
Combination of noncovalent conformational locks and side chain engineering to tune the crystallinity of nonfullerene acceptors for high-performance P3HT based organic solar cells†
Pan
Ye‡
a,
Yusheng
Chen‡
a,
Jianfei
Wu‡
ab,
Xiaoxi
Wu
a,
Yunxiao
Xu
ac,
Zijie
Li
a,
Shikai
Hong
a,
Ming
Sun
*d,
Aidong
Peng
*a and
Hui
Huang
 *a
*a
aCollege of Materials Science and Opto-Electronic Technology & Key Laboratory of Vacuum Physic, University of Chinese Academy of Sciences, Beijing, 100049, P. R. China. E-mail: huihuang@ucas.ac.cn
bBeijing Key Laboratory of Function Materials for Molecular & Structure Construction School of Materials Science and Engineering University of Science and Technology Beijing, Beijing, 100083, P. R. China
cKey Laboratory of Flexible Electronics (KLOFE) & Institute of Advanced Materials (IAM), Jiangsu National Synergetic Innovation Center for Advanced Materials (SICAM), Nanjing Tech University, 30 South Puzhu Road, Nanjing, 211816, Jiangsu, P. R. China
dSchool of Electronic and Optical Engineering, Nanjing University of Science and Technology, Nanjing, Jiangsu 210094, P. R. China
First published on 15th October 2018
Abstract
P3HT-based organic solar cells (OSCs) have great advantages for commercialization including straightforward and scalable synthesis and well-developed roll-to-roll manufacturing technology. However, it is difficult to control the morphology of P3HT:acceptor blend films due to their highly crystalline characteristics. In this work, we designed and synthesized two thiazole (Tz) containing small molecular acceptors with an A–π–D–π–A type structure for the P3HT donor material. Both small molecules exhibit a good planar configuration due to incorporation of S⋯N noncovalent conformational locks. Upon changing the side chains, the interchain π–π stacking and the crystallinity of the small molecules were fine-tuned. Interestingly, P-IDTzR with bulky side chains exhibits suitable crystallinity, which matches well with P3HT. As a result, the P3HT:P-IDTzR blend films demonstrate optimal morphology, leading to a larger short circuit current (JSC), an enhanced fill factor (FF), and thus a larger power conversion efficiency (5.01%). This contribution provides important guidance in designing nonfullerene acceptors for high-performance P3HT based OSCs.
Introduction
Organic solar cells (OSCs) have been widely investigated due to their advantages of ease of processing, flexibility, light weight and low cost for scale-up production.1–8 Recently, the power conversion efficiencies (PCEs) of solution-processed bulk heterojunction (BHJ) OSCs have moved beyond 13%, which is driven by high-performance donor and non-fullerene acceptor materials.9–11 However, the most successful donor materials,12–16 such as poly[4-(5-(4,8-bis(3-butylnonyl)benzo[1,2-b:4,5-b′]dithiophen-2-yl)thiophen-2-yl)-2-(2-butyloctyl)-5,6-difluoro-7-(thiophen-2-yl)-2H-benzo[d][1,2,3]triazole] (PBnDT-FTAZ)14 and poly[2,6-(4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)benzo[1,2-b:4,5-b′]dithiophene)-co-(1,3-di(5-thiophene-2-yl)-5,7-bis(2-ethylhexyl)-benzo[1,2-c:4,5-c′]dithiophene-4,8-dione)] (PBDB-T),15 usually presented some intrinsic drawbacks, including complicated synthesis, difficulty in scaling up for large-scale fabrication, and low device reproducibility, which hindered the commercialization of OSC technology.17–19 On the other hand, poly(3-hexylthiophene) (P3HT), as one kind of cheap material with a regular structure, has significant advantages for industrialization due to its easy synthesis methods, scalable quantity, well-developed manufacturing technologies, and good charge transport properties.20–22 Thus, P3HT-based OSCs have attracted considerable attention in the past several years, showing a promising future in commercialization.23,24Though P3HT-based OSCs developed fast,25–31 only a few examples afforded efficiencies over 5%,25,26,32–37 which may be ascribed to several reasons. First, P3HT possesses a large bandgap (∼1.9 eV), which exhibits negligible absorption in the wavelength range of over 650 nm. Second, the high-lying HOMO energy levels of P3HT lead to relatively low VOC of OSCs. Third, highly crystalline characteristics of P3HT raised the difficulty of controlling the morphology of P3HT:acceptor blend films. In order to achieve high-efficiency P3HT-based OSCs, many efforts have been made to tune the crystallinities of acceptors to fit P3HT for optimal morphology.33,34
The noncovalent conformational lock (NCL) strategy is an efficient method to enhance the planarity and rigidity of conjugated systems, and thus tune the intermolecular packing, crystallinity and charge transport properties of the organic semiconductors.38,39 In the past few years, various types of NCLs including S⋯X (X = F, Cl, Br),40,41 S⋯O,42,43 and Se⋯O,44etc. have been used for building high-performance organic semiconductors for different organic electronics, including OSCs45–47 and OTFTs.41,48 Yu and coworkers employed S⋯O NCLs to construct polymeric semiconductors for OTFTs with a high charge transport mobility of 5.37 cm2 V−1 s−1.49 Bo et al. used S⋯O NCls to build a nonfullerene acceptor for OSCs, yielding an efficiency of 8.41%, much higher than the analogous one without S⋯O NCLs (2.17%).50
Side chain engineering has been employed as an effective strategy to modulate intermolecular interactions of organic semiconductors and the morphology of donor:acceptor blend films of OSCs.51–57 For example, Wang and coworkers replaced the conjugated 5-(2-ethyl-hexyl)thiophene side chain with the non-conjugated 4-hexylbenzene side chain of the 4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)benzo[1,2-b:4,5-b′]di(cyclopenta-dithiophene) electron-donating core, resulting in a deeper absorption, slightly higher-lying lowest unoccupied molecular orbital (LUMO) and faster electron mobility. Thus, the PCE was dramatically increased from 8.54% to 11.0%.52 Yao et al. studied the effects of the molar ratios of the urea-containing alkyl chains vs. branching alkyl chains on diketopyrrolopyrrole (DPP)–quarterthiophene systems. Interestingly, the side chains with urea groups can form hydrogen bonds, which facilitates the lamellar stacking of alkyl chains and enhances the π–π stacking of neighboring conjugated backbones. Thus, the optimal systems showed excellent hole mobilities (∼13.1 cm2 V−1 s−1) and photovoltaic performances (PCE > 6%).53
Herein, two thiazole (Tz) based small molecule acceptors (H-IDTzR and P-IDTzR) with N⋯S NCLs were designed and synthesized for P3HT-based OSCs. The NCLs ensured the high planarity of the backbone and high crystallinity of the acceptors. Upon changing the alkyl side chains from n-hexyl to phenyl-hexyl, the crystallinity of the acceptors was tuned. Thus, the P-IDTzR molecules exhibited low but suitable crystallinity for P3HT, affording optimal morphology for the blend films. As a result, P3HT:P-IDTzR based OSCs afforded a maximum PCE of 5.01%, much higher than that of P3HT:H-IDTzR based ones (3.53%).
Results and discussion
The molecular structures of H-IDTzR and P-IDTzR are shown in Fig. 1, and the synthetic routes are described in the ESI.† These two compounds were synthesized by the Stille coupling reaction, followed by Knoevenagel condensation with moderate yields. The two compounds were fully characterized by 1H and 13C NMR and elementary analysis. Both compounds are soluble in common organic solvents such as chloroform (CF), chlorobenzene (CB), and o-dichlorobenzene (DCB) at room temperature. Thermogravimetric analysis (TGA) showed that these two small molecules exhibit good thermal stability with decomposition temperatures (5% weight loss) about 400 °C under a N2 atmosphere (Fig. S1, ESI†). Furthermore, the differential scanning calorimetry (DSC) measurement demonstrates that H-IDTzR exhibits obvious melting and crystallization peaks at 268 and 238 °C, respectively. However, H-IDTzR has no melting or crystallization peaks (see Fig. 2c). These observations reveal that H-IDTzR may adopt a tighter interchain packing due to less bulky side chains, resulting in higher crystallization in comparison to P-IDTzR.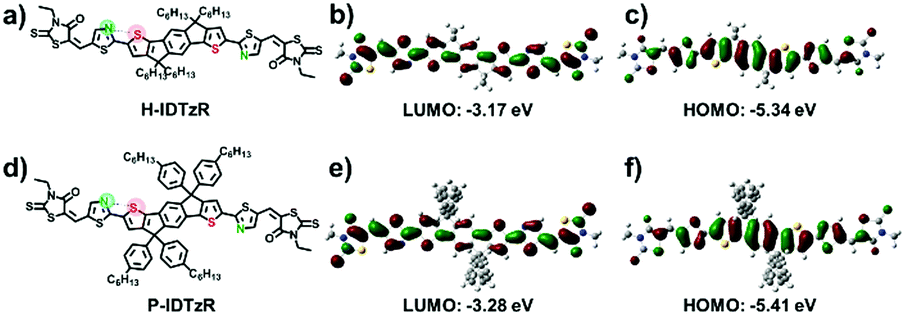 | ||
| Fig. 1 (a and d) Chemical structures of H-IDTzR and P-IDTzR. (b and c) and (e and f) LUMO and highest occupied molecular orbital (HOMO) distribution of H-IDTzR and P-IDTzR calculated using the Gaussian 09 program at the B3LYP/6-31G (d, p) level. | ||
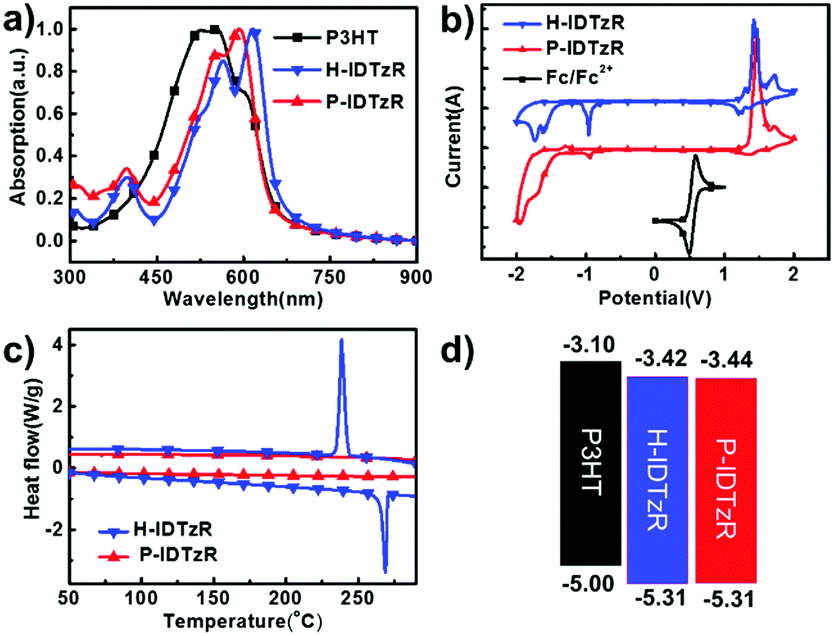 | ||
| Fig. 2 (a) UV-vis absorption spectra for P3HT, H-IDTzR and P-IDTzR film. (b) CV curves of H-IDTzR and P-IDTzR measured in acetonitrile solution with 0.1 M Bu4PF6 at a scan rate of 50 mV s−1. (c) DSC thermogram of H-IDTzR and P-IDTzR during the second heating and cooling cycle, measured at 10 °C min−1 under nitrogen. (d) Energy level diagram of the materials. | ||
Density functional theory (DFT) calculations were performed to investigate the chemical geometry structure and molecular frontier orbitals of H-IDTzR and P-IDTzR at the B3LYP/6-31G(d,p) level. The backbones of the two molecules exhibit highly planar conformations due to the existence of S⋯N noncovalent conformational locks between the sulfur of the IDT core and the nitrogen of the thiazole bridging group (Fig. S2, ESI†), which is beneficial for intermolecular π–π stacking and charge hopping.39,42,44–46 Furthermore, the bulky hexylphenyl chains on the IDT core of P-IDTzR are more stereo-demanding than the straight hexyl chains of H-IDTzR, which may restrain excessive self-aggregation and over-sized phase separation in the blend films of P3HT:P-IDTzR. In addition, the molecular frontier orbitals of these two molecules are well distributed along the conjugated backbone, which is beneficial for electron transport.
Cyclic voltammetry (CV) measurement was employed to investigate the electrochemical properties of the molecular acceptors. The energy levels of P3HT, H-IDTzR and P-IDTzR are shown in Fig. 2b. According to the formula ELUMO = –(ϕred + 4.80 – ϕFc/Fc+), the LUMO energy levels of the H-IDTzR and P-IDTzR films are −3.42 and −3.44 eV, respectively. The HOMO energy levels are estimated by the formula EHOMO = ELUMO − Eg to afford −5.31 and −5.31 eV for H-IDTzR and P-IDTzR, respectively. The LUMO and HOMO energy levels of P-IDTzR and H-IDTzR are similar, which suggests that the hexyl and phenyl-hexyl chains possess similar electron donating properties. Note that the HOMO/LUMO energy levels of P3HT are −5.0/−3.1 eV.58 Thus, the ΔHOMO/ΔLUMO for P3HT:H-IDTzR and P3HT:P-IDTzR are calculated to be 0.31/0.32 eV and 0.31/0.34 eV, respectively, which affords suitable energy offsets for efficient exciton dissociation (see Table 1).
| Compound | λ max [nm] | λ max [nm] | λ onset [nm] | E optg [eV] | LUMO [eV] | HOMO [eV] |
|---|---|---|---|---|---|---|
| a The λmax values were tested in solution. b The λmax and λonset values were obtained using the film form. c Optical bandgaps were calculated by the equation Eoptg = 1240/λonset. d Calculated from the cyclic voltammogram. e Calculated using the formula EHOMO = ELUMO – Eg. | ||||||
| H-IDTzR | 582a | 616b | 661b | 1.87c | −3.44d | −5.31e |
| P-IDTzR | 571a | 592b | 654b | 1.89c | −3.42d | −5.31e |
The normalized UV-vis absorption spectra of P3HT, H-IDTzR and P-IDTzR in CHCl3 solutions and as thin films are shown in Fig. 2a and Fig. S3a (ESI†). In the CHCl3 solution, H-IDTzR and P-IDTzR have similar absorption profiles with maximum absorption at 582 nm and 571 nm, respectively. Not surprisingly, both molecules show an obvious red-shift in the thin films in comparison to the absorption of solution, suggesting the strong intermolecular interactions in the solid state. Moreover, the maximum absorption of H-IDTzR exhibits a relatively larger bathochromic shift of 34 nm than that of P-IDTzR (21 nm), indicating that the former one possesses stronger π–π stacking. It should be pointed out that the shoulder peak of H-IDTzR is stronger in comparison to P-IDTzR, which also indicates that H-IDTzR exhibits stronger intermolecular π–π interactions and self-organization behaviour in the thin film due to the smaller steric hindrance of hexyl chains. Based on the onset wavelength of the thin films, the optical bandgaps of H-IDTzR and P-IDTzR were estimated to be 1.87 and 1.89 eV, respectively (see Table 1). These results indicate that the side chains have significant effects on the molecular self-aggregation and optical properties. As shown in Fig. S3b (ESI†), the absorption coefficient of the P3HT:H-IDTzR blend film is higher than that of P3HT:P-IDTzR, which reveals that more photons may be absorbed in the H-IDTzR based devices.
The photovoltaic performance based on these two molecules was investigated with an inverted structure of ITO/ZnO/P3HT:acceptor/MoO3/Ag. The typical J–V curves of devices under 100 mW cm−2 AM 1.5G illumination are shown in Fig. 3a, and the photovoltaic performances are summarized in Table 2 and Tables S1–S3 (ESI†). The P3HT:H-IDTzR based devices exhibit a moderate efficiency of 3.53% with a VOC of 1.04 V, JSC of 6.67 mA cm−2, and FF of 52.5%. Replacing H-IDTzR with P-IDTzR, P3HT:P-IDTzR based OSCs afford a comparable VOC of 1.02 V, a slightly higher FF of 54.6%, and a significantly higher JSC of 9.00 mA cm−2, resulting in a promising PCE of 5.01%, which is much higher than that of the P3HT:PC71BM-based devices (3.55%). Obviously, the comparable VOC is reasonably ascribed to the similar LUMO energy levels of the two nonfullerene acceptors.
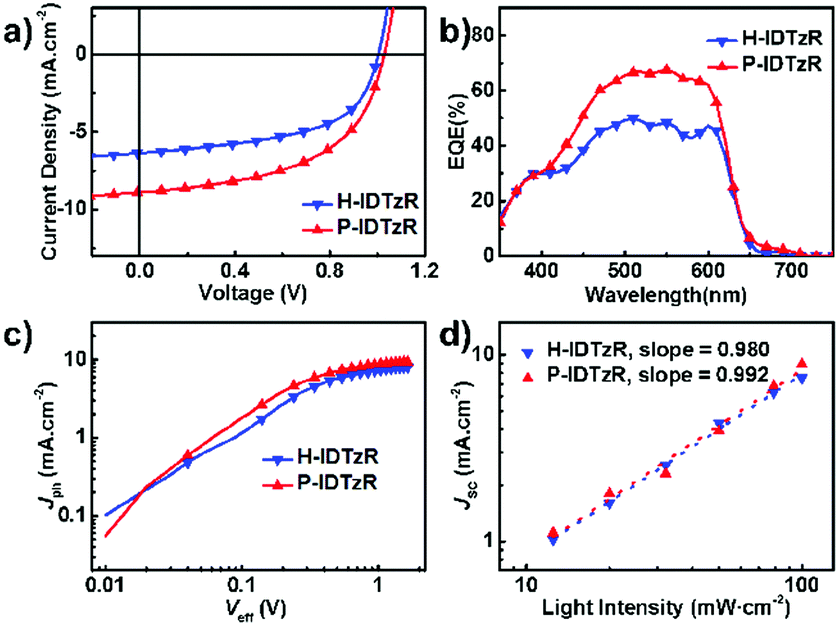 | ||
| Fig. 3 (a) J–V and (b) EQE curves for the H-IDTzR and P-IDTzR-based devices. (c) Photocurrent dependences on the effective voltage and (d) light intensity for the H-IDTZR and P-IDTzR-based devices. | ||
| Device | V OC [V] | J SC [mA cm−2] | J SC [mA cm−2] | FF [%] | PCEb [%] |
|---|---|---|---|---|---|
| a Integrated current densities were calculated from the EQE spectrum. b Average PCE values determined by individual 20 devices are shown in parentheses. c Thermal annealing at 135 °C for 10 min. | |||||
| P3HT:H-IDTzR | 1.04 | 6.67 | 6.29 | 52.5 | 3.53 (3.41) |
| P3HT:P-IDTzR | 1.02 | 9.00 | 8.36 | 54.6 | 5.01 (4.88) |
| P3HT:PC71BMc | 0.53 | 11.2 | 10.8 | 59.7 | 3.55 (3.47) |
External quantum efficiency (EQE) spectra of the P3HT:H-IDTzR and P3HT:P-IDTzR based devices are shown in Fig. 3b. The JSC values of the P3HT:H-IDTzR and P3HT:P-IDTzR devices calculated from integration of the EQE spectrum with the AM 1.5G reference spectra are 6.29 mA cm−2 and 8.36 mA cm−2, respectively, which are in agreement with the values from the J–V curves with a small deviation. Moreover, the shape of the EQE spectra is consistent with that of the UV-vis absorption spectra of the blend films, revealing that the light absorption contributed to the difference in the Jsc value.
To further probe the difference in the Jsc values, steady-state photoluminescence spectroscopy (PL) of the pure films and blend films was performed to investigate the exciton split process. As shown in Fig. S4 (ESI†), the fluorescence spectra of the P3HT:H-IDTzR and P3HT:P-IDTzR blend films show a negligible emission peak in comparison to pure films when the films are excited at 590 nm, which indicates that excitons generated by acceptors and donors can be split efficiently. Note that the blended film of P3HT:P-IDTzR demonstrates a higher quenching efficiency than that of P3HT:H-IDTzR, suggesting more efficient exciton split and thus a higher JSC value.
To understand the influence of side chains on the charge carrier transport mobility and thus the photovoltaic performance, the electron and hole mobilities of P3HT:H-IDTzR and P3HT:P-IDTzR blend films were measured by using the space charge limited current (SCLC) method with a device structure of ITO/PEDOT:PSS/active layer/MoO3/Ag for holes and ITO/Al/active layer/Al for electrons (see Fig. S5a and b, ESI†). The hole and electron mobilities of P3HT:H-IDTzR blend films are estimated to be 1.04 × 10−4 and 1.83 × 10−6 cm2 V−1 s−1, respectively, while those of the blend films of P3HT:P-IDTzR are 0.90 × 10−4 and 7.08 × 10−6 cm2 V−1 s−1, respectively. The P3HT:P-IDTzR blend films showed a higher electron transport property and thus more balanced hole/electron mobility, which may facilitate charge transfer and reduce charge recombination, resulting in higher JSC and FF in devices.45 Note the charge transport mobility is pretty low in these systems, which may account for the low FF in device performance.
To further investigate the charge generation and extraction of the P3HT:H-IDTzR and P3HT:P-IDTzR devices, the dependence of photocurrent density (Jph) on the effective voltage (Veff) and the correlation of JSC with light density (Plight) were measured. Fig. 3c displays the curves of Jphvs. Veff of both devices, where the Jph is defined as Jph = JLight − JDark, and Veff as Veff = V0 – Va, and Va is the applied voltage and V0 is the voltage at which Jph = 0.59 In comparison to P3HT:H-IDTzR devices, the P3HT:P-IDTzR devices exhibit a higher charge separation efficiency and transport efficiency, which is the reason for the higher Jph/Jsat at the low electric field. Fig. 3d shows the curves of JSCvs. Plight, which was described as JSC ∝ Plightk. When k is close to 1.00, the charge recombination is negligible.47 The k value of the P3HT:P-IDTzR devices is 0.992, while that of the P3HT:H-IDTzR devices is 0.98, suggesting that the former one demonstrates more efficient exciton dissociation and weaker recombination, which supports the fact that the former one possesses higher JSC values.60
The crystallinity of the acceptors has an important influence on the morphology of blend films, thus atomic force microscopy (AFM) and transmission electron microscopy (TEM) were employed to study the morphology of the blend films. As shown in Fig. 4a and b, the P3HT:H-IDTzR blend film demonstrates obvious aggregation features with a large root-mean-square (RMS) roughness value of 10.7 nm, whereas the P3HT:P-IDTzR blend film exhibits a much more uniform and smooth surface with a RMS value of 0.6 nm. The phase image of the blend films (Fig. S6, ESI†) also showed that the P3HT:H-IDTzR blend film possesses crystalline features, while the P3HT:P-IDTzR film exhibits featureless homogeneous characteristics. Obviously, the highly crystalline properties of H-IDTzR may contribute to the rougher surface and higher crystallinity of the blend films. Thus, the P3HT:P-IDTzR blend film possesses excellent contact between the active layer and interfacial layer, beneficial for charge collection from the active layer to the electrode. TEM images (Fig. 4c and d) show that the P3HT:H-IDTzR blend film clearly exhibits a fiber shape with extra-large domains, which is very unfavourable for exciton diffusion and separation. This may also be attributed to the over-strong crystallinity of H-IDTzR, forming large self-aggregates in the blended film. In contrast, the P3HT:P-IDTzR blend film presents a homogeneous morphology with small domains, which suggests that the strong self-aggregation is prevented efficiently upon replacing hexyl side chains with the more sterically hindering hexylphenyl side chains. Obviously, this appropriate phase-separation facilitates efficient exciton diffusion and dissociation, resulting in high JSC, consistent with the photovoltaic performances.
 | ||
| Fig. 4 (a and c) AFM height and TEM images for P3HT:H-IDTzR blend films and (b and d) P3HT:P-IDTzR blend films. | ||
Conclusions
In this work, we designed and synthesized two new Tz-containing non-fullerene small molecule acceptors with an A–π–D–π–A structure with appropriate LUMO and HOMO energy levels matching well with P3HT. The two small molecules exhibit a highly planar conformation with N⋯S noncovalent conformational locks. Upon turning the side chains to modulate the interchain π–π stacking of the acceptors and the morphology of the P3HT:acceptor blend films, the photovoltaic performances of the P3HT based OSCs were systematically tuned. The linear hexyl chain on the IDT group (H-IDTzR) promotes a stronger intermolecular packing, which causes severe self-aggregation and large domains when blended with P3HT. In comparison, the hexylphenyl chain on the IDT group (P-IDTzR) can form favorable phase domains without severe self-aggregation due to its steric hindrance. As a result, the P-IDTzR based OSCs exhibited a much higher JSC of 9.00 mA cm−2 and an enhanced PCE of 5.01%. These results provide an important guidance to tune the crystallinity of acceptors for high-performance P3HT OSCs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the NSFC (21774130 and 21574135), Municipal Natural Science Foundation (2162043), the Key Research Program of Frontier Sciences, CAS (QYZDB-SSW-JSC046), the Key Research Program of the Chinese Academy of Sciences (XDPB08-2), One Hundred Talents Program of the Chinese Academy of Sciences, and the University of the Chinese Academy of Sciences. DFT results described in this communication were obtained from the National Supercomputing Center in Shenzhen (Shenzhen Cloud Computing Center).Notes and references
- L. Huo, T. Liu, X. Sun, Y. Cai, A. J. Heeger and Y. Sun, Adv. Mater., 2015, 27, 2938–2944 CrossRef CAS.
- J. Zhao, Y. Li, G. Yang, K. Jiang, H. Lin, H. Ade, W. Ma and H. Yan, Nat. Energy, 2016, 1, 15027 CrossRef CAS.
- J. E. Anthony, Chem. Mater., 2011, 23, 583–590 CrossRef CAS.
- C. Cui, X. Guo, J. Min, B. Guo, X. Cheng, M. Zhang, C. J. Brabec and Y. Li, Adv. Mater., 2015, 27, 7469–7475 CrossRef CAS.
- F. Liu, Z. Zhou, C. Zhang, T. Vergote, H. Fan, F. Liu and X. Zhu, J. Am. Chem. Soc., 2016, 138, 15523–15526 CrossRef CAS.
- Y. Yang, Z.-G. Zhang, H. Bin, S. Chen, L. Gao, L. Xue, C. Yang and Y. Li, J. Am. Chem. Soc., 2016, 138, 15011–15018 CrossRef CAS PubMed.
- X. Liu, J. Wang, J. Peng and Z. Liang, Macromolecules, 2017, 50, 6954–6960 CrossRef CAS.
- L. Yang, W. Gu, L. Lv, Y. Chen, Y. Yang, P. Ye, J. Wu, L. Hong, A. Peng and H. Huang, Angew. Chem., Int. Ed., 2018, 57, 1096–1102 CrossRef CAS.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148–7151 CrossRef CAS.
- X. Xu, T. Yu, Z. Bi, W. Ma, Y. Li and Q. Peng, Adv. Mater., 2018, 30, 1703973 CrossRef.
- S. Li, L. Ye, W. Zhao, H. Yan, B. Yang, D. Liu, W. Li, H. Ade and J. Hou, J. Am. Chem. Soc., 2018, 140, 7159–7167 CrossRef CAS.
- W. Chen, T. Xu, F. He, W. Wang, C. Wang, J. Strzalka, Y. Liu, J. Wen, D. J. Miller, J. Chen, K. Hong, L. Yu and S. B. Darling, Nano Lett., 2011, 11, 3707–3713 CrossRef CAS.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 5293 CrossRef CAS.
- S. C. Price, A. C. Stuart, L. Yang, H. Zhou and W. You, J. Am. Chem. Soc., 2011, 133, 4625–4631 CrossRef CAS.
- D. Qian, L. Ye, M. Zhang, Y. Liang, L. Li, Y. Huang, X. Guo, S. Zhang, Z. A. Tan and J. Hou, Macromolecules, 2012, 45, 9611–9617 CrossRef CAS.
- L. Gao, Z. Zhang, L. Xue, J. Min, J. Zhang, Z. Wei and Y. Li, Adv. Mater., 2016, 28, 1884–1890 CrossRef CAS.
- A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS.
- Q. Zhang, B. Kan, F. Liu, G. Long, X. Wan, X. Chen, Y. Zuo, W. Ni, H. Zhang, M. Li, Z. Hu, F. Huang, Y. Cao, Z. Liang, M. Zhang, T. P. Russell and Y. Chen, Nat. Photonics, 2014, 9, 35–41 CrossRef.
- Y. Lin and X. Zhan, Mater. Horiz., 2014, 1, 470–488 RSC.
- W. Chen and Q. Zhang, J. Mater. Chem. C, 2017, 5, 1275–1302 RSC.
- M. T. Dang, L. Hirsch and G. Wantz, Adv. Mater., 2011, 23, 3597–3602 CrossRef CAS.
- W. Han, M. He, M. Byun, B. Li and Z. Lin, Angew. Chem., Int. Ed., 2013, 52, 2564–2568 CrossRef CAS.
- A. Tournebize, P.-O. Bussière, A. Rivaton, J.-L. Gardette, H. Medlej, R. C. Hiorns, C. Dagron-Lartigau, F. C. Krebs and K. Norrman, Chem. Mater., 2013, 25, 4522–4528 CrossRef CAS.
- R. Søndergaard, M. Manceau, M. Jørgensen and F. C. Krebs, Adv. Energy Mater., 2012, 2, 415–418 CrossRef.
- Y. Wu, H. Bai, Z. Wang, P. Cheng, S. Zhu, Y. Wang, W. Ma and X. Zhan, Energy Environ. Sci., 2015, 8, 3215–3221 RSC.
- S. Li, W. Liu, M. Shi, J. Mai, T.-K. Lau, J. Wan, X. Lu, C.-Z. Li and H. Chen, Energy Environ. Sci., 2016, 9, 604–610 RSC.
- F. Liu, J. Zhang, Z. Zhou, J. Zhang, Z. Wei and X. Zhu, J. Mater. Chem. A, 2017, 5, 16573–16579 RSC.
- C. Yuan, W. Liu, M. Shi, S. Li, Y. Wang, H. Chen, C.-Z. Li and H. Chen, Dyes Pigm., 2017, 143, 217–222 CrossRef CAS.
- S. Holliday, R. S. Ashraf, C. B. Nielsen, M. Kirkus, J. A. Rohr, C. H. Tan, E. Collado-Fregoso, A. C. Knall, J. R. Durrant, J. Nelson and I. McCulloch, J. Am. Chem. Soc., 2015, 137, 898–904 CrossRef CAS.
- B. Xiao, A. Tang, J. Yang, A. Mahmood, X. Sun and E. Zhou, ACS Appl. Mater. Interfaces, 2018, 10, 10254–10261 CrossRef CAS.
- B. Xiao, A. Tang, J. Yang, Z. Wei and E. Zhou, ACS Macro Lett., 2017, 6, 410–414 CrossRef CAS.
- D. Baran, R. S. Ashraf, D. A. Hanifi, M. Abdelsamie, N. Gasparini, J. A. Röhr, S. Holliday, A. Wadsworth, S. Lockett, M. Neophytou, C. J. M. Emmott, J. Nelson, C. J. Brabec, A. Amassian, A. Salleo, T. Kirchartz, J. R. Durrant and I. McCulloch, Nat. Mater., 2016, 16, 363–369 CrossRef.
- B. Xiao, A. Tang, L. Cheng, J. Zhang, Z. Wei, Q. Zeng and E. Zhou, Sol. RRL, 2017, 1, 1700166 CrossRef.
- S. Holliday, R. S. Ashraf, A. Wadsworth, D. Baran, S. A. Yousaf, C. B. Nielsen, C. H. Tan, S. D. Dimitrov, Z. Shang, N. Gasparini, M. Alamoudi, F. Laquai, C. J. Brabec, A. Salleo, J. R. Durrant and I. McCulloch, Nat. Commun., 2016, 7, 11585 CrossRef CAS.
- B. Xiao, A. Tang, J. Zhang, A. Mahmood, Z. Wei and E. Zhou, Adv. Energy Mater., 2017, 7, 1602269 CrossRef.
- B. Xiao, A. Tang, Q. Zhang, G. Li, X. Wang and E. Zhou, ACS Appl. Mater. Interfaces, 2018, 10, 10254–10261 CrossRef CAS.
- Q. Zhang, B. Xiao, M. Du, G. Li, A. Tang and E. Zhou, J. Mater. Chem. C, 2018, 6, 10902–10909 RSC.
- S. Yu, A. Peng, S. Zhang and H. Hang, Sci. China: Chem., 2018 DOI:10.1007/s11426-018-9315-2.
- H. Huang, L. Yang, A. Facchetti and T. J. Marks, Chem. Rev., 2017, 117, 10291–10318 CrossRef CAS PubMed.
- S. Zhang, Y. Qin, M. A. Uddin, B. Jang, W. Zhao, D. Liu, H. Y. Woo and J. Hou, Macromolecules, 2016, 49, 2993–3000 CrossRef CAS.
- J. Yang, Z. Zhao, H. Geng, C. Cheng, J. Chen, Y. Sun, L. Shi, Y. Yi, Z. Shuai, Y. Guo, S. Wang and Y. Liu, Adv. Mater., 2017, 29, 1702115 CrossRef PubMed.
- L. Lv, X. Wang, T. Dong, X. Wang, X. Wu, L. Yang and H. Huang, Mater. Chem. Front., 2017, 1, 1317–1323 RSC.
- H. Huang, N. Zhou, R. P. Ortiz, Z. Chen, S. Loser, S. Zhang, X. Guo, J. Casado, J. T. López Navarrete, X. Yu, A. Facchetti and T. J. Marks, Adv. Funct. Mater., 2014, 24, 2782–2793 CrossRef CAS.
- T. Dong, L. Lv, L. Feng, Y. Xia, W. Deng, P. Ye, B. Yang, S. Ding, A. Facchetti, H. Dong and H. Huang, Adv. Mater., 2017, 29, 1606025 CrossRef PubMed.
- S. Yu, Y. Chen, L. Yang, P. Ye, J. Wu, J. Yu, S. Zhang, Y. Gao and H. Huang, J. Mater. Chem. A, 2017, 5, 21674–21678 RSC.
- P. Ye, Y. Chen, J. Wu, X. Wu, S. Yu, W. Xing, Q. Liu, X. Jia, A. Peng and H. Huang, J. Mater. Chem. C, 2017, 5, 12591–12596 RSC.
- L. Yang, W. Gu, Y. Yang, L. Hong, X. Zhang, Y. Xiao, X. Wu, A. Peng and H. Huang, Small Methods, 2018, 2, 1700330 CrossRef.
- C. Zhu, Z. Zhao, H. Chen, L. Zheng, X. Li, J. Chen, Y. Sun, F. Liu, Y. Guo and Y. Liu, J. Am. Chem. Soc., 2017, 139, 17735–17738 CrossRef CAS.
- W. Zhang, Z. Mao, J. Huang, D. Gao and G. Yu, Macromolecules, 2016, 49, 6401–6410 CrossRef CAS.
- Y. Liu, Z. Zhang, S. Feng, M. Li, L. Wu, R. Hou, X. Xu, X. Chen and Z. Bo, J. Am. Chem. Soc., 2017, 139, 3356–3359 CrossRef CAS.
- L. Fang, Y. Zhou, Y.-X. Yao, Y. Diao, W.-Y. Lee, A. L. Appleton, R. Allen, J. Reinspach, S. C. B. Mannsfeld and Z. Bao, Chem. Mater., 2013, 25, 4874–4880 CrossRef CAS.
- W. Jiayu, W. Wei, W. Xiaohui, W. Yang, Z. Qianqian, Y. Cenqi, M. Wei, Y. Wei and Z. Xiaowei, Adv. Mater., 2017, 29, 1702125 CrossRef PubMed.
- J. Yao, C. Yu, Z. Liu, H. Luo, Y. Yang, G. Zhang and D. Zhang, J. Am. Chem. Soc., 2016, 138, 173–185 CrossRef CAS.
- H. Huang, J. Youn, R. Ponce Ortiz, Y. Zheng, A. Facchetti and T. Marks, Chem. Mater., 2011, 23, 2185–2200 CrossRef CAS.
- X. Liu, C. Zhang, C. Duan, M. Li, Z. Hu, J. Wang, F. Liu, N. Li, C. J. Brabec, R. A. J. Janssen, G. C. Bazan, F. Huang and Y. Cao, J. Am. Chem. Soc., 2018, 140, 8934–8943 CrossRef CAS PubMed.
- Z. Fei, F. D. Eisner, X. Jiao, M. Azzouzi, J. A. Röhr, Y. Han, M. Shahid, A. S. R. Chesman, C. D. Easton, C. R. McNeill, T. D. Anthopoulos, J. Nelson and M. Heeney, Adv. Mater., 2018, 30, 1705209 CrossRef PubMed.
- S. Liu, Y. Firdaus, S. Thomas, Z. Kan, F. Cruciani, S. Lopatin, J.-L. Bredas and P. M. Beaujuge, Angew. Chem., Int. Ed., 2018, 57, 531–535 CrossRef CAS.
- A. A. Bakulin, A. Rao, V. G. Pavelyev, P. H. M. van Loosdrecht, M. S. Pshenichnikov, D. Niedzialek, J. Cornil, D. Beljonne and R. H. Friend, Science, 2012, 335, 1340–1344 CrossRef CAS.
- L. Lv, X. Wang, X. Wang, L. Yang, T. Dong, Z. Yang and H. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 34620–34629 CrossRef CAS.
- Y. Chen, P. Ye, Z.-G. Zhu, X. Wang, L. Yang, X. Xu, X. Wu, T. Dong, H. Zhang, J. Hou, F. Liu and H. Huang, Adv. Mater., 2017, 29, 1603154 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00461g |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2019 |