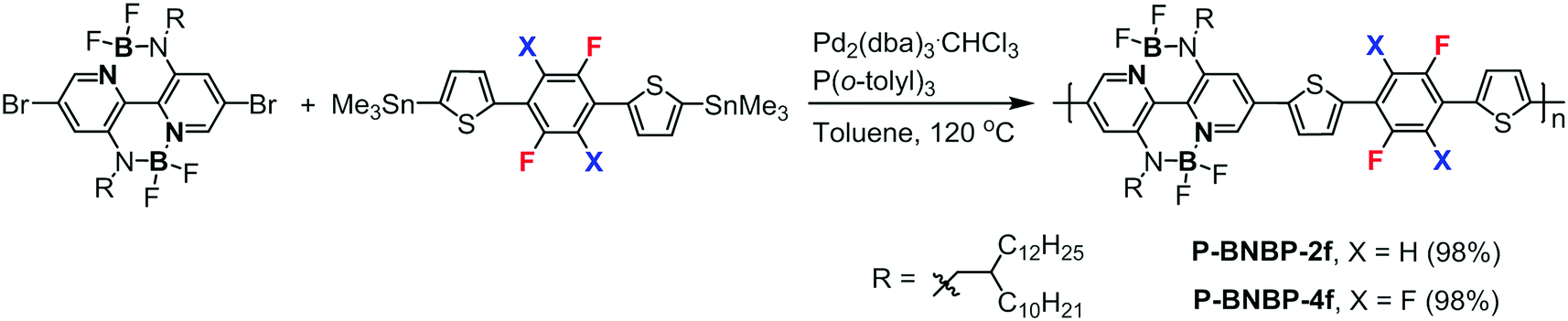DOI:
10.1039/C8QM00492G
(Research Article)
Mater. Chem. Front., 2019,
3, 70-77
Double B←N bridged bipyridine-containing polymer acceptors with enhanced electron mobility for all-polymer solar cells†
Received
28th September 2018
, Accepted 23rd October 2018
First published on 25th October 2018
Abstract
With the aim of developing polymer electron acceptors with high electron mobilities (μe) for all-polymer solar cells (all-PSCs), we synthesized two novel polymer acceptors (P-BNBP-2f and P-BNBP-4f) containing alternate double B←N bridged bipyridine (BNBP) and 2,2′-(2,5-difluoro-1,4-phenylene)dithiophene/2,2′-(perfluoro-1,4-phenylene)dithiophene with extended conjugated structures. In comparison to P-BNBP-4f, P-BNBP-2f exhibits a small π–π stacking distance of 3.60 Å, leading to a sufficient electron mobility of 5.40 × 10−4 cm2 V−1 s−1 (measured by the space-charge-limited current method). This μe value is among the highest values of the conventional polymer acceptors, and is close to the hole mobilities (μh) of high-efficiency polymer donors. Moreover, P-BNBP-2f possesses high-lying LUMO/HOMO energy levels of −3.42/−5.81 eV, which match well with that of the reported excellent polymer donors of 2D-conjugated bithienyl-benzodithiophene-alt-fluorobenzotriazole (J61) and thienyl-substituted BDT with alkoxycarbonyl-substituted thieno[3,4-b]thiophene (PBDTTT-E-T). In addition, the blend films based on P-BNBP-2f exhibit intermixed morphologies, which are beneficial for efficient excition dissociation. As a result, all-PSCs with P-BNBP-2f as an electron acceptor work very well and exhibit a power conversion efficiency of 5.46%. These results demonstrate that high electron mobility of a polymer electron acceptor is very important to produce efficient all-PSCs.
Introduction
All-polymer solar cells (all-PSCs), comprising a binary blend of conjugated polymer electron donor and polymer electron acceptor as the photoactive layer, have recently attracted great attention. Compared with fullerene-based polymer solar cells, all-PSCs provide many advantages including improved light absorption, tunable energy levels, and enhanced thermal and mechanical stability.1–4 Great efforts have been made to improve the power conversion efficiency (PCE) of all-PSCs. One efficient approach is to develop new polymer donors that show complementary absorption spectra and matched energy levels with that of conventional polymer acceptors. For example, Huang et al. have developed a series of wide-bandgap polymer donors containing electron-deficient 5H-pyrrolo[3,4-f]benzotriazole-5,7(6H)-dione units that have exhibited a champion PCE of over 10%.5 Recently, the design of new polymer acceptors has promoted the development of all-PSCs. However, excellent polymer acceptors are still very limited, and are mostly based on the electron-deficient naphthalene diimide (NDI) unit and perylenediimide (PDI) unit.6–14 For example, Zhao et al. reported a family of polymer acceptors based on fused PDI units with a PCE of over 8%.15 Beaujuge et al. demonstrated the use of 3,4-difluorothiophene to design polymer acceptors for efficient all-PSCs.16,17 Guo et al. developed bithiophene imide as a new electron-deficient building block for polymer acceptors.18 Li et al. reported a novel polymer acceptor containing a dicyanovinylindan-1-one unit that exhibited a PCE of over 9%.19
In the design of polymer acceptors, attention needs to be paid to their key properties: (1) low-lying lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) energy levels (ELUMO/EHOMO) for efficient charge separation; (2) wide absorption spectra for strong sunlight harvesting; (3) high electron mobilities for efficient electron transport from the polymer acceptor to the cathode to improve the charge extraction.20–23 Although intensive studies have been focused on optimizing these properties, the electron mobilities (μe) of polymer acceptors are usually lower than the hole mobilities (μh) of polymer donors by one order of magnitude.24,25 The low μe seriously influences the charge recombination and device performance of all-PSCs. Thus, it is of great importance to develop novel polymer acceptors with good electron mobilities.
A novel strategy to develop polymer acceptors using a boron–nitrogen coordination bond (B←N) has been proposed.26,27 A family of polymer acceptors were developed based on double B←N bridged bipyridine (BNBP) as the electron-deficient building block.28–34 Their ELUMO has been precisely tuned in a wide range because of the delocalized LUMOs over the polymer backbones. Moreover, their absorption spectra have been broadened by incorporating low-bandgap comonomers. In this manuscript, we aim to develop polymer acceptors containing BNBP units with good electron mobilities for all-PSCs. It is well-known that fluorination of the polymer backbones could improve the charge carrier mobility by enhancing main chain planarity and impact the molecular energy levels for charge carrier generation and recombination, as well as optimize the blend morphology.35 Therefore, with the introduction of 2,2′-(2,5-difluoro-1,4-phenylene)dithiophene (2fPT) and 2,2′-(perfluoro-1,4-phenylene)dithiophene (4fPT) units with extended conjugated structures, two novel BNBP-based polymer acceptors (P-BNBP-2f and P-BNBP-4f) were synthesized. In comparison to P-BNBP-4f, P-BNBP-2f exhibits a small π–π stacking distance (dπ–π) of 3.60 Å and a sufficient electron mobility of 5.40 × 10−4 cm2 V−1 s−1. This μe value is among the highest values of the conventional polymer acceptors.36–41 The resulting all-PSCs with P-BNBP-2f as an electron acceptor work very well and exhibit a power conversion efficiency (PCE) of 5.46% with a high open-circuit voltage (VOC) of 1.27 V. We comprehensively investigate the molecular configurations, polymer packing structures and electron transporting properties, as well as the electronic properties and all-PSC device applications of these two polymers.
Results and discussion
Synthesis and characterization
Scheme 1 shows the synthetic routes of P-BNBP-2f and P-BNBP-4f. The monomers were prepared following the previous ref. 33, 42 and 43. The two polymers were synthesized via Stille-polymerization with Pd2(dba)3·CHCl3/P(o-tolyl)3 as a catalyst/ligand. Their chemical structures were confirmed by 1H NMR and elemental analysis. According to gel permeation chromotography (GPC) at 150 °C with 1,2,4-trichlorobenzene as the eluent, the number-average molecular weight (Mn) and polydispersity (PDI) are 43.4 kDa and 1.88 for P-BNBP-2f and 40.8 kDa and 1.95 for P-BNBP-4f, respectively. According to thermogravimetric analysis (TGA) (Fig. S1, ESI†), P-BNBP-2f and P-BNBP-4f both show good thermal stability with thermal decomposition temperatures (Td, 5% weight loss) of over 350 °C.
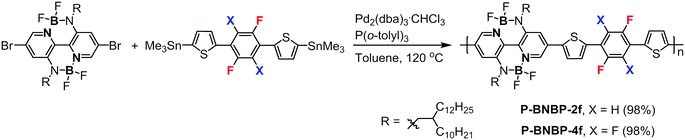 |
| | Scheme 1 Synthesis of P-BNBP-2f and P-BNBP-4f. | |
Theoretical calculations
To elucidate the molecular configurations and electronic structures of the polymers, density functional theory (DFT) calculations at the B3LYP/6-31G(d,p) level of theory were performed on the model compounds containing four repeating units with long alkyl chains replaced by methyl groups (Fig. S2, ESI†). Both of them exhibit pseudo-straight polymer backbone configurations. The torsional angles between the BNBP and 2fPT/4fPT units in P-BNBP-2f and P-BNBP-4f are around 22°, which enables efficient conjugation in the polymers. In addition, the average torsional angles between the thiophene and phenylene rings in the 2fPT and 4fPT units are calculated to be about 8.5° and 3.0°, respectively. The relatively smaller torsional angle in P-BNBP-4f is due to the F–H and F–S interactions that doubly “lock” the planar conformation,44,45 resulting in its higher rigidity than P-BNBP-2f. Their different configurations would impact on the molecular packing structures in thin films. Moreover, in comparison to the 3,3′-difluoro-2,2′-bithiophene (fBT) unit in our reported polymer,29 the longer 2fPT and 4fPT units are expected to reduce the density of the fluorine atoms on the boron atom of the polymers, thus leading to decreased steric hindrance of these fluorine atoms and smaller π–π stacking distances of the polymers. In addition, while the LUMOs of the two polymers are delocalized over the conjugated backbones, their HOMOs are mainly localized on the BNBP building blocks.
Molecular stacking and charge transporting properties
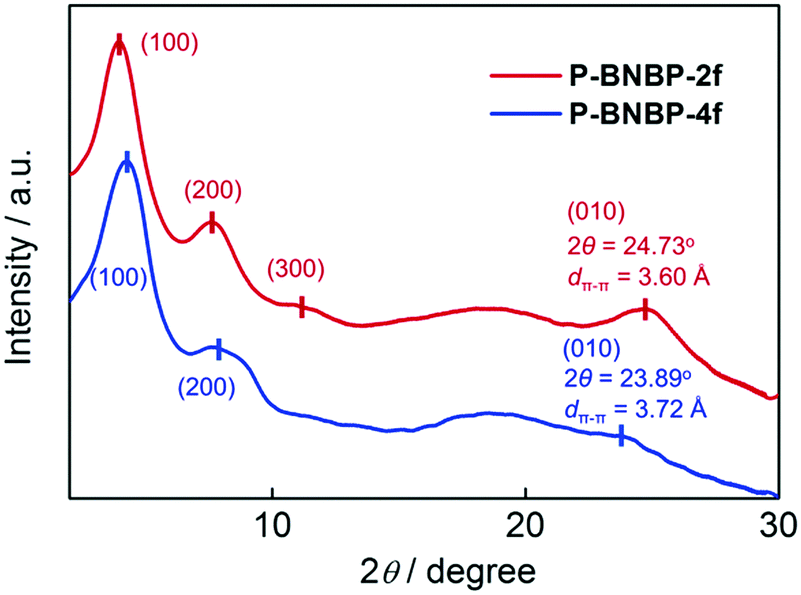
Grazing incident X-ray diffraction (GI-XRD) measurements were performed on the drop-cast films of the two polymers. As shown in Fig. 1, the P-BNBP-2f film gives a (100) diffraction peak at 2θ = 3.95° along with the (200) and (300) diffraction peaks, while the P-BNBP-4f film exhibits a (100) diffraction peak at 2θ = 4.20° along with the (200) diffraction peak. The three-order diffraction signals of P-BNBP-2f indicate its better crystallinity. Moreover, the P-BNBP-2f film possesses an intense (010) diffraction peak at 24.73°, corresponding to a π–π stacking distance of 3.60 Å of the polymer backbones. In contrast, P-BNBP-4f exhibits a weak diffraction peak at 23.89° that is corresponding to a dπ–π of 3.72 Å. The smaller dπ–π of P-BNBP-2f is probably due to its less rigid conjugated backbone, which is very beneficial for polymer stacking.46–48 This small dπ–π and dense molecular stacking will facilitate electron hopping between polymer backbones and lead to high electron mobility.
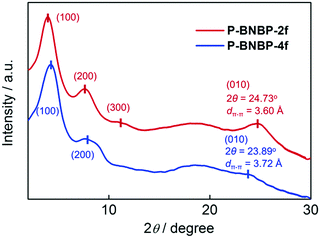 |
| | Fig. 1 GI-XRD patterns of the drop-cast films of P-BNBP-2f and P-BNBP-4f. | |
The electron mobilities of the two polymers were estimated using the space-charge-limited current (SCLC) method with the current density/voltage (J–V) curves of the electron-only devices (Fig. S3, ESI†). The electron mobility is 5.40 × 10−4 cm2 V−1 s−1 for P-BNBP-2f and 1.42 × 10−4 cm2 V−1 s−1 for P-BNBP-4f. The electron mobility of P-BNBP-2f is higher than that (∼1 × 10−4 cm2 V−1 s−1) of our previously developed BNBP-based polymer acceptors.28,33 It is also among the highest values of the conventional polymer acceptors, and is comparable to the hole mobilities of high-efficiency polymer donors.24,38 Thus, the high electron mobility of P-BNBP-2f will be desirable for its application as a polymer acceptor in all-PSCs.
Photophysical properties
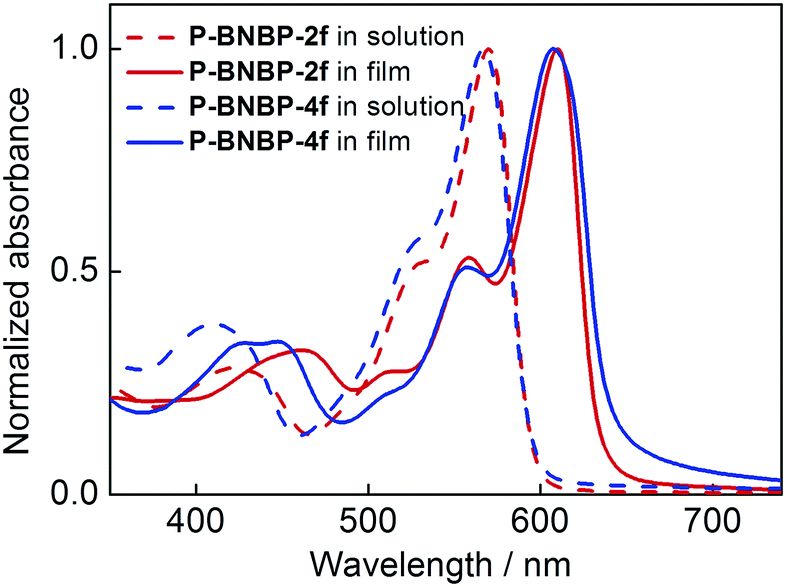
UV/vis absorption spectra of the two polymers in o-dichlorobenzene (o-DCB) solutions at 90 °C and 25 °C and in thin films were measured. The corresponding data are summarized in Table 1. As shown in Fig. 2, they exhibit almost identical absorption spectra at 90 °C with the maximum absorption peak (λmax) of 570 nm for P-BNBP-2f and 567 nm for P-BNBP-4f. During cooling to 25 °C (Fig. S4a, ESI†), both of them display red-shifted absorption bands with the λmax of 614 nm for P-BNBP-2f and 600 nm for P-BNBP-4f. This is in accordance with their aforementioned pseudo-straight polymer backbone configurations, which can promote their aggregations in the cool solutions. Moreover, the absorption spectra of P-BNBP-2f from in solution (25 °C) to in film are blue-shifted by 4 nm, which is probably because of its relatively strong pre-aggregation in cool solution. The absorption coefficients of P-BNBP-2f and P-BNBP-4f in films are 1.38 × 105 cm−1 and 1.26 × 105 cm−1, respectively, along with similar optical band gaps (Eg) of ca. 1.95 eV.
Table 1 Photophysical and electrochemical properties, π–π stacking distances and electron mobilities of P-BNBP-2f and P-BNBP-4f
| Polymer |
λ
max
(nm) |
ε
max
(cm−1) |
λ
max
(nm) |
E
optg
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (eV)
(eV) |
E
oxonset
(V) |
E
redonset
(V) |
E
HOMO
(eV) |
E
LUMO
(eV) |
d
π–π (Å) |
μ
e (cm2 V−1 s−1) |
|
Measured in o-DCB solution at 90 °C.
Measured in thin film.
Onset potential vs. Fc/Fc+.
Calculated using the equation of ELUMO/EHOMO = −(4.80 + Eredonset/Eoxonset) eV.
|
|
P-BNBP-2f
|
570 |
1.38 × 105 |
610 |
1.96 |
+1.01 |
−1.38 |
−5.81 |
−3.42 |
3.60 |
(5.40 ± 1.1) × 10−4 |
|
P-BNBP-4f
|
567 |
1.26 × 105 |
607 |
1.93 |
+1.16 |
−1.34 |
−5.96 |
−3.46 |
3.72 |
(1.42 ± 0.9) × 10−4 |
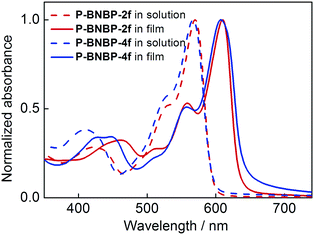 |
| | Fig. 2 UV/vis absorption spectra of P-BNBP-2f and P-BNBP-4f in solutions at 90 °C and in thin films. | |
Electrochemical properties
Cyclic voltammetry (CV) was employed to investigate their electrochemical properties. The measurements were carried out using their thin films. As shown in Fig. S4b (ESI†), while P-BNBP-2f exhibits the reduction and oxidation onset potentials (vs. Fc/Fc+) of Eredonset = −1.38 V and Eoxonset = +1.01 V, P-BNBP-4f shows a similar Eredonset of −1.34 V but more positive Eoxonset of +1.16 V. According to the equations of ELUMO/EHOMO = −(4.80 + Eredonset/Eoxonset) eV, their ELUMO/EHOMO are calculated to be −3.42 eV/−5.81 eV for P-BNBP-2f and −3.46 eV/−5.96 eV for P-BNBP-4f, respectively. It is known that the difference between the ELUMO of the electron acceptor and the EHOMO of the electron donor is related to VOC in PSCs. The ELUMO values of P-BNBP-2f and P-BNBP-4f are higher than that of the reported BNBP-based polymer acceptors by ca. 0.2 eV and higher than those of the reported excellent polymer acceptors by ca. 0.4 eV,49–51 which is expected to improve the VOC of all-PSCs.
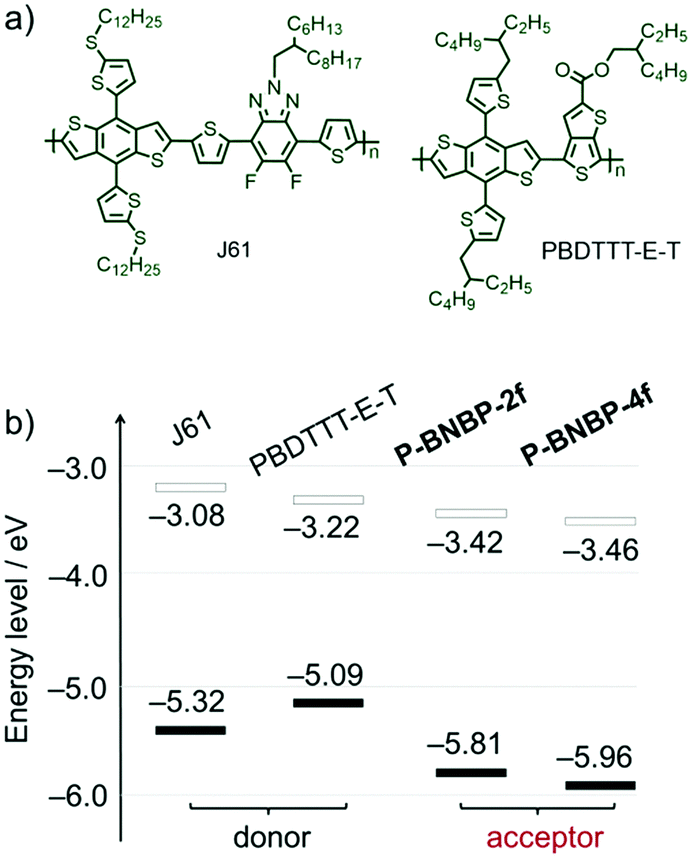
In addition, the LUMO/HOMO energy level alignments of our polymer acceptors along with two typical polymer donors 2D-conjugated bithienyl-benzodithiophene-alt-fluorobenzotriazole (J61) and thienyl-substituted BDT with alkoxycarbonyl-substituted thieno[3,4-b]thiophene (PBDTTT-E-T)52,53 are provided in Fig. 3b. In comparison to J61 and PBDTTT-E-T, P-BNBP-2f and P-BNBP-4f both possess obviously lower ELUMO/EHOMO. This means that the photo-induced charge separation would be very efficient between the polymer donor and polymer acceptor because of the sufficient driving forces.
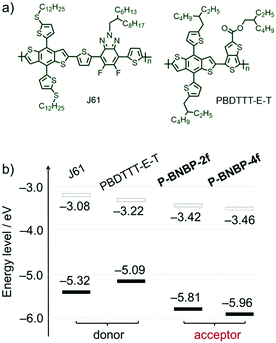 |
| | Fig. 3 (a) Chemical structures of polymer donors J61 and PBDTTT-E-T. (b) The LUMO/HOMO energy level alignments of J61, PBDTTT-E-T, P-BNBP-2f and P-BNBP-4f. | |
All-polymer solar cells
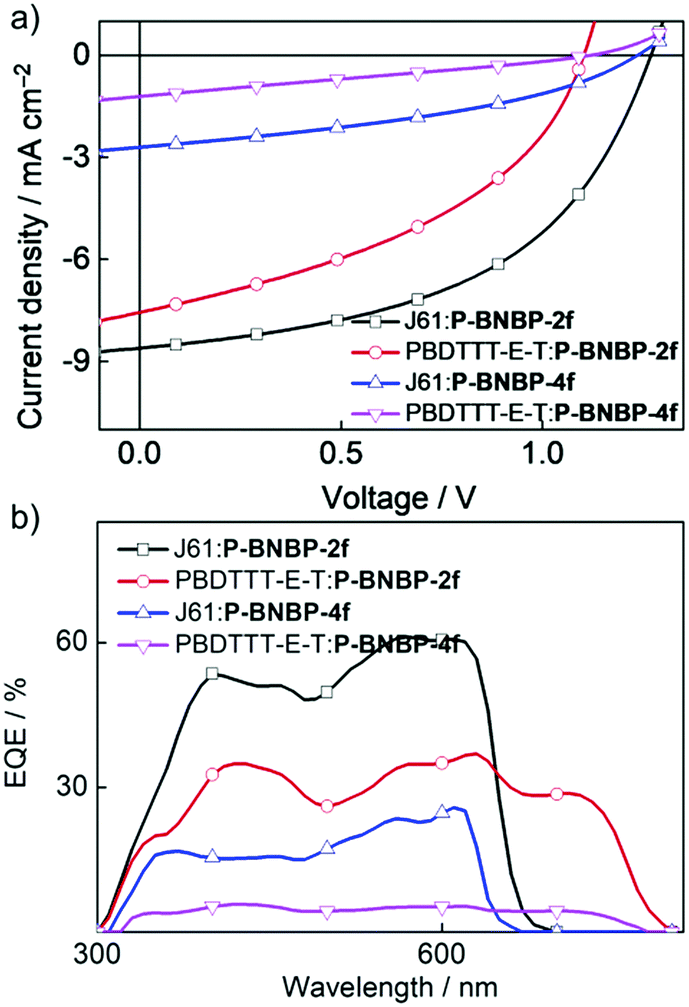
To investigate the photovoltaic properties of P-BNBP-2f and P-BNBP-4f as polymer acceptors, we fabricated all-PSC devices with the device configuration of ITO/PEDOT:PSS/polymer donor:polymer acceptor/Ca/Al. The active layer was spin-coated from the blend of the polymer acceptor and polymer donor with a total blend polymer concentration of 7 mg mL−1 in chloroform solution. The spin-coating speed was 1600 rpm and the active layer thickness was ca. 80 nm. Fig. 4 shows the current density–voltage (J–V) curves under AM 1.5G illumination (100 mW cm−2) and the external quantum efficiency (EQE) spectra of the optimized all-PSC devices. The corresponding device data are summarized in Table 2. The J61:P-BNBP-4f device exhibits a PCE of 1.30% with a VOC of 1.24 V, a short-circuit current (JSC) of 2.71 mA cm−2 and a fill factor (FF) of 0.39. The J61:P-BNBP-2f device shows a VOC of 1.27 V, a JSC of 8.61 mA cm−2 and a FF of 0.50, corresponding to an average PCE of 5.46%. The VOC values of the two devices are among the highest values of the reported all-PSCs, and are also comparable with the polymer donor/small molecular acceptor type PSCs.54–57 They are ascribed to the large offsets between the EHOMO of the polymer donors and the ELUMO of the polymer acceptors. On the other hand, the PCE of the J61:P-BNBP-2f device is much higher than that of the J61:P-BNBP-4f device. This is attributed to the enhanced JSC and FF values, which are probably because of the high electron mobility of P-BNBP-2f and the optimal morphology of the active layer. Similarly, while the PBDTTT-E-T:P-BNBP-4f device only shows a photovoltaic response, the PBDTTT-E-T:P-BNBP-2f device displays a PCE of 3.51%. The device statistics are provided in Table S9 in the ESI.†
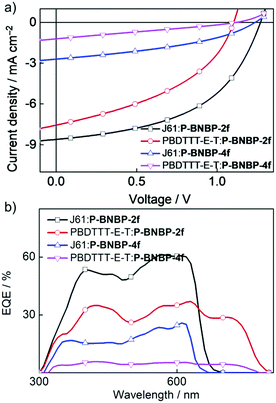 |
| | Fig. 4 (a) J–V curves and (b) EQE spectra of the four all-PSC devices. | |
Table 2 Summary of the all-PSC device performance
| Donor:acceptor |
V
OC (V) |
J
SC (mA cm−2) |
FF |
PCEa (%) |
EQE |
μ
e (cm2 V−1 s−1) |
μ
h (cm2 V−1 s−1) |
Thickness (nm) |
α
|
|
The PCEs were the average values based on eight devices.
|
| J61:P-BNBP-2f |
1.27 |
8.61 |
0.50 |
5.46 |
0.61 |
(9.72 ± 0.5) × 10−4 |
(2.36 ± 0.8) × 10−4 |
84 |
0.96 |
| PBDTTT-E-T:P-BNBP-2f |
1.10 |
7.56 |
0.42 |
3.51 |
0.37 |
(1.39 ± 0.4) × 10−4 |
(4.92 ± 0.5) × 10−4 |
75 |
0.96 |
| J61:P-BNBP-4f |
1.24 |
2.71 |
0.39 |
1.30 |
0.26 |
(2.39 ± 1.0) × 10−4 |
(4.06 ± 1.3) × 10−5 |
86 |
0.92 |
| PBDTTT-E-T:P-BNBP-4f |
1.11 |
1.18 |
0.28 |
0.36 |
0.06 |
(5.36 ± 0.7) × 10−5 |
(2.16 ± 0.6) × 10−4 |
72 |
0.89 |
The maximum EQE value is 0.61 at 567 nm for the J61:P-BNBP-2f device and 0.37 at 627 nm for the PBDTTT-E-T:P-BNBP-2f device, which are significantly higher than that of the corresponding devices based on P-BNBP-4f. The JSC values calculated from the integration of the EQE spectra with the AM1.5G spectrum agree well with the JSC values obtained from the J–V scans within an error of 5%.
Charge transporting properties of the active layers
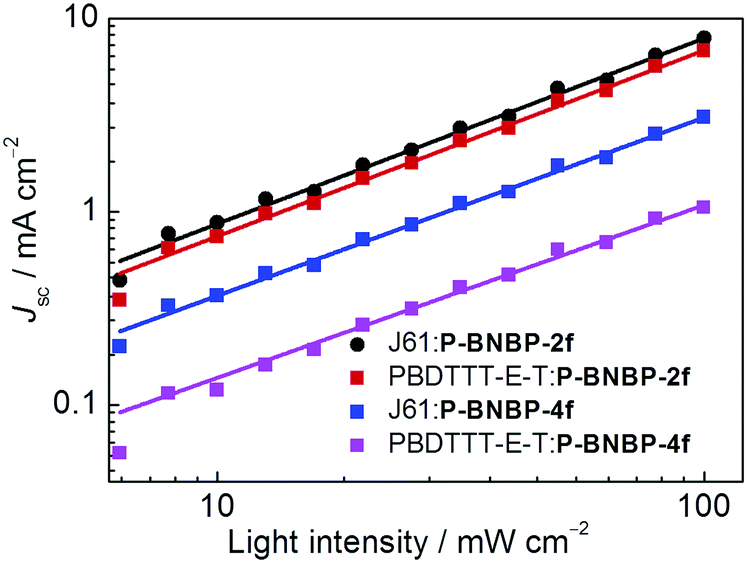
We investigated charge transport properties using the SCLC method with electron-only (ITO/PEIE/active layer/Ca/Al) and hole-only devices (ITO/PEDOT:PSS/active layer/MoO3/Al).58 The results are listed in Table 2. The hole mobility and electron mobility are 2.36 × 10−4 cm2 V−1 s−1 and 9.72 × 10−4 cm2 V−1 s−1 for the J61:P-BNBP-2f blend film, and 4.06 × 10−5 cm2 V−1 s−1 and 2.39 × 10−4 cm2 V−1 s−1 for the J61:P-BNBP-4f blend film, respectively. The improved charge mobilities of the J61:P-BNBP-2f blend film contribute to the enhanced JSC and FF values of this all-PSC device. Similar charge transport properties are also observed in the all-PSC devices using PBDTTT-E-T as a polymer donor. The charge recombination mechanism in these all-PSCs was studied by measuring the light-intensity dependent JSC characteristics.59–62 The correlation between JSC and light intensity follows a power-law dependence (JSC ∝ Plightα, where Plight is light intensity and α is the calculated power-law exponent). The bimolecular recombination can be negligible under short circuit conditions when α is unity. As shown in Fig. 5, the fitted α value is 0.96 for the J61:P-BNBP-2f blend and 0.92 for the J61:P-BNBP-4f blend, respectively. The α value of the J61:P-BNBP-2f blend is closer to unity, indicating the more significantly suppressed bimolecular charge recombination in this all-PSC device. The all-PSC devices using PBDTTT-E-T as a polymer donor show similar bimolecular charge recombination to that of the devices with J61, respectively. Therefore, the suppressed charge recombination benefits the better device performance of all-PSCs with P-BNBP-2f as an electron acceptor.
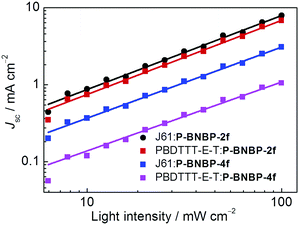 |
| | Fig. 5
J
SC
versus light intensity for the four all-PSC devices. | |
Blend morphology analysis
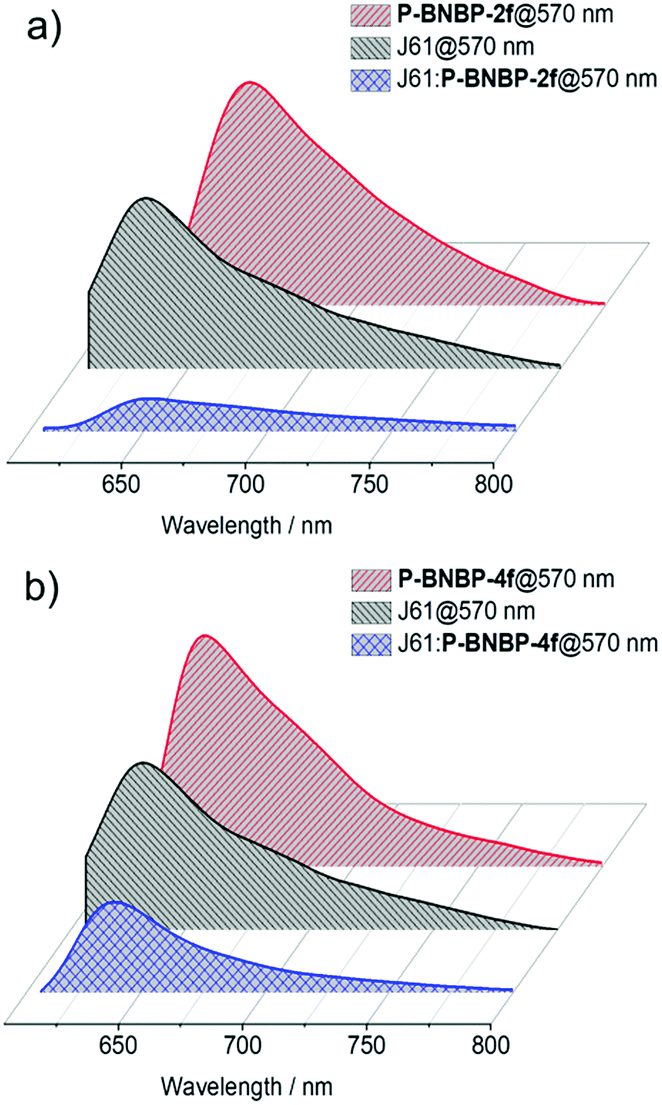
We then fully investigated the blend films based on a J61 polymer donor. The photoluminescence (PL) spectra of J61 and the polymer blends were measured to study the exciton dissociation and charge transfer in the blend films. As shown in Fig. 6, the PL emissions of the pristine P-BNBP-2f, P-BNBP-4f and J61 films are all in the range of 600–800 nm with the main peaks at 657, 640 and 633 nm (excited at 570 nm), respectively. The PL quenching efficiencies are 82% and 80% for P-BNBP-2f and J61 in the blend film, respectively. They are 74% and 43% for P-BNBP-4f and J61, respectively. Thus, the higher PL quenching efficiencies for the P-BNBP-2f-based blend film indicate that they have more intermixed morphologies, which are beneficial for exciton dissociations in the devices.
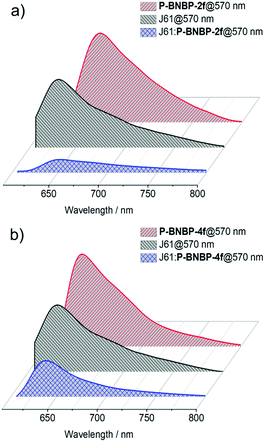 |
| | Fig. 6 Photoluminescence spectra of the polymer pristine films and the polymer blend films based on (a) P-BNBP-2f and J61 and (b) P-BNBP-4f and J61. | |
The morphologies of the J61:P-BNBP-2f and J61:P-BNBP-4f blend films were characterized by atomic force microscopy (AFM). Both of them exhibit fibrillar nanostructures in the AFM images. The surface of the J61:P-BNBP-2f blend shows a root-mean-square (RMS) of about 0.90 nm, which is slightly lower than that of the J61:P-BNBP-4f blend (1.02 nm) (Fig. S14, ESI†). The lower surface roughness of the J61:P-BNBP-2f blend is coincident with the intermixed morphology demonstrated in the PL quenching measurements. We then used GI-XRD and in-plane X-ray diffraction (IP-XRD) measurements to study polymer stacking modes in the spin-coated pristine and blend films. As shown in Fig. 7, the GI-XRD patterns display the (100) and (010) diffractions at 2θ = 3.91° and 24.40° for the pristine P-BNBP-2f film and the (100) diffraction at 2θ = 3.90° for the P-BNBP-4f film in the out-of-plane direction, respectively. It is suggested that the π–π stacking is strong for the spin-coated P-BNBP-2f film, and is absent for the spin-coated P-BNBP-4f film. More importantly, their molecular packing modes are partially maintained in the blend films. Both of P-BNBP-2f and J61 in the blend films exhibit strong π–π stacking in the out-of-plane direction, which leads to the good charge transport in the active layers. This is consistent with the high electron/hole mobilities of the J61:P-BNBP-2f active layer.
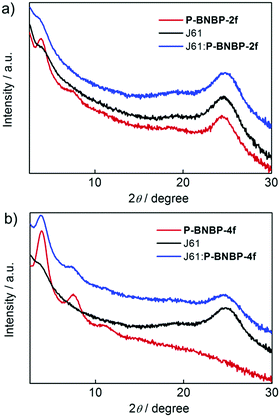 |
| | Fig. 7 GI-XRD patterns of the polymer pristine films and the polymer blend films based on (a) P-BNBP-2f and J61 as well as (b) P-BNBP-4f and J61. | |
Conclusions
In summary, we synthesized two novel polymer acceptors based on a BNBP unit for all-PSCs. In comparison to P-BNBP-4f, P-BNBP-2f has shown dense molecular stacking in the thin films and thus a good electron mobility. Using P-BNBP-2f as an electron acceptor, the all-PSC devices work very well and a PCE of 5.46% was obtained. The optimal device performance can be attributed to improved charge transport and more intermixed morphologies in the active layers. This study not only provides an excellent polymer acceptor material, but also demonstrates that high electron mobilities are very important for polymer acceptors to produce efficient all-PSCs.
Experimental section
Materials
Polymer donors PBDTTT-E-T and J61 were purchased from Solarmer Energy Inc. 5,5′-(Difluoro-1,4-phenylene)bis(thiophene-5,2-diyl)-bis(trimethylstannane) and 5,5′-(perfluoro-1,4-phenylene)bis(thiophene-5,2-diyl)-bis(trimethylstannane) monomer and dibromo-substituted BNBP monomer were prepared according to the reported literature procedures. Commercially available solvents and reagents were used without further purification unless stated otherwise.
Measurements and characterization
1H NMR spectra were measured using a Bruker AV-400 (400 MHz for 1H) spectrometer in C6H4Cl2 at 100 °C. Chemical shift is reported in δ ppm using C6H4Cl2 (7.29 ppm) for 1H NMR. Elemental analysis was performed on a VarioEL elemental analyzer. The molecular weight was determined by gel permeation chromatography (GPC) on a Waters 410 instrument. Thermal analysis was performed on a Perkin-Elmer 7 instrument under nitrogen flow at a heating rate of 10 °C min−1. UV/vis absorption spectra were measured using a Shimadzu UV-3600 spectrometer. Fluorescence spectra were measured using a Hitachi F-4500 spectrometer in spectral grade solvents. Cyclic voltammetry (CV) was performed on a CHI660a electrochemical workstation using Bu4NClO4 (0.1 M) in acetonitrile as electrolyte solution and ferrocene as an internal reference at a scan rate of 100 mV s−1. The CV cell consisted of a glassy carbon electrode, a Pt wire counter electrode, and a standard Ag/AgCl reference electrode. The polymer was cast on the working electrode for measurements. The redox potentials were calibrated with ferrocene as an internal standard. The molecular packing was investigated by grazing incidence X-ray diffraction (GI-XRD) and in-plane X-ray diffraction (IP-XRD). GI-XRD data were obtained on a Bruker D8 Discover reflector (Cu Kα, λ = 1.54056 Å) under 40 kV and 40 mA tube current. The IP-XRD data were obtained by using a RigakuSmartLab X-ray diffractometer with an X-ray generation power of 40 kV tube voltage and 30 mA tube current. Atomic force microscopy (AFM) measurement was performed with a SPA300HV.
Synthesis
Polymer P-BNBP-2f.
Starting materials of dibromo-substituted BNBP monomer (110.9 mg, 0.10 mmol), 5,5′-(difluoro-1,4-phenylene)bis(thiophene-5,2-diyl)-bis(trimethylstannane) (60.2 mg, 0.10 mmol), Pd2(dba)3·CHCl3 (2.1 mg, 0.002 mmol), and P(o-tolyl)3 (4.9 mg, 0.016 mmol) were placed in a two-necked flask under argon, and then dried toluene (30 mL) was added. After the mixture was stirred at 120 °C for 24 h, an end-capping reaction was carried out by adding 5,5′-bis(trimethylstannyl)-2,2′-bithiophene (5.93 mg, 0.012 mmol) and then bromobenzene (200.0 mg, 1.28 mmol). After cooling, the resulting organic phase was extracted with chloroform (120 mL) and washed with water. After the solvents were removed, the residue was dispersed in acetonitrile and the precipitate was collected. The polymer was purified by Soxhlet extraction with acetone, hexane, and THF to remove impurities and catalysts as well as the residual monomers. The left residual was finally dissolved in CHCl3, which was then dispersed in acetonitrile. The polymer P-BNBP-2f was collected and dried under vacuum overnight. Yield: 120 mg (98%). 1H NMR (400 MHz, C6D4Cl2, 100 °C): δ 8.74 (s, 1H), 7.99 (s, 1H), 7.72 (s, 1H), 7.62 (s, 1H), 3.98 (s, 2H), 2.31 (s, 1H), 1.81–1.36 (m, 33H), 1.08 (s, 6H). GPC (TCB, polystyrene standard, 150 °C): Mn = 43.4 kDa, PDI = 1.88. Anal. calcd for C72H108B2F6N4S2: C, 70.34; H, 8.85; B, 1.76; F, 9.27; N, 4.56; S, 5.22. Found: C, 69.97; H, 8.67; N, 4.68; S, 5.06.
Polymer P-BNBP-4f.
P-BNBP-4f was synthesized from dibromo-substituted BNBP monomer and 5,5′-(perfluoro-1,4-phenylene)bis(thiophene-5,2-diyl)-bis(trimethylstannane) following the synthesis procedure of P-BNBP-2f. Yield: 110 mg (98%). 1H NMR (400 MHz, C6D4Cl2, 100 °C): δ 8.76 (s, 1H), 8.02 (s, 1H), 7.93 (s, 1H), 7.67 (s, 1H), 3.98 (s, 2H), 2.31 (s, 1H), 1.81–1.36 (m, 42H), 1.08 (s, 6H). GPC (TCB, polystyrene standard, 150 °C): Mn = 40.8 kDa, PDI = 1.95. Anal. calcd for C72H106B2F8N4S2: C, 68.34; H, 8.44; B, 1.71; F, 12.01; N, 4.43; S, 5.07. Found: C, 68.28; H, 8.25; N, 4.29; S, 4.90.
All-PSC device fabrications and measurements
Indium tin oxide (ITO) glass substrates were cleaned by sequential ultrasonication in detergent, deionized water, acetone, and isopropyl alcohol, followed by being dried at 120 °C for 30 min and treated with UV-ozone for 30 min. Then PEDOT:PSS (CleviosPVP Al4083 from H. C. Starck Inc.) was spin-coated on the ITO glass substrates at 5000 rpm for 40 s to give a thickness of 35 nm, followed by baking at 120 °C for 30 min. The substrates were transferred to a nitrogen-filled glovebox. The blend solutions were spin-coated onto the PEDOT:PSS layer to produce the active layer (80–120 nm). The J61:P-BNBP-2f, J61:P-BNBP-4f, PBDTTT-E-T:P-BNBP-2f, and PBDTTT-E-T:P-BNBP-4f blends (w:w, 1:1) were prepared in CF solutions. The J61:P-BNBP-2f or P-BNBP-4f blends in different ratios (w:w, 1:2, 1:1 and 2:1) and with/without additives (CN) were also used. The obtained active layers were annealed at room temperature, 80 °C, 100 °C, and 120 °C for 10 min. Finally, the active layers were transferred to a vacuum chamber, and Ca (20 nm) and Al (100 nm) were sequentially deposited by thermal evaporation at the pressure of about 2 × 10−4 Pa. The active area of each device was 8 mm2. The current density–voltage (J–V) curves of the devices were measured using a computer-controlled Keithley 2400 source meter under AM 1.5G simulated solar light illumination (100 mW cm−2) provided by a XES-40S2-CE Class Solar Simulator (SAN-EI Electric Co., Ltd, Japan). The EQE spectrum was measured using a Solar Cell Spectral Response Measurement System QE-R3011 (Enlitech Co., Ltd). The light intensity at each wavelength was calibrated using a calibrated monosilicon diode.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by the National Natural Science Foundation of China (No. 21805148 and 21574129), the Youth Innovation Promotion Association of Chinese Academy of Sciences (No. 2017265), and the Open Project of State Key Laboratory of Supramolecular Structure and Materials (No. sklssm201803).
Notes and references
- T. Kim, J.-H. Kim, T. E. Kang, C. Lee, H. Kang, M. Shin, C. Wang, B. Ma, U. Jeong, T.-S. Kim and B. J. Kim, Nat. Commun., 2015, 6, 8547–8553 CrossRef CAS.
- A. Facchetti, Mater. Today, 2013, 16, 123–132 CrossRef CAS.
- C. R. McNeill, Energy Environ. Sci., 2012, 5, 5653–5667 RSC.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CrossRef CAS.
- B. Fan, L. Ying, P. Zhu, F. Pan, F. Liu, J. Chen, F. Huang and Y. Cao, Adv. Mater., 2017, 29, 1703906 CrossRef.
- M. Schubert, D. Dolfen, J. Frisch, S. Roland, R. Steyrleuthner, B. Stiller, Z. Chen, U. Scherf, N. Koch, A. Facchetti and D. Neher, Adv. Energy Mater., 2012, 2, 369–380 CrossRef CAS.
- M. Yuan, M. M. Durban, P. D. Kazarinoff, D. F. Zeigler, A. H. Rice, Y. Segawa and C. K. Luscombe, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4061–4069 CrossRef CAS.
- Y.-J. Hwang, B. A. Courtright, A. S. Ferreira, S. H. Tolbert and S. A. Jenekhe, Adv. Mater., 2015, 27, 4578–4584 CrossRef CAS.
- J. W. Jung, J. W. Jo, C. C. Chueh, F. Liu, W. H. Jo, T. P. Russell and A. K. Jen, Adv. Mater., 2015, 27, 3310–3317 CrossRef CAS.
- H.-H. Cho, T. Kim, K. Kim, C. Lee, F. S. Kim and B. J. Kim, J. Mater. Chem. A, 2017, 5, 5449–5459 RSC.
- X. Zhan, Z. Tan, B. Domercq, Z. An, X. Zhang, S. Barlow, Y. Li, D. Zhu, B. Kippelen and S. R. Marder, J. Am. Chem. Soc., 2007, 129, 7246–7247 CrossRef CAS.
- J. Yang, B. Xiao, K. Tajima, M. Nakano, K. Takimiya, A. Tang and E. Zhou, Macromolecules, 2017, 50, 3179–3185 CrossRef CAS.
- E. Zhou, J. Cong, K. Hashimoto and K. Tajima, Adv. Mater., 2013, 25, 6991–6996 CrossRef CAS.
- J. Yang, F. Chen, B. Xiao, S. Sun, X. Sun, K. Tajima, A. Tang and E. Zhou, Sol. RRL, 2018, 2, 1700230 CrossRef.
- Y. Guo, Y. Li, O. Awartani, H. Han, J. Zhao, H. Ade, H. Yan and D. Zhao, Adv. Mater., 2017, 29, 1700309 CrossRef.
- S. Liu, Y. Firdaus, S. Thomas, Z. Kan, F. Cruciani, S. Lopatin, J.-L. Bredas and P. M. Beaujuge, Angew. Chem., Int. Ed., 2018, 57, 531–535 CrossRef CAS PubMed.
- S. Liu, Z. Kan, S. Thomas, F. Cruciani, J.-L. Brédas and P. M. Beaujuge, Angew. Chem., Int. Ed., 2016, 55, 12996–13001 CrossRef CAS.
- Y. Wang, Z. Yan, H. Guo, M. A. Uddin, S. Ling, X. Zhou, H. Su, J. Dai, H. Y. Woo and X. Guo, Angew. Chem., Int. Ed., 2017, 56, 15304–15308 CrossRef CAS.
- Z. Zhang, Y. Yang, J. Yao, L. Xue, S. Chen, X. Li, W. Morrison, C. Yang and Y. Li, Angew. Chem., Int. Ed., 2017, 56, 13503–13507 CrossRef CAS PubMed.
- J. Choi, K.-H. Kim, H. Yu, C. Lee, H. Kang, I. Song, Y. Kim, J. H. Oh and B. J. Kim, Chem. Mater., 2015, 27, 5230–5237 CrossRef CAS.
- J. E. Anthony, A. Facchetti, M. Heeney, S. R. Marder and X. Zhan, Adv. Mater., 2010, 22, 3876–3892 CrossRef CAS.
- X. Zhao and X. Zhan, Chem. Soc. Rev., 2011, 40, 3728–3743 RSC.
- N. Zhou, H. Lin, S. J. Lou, X. Yu, P. Guo, E. F. Manley, S. Loser, P. Hartnett, H. Huang, M. R. Wasielewski, L. X. Chen, R. P. H. Chang, A. Facchetti and T. J. Marks, Adv. Energy Mater., 2014, 4, 1300785 CrossRef.
- L. Ye, S. Zhang, L. Huo, M. Zhang and J. Hou, Acc. Chem. Res., 2014, 47, 1595–1603 CrossRef CAS.
- H. Bin, L. Gao, Z. Zhang, Y. Yang, Y. Zhang, C. Zhang, S. Chen, L. Xue, C. Yang, M. Xiao and Y. Li, Nat. Commun., 2016, 7, 13651–13661 CrossRef CAS.
- C. Dou, Z. Ding, Z. Zhang, Z. Xie, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2015, 54, 3648–3652 CrossRef CAS.
- R. Zhao, C. Dou, Z. Xie, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2016, 55, 5313–5317 CrossRef CAS.
- C. Dou, X. Long, Z. Ding, Z. Xie, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2016, 55, 1436–1440 CrossRef CAS.
- X. Long, Z. Ding, C. Dou, J. Zhang, J. Liu and L. Wang, Adv. Mater., 2016, 28, 6504–6508 CrossRef CAS.
- Z. Ding, X. Long, C. Dou, J. Liu and L. Wang, Chem. Sci., 2016, 7, 6197–6202 RSC.
- R. Zhao, Z. Bi, C. Dou, W. Ma, Y. Han, J. Liu and L. Wang, Macromolecules, 2017, 50, 3171–3178 CrossRef CAS.
- R. Zhao, C. Dou, J. Liu and L. Wang, Chin. J. Polym. Sci., 2017, 35, 198–206 CrossRef CAS.
- X. Long, C. Dou, J. Liu and L. Wang, Macromolecules, 2017, 50, 8521–8528 CrossRef CAS.
- X. Long, N. Wang, Z. Ding, C. Dou, J. Liu and L. Wang, J. Mater. Chem. C, 2017, 4, 9961–9967 RSC.
- N. Leclerc, P. Chávez, O. Ibraikulov, T. Heiser and P. Lévêque, Polymers, 2016, 8, 11–37 CrossRef.
- L. Gao, Z. Zhang, L. Xue, J. Min, J. Zhang, Z. Wei and Y. Li, Adv. Mater., 2016, 28, 1884–1890 CrossRef CAS.
- Z. Li, X. Xu, W. Zhang, X. Meng, Z. Genene, W. Ma, W. Mammo, A. Yartsev, M. R. Andersson, R. A. J. Janssen and E. Wang, Energy Environ. Sci., 2017, 10, 2212–2221 RSC.
- Y. Yang, Z. Zhang, H. Bin, S. Chen, L. Gao, L. Xue, C. Yang and Y. Li, J. Am. Chem. Soc., 2016, 138, 15011–15018 CrossRef CAS.
- G. Yang, J. Liu, L.-K. Ma, S. Chen, J. Y. L. Lai, W. Ma and H. Yan, Mater. Chem. Front., 2018, 2, 1360–1365 RSC.
- T. Li, H. Zhang, Z. Xiao, J. J. Rech, H. Niu, W. You and L. Ding, Mater. Chem. Front., 2018, 2, 700–703 RSC.
- K. Kawashima, Y. Tamai, H. Ohkita, I. Osaka and K. Takimiya, Nat. Commun., 2015, 6, 10085–10093 CrossRef CAS.
- P. Sonar, J. Chang, Z. Shi, J. Wu and J. Li, J. Mater. Chem. C, 2015, 3, 2080–2085 RSC.
- J. H. Park, E. H. Jung, J. W. Jung and W. H. Jo, Adv. Mater., 2013, 25, 2583–2588 CrossRef CAS.
- Y. Wang, A. T.-R. Tan, T. Mori and T. Michinobu, J. Mater. Chem. C, 2018, 6, 3593–3603 RSC.
- M. Wang, M. J. Ford, A. T. Lill, H. Phan, T.-Q. Nguyen and G. C. Bazan, Adv. Mater., 2017, 29, 1603830 CrossRef.
- Z. Fei, P. Boufflet, S. Wood, J. Wade, J. Moriarty, E. Gann, E. L. Ratcliff, C. R. McNeil, H. Sirringhaus, J.-S. Kim and M. Heeney, J. Am. Chem. Soc., 2015, 137, 6866–6879 CrossRef CAS.
- M. Wang, M. J. Ford, C. Zhou, M. Seifrid, T.-Q. Nguyen and G. C. Bazan, J. Am. Chem. Soc., 2017, 139, 17624–17631 CrossRef CAS.
- G. Conboy, H. J. Spencer, E. Angioni, A. L. Kanibolotsky, N. J. Findlay, S. J. Coles, C. Wilson, M. B. Pitak, C. Risko, V. Coropceanu, J.-L. Brédas and P. J. Skabara, Mater. Horiz., 2016, 3, 333–339 RSC.
- G. Wang, N. D. Eastham, T. J. Aldrich, B. Ma, E. F. Manley, Z. Chen, L. X. Chen, M. O. de la Cruz, R. P. H. Chang, F. S. Melkonyan, A. Facchetti and T. J. Marks, Adv. Energy Mater., 2018, 1702173 CrossRef.
- J. Yuan, M. J. Ford, Y. Xu, Y. Zhang, G. C. Bazan and W. Ma, Adv. Energy Mater., 2018, 1703291 CrossRef.
- J. Jung, W. Lee, C. Lee, H. Ahn and B. J. Kim, Adv. Energy Mater., 2016, 6, 1600504 CrossRef.
- H. Bin, Z. Zhang, L. Gao, S. Chen, L. Zhong, L. Xue, C. Yang and Y. Li, J. Am. Chem. Soc., 2016, 138, 4657–4664 CrossRef CAS.
- L. Huo, S. Zhang, X. Guo, F. Xu, Y. Li and J. Hou, Angew. Chem., Int. Ed., 2011, 50, 9697–9702 CrossRef CAS PubMed.
- Y. Fu, B. Wang, J. Qu, Y. Wu, W. Ma, Y. Geng, Y. Han and Z. Xie, Adv. Funct. Mater., 2016, 26, 5922–5929 CrossRef CAS.
- Z. Ding, X. Long, B. Meng, K. Bai, C. Dou, J. Liu and L. Wang, Nano Energy, 2017, 32, 216–224 CrossRef CAS.
- A. Tang, B. Xiao, Y. Wang, F. Gao, K. Tajima, H. Bin, Z. Zhang, Y. Li, Z. Wei and E. Zhou, Adv. Funct. Mater., 2018, 28, 1704507 CrossRef.
- B. Xiao, A. Tang, J. Yang, Z. Wei and E. Zhou, ACS Macro Lett., 2017, 6, 410–414 CrossRef CAS.
- V. Mihailetchi, J. Wildeman and P. Blom, Phys. Rev. Lett., 2005, 94, 126602 CrossRef CAS.
- L. J. A. Koster, V. D. Mihailetchi, H. Xie and P. W. M. Blom, Appl. Phys. Lett., 2005, 87, 203502 CrossRef.
- L. Lu, W. Chen, T. Xu and L. Yu, Nat. Commun., 2015, 6, 7327–7333 CrossRef CAS.
- J.-L. Wu, F.-C. Chen, Y.-S. Hsiao, F.-C. Chien, P. Chen, C.-H. Kuo, M. H. Huang and C.-S. Hsu, ACS Nano, 2011, 5, 959–967 CrossRef CAS.
- S. R. Cowan, A. Roy and A. J. Heeger, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 245207 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization and thermal properties of the polymers, as well as all-PSC device performance and charge-transporting properties. See DOI: 10.1039/c8qm00492g |
|
| This journal is © the Partner Organisations 2019 |
Click here to see how this site uses Cookies. View our privacy policy here.