
Influence of halogen substitution on aggregation-induced near infrared emission of borondifluoride complexes of 2′-hydroxychalcones†
Anthony
D’Aléo
*ab,
Andres
Saul
 b,
Claudio
Attaccalite
b and
Frédéric
Fages
*b
b,
Claudio
Attaccalite
b and
Frédéric
Fages
*b
aBuilding Blocks for Future Electronics Laboratory (2-B FUEL), The joint CNRS-Ewha-Yonsei Laboratory, UMI 2002, Seoul, Republic of Korea
bAix Marseille Univ, CNRS, CINaM UMR 7325, Campus de Luminy Case 913, 13288 Marseille, France. E-mail: daleo@cinam.univ-mrs.fr; frederic.fages@univ-amu.fr
First published on 31st October 2018
Abstract
The generation of organic dyes displaying intense red and near infrared fluorescence emission in solution and in the solid state is a topic of intense current interest. In this study, we present the synthesis and investigation of nine new derivatives of borondifluoride complexes of 2′-hydroxychalcones: four homo- and five hetero-substituted compounds X–X and X–Y (X, Y = F, Cl, Br, and I), where two halogen atoms are attached ortho and para (X and Y respectively in X–Y) to the 2′-hydroxyl group. UV-vis absorption fluorescence spectra in solution and in the solid state, DFT calculations, and analyses of single-crystal structures were performed. For all compounds, a fluorescence emission enhancement was clearly observed when passing from the solution (DCM) to the crystal (up to more than 10 fold). These molecules thus exhibit aggregation-induced enhanced emission (AIEE) with fluorescence maxima ranging from 654 nm (F–F) to 807 nm (I–I) in the solid state. The highest fluorescence quantum yield value within the series is reached for Cl–Cl (24% at 730 nm) whilst I–Br shows a rather good efficiency (2.5% at 806 nm) despite the presence of the two heavy iodine and bromine atoms. This study sheds light on the role of the halogen substitution on crystalline packing and solid-state emission properties of the chalcone–BF2 dyes. We outline that using halogen atoms provides a useful cocktail of steric, electrostatic and photophysical ingredients upon which to base the generation of near infrared emitting organic solids.
Introduction
Organic dyes displaying bright fluorescence in the solid state are the subject of intense research interest because of their many applications in bioimaging,1,2 chemical sensing,3,4 solid-state lasing,5–7 telecommunications,8 and information display technologies.9–11 In particular, their use in organic electronics has led to far-reaching advances in the generation of high performance organic light-emitting diodes (OLEDs).12,13 However, most organic chromophores suffer from severe fluorescence quenching in the solid state and thus display low emission efficiency whereas they are highly emissive in solution.14 The design of dyes that emit efficiently in the solid state is a difficult task because methods for predicting crystalline structures of organic molecules still need to be improved.15 Nonetheless, various strategies can be applied such as the introduction of bulky groups or quaternary centres in the vicinity of aromatic rings that hamper close π-stacking.16 Another approach is based on restricted intramolecular rotation and has been demonstrated by Tang et al. to yield a variety of fluorophores displaying the so-called aggregation induced emission (AIE) process.17–19 Enhancement of fluorescence properties in neat film states has been first reported by Hanack and Oelkrug,20 which led to the introduction of the terms “aggregation-induced enhanced emission” (AIEE)21 and more recently “solid-state luminescence enhancement” (SLE)22 by Park and Gierschner.Previously we reported on a series of borondifluoride complexes of 2′-hydroxychalcone and curcuminoid dyes that display fluorescence in the near infrared (NIR) in the solid state whilst only weak emission was detected in solution.23–26 Chalcone dyes containing a donor group appended to the strong electron withdrawing borondifluoride (BF2) complex of acetylphenolate or acetylnaphtholate groups have a push–pull structure and provided access to efficient red to NIR-emitting organic solids.23,27 Here, we report the synthesis and photophysical investigation of a series of nine related derivatives (Chart 1) bearing the anisole ring as an electron donor group and a dihalogeno-substituted acetylphenolate moiety. Terminal halides are known to direct the self-assembly of organic molecules, including dyes, via a wide variety of intermolecular interactions such as H-bonds, halogen–halogen and halogen–arene interactions whose strengths depend on differences in their electrostatic potentials and size.28–31 Halogen atoms, especially the heavier ones (Br, I), increase spin orbit coupling via the heavy atom effect and thus induce efficient quenching of singlet excited states.32 However, our previous work showed that the BF2 complexes of two monohalogenated hydroxychalcones displayed NIR emission beyond 850 nm.23 These considerations prompted us to further investigate the halogen effect on solid-state optical properties of this class of dyes.
 | ||
| Chart 1 | ||
The series investigated here is composed of four homo-substituted compounds X–X (X = F, Cl, Br, and I), and five hetero-substituted compounds X–Y (Cl–Br, Br–Cl, I–Cl, I–Br, and I–F), respectively, where the two halogen atoms are attached ortho (X) and para (Y in X–Y) to the 2′-hydroxyl group. Their behaviour is compared to that of the previously published H–H parent compound lacking halogen atoms.23 The new compounds display far red and NIR emissions with fluorescence quantum yields that are higher than those in solution and rather high in that spectral range and a fortiori in the condensed phase. The results show that a process of AIEE underpins this behaviour. Furthermore, we discuss the trends in emission redshift in terms of interchromophoric interactions in relation to the solid-state packing and attempt to evaluate the role of the halogen nature in all compounds and substitution positions in X–Y on the solid-state optical properties.
Results and discussion
Synthesis and chemical stability
The 2′-hydroxychalcone ligands were synthetized in a one-step reaction via a Claisen–Schmidt condensation between p-anisaldehyde and the appropriate acetophenone derivative in ethanol as a solvent using sodium hydroxide as a base.23 Complexation of the BF2 group was achieved by reacting the free ligands with borontrifluoride etherate in dichloromethane (DCM). The detailed synthesis and characterization of the nine compounds are described in the ESI.† Depending on their structure, the dyes were observed to be more or less stable in protic or polar media and this behaviour can be ascribed to the negative inductive effect of the halogen atoms. A decrease of the electronic charge density at the meta-carbonyl oxygen atom is noticed in the HOMO of the halogenated derivatives (Fig. 1), which weakens the B–O bond. The boron chelate is thus less stable than BF2 complexes of other chalcone derivatives reported previously.23,27 In order to assess the extent of solvolytic B–O bond cleavage, we monitored spectrospically the kinetics of decomplexation of the boron difluoride moiety in dichloromethane solution containing 0.5 mol% of ethanol. As can be seen for homo-substituted compounds (X–X, X = F, Cl, Br, and I) (Table S1 in ESI†), the presence of the halogen atoms reduces the stability of the dyes versus solvolysis. Except for F–F, the average kobs values are in the same range (ca. 2 × 10−4 s−1) and almost one order of magnitude larger than that of H–H. Going from Cl–Cl to I–I, a gradual decrease (increase) of kobs (stability) is observed. Dye F–F is the less stable compound, consistent with the high electronegativity of the fluorine atom. Using dry DCM, we could record spectra safely without detecting any noticeable degradation of the borondifluoride complexes. However, the use of more polar and more coordinating solvents, such as acetonitrile, even when dried carefully, induced boron release, which made measurements hardly reliable in these cases. Importantly, all the compounds are stable in the solid state.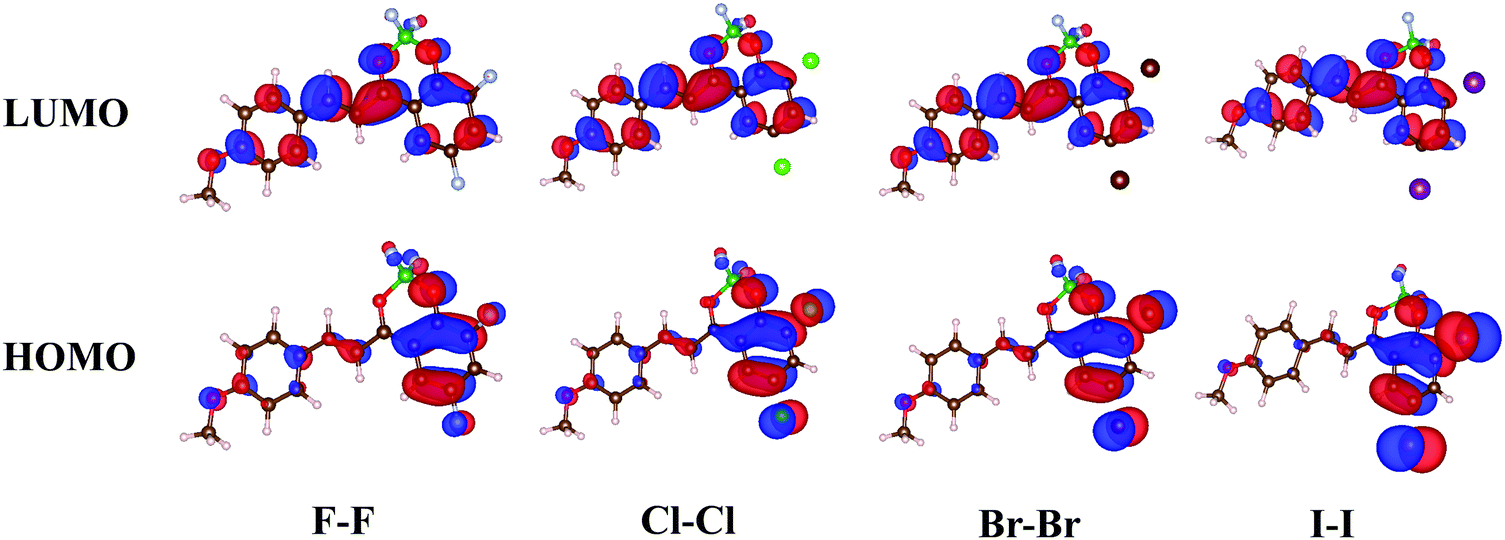 | ||
| Fig. 1 HOMO and LUMO molecular orbitals of F–F, Cl–Cl, Br–Br and I–I. | ||
Quantum mechanical calculations
In order to get valuable insights into the aggregation behaviour of the halogen-substituted dyes, calculations based on density functional theory (DFT) were performed on optimized geometries to obtain electron density distribution in frontier molecular orbitals (FMOs) and calculate the ground-state dipole moment intensity and orientation (details in ESI†). Fig. 1 shows similar FMO patterns for X–X (X = F, Cl, Br, and I). The highest occupied molecular orbital (HOMO) is mainly localized on the halogen-substituted acetylphenolate moiety, the contribution of the anisole ring to the electron delocalization being minor. The situation is reversed in the lowest unoccupied molecular orbital (LUMO). Ground-state dipole moment values of these four dyes, μgs, were found to be very similar (i.e. 11.6–11.8 D) suggesting that the nature of the halogen atom has little influence on this parameter. Such large values of μgs are in line with those obtained for related dyes.27 The dipole moment makes an angle of ca. 40°–50° with the molecular main axis (Fig. S1 in ESI†), which is also consistent with the previous data. The same conclusions can be drawn for the hetero-substituted compounds Cl–Br and Br–Cl (Fig. S2 in ESI†).Single-crystal X-ray diffraction
The X-ray crystal structures of Cl–Cl, Br–Br, I–I, Br–Cl and Cl–Br were obtained by slow evaporation of DCM and they are compared to that published for H–H.23 The crystal data are reported in Table S2 (ESI†). The five molecules crystallized in the same system (monoclinic) and same space groups (P21/c). When comparing Cl–Cl, Br–Br and I–I, it is interesting to notice that the size of the halogen induces an increase of the elementary unit cell dimension. Br–Cl and Cl–Br present elementary unit cell volumes intermediate between those of Cl–Cl and Br–Br. The X-ray powder diffraction patterns were calculated from the single crystal structures and compared to the experimental ones (see Fig. S3–S7 in ESI†). A good agreement was found for all the reported dyes.The three homo-substituted compounds (X–X, X = Cl, Br, and I) display a nearly planar π-conjugated system in the crystal (Fig. S8 and S9 in ESI†). The crystal structures of Cl–Cl and Br–Br are similar while I–I adopts a different packing mode. As shown in Fig. 2a and Fig. S10 (ESI†), Cl–Cl molecules align their long axis into parallel linear strings that are contained in planes parallel to (−101). In a string, Cl–Cl molecules have the same orientation symbolized by a blue arrow (arbitrarily oriented from dioxaborinine moiety toward anisole) in Fig. 2a. Two adjacent strings in the same plane adopt an antisense sequence. Strings of two adjacent planes stack parallel with the same molecular orientation but they are longitudinally displaced with respect to each other (Fig. 2b). As a result, Cl–Cl molecules interact in a parallel fashion with the dichlorobenzene moiety of one molecule in one plane stacking over the anisole ring of the closest molecule in the adjacent plane. This assembling mode can be rationalized in terms of the electron density features found in the HOMOs which favours intermolecular donor–acceptor interactions between the electron rich dichlorobenzene moiety and electron poor anisole ring. As a result of this arrangement, intermolecular dipole–dipole interactions cancel out within planes parallel to (−101). This situation is very different from that prevailing for compound H–H that was shown to experience antiparallel pairwise association. In this case, the dipole–dipole interactions are minimized within intermolecular dimers.23,33 To complete the description of the linear strings, it is observed that molecular planes of neighbouring molecules are slightly offset (Fig. 2c). As a result, two characteristic distances separating the average molecular planes of stacking molecules belonging to adjacent crystallographic planes can be characterized (d = 6.929 Å and d′ = 7.113 Å, Scheme 1) for Cl–Cl, which means that molecular overlap occurs in any case at the van der Waals contact. Along the same direction perpendicular to the (−101) plane, Cl–Cl molecules stack up in piles generating a brick-wall like pattern (Fig. 2d). The X-ray structure of Br–Br can be described exactly in the same manner (Fig. S11 in ESI†) but the interplanar distances are slightly shorter (d = 6.889 Å, d′ = 7.120 Å).
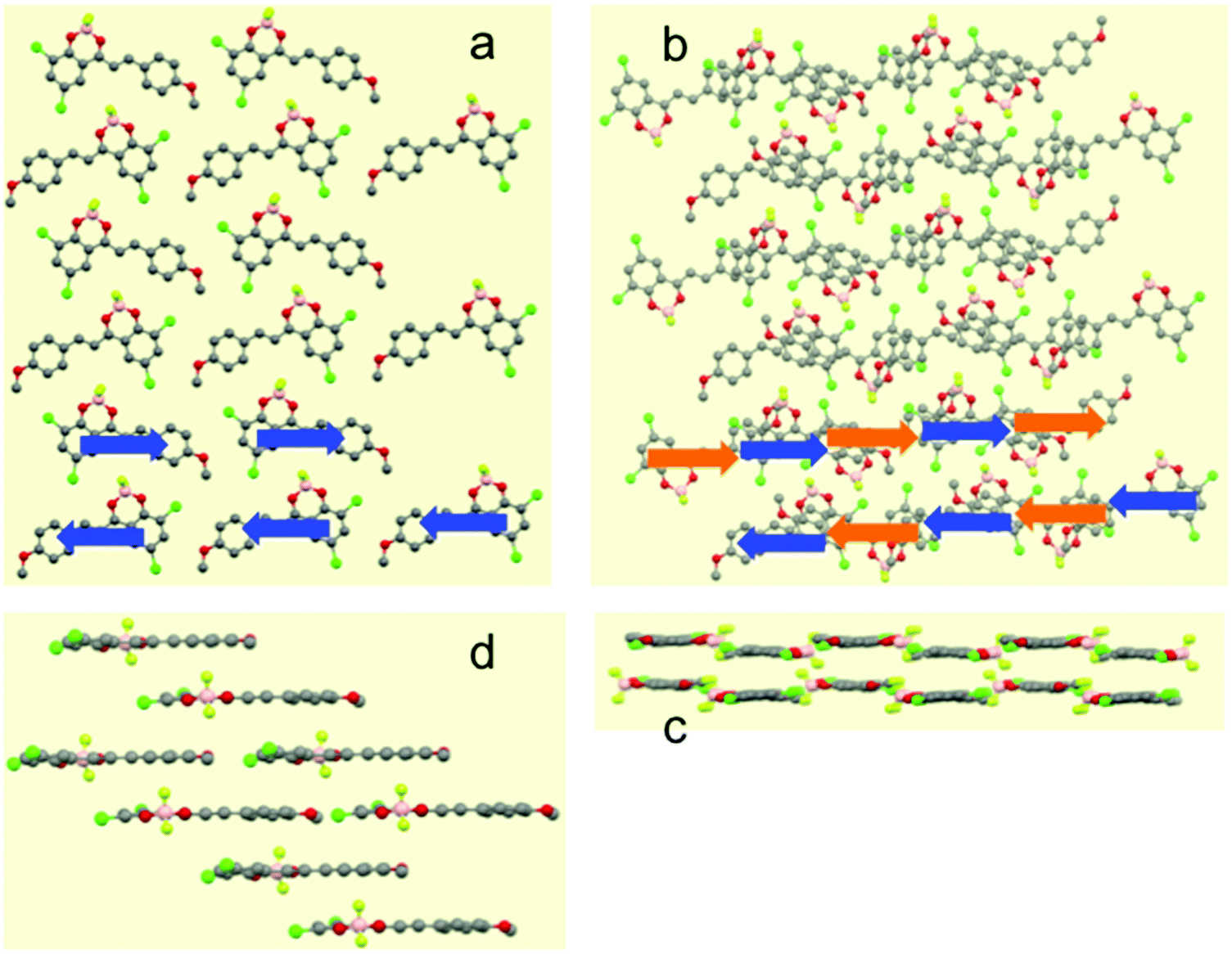 | ||
| Fig. 2 Selected views of the single-crystal structure of compound Cl–Cl. (a) Single sheet formed by antiparallel strings of molecules in planes parallel to (−101); (b) stack of the former sheet (upper layer, blue arrows) and an adjacent sheet (bottom layer, orange arrows); (c) view along [101] of the two former layers; (d) view along the b axis of the molecular stacks. | ||
 | ||
| Scheme 1 Side view of the stacks formed by Cl–Cl. Arrows are oriented from the dioxaborinine moiety toward the anisole ring. | ||
In contrast, the single-crystal structure of compound I–I shows the typical pairwise association of antiparallel molecules (Fig. 3 and Fig. S12 in ESI†) mentioned previously for H–H and other related compounds.23,27 As already reported, this molecular packing is characterized by the occurrence of cofacial tight dimers with a short interplanar distance (d1 = 3.31 Å, Scheme 2). These dimers form 1D π-stacks along the crystallographic c axis. Two other characteristic distances between the average planes of adjacent molecules of neighbouring dimers are found. A long distance (d2 = 3.55 Å) is measured when the inter-dimer contact occurs via two anisole rings, and a shorter one (d3 = 3.17 Å) that stems from the interaction between two diiodophenolate rings (Fig. 3).
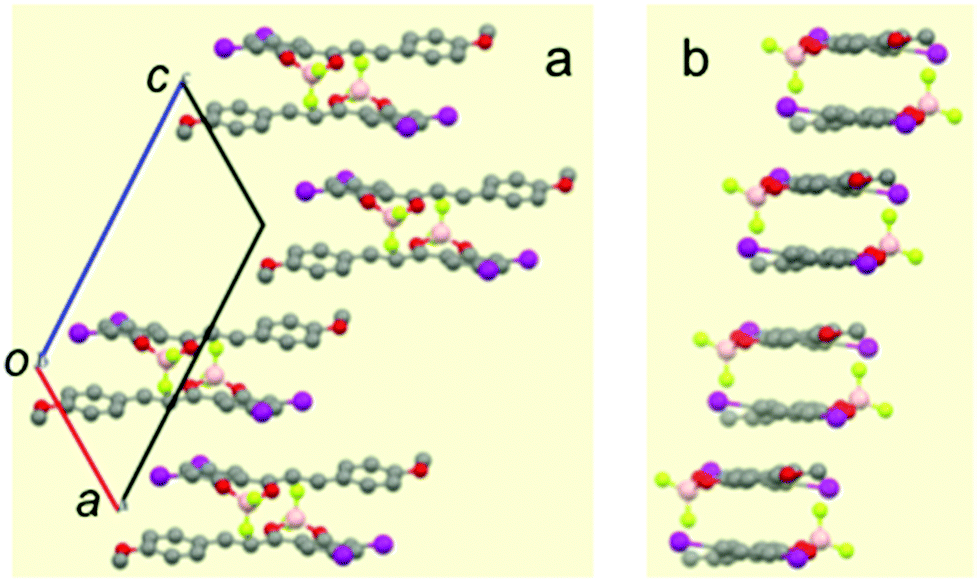 | ||
| Fig. 3 Selected views of the single-crystal structure of compound I–I. (a) View of the offset dimers forming 1D arrays along the crystallographic b axis; (b) view of the same stacks along the molecular long axis. | ||
 | ||
| Scheme 2 Side view of the stacks formed by I–I. Arrows are oriented from the dioxaborinine moiety toward the anisole ring. | ||
Like the homo-substituted compounds, the hetero-substituted Cl–Br and Br–Cl molecules are also planar in the crystal (Fig. S9 in ESI†). They display the same packing arrangement as well but slightly different characteristic distances (Cl–Br: d = 6.845 Å, d′ = 7.017 Å; Br–Cl: d = 6.907 Å, d′ = 7.156 Å) are measured relative to Cl–Cl and Br–Br (Fig. S13 and S14 in ESI†). In particular, one finds the brick-wall arrangement, which is due to the slipped cofacial interaction between adjacent molecules leading to a common value of ca. 69° of the pitch angle for the four compounds Cl–Cl, Br–Br, Cl–Br and Br–Cl.34,35
For all compounds we do not observe any occurrence of halogen–halogen interactions between halides bound to the acetylphenolate moiety. However, intermolecular distances shorter than the sum of the van der Waals radii (3.45 Å) are noticed between fluorine atoms of BF2 groups and iodine atoms in I–I (3.207 Å). Except for Br–Br, chlorine, bromine and iodine atoms in the four other compounds form short contacts with hydrogens of olefinic (Cl–Cl, Cl–Br, and Br–Cl), anisole (I–I) and methyl (Cl–Br) groups. These interactions develop within and between linear strings belonging to the same plane. We also observe F–H intermolecular bonds between neighbouring molecules of adjacent planes. All together, these features do not allow us to figure out whether dispersive interactions of halogen atoms play an active role in the crystalline organization. It seems more likely that the latter is ascribable to a combination of the steric effect of the halogens, and of electrostatic and π–π stacking interactions between the dipolar π-conjugated dye molecules.
UV-visible absorption and fluorescence emission in solution
Because the borondifluoride complexes are stable in DCM, their solution-phase optical properties were investigated in this solvent. The electronic absorption spectra are displayed in Fig. 4 and Fig. S15 (ESI†). These consist mainly of a broad transition band in the visible region.23,27 This transition is located at ca. 480 nm for all the dihalogenated compounds. The redshift of 25 nm relative to H–H can be attributed to the auxochromic effect of halogens (Table 1). | ||
Fig. 4 UV-visible absorption spectra of thin films (—) and solutions in DCM ( ), and fluorescence spectra of powder samples (----) and solutions in DCM ( ), and fluorescence spectra of powder samples (----) and solutions in DCM ( ) for (a) F–F; (b) Cl–Cl; (c) Br–Br and (d) I–I. ) for (a) F–F; (b) Cl–Cl; (c) Br–Br and (d) I–I. | ||
| Compound | Solution | Solid state | |||||||
|---|---|---|---|---|---|---|---|---|---|
| λ abs | ε max | λ em | ΔνST | Φ f | λ abs | λ em | ΔνST | Φ f | |
| a Absorption maximum wavelengths λabs (nm), molar absorption coefficients εmax (M−1 cm−1), fluorescence maximum wavelengths λem (nm), Stokes shifts ΔνST (cm−1), fluorescence quantum yields Φf (%). | |||||||||
| H–H | 455 | 41![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 190 190 |
556 | 3992 | 2.0 | 351 | 627 | 12541 |
21.0 |
| F–F | 480 | 43160 |
584 | 3710 | 1.1 | 359 | 654 | 12565 |
10 |
| Cl–Cl | 481 | 44630 |
596 | 4011 | 1.75 | 475 | 730 | 7354 | 24.0 |
| Br–Br | 482 | 45655 |
598 | 4024 | 1.2 | 567 | 749 | 4286 | 5.0 |
| I–I | 482 | 42980 |
607 | 4272 | 0.25 | 510 | 807 | 7216 | 1.0 |
| Cl–Br | 480 | 44030 |
598 | 4111 | 1.4 | 561 | 690 | 3333 | 16.0 |
| Br–Cl | 480 | 42980 |
601 | 4194 | 1.3 | 546 | 742 | 4838 | 13.0 |
| I–F | 482 | 43810 |
607 | 4272 | 0.5 | 456 | 710 | 7845 | 3.0 |
| I–Cl | 481 | 41920 |
604 | 4234 | 0.35 | 598 | 761 | 3582 | 5.0 |
| I–Br | 483 | 46100 |
607 | 4229 | 0.1 | 585 | 806 | 4687 | 2.5 |
The borondifluoride complexes show very weak emission in DCM with fluorescence quantum yields lower than 2%. Such low values have been demonstrated to arise from free rotations of the molecule backbone in fluid solution resulting in highly efficient nonradiative decay rate, which is consistent with the behaviour of chalcone derivatives reported by Rettig et al.36 The presence of iodine atoms and to a lower extent Br atoms results in a further reduction of fluorescence quantum yields (<1%), which can be explained by the internal heavy atom effect. One observes that the emission maximum wavelength is red-shifted by 28 nm when going from H–H to F–F. Such shift correlates with the bathochromic shift of the electronic UV-vis absorption spectrum resulting in similar Stokes shift for H–H and F–F (3992 cm−1vs. 3710 cm−1, respectively). However, a larger redshift of fluorescence is observed when decreasing the electronegativity of the halogen with an emission going from 584 nm for F–F to 607 nm for I–I. This redshift of the emission leads to a slight increase of the Stokes shift spanning from 4011 cm−1 for Cl–Cl to 4272 cm−1 for I–I.
Solid-state absorption and fluorescence properties
The solid-state absorption spectra were measured on drop cast films. As we noticed in an earlier publication,27 the tentative correlation between the single crystal data and the absorption spectra of thin films can afford valuable insights.21 This approach is based on the consideration of cofacial interactions between nearest neighbours, which represents an oversimplified approach given the manifold interactions in the crystal, yet a reasonable one because the effect of cofacial π–π stacking is supposed to dominate spectral changes. We see from Fig. 4 and Fig. S15 (ESI†) that the absorption spectra reveal rather complex features characteristic of excitonically coupled chromophores.37 None of the spectra measured resemble that of H–H where a strong H-like aggregation was found to occur (Fig. 3a), which is consistent with the X-ray diffraction analysis. Among the X–X series (X = F, Cl, Br, and I), the UV-vis spectrum of Br–Br is interesting because it features a sharp J-like transition band appearing at lower energy. This observation is in agreement with the single-crystal structure of Br–Br that shows brick-wall patterns. The UV-vis spectrum of Cl–Cl, however, is different although this dye has a crystal organization similar to that of the bromo-analogue Br–Br. I–I does not exhibit a J-type aggregation optical signature either, but given the fact that the type of solid-state packing resembles that of H–H this is not surprising. In the case of the hetero-substituted compounds X–Y, it is worth noting that compounds containing the larger halogen atom at the X position (Br–Cl, I–Cl, and I–Br) all display absorption spectra characteristic of J-type aggregates (Fig. S15 in ESI†).Like many borondifluoride complexes of hydroxychalcones,23,27 curcuminoids25,26 and other acetylacetonate derivatives,16 the halogenated dyes investigated here are emissive in the solid state. The fluorescence emission profiles recorded for all dyes were found identical for drop cast polycrystalline thin films and microcrystalline powders (Table 1). The spectra consist of a single broad band maximum spanning from 650 nm to 807 nm. For all the dyes investigated, the solid-state photoluminescence quantum yields determined on polycrystalline samples using a calibrated integration sphere were larger than the values measured in DCM solution (Table 1). We showed previously that restricted intramolecular rotations could be substantially suppressed in related chalcone–BF2 dyes when they were diluted in a PMMA film, which indicated that AIEE could account for the increased emission in the solid state with respect to solution behaviour.27 We thus believe that AIEE is also likely to occur in the di-halogenated analogues investigated here. Unfortunately, this experiment was not possible here because we observed some degradation of the dyes in PMMA solution during the film fabrication process. Within the X–X series, the heavier the halogen element, the longer the emission maximum wavelength. In the case of Cl–Cl, emission occurs in the NIR with a notable quantum yield (24%). Br–Br and I–I are less emissive which is due to quenching by the heavy atom effect. These trends are also observed for X–Y. The comparison between Br–Br, Br–Cl and Cl–Br is interesting because the number and substitution position of the bromine atom determine both the fluorescence quantum yield and the emission maximum wavelength. As described above, the crystal organization is similar for the three compounds but lattice parameters and characteristic distances differ. The replacement of one bromine atom in Br–Br by one chlorine atom at whatever position leads to a more than 2.5-fold increase of fluorescence quantum yield, which is due to a decreased heavy atom effect. However, only Br–Cl emits in the NIR at a wavelength (742 nm) close to that of Br–Br, while Cl–Br fluoresces in the far red (690 nm).
It is also noteworthy that the photoluminescence quantum yields of the borondifluoride complex 2′-hydroxychalcone dyes containing iodine atoms are still high (Table 1) despite the fact they are likely to be subject to intramolecular and intermolecular internal heavy atom effects in the solid state. This is the case of I–Cl and I–Br which display the most red-shifted emissions with acceptable efficiencies despite the cumulative influence of the heavy atom effect and energy gap law. As noticed in the case of UV-vis absorption, an iodine atom located ortho to the 2′-hydroxyl group induces J-aggregation and it is likely that this feature compensates, at least in part, the impact of the two possible quenching channels. In that respect, compound I–Cl is remarkable with a Φf = 5% at 761 nm.
Conclusions
In summary, a series of new solid-state far-red and NIR fluorophores were synthesized. These molecules consist of borondifluoride complexes of 2′-hydroxychalcone cores which contain two halogen atoms on the acetylphenolate moiety. The effect of the nature and position of the halogen on the optical properties was investigated for homo- (X–X) and hetero- (X–Y) disubstituted derivatives, respectively, using UV-vis absorption spectra, fluorescence spectra, DFT calculations, and analyses of single-crystal structures. For all compounds, a fluorescence emission enhancement was clearly observed when passing from the solution (DCM) to the crystal (up to more than 10 fold). Given the known propensity of chalcone derivatives to undergo excited-state quenching via internal conversion in solution, this observation is consistent with the situation in the crystal where the planar structure of the dye molecules and their tight packing restrict molecular rotations and in turn increase the efficiency of radiative deactivation channels. The X-ray structures do not reveal the occurrence of intermolecular halogen–halogen bonds which are expected to further decrease the degrees of rotational freedom in the solid state. As a result, the AIEE process enables fluorescence emission in the red and the NIR with significant fluorescence quantum yields. As such, this can be seen as an aggregation induced NIR emission (AI-NIR-E). However, we also show that the halogenated dyes adopt packing arrangements that differ from those previously reported for boron complexes of 2′-hydroxychalcones. A steric effect associated with the larger size of the halogen atoms results in the formation of J-aggregates which certainly contribute to the emission increase. Another important conclusion of this study is the fact that the use of the heavier halogen atoms (Br, I) is not prohibited in the design of solid-state fluorophores with reasonable emission efficiency.38 This is quite counterintuitive since these heavy atoms are prone to induce efficient spin orbit coupling and to depopulate singlet excited states non-radiatively. The case of Cl–Br and Br–Cl is amazing in the sense that it illustrates how simply switching bromine and chlorine atom positions makes it possible to increase the efficiency of emission in the NIR. Therefore, the solid-state fluorescence properties are not only sensitive to the nature of the halogen atom but also to its position in the molecular backbone. This means that a subtle chemical change strongly affects the solid-state optical properties. Despite the fact that crystal structures of organic molecules are difficult to predict,15 this study provides valuable clues toward the fundamental knowledge of other kinds of fluorophores in which halogen functionalization may help to obtain enhanced solid-state emission in the search for advanced functional materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr M. Giorgi and Dr V. Heresanu for their valuable help in X-ray diffraction investigation of single-crystal and powder samples, respectively.References
- V. Parthasarathy, S. Fery-Forgues, E. Campioli, G. Recher, F. Terenziani and M. Blanchard-Desce, Small, 2011, 7, 3219–3229 CrossRef CAS.
- J. Geng, K. Li, D. Ding, X. Zhang, W. Qin, J. Liu, B. Z. Tang and B. Liu, Small, 2012, 8, 3655–3663 CrossRef CAS.
- M. Wang, G. Zhang, D. Zhang, D. Zhu and B. Z. Tang, J. Mater. Chem., 2010, 20, 1858–1867 RSC.
- Y. Dong, J. W. Y. Lam, A. Qin, J. Liu, Z. Li and B. Z. Tang, Appl. Phys. Lett., 2007, 91, 011111 CrossRef.
- H.-H. Fang, Q.-D. Chen, J. Yang, H. Xia, Y.-G. Ma, H.-Y. Wang and H.-B. Sun, Opt. Lett., 2010, 35, 441–443 CrossRef CAS.
- A. S. D. Sandanayaka, T. Matsushima, F. Bencheikh, K. Yoshida, M. Inoue, T. Fujihara, K. Goushi, J.-C. Ribierre and C. Adachi, Sci. Adv., 2017, 3, e1602570 CrossRef.
- D. H. Kim, A. S. D. Sandanayaka, L. Zhao, D. Pitrat, J. C. Mulatier, T. Matsushima, C. Andraud, J. C. Ribierre and C. Adachi, Appl. Phys. Lett., 2017, 110, 023303 CrossRef.
- M. Shimizu and T. Hiyama, Chem. – Asian J., 2010, 5, 1516–1531 CrossRef CAS.
- K. Albrecht, K. Matsuoka, D. Yokoyama, Y. Sakai, A. Nakayama, K. Fujita and K. Yamamoto, Chem. Commun., 2017, 53, 2439–2442 RSC.
- D. H. Kim, K. Inada, L. Zhao, T. Komino, N. Matsumoto, J. C. Ribierre and C. Adachi, J. Mater. Chem. C, 2017, 5, 1216–1223 RSC.
- C. Li, R. Duan, B. Liang, G. Han, S. Wang, K. Ye, Y. Liu, Y. Li and Y. Wang, Angew. Chem., Int. Ed., 2017, 56, 11525–11529 CrossRef CAS.
- D.-H. Kim, A. D’Aléo, X.-K. Chen, A. S. D. Sandanayaka, D. Yao, L. Zhao, T. Komino, E. Zaborova, G. Canard, Y. Tsuchiya, E. Choi, J. W. Wu, F. Fages, J.-L. Brédas, J.-C. Ribierre and C. Adachi, Nat. Photonics, 2018, 12, 98–104 CrossRef CAS.
- A. D'Aléo, H. M. Sazzad, D. H. Kim, E. Y. Choi, J. W. Wu, C. Canard, F. Fages, J.-C. Ribierre and C. Adachi, Chem. Commun., 2017, 53, 7003–7006 RSC.
- X. Ma, R. Sun, J. Cheng, J. Liu, F. Gou, H. Xiang and X. Zhou, J. Chem. Educ., 2016, 93, 345–350 CrossRef CAS.
- G. M. Day, Crystallogr. Rev., 2011, 11, 3–52 CrossRef.
- A. D’Aléo and F. Fages, Disp. Imaging, 2014, 1, 149–174 Search PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- Y. Hong, J. W. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS.
- D. Oelkrug, A. Tompert, J. Gierschner, H.-J. Egelhaaf, M. Hanack, M. Hohloch and E. Steinhuber, J. Phys. Chem. B, 1998, 102, 1902–1907 CrossRef CAS.
- B.-K. An, J. Gierschner and S. Y. Park, Acc. Chem. Res., 2012, 45, 544–554 CrossRef CAS.
- J. Shi, L. E. Aguilar Suarez, S.-J. Yoon, S. Varghese, C. Serpa, S. Y. Park, L. Lüer, D. Roca-Sanjuán, B. Milián-Medina and J. Gierschner, J. Phys. Chem. C, 2017, 121, 23166–23183 CrossRef CAS.
- A. D'Aléo, D. Gachet, V. Heresanu, M. Giorgi and F. Fages, Chem. – Eur. J., 2012, 18, 12764–12772 CrossRef.
- A. D'Aléo and F. Fages, Photochem. Photobiol. Sci., 2013, 12, 500–510 RSC.
- A. Felouat, A. D'Aléo and F. Fages, J. Org. Chem., 2013, 78, 4446–4455 CrossRef CAS.
- A. D'Aléo, A. Felouat, V. Heresanu, A. Ranguis, D. Chaudanson, A. Karapetyan, M. Giorgi and F. Fages, J. Mater. Chem. C, 2014, 2, 5208–5215 RSC.
- A. D’Aléo, V. Heresanu, M. Giorgi, B. Le Guennic, D. Jacquemin and F. Fages, J. Phys. Chem. C, 2014, 118, 11906–11918 CrossRef.
- U. Mayerhöffer and F. Würthner, Angew. Chem., Int. Ed., 2012, 51, 1–6 CrossRef.
- M. Fourmigué and P. Batail, Chem. Rev., 2004, 104, 5379–5418 CrossRef.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748–7757 RSC.
- P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114–6127 CrossRef CAS.
- J. B. Birks, Photophysics of Aromatic Molecules. Wiley Monographs in Chemical Physics, Wiley-Interscience, New York, 1970 Search PubMed.
- P. Alemany, A. D'Aléo, M. Giorgi, E. Canadell and F. Fages, Cryst. Growth Des., 2014, 14, 3700–3703 CrossRef CAS.
- M. D. Curtis, J. Cao and J. W. Kampf, J. Am. Chem. Soc., 2004, 124, 4318–4328 CrossRef.
- J. Gierschner and S. Y. Park, J. Mater. Chem. C, 2013, 1, 5818–5832 RSC.
- K. Rurack, M. L. Dekhtyar, J. L. Bricks, U. Resch-Genger and W. Rettig, J. Phys. Chem. A, 1999, 103, 9626–9635 CrossRef CAS.
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS.
- U. Mayerhöffer, B. Fimmel and F. Würthner, Angew. Chem., Int. Ed., 2011, 50, 1–5 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1578963–1578967. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8qm00478a |
| This journal is © the Partner Organisations 2019 |