 2.70–1.98 (m), 1.97–1.75 (m), 1.75–1.46 (m), 1.46–1.31 (m), 1.31–1.24 (m), 1.22–0.96 (m).
3.9 Synthesis of P-Red&Blue polymers16
TPA-VPy-CVB (0.02 mmol, 0.018 mmol, 0.014 mmol, 0.012 mmol, 0.009 mmol, 0.0072 mmol, 0.006 mmol, 0 mmol) and TFBP-VB (0 mmol, 0.036 mmol, 0.036 mmol, 0.036 mmol, 0.036 mmol, 0.036 mmol, 0.036 mmol, 0.02 mmol, respectively) were added to eight flasks. N-Isopropylacrylamide (1.0 g, 8.85 mmol), AIBN (50 mg) and DMSO (20 mL) were added and the reaction mixtures were stirred at 90 °C under N2 for 9 h. Then the DMSO was evaporated and the resulting precipitates were washed several times with n-hexane to obtain solid powders. 1H NMR (600 MHz, CDCl3) δ (TMS, ppm): 8.39–8.22 (m), 7.66 (m), 7.56–7.40 (m), 6.98–5.78 (m), 5.60–5.43 (m), 5.35–5.19 (m), 4.14–3.80 (m), 3.61 (m), 3.50–3.44 (m), 3.25–2.54 (m), 2.61–2.58 (m), 2.41–1.95 (m), 1.92–1.71 (m), 1.71–1.47 (m), 1.45–1.23 (m), 1.23–0.68 (m).
3.10 Preparation of gels17
Polymers (50 mg) were dissolved in aqueous solutions (0.5 mL) in sealed test tubes and kept cool until dissolution was complete. They were then heated to 50 °C in a water bath to form the hydrogels. Gel formation was evaluated by testing for stability to the inversion of the test tube. Then, the xerogels were produced by solvent evaporation.
3.11 Fluorescence quantum yield (ΦF) measurements
Fluorescence quantum yield measurements were obtained using a FluoroSENS 9003 fluorescence spectrophotometer. The fluorescence quantum yields were calculated using an integrated sphere. The fluorescence quantum yields were collected at different excitation wavelengths as follows: 440 nm for TPA-Py-CVB and P-Orange, 485 nm for TPA-VPy-CVB and P-Red, 335 nm for TFBP-VB, and 350 nm for P-Orange&Blue and P-Red&Blue. Each emission spectrum was collected from 370 to 700 nm with 1 nm steps and 100 ms dwell times.
4. Conclusions
In conclusion, our research results firmly verified the general strategy of generating white light emission from two complementary colored luminescent dyes. It was important to have equal emission intensities from the two cores for the synthesis of white-light emissive materials. This was necessary for polymers and small molecules. However, compared to the white-light emissive small molecules, our copolymers offered the following additional advantages: white light emission in a versatile range of states, water miscibility, solubility in organic solvents, sensitivity to polarity and electrical conductivity, and large-scale production capacity. These advantages are very beneficial for extensive applications in the fields of soft matter, smart materials, biological sensors, etc. In this study, a preliminary application of the copolymers for polarity and electrical conductivity sensing was explored. The behavior of the polymers was largely due to the different emission performances of the two complementary colored luminescent dyes in polar and non-polar solutions. In general, our research results highlight various possibilities for designing white-light emissive materials from colored luminescent dyes with neutral, positive, or negative charges. The cooperation of these dyes deserves to be the focus of novel research and future studies.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (no. 21764012). We thank the Key Laboratory of Eco-Environment-Related Polymer Materials (Northwest Normal University) and the Ministry of Education Scholars Innovation Team (IRT 1177) for financial support.
Notes and references
-
(a) X. L. Ni, S. Chen, Y. Yanga, S. Reineke, F. Lindner, G. Schwartz, N. Seidler, K. Walzer, B. Lüssem and K. Leo, Nature, 2009, 459, 234–239 CrossRef PubMed;
(b) M. C. Gather, A. Koehnen, A. Falcou, H. Becker and K. Meerholz, Adv. Funct. Mater., 2007, 17, 191–200 CrossRef CAS;
(c) G. M. Farinola and R. Ragni, Chem. Soc. Rev., 2011, 40, 3467–3482 RSC;
(d) D. Li, J. Wang and X. Ma, Adv. Opt. Mater., 2018, 6, 1800273 CrossRef;
(e) H. Wu, G. Zhou, J. Zou, C. L. Ho, W. Y. Wong, W. Yang, J. Peng and Y. Cao, Adv. Mater., 2009, 21, 4181–4184 CrossRef CAS.
-
(a) Z. Tao, J. Am. Chem. Soc., 2016, 138, 6177–6183 CrossRef PubMed;
(b) J. Liang, L. Ying, F. Huang and Y. Cao, J. Mater. Chem. C, 2016, 4, 10993–11006 RSC;
(c) P. Kumar, S. Soumya and E. Prasad, ACS Appl. Mater. Interfaces, 2016, 8, 8068–8075 CrossRef CAS PubMed;
(d) L. Mao, Y. Wu, C. C. Stoumpos, M. R. Wasielewski and M. G. Kanatzidis, J. Am. Chem. Soc., 2017, 139, 5210–5215 CrossRef CAS PubMed.
-
(a) D. K. Maiti, R. Bhattacharjee, A. Datta and A. Banerjee, J. Phys. Chem. C, 2013, 117, 23178–23189 CrossRef CAS;
(b) C. Vijayakumar, V. K. Praveen and A. Ajayaghosh, Adv. Mater., 2009, 21, 2059–2063 CrossRef CAS;
(c) N. Thejokalyani and S. J. Dhoble, Renewable Sustainable Energy Rev., 2014, 32, 448–467 CrossRef CAS.
-
(a) S. Shinde and S. K. Asha, Macromolecules, 2016, 49, 8134–8145 CrossRef CAS;
(b) H. Wu, L. Ying, W. Yang and Y. Cao, Chem. Soc. Rev., 2009, 38, 3391–3400 RSC;
(c) H. Y. Lin, C. W. Sher, C. H. Lin, H. H. Tu, X. Y. Chen, Y. C. Lai, C. C. Lin, H. M. Chen, P. Yu, H. F. Meng, H. C. Kuo and G. C. Chi, ACS Appl. Mater. Interfaces, 2017, 9, 35279–35286 CrossRef CAS PubMed.
-
(a) J. Malinge, C. Allain, A. Brosseau and P. Audebert, Angew. Chem., Int. Ed., 2012, 124, 8662–8665 CrossRef;
(b) A. Layek, P. C. Stanish, V. Chirmanov and P. V. Radovanovic, Chem. Mater., 2015, 27, 1021–1030 CrossRef CAS;
(c) X. Zhang, S. Rehm, M. M. Safont-Sempere and F. Würthner, Nat. Chem., 2009, 1, 623–629 CrossRef CAS PubMed;
(d) V. K. Sharma, B. Guzelturk, T. Erdem, Y. Kelestemur and H. V. Demir, ACS Appl. Mater. Interfaces, 2014, 6, 3654–3660 CrossRef CAS PubMed.
-
(a) Y. Sun, Y. Lei, L. Liao and W. Hu, Angew. Chem., Int. Ed., 2017, 56, 10352–10356 CrossRef CAS PubMed;
(b) Q. W. Zhang, D. Li, X. Li, P. B. White, J. Mecinovic, X. Ma, A. Hans, J. M. N. Roeland and H. Tian, J. Am. Chem. Soc., 2016, 138, 13541–13550 CrossRef CAS PubMed;
(c) Y. Xia, S. Chen and X. L. Ni, ACS Appl. Mater. Interfaces, 2018, 10, 13048–13052 CrossRef CAS PubMed;
(d) D. Li, F. Lu, J. Wang, W. Hu, X. M. Cao, X. Ma and H. Tian, J. Am. Chem. Soc., 2018, 140, 1916–1923 CrossRef CAS PubMed;
(e) H. T. Feng, X. Zheng, X. Gu, M. Chen, J. W. Lam, X. Huang and B. Z. Tang, Chem. Mater., 2018, 30, 1285–1290 CrossRef CAS.
-
(a) J. Wang, X. Yao, Y. Liu, H. Zhou, W. Chen, G. Sun, J. Sun, X. Ma and H. Tian, Adv. Opt. Mater., 2018, 6, 1800074 CrossRef;
(b) P. Bairi, B. Roy, P. Chakraborty and A. K. Nandi, ACS Appl. Mater. Interfaces, 2013, 5, 5478–5485 CrossRef CAS PubMed;
(c) Q. Zhao, Y. Chen, S. H. Li and Y. Liu, Chem. Commun., 2018, 54, 200–203 RSC;
(d) P. Pallavi, S. Bandyopadhyay, J. Louis, A. Deshmukh and A. Patra, Chem. Commun., 2017, 53, 1257–1260 RSC;
(e) V. K. Praveen, C. Ranjith and N. Armaroli, Angew. Chem., Int. Ed., 2014, 53, 365–368 CrossRef CAS PubMed.
-
(a) Z. He, W. Zhao, J. W. Lam, Q. Peng, H. Ma, G. Liang, Z. Shuai and B. Z. Tang, Nat. Commun., 2017, 8, 416 CrossRef PubMed;
(b) Z. Xie, C. Chen, S. Xu, J. Li, Y. Zhang, S. Liu, J. Xu and Z. Chi, Angew. Chem., Int. Ed., 2015, 54, 7181–7184 CrossRef CAS PubMed;
(c) D. Tu, P. Leong, S. Guo, H. Yan, C. Lu and Q. Zhao, Angew. Chem., Int. Ed., 2017, 129, 11528–11532 CrossRef;
(d) J. Wang, X. Gu, H. Ma, Q. Peng, X. Huang, X. Zheng, S. H. P. Sung, G. Shan, J. W. Y. Lam, Z. Shuai and B. Z. Tang, Nat. Commun., 2018, 9, 2963 CrossRef PubMed;
(e) D. Li, W. Hu, J. Wang, Q. Zhang, X. M. Cao, X. Ma and H. Tian, Chem. Sci., 2018, 9, 5709 RSC.
-
(a) X. Wang, Y. Xu, X. Ma and H. Tian, Dyes Pigm., 2014, 110, 2–27 CrossRef;
(b) Q. Y. Yang and J. M. Lehn, Angew. Chem., Int. Ed., 2014, 53, 4572–4577 CrossRef CAS PubMed.
-
(a) P. Chen, Q. Li, S. Grindy and N. Holten-Andersen, J. Am. Chem. Soc., 2015, 137, 11590–11593 CrossRef CAS PubMed;
(b) X. K. Liu, C. J. Zheng, M. F. Lo, J. Xiao, Z. Chen, C. L. Liu, C. S. Lee, M. K. Fung and X. H. Zhang, Chem. Mater., 2013, 25, 4454–4459 CrossRef CAS.
-
(a) D. Li, J. Wang and X. Ma, Adv. Opt. Mater., 2018, 1800273 CrossRef;
(b) T. Wang, P. Li and H. Li, ACS Appl. Mater. Interfaces, 2014, 6, 12915–12921 CrossRef CAS PubMed.
- Z. Zhang, D. Liu, D. Li, K. Huang, Y. Zhang, Z. Shi, R. Xie, M. Y. Han, Y. Wang and W. Yang, Chem. Mater., 2015, 27, 1405–1411 CrossRef CAS.
-
(a) L. A. Jones, J. Opt. Soc. Am., 1943, 33, 534–543 CrossRef;
(b) W. S. Stiles and J. M. Burch, Opt. Acta, 1995, 2, 168–181 CrossRef.
- H. Ma, M. Yang, C. Zhang, Y. Ma, Y. Qin, Z. Lei, L. Chang, L. Lei, T. Wang and Y. Yang, J. Mater. Chem. B, 2017, 5, 8525–8531 RSC.
- C. Allain, F. Schmidt, R. Lartia, G. Bordeau, C. Fiorini-Debuisschert, F. Charra, P. Tauc and M. P. Teulade-Fichou, ChemBioChem, 2007, 8, 424–433 CrossRef CAS PubMed.
- H. Ma, C. Qi, C. Cheng, Z. Yang, H. Cao, Z. Yang, J. Tong, X. Yao and Z. Lei, ACS Appl. Mater. Interfaces, 2016, 8, 8341–8348 CrossRef CAS PubMed.
- H. Ma, Y. Qin, Z. Yang, M. Yang, Y. Ma, P. Yin, Y. Yang, T. Wang, Z. Lei and X. Yao, ACS Appl. Mater. Interfaces, 2018, 10, 20064–20072 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00599k |
|
| This journal is © the Partner Organisations 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*