
Simple and sensitive colorimetric detection of a trace amount of 2,4,6-trinitrotoluene (TNT) with QD multilayer-modified microchannel assays†
Tao
Hu
a,
Wen
Sang
a,
Ke
Chen
a,
Hongxi
Gu
b,
Zhonghua
Ni
*a and
Shaoqin
Liu
 *b
*b
aSchool of Mechanical Engineering, and Jiangsu Key Laboratory for Design and Manufacture of Micro–Nano Biomedical Instruments, Southeast University, Nanjing 211189, China. E-mail: nzh2003@seu.edu.cn
bKey Laboratory of Micro-systems and Micro-structures Manufacturing, Ministry of Education, Harbin Institute of Technology, Harbin 150080, China. E-mail: shaoqinliu@hit.edu.cn
First published on 19th October 2018
Abstract
Rapid and selective detection of trace levels of nitroaromatic explosives, in particular, 2,4,6-trinitrotoluene (TNT), is a key challenge for both public security and environmental monitoring. In the present study, we fabricated two colored quantum dot (QD) multilayer-decorated microchannel assays via a layer-by-layer assembly technique. Due to the energy transfer between red QDs and green QDs, the fluorescence intensity of the red QDs significantly enhanced. When the assays were exposed to TNT aqueous solution, the formation of Meisenheimer complexes between the electron-poor aromatic groups of TNT and electron-rich amines on the surface of the red QDs caused fluorescence quenching of the red QDs and partial recovery of the green QDs, thus resulting in multicolor changes of the multilayers from red to yellow to green and then to colorless. The assay has a broad linear range of 10 ppt to 107 ppt and low detection limit (5.24 ppt) for TNT. The specificity of this assay for the detection of TNT is demonstrated against several TNT analogs. Additionally, this technique was successfully applied to detect TNT concentration in different water samples. Due to its simplicity, short detection time, high sensitivity and low cost, the assay is well suited for the detection of ultratrace levels of TNT.
Introduction
The rapid, sensitive and selective detection of small amounts of nitroaromatic explosives, in particular, 2,4,6-trinitrotoluene (TNT), has been a key challenge due to the increasing threat of explosive-based terrorism and the need of environmental monitoring of drinking and waste water.1–3 A great number of elegant methods, including chromatographic separation techniques,4 mass spectrometry,5,6 ion mobility spectrometry,7 electrochemical strategies,8–11 electrochemiluminescence12 and spectroscopic approaches,13–15 have been successfully applied for explosive detection. Although these techniques have some advantages, most of them suffer from limitations such as the requirements for expensive equipment, complex operation, and lack of portability. Furthermore, sophisticated vapor sampling, instrument calibration and pre-concentration procedures have greatly hampered wide applications of the abovementioned methods.16,17 To overcome these limitations, many efforts have been paid to develop sensitive, real-time, portable and cheap detection systems to monitor ultratrace levels of explosives. Among these efforts, fluorescence-quenching detection of nitroaromatic explosives has become a powerful tool due to its low cost, portability and ability for specific and rapid identification.18–30 The fluorescence-quenching technique relies on photo-induced electron transfer from the fluorophore to the nitroaromatic explosive, most of which are strong fluorescence quenchers, causing electron-transfer-mediated fluorescence quenching.25 Due to the high fluorescence yield and the ability to form donor–acceptor complexes with nitroaromatic explosives, pyrene derivatives,31,32 terthiophene derivatives,33,34 and their hybrid materials have been used as fluorophores for the visual detection of nitroaromatic explosives. Moreover, to overcome the disadvantages of organic dyes, fluorescent nanoparticles such as quantum dots (QDs)19,20,22,24,26,29,30 and graphene35 have been used for designing new fluorescence-quenching sensing systems for nitroaromatic explosives. Due to their variable and sharp emission peaks, QDs-based sensing systems lend themselves to multichannel detection of multiple types of explosives in a rapid single fluorometric test.36 However, most fluorescence-quenching sensing systems use changes in the fluorescence intensity of the fluorophores as the indicator. Therefore, when the concentration of explosives is low or the quenching response of explosives toward fluorophores is poor, fluorescence-quenching methods do not have sensitive color changes and are thus semi-quantitative.27Herein, we describe a novel microfluidic platform modified with a QD multilayer for the visual detection of ultratrace levels of TNT. It is based on multilayers comprising two colored QDs: water-soluble 3-mercaptopropionic acid (MPA)-capped CdTe QDs with green emission (green QDs) and L-cysteine-capped CdTe QDs with red emission (red QDs). As shown in Fig. 1a, green QDs and red QDs were sequentially deposited on glass slides using a layer-by-layer (LbL) assembly technique. Due to the Förster resonance energy transfer (FRET) that occurs between the red and green QDs,37–39 the fluorescence intensity of the red QDs was markedly enhanced. Upon exposure to low-concentration nitroaromatic explosives, the electro-poor nitroaromatic groups of the explosives initially react with the electron-rich amines on the surface of the red QDs in the outer layer of the two-colored QD multilayers to form Meisenheimer complexes,40 causing fluorescence quenching of the red QDs and partial recovery of the green QDs (Fig. 1b). By measuring the ratio of the green and red emission intensities, detection limits in parts-per-trillion (ppt) can be achieved. Further increasing the concentration results in the penetration of the nitroaromatic explosives into the inner layer of two-colored QD multilayers. Electron deficient nitro-aromatic explosives can act as electron acceptors for photoexcited electrons of the green QDs, leading to fluorescence quenching. The proposed sensing concept was further integrated with the microchannel array analysis method to generate a visual assay. The as-obtained microfluidic assay can be easily integrated with devices such as cameras and UV lamps, and are highly promising for the realization of portable, real-time, and cheap platforms for the visual monitoring of ultratrace levels of explosives. Thus, such microfluidic chips can be conveniently utilized for rapid, in-field water examination and monitoring.
 | ||
| Fig. 1 (a) Scheme for fabrication of QD multilayers-coated film for visual determination of TNT and the detection mechanism. (b) Schematic illustrations for the quenching mechanism of fluorescence by TNT analyte. (c) Fluorescence spectra of (PEI/PSS)3(PAH/green QDs)10(PAH/PSS)3(PAH/red QDs)5 multilayers deposited on glass substrates. | ||
Experimental section
Materials
TNT at a concentration of 1 mg mL−1 in acetonitrile was purchased from Aladdin (Caution: All nitroaromatic compounds used in the present study are highly explosive materials and should be handled only in small quantities). Cd(ClO4)2·6H2O, Al2Te3 powders and mercaptopropionic acid (MPA) were purchased from Alfa Aesar. L-Cysteine hydrochloride, poly(ethylenimine) (PEI, MW 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), sodium polystyrenesulfonate (PSS, MW 70000), poly(allylamine hydrochloride) (PAH, MW 8000–11000) were purchased from Sigma-Aldrich. Other reagents were commercially available and of analytical reagent grade or the highest available purity and used without further purification. Ultrapure water obtained from a Millipore water purification system (≧18 MΩ, Milli-Q, Millipore) was used in the entire study.
000), sodium polystyrenesulfonate (PSS, MW 70000), poly(allylamine hydrochloride) (PAH, MW 8000–11000) were purchased from Sigma-Aldrich. Other reagents were commercially available and of analytical reagent grade or the highest available purity and used without further purification. Ultrapure water obtained from a Millipore water purification system (≧18 MΩ, Milli-Q, Millipore) was used in the entire study.
Apparatus
UV-vis-NIR absorption measurements were performed using a Hitachi U4100 spectrometer. Fluorescence spectra were recorded using a Fluoromax-4 spectrometer (Horiba Jobin Yvon) with an excitation wavelength of 420 nm. All solutions were loaded in a 10 mL plastic syringe and injected into a microchannel by a programmable syringe pump (KDS201, KD Scientific, Inc.). Transmission electron microscopy (TEM) images were taken by FEI Tecnai G220 S-TWIN transmission electron microscope.Microfluidic chips fabrication
A standard soft lithography technique was used to fabricate the PDMS microfluidic chip. The silicon substrate was prepared by cleaning in a solution of H2SO4/H2O2 (3/1, v/v) at 120 °C for 20 minutes and then dehydrating on a hot plate at 200 °C for 30 minutes. A 50 μm-thick SU-8 2050 layer was spun onto the silicon substrate with a spin speed of 500 rpm for 10 seconds, followed by a spin speed of 3000 rpm for 30 seconds. The substrate was subsequently baked on a hot plate at 65 °C for 2 minutes and at 95 °C for 7 minutes after quiescence for 30 minutes. After the substrate was cooled to room temperature, UV exposure was performed in a UV mask aligner with an exposure power density of 18 mW cm−2 for 50 seconds. The silicon substrate after exposure was treated at 65 °C for 2 minutes and at 95 °C for 7 minutes. After cooling to room temperature, the substrate was immersed in SU-8 Developer with ultrasonic cleaning for 7 minutes to remove the unexposed photoresist. After cleaning with deionized water, the male mold was treated at 200 °C for 30 minutes.PDMS prepolymer was thoroughly mixed with a curing agent at a weight ratio of 10:1 and poured onto the prepared male mold in a petri dish. After curing at 80 °C for 2 hours, the PDMS layer was detached from the silicon wafer and cut into 1.5 cm wide pieces. Two holes with a 0.75 mm diameter were perforated on both ends of each channel successively. After treatment in a plasm cleaner for 5 minutes, both the PDMS layer and glass substrate were brought into contact and bonded together to enclose the microfluidic channel and complete the chip. Finally, the chip was put in an oven at 120 °C for 3 hours to enhance the bonding strength of PDMS and glass substrate.
TNT determination
The procedure performed for TNT determination on a glass substrate sensor was as follows. (1) The sensor was first incubated with aqueous TNT solutions of different concentration for 10 min. (2) The sensor was taken off, dried by N2 flow and then observed via fluorescence spectroscopy or directly by the naked eye under the UV light.The procedure for TNT determination using a microfluidic chips was almost same as that with glass substrates: TNT aqueous solutions of different concentration were filled in different microchannels using an injection pump or an injection syringe and incubated (no flow) for 5 min, and then observed by naked eyes under UV light.
Results and discussion
The TNT visual microfluidic platform, illustrated in Fig. 1a, consists of two colored QD multilayer-coated microchannels, which were fabricated through the stepwise deposition of green QDs or red QDs with polyelectrolyte (the detailed information of green QDs or red QDs are shown in Fig. S1 and S2 (ESI†); green QDs show a fluorescence maximum at 520 nm and red QDs show a fluorescence peak at 620 nm). We initially assembled 10 bilayers of PAH/green QDs unit, and then assembled 3 bilayers of PAH/PSS and 5 bilayers of PAH/red QDs onto it to obtain a two color-emitting thin film. In order to control the response of the two color-emitting thin film toward different concentrations of TNT, the PAH/green QD and PAH/PSS bilayers were built in 1 mg mL−1 positively charged PAH containing 1 M NaCl, while the PAH/red QD bilayers were fabricated in 1 mg mL−1 PAH without NaCl. Moreover, 3 bilayers of PAH/PSS were further introduced between the 10 bilayers of PAH/green QDs and 5 bilayers of PAH/red QDs to avoid red QD penetration into the PAH/green QD multilayers. Therefore, the inner PAH/green QD layers are thicker and denser than the outer PAH/red QDs layers. Thus, the permeability of TNT through the inner layers could be limited.41,42The assembly processes were monitored by UV-vis absorption spectroscopy and fluorescence spectroscopy (Fig. S3 in ESI†). UV-vis absorption spectra show that the intensity of the absorption band at 500 nm correlates linearly with the bilayer number x. The intensity of the luminescence peak with a maximum at ∼520 nm, attributed to the green QDs, displays a linear increase as a function of x for the (PEI/PSS)3(PAH/green QDs)x (solid line in Fig. S3b, ESI†). Both UV-vis absorption and fluorescence spectra are indicative of a stepwise and regular deposition procedure with almost equal amounts of green CdTe QDs incorporated in each cycle. However, fluorescence spectra of (PEI/PSS)3(PAH/green QDs)10(PAH/PSS)3(PAH/red QDs)y multilayers reveal that the intensity of the sharp peak at ∼520 nm, attributed to the (PAH/green QDs)10 unit, exhibits a consistent decrease, while that at ∼620 nm, attributed to red CdTe QDs shows, a gradual increase along with y (dashed line in Fig. S3b, ESI†). It should be noted that the fluorescence intensity at ∼620 nm for the (PEI/PSS)3(PAH/green QDs)10(PAH/PSS)3(PAH/red QDs)5 multilayers is almost 2.5 times that at ∼520 nm (Fig. 1b), which is probably attributed to energy transfer from the PAH/green QDs unit to the PAH/red QDs.43 The FRET efficiency of (PEI/PSS)3(PAH/green QDs)10(PAH/PSS)3(PAH/red QDs)y increases with y, and is calculated as 11.7%, 21.6%, 31.7%, 42.4% and 45.6% for y = 1–5, respectively. The thickness of the multilayer film was characterized by atomic force microscopy (AFM). As shown in Fig. S4 (ESI†), the thickness of the QD multilayers-coated film was about 45 nm, which is consistent with the theoretically calculated values.
We first examined the response of (PEI/PSS)3(PAH/green QDs)10(PAH/PSS)3(PAH/red QDs)5 multilayers deposited on glass substrates toward low concentration TNT. TNT itself does not exhibit fluorescence in the wavelength range of 400–700 nm with 365 nm as the excitation wavelength (Fig. S5, ESI†). Fig. 2a shows the fluorescence emission spectra of (PEI/PSS)3(PAH/green QDs)10(PAH/PSS)3(PAH/red QDs)5 multilayers deposited on glass substrates in the presence of different concentrations of TNT at 365 nm excitation. It can be seen that the multilayers exhibit a fast and sensitive response to TNT. As shown by the green curve in Fig. 2a, after the multilayer is exposed to 10 ppt TNT for 5 min, a sharp quenching in fluorescence intensity at ∼620 nm (18%) is observed, whereas the intensity at ∼520 nm increases. Upon increasing the concentration of TNT to 100 ppt, the fluorescence intensity at ∼620 nm of the multilayers gradually quenches, showing 40% quenching. At the same time, the fluorescence intensity at ∼520 nm shows a consistent increase. The fluorescence quenching of the red emission can be attributed to the formation of Meisenheimer complexes, arising from covalent bonding between the nucleophilic cysteine grafted on the surface of red QDs and electron-deficient nitroaromatic compounds.44 Analysis of 13C NMR spectra of a mixture of 2 mM TNT and 6 mM cysteine showed that the 12C signals of cysteine are shifted upfield by approximately 8.5 ppm and the C1 signal of TNT moves from 98.7 to 73 ppm (Fig. S6, ESI†), confirming that TNT forms a Meisenheimer complex with cysteine.40 The change in the luminescence color under UV irradiation of the multilayers after exposure to TNT are displayed in Fig. 2b. Even ultratrace levels of TNT result in a distinct color change, and the color of the multilayers varies from red (10 ppt) to yellow (104 ppt) and then to green (105 ppt) with the increase in TNT concentration. Furthermore, it can be found that the intensity ratio of green QDs to red QDs increases linearly with the logarithm of the TNT concentration in the range of 10–105 ppt (Fig. 2c). The limit of detection was calculated to be 5.24 ppt using the formula 3σ/slope. The fluorescence quenching efficiency of TNT on (PEI/PSS)3(PAH/green QDs)10(PAH/PSS)3(PAH/red QDs)5 multilayers was analyzed using the Stern–Volmer equation:
| I0/I = KS–V[TNT] + 1 | (1) |
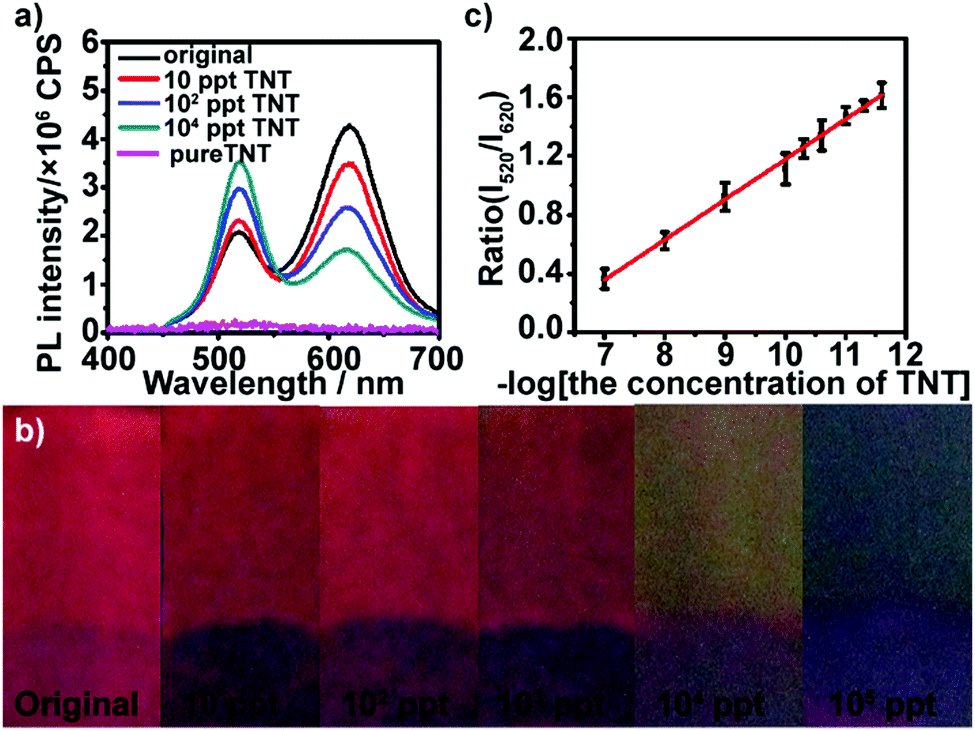 | ||
| Fig. 2 (a) FL spectra of (PEI/PSS)3(PAH/green QDs)10(PAH/PSS)3(PAH/red QDs)5 multilayers before and after the addition of different concentrations of TNT: 0 (black curve), 10 (red curve), 102 (blue curve) and 104 ppt (dark cyan curve) and pure TNT solution (magenta curve). (b) Photo images of (PEI/PSS)3(PAH/green QDs)10(PAH/PSS)3(PAH/red QDs)5 multilayers before and after the addition of different concentrations of TNT. (c) Dependence of the fluorescence emission intensity ratio (I520/I620) of QD multilayers on the logarithm of TNT concentration. | ||
The multilayers also respond to TNT in high concentration. At high concentration conditions, the red QDs were almost quenched by TNT, and more TNT could permeate the film and continue quenching the green QDs gradually with the increase in concentration. As shown in Fig. 3a, after the multilayer is exposed to 5 × 105–107 ppt TNT for 5 min, the fluorescence at ∼520 nm is gradually quenched, while the fluorescence intensity at ∼620 nm of the multilayers remains constant. Furthermore, it can be found that the color of the multilayers varies from dark green (5 × 105 ppt) until total quenching (107 ppt) with the increase in TNT concentration (Fig. 3b). The green fluorescence quenching can be attributed to electron-transfer between TNT and photoexcited electrons of the green QDs. The reduction potential of TNT is −0.7 V versus NHE,45 which makes it a good electron acceptor for photoexcited electrons from the green QDs (Fig. S7, ESI†). The intensity ratio of the red QDs to green QDs also had a linear relationship with the logarithm of TNT concentration in the range from 5 × 105 to ∼107 ppt (Fig. 3c).
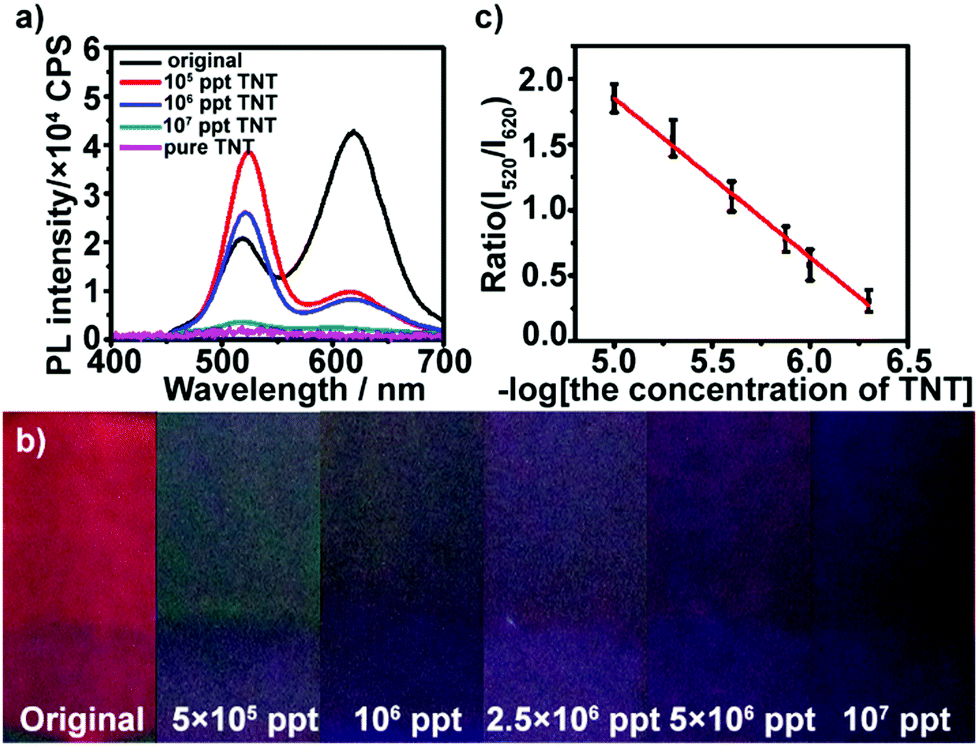 | ||
| Fig. 3 (a) FL spectra of (PEI/PSS)3(PAH/green QDs)10(PAH/PSS)3(PAH/red QDs)5 multilayers before and after addition of TNT with different concentration: 0 (black curve), 105 (red curve), 106 (blue curve) and 107 ppt (dark cyan curve) and pure TNT solution (magenta curve). (b) Photo images of (PEI/PSS)3(PAH/green QDs)10(PAH/PSS)3(PAH/red QDs)5 multilayers before and after the addition of different concentration of TNT. (c) Dependence of the fluorescence emission intensity ratio (I520/I620) of QD multilayers on the logarithm of TNT concentration. | ||
Encouraged by the higher response and distinct color change of (PEI/PSS)3(PAH/green QDs)10(PAH/PSS)3(PAH/red QDs)5 multi-layers toward TNT, we fabricated QD multilayer-modified microchannel visual assays. The combination of QD multilayers with microchannel chips could not only decrease the sample volume and interference from operation, but also achieve multiplexing, fast analysis and improved storage stability of the sensing system.46–50 As depicted in Fig. 4a, the (PEI/PSS)3(PAH/green QDs)10(PAH/PSS)3(PAH/red QDs)5 multilayers was deposited stepwise on the microchannel chips using a similar approach as the glass substrates. The diameter of microchannels is 100 μm. The as-prepared QD multilayer-coated microchannel fluoresces red color (Fig. S8, ESI†). However, after the QD multilayers-coated microchannels were impregnated with TNT aqueous solution of different concentrations, the luminescence color change of the microchannels was triggered in 5 min. As shown in Fig. 4b, the addition of TNT solution leads to a luminescence color shift from red to orange to green and then to colorless as compared with that observed for the control. The distinct multicolor changes at a low concentration of 10 ppt can be easily identified visually. It should be pointed out that the limit of detection obtained with the (PEI/PSS)3(PAH/green QDs)10(PAH/PSS)3(PAH/red QDs)5 multilayer-coated microfluidic chips is much lower than that obtained with previously reported QD systems (Table S1 in ESI†). The above results indicated that that the ability to monitor TNT was improved in terms of the limit of detection, linear range and colorimetric output using dual-color QD multilayers.
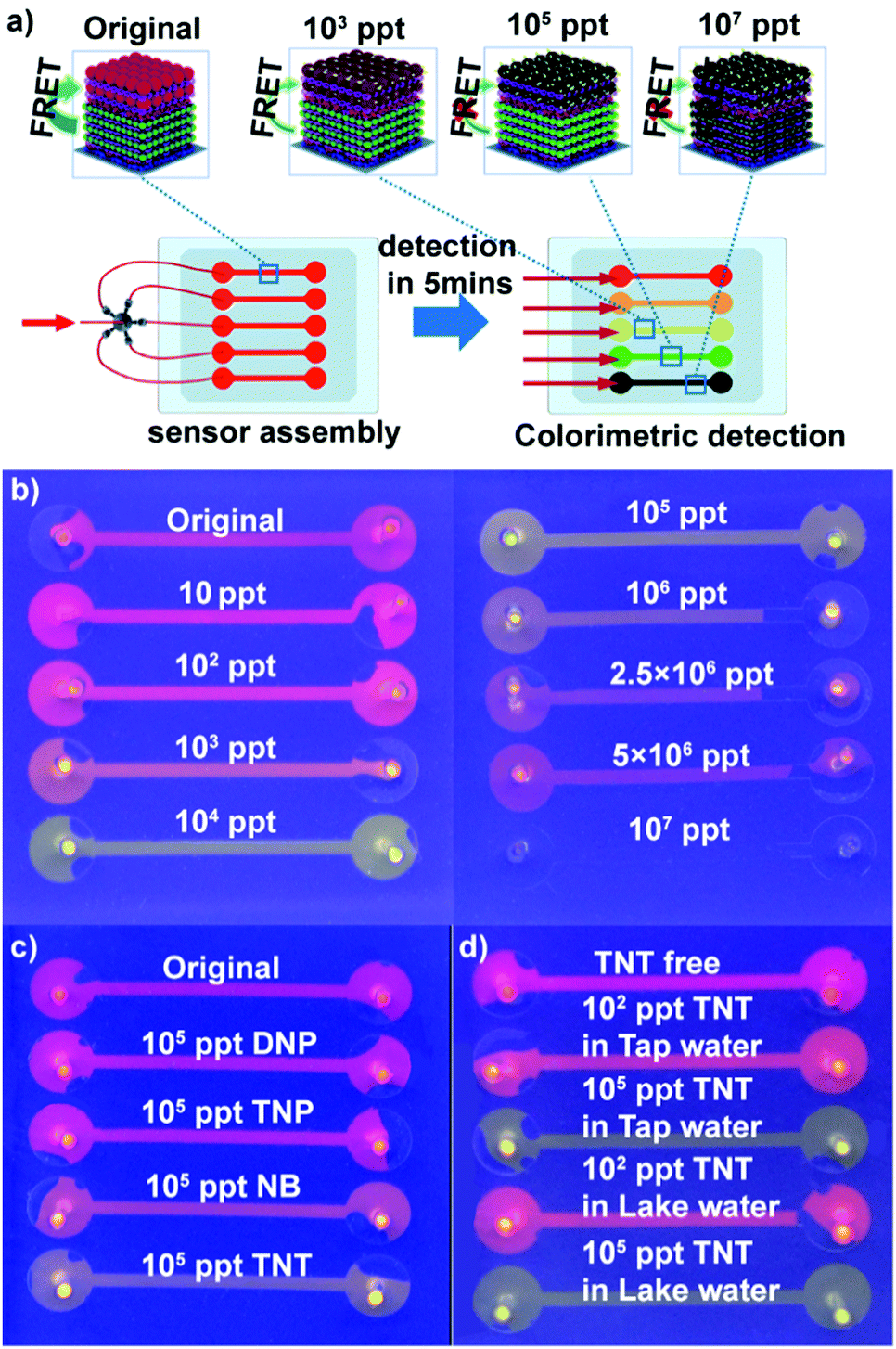 | ||
| Fig. 4 (a) Assembly process of (PEI/PSS)3(PAH/green QDs)10(PAH/PSS)3(PAH/red QDs)5 multilayers-based microchannel assays. (b) Photographs of microchannel assays before and after exposure to TNT of difference concentration. The photo was taken under a 365 nm UV lamp. (c) Response of (PEI/PSS)3(PAH/green QDs)10(PAH/PSS)3(PAH/red QDs)5 multilayers-based microchannel assays toward different TNT analogs. The concentration of TNT analogs is 105 ppt. (d) (PEI/PSS)3(PAH/green QDs)10(PAH/PSS)3(PAH/red QDs)5 multilayers-based microchannel assays for visual determination of TNT in real samples. | ||
To evaluate the specificity of the obtained QD multilayers-coated microchannel assays for TNT detection, control experiments were conducted using other nitro compounds. The experimental results show that in contrast to TNT, 2,4-dinitropheno (DNP), 2,4,6-trinitrophenol (TNP) and nitro-benzene (NB) exhibit negligible color change, even when their concentration is as high as 105 ppt (Fig. 4c). The lower quenching efficiency of DNP and NB could be explained by their lower electron accepting character due to the presence of nitro groups.45 However, although 2,4,6-TNP had enough nitro groups, the formation of intermolecular hydrogen bonding between the hydroxyl and nitro groups may prevent nitrophenol bonding to the cysteine monolayer grafted on the red QDs; hence, TNP had lower quenching efficiency.9,19,24,51 This experiment confirms the very high specificity of the QD multilayers-coated microchannel assay toward TNT.
Moreover, the storage stability of our QD multilayers-coated microchannel assays was investigated. After the assays were stored in air for 4 months, we did not observe any change in the fluorescence color of the assay (Fig. S9, ESI†). This result indicates that the LbL structure in the microfluidic device provides a favorable microenvironment to maintain the photochemical properties of QDs, which will be beneficial for the commercial application of the QD multilayers-coated microchannel assays.
We next used the QD multilayers-coated microchannel assays to detect TNT in different environmental water samples. TNT of a wide range of concentration was spiked into tap water and Jiulong Lake water (Nanjing, JiangSu, China). As shown in Fig. 4d, it is clearly found that TNT free tap and lake water did not cause any color change, indicating that the real water samples had no quenching effect on the assays. Moreover, the presence of common metal ions has a negligible effect on the fluorescence of the QD multilayers-coated microchannel assays. Fig. S10 (ESI†) shows the response of (PEI/PSS)3(PAH/green QDs)10(PAH/PSS)3(PAH/red QDs)5 multilayers-based microchannel assays toward different common ions. It can be found that the presence of 50 ppm K+, 50 ppm Na+, 10 ppm Cu2+, 10 ppm Ca2+, 10 ppm Fe3+, 10 ppm Mg2+, 1 ppm Cd2+, 1 ppm Pd2+, and 1 ppm Hg2+ did not cause distinct fluorescent quenching. In contrast, the presence of 105 ppt TNT causes a color shift from red to green. It is noteworthy that the color change of the assays for TNT solution and different TNT analogs of the same concentration is identical, regardless of whether pure water, tap water or lake water (Fig. 4d and Fig. S11, ESI†). Thus, these results demonstrate that our present assay can successfully detect TNT.
Conclusions
In conclusion, a simple and rapid QDs multilayers-coated microchannel assay has been successfully developed to monitor and measure TNT. Compared with conventional QD-based fluorescence quenching systems, our proposed assay was constructed from two colored QDs. This system provided a rapid and distinctive colorimetric readout for visual detection of ultratrace levels of TNT, with a limit of detection of 5.24 ppt. The microchannel nature of the assay has advantages of less sample volume, multiple analysis and good storage stability. The proposed technology has the ability to be fabricated into inexpensive, portable, easy-to-use and disposable monitoring systems for explosive residues in water, with environmental and security applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grant No. 51605088, 51372054, 51505083), the Natural Science Foundation of Jiangsu Province (No. BK20150600), and the Fundamental Research Funds for the Central Universities (2242016K41022).Notes and references
- R. J. Colton and J. N. Russell, Science, 2003, 299, 1324–1325 CrossRef CAS.
- J. I. Steinfeld and J. Wormhoudt, Annu. Rev. Phys. Chem., 1998, 49, 203–232 CrossRef CAS PubMed.
- J. Yinon, TrAC, Trends Anal. Chem., 2002, 21, 292–301 CrossRef CAS.
- D. S. Moore, Rev. Sci. Instrum., 2004, 75, 2499–2512 CrossRef CAS.
- N. L. Sanders, S. Kothari, G. Huang, G. Salazar and R. G. Cooks, Anal. Chem., 2010, 82, 5313–5316 CrossRef CAS.
- Y. Zhang, X. Ma, S. Zhang, C. Yang, Z. Ouyang and X. Zhang, Analyst, 2009, 134, 176–181 RSC.
- M. Tam and H. H. Hill, Anal. Chem., 2004, 76, 2741–2747 CrossRef CAS.
- S. Hrapovic, E. Majid, Y. Liu, K. Male and J. H. Luong, Anal. Chem., 2006, 78, 5504–5512 CrossRef CAS.
- M. Riskin, R. Tel-Vered, T. Bourenko, E. Granot and I. Willner, J. Am. Chem. Soc., 2008, 130, 9726–9733 CrossRef CAS.
- J. Wang, S. B. Hocevar and B. Ogorevc, Electrochem. Commun., 2004, 6, 176–179 CrossRef CAS.
- H. X. Zhang, A. M. Cao, J. S. Hu, L. J. Wan and S. T. Lee, Anal. Chem., 2006, 78, 1967–1971 CrossRef CAS.
- W. Qi, M. Xu, L. Pang, Z. Liu, W. Zhang, S. Majeed and G. Xu, Chem. – Eur. J., 2014, 20, 4829–4835 CrossRef CAS.
- D. R. Shankaran, K. V. Gobi, T. Sakai, K. Matsumoto, K. Toko and N. Miura, Biosens. Bioelectron., 2005, 20, 1750–1756 CrossRef CAS.
- J. M. Sylvia, J. A. Janni, J. Klein and K. M. Spencer, Anal. Chem., 2000, 72, 5834–5840 CrossRef CAS.
- B. Zachhuber, G. Ramer, A. Hobro and B. Lendl, Anal. Bioanal. Chem., 2011, 400, 2439–2447 CrossRef CAS.
- D. S. Moore, Rev. Sci. Instrum., 2004, 75, 2499–2512 CrossRef CAS.
- S. J. Toal and W. C. Trogler, J. Mater. Chem., 2006, 28, 2871–2883 RSC.
- J. S. Yang and T. M. Swager, J. Am. Chem. Soc., 1998, 120, 11864–11873 CrossRef CAS.
- C. Carrillo-Carrión, B. M. Simonet and M. Valcárcel, Anal. Chim. Acta, 2013, 792, 93–100 CrossRef PubMed.
- E. R. Goldman, I. L. Medintz, J. L. Whitley, A. Hayhurst, A. R. Clapp, H. T. Uyeda, J. R. Deschamps, M. E. Lassman and H. Mattoussi, J. Am. Chem. Soc., 2005, 127, 6744–6751 CrossRef CAS.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC.
- B. Liu, C. Tong, L. Feng, C. Wang, Y. He and C. Lü, Chem. – Eur. J., 2014, 20, 2132–2137 CrossRef CAS.
- Y. Ma, S. Xu, S. Wang and L. Wang, TrAC, Trends Anal. Chem., 2015, 67, 209–216 CrossRef CAS.
- R. Tu, B. Liu, Z. Wang, D. Gao, F. Wang, Q. Fang and Z. Zhang, Anal. Chem., 2008, 80, 3458–3465 CrossRef CAS.
- Y. Salinas, R. Martínez-Máñez, M. D. Marcos and F. Sancenón, Chem. Soc. Rev., 2012, 41, 1261–1296 RSC.
- R. C. Stringer, S. Gangopadhyay and S. A. Grant, Anal. Chem., 2010, 82, 4015–4019 CrossRef CAS.
- X. Sun, Y. Wang and Y. Lei, Chem. Soc. Rev., 2015, 44, 8019–8061 RSC.
- S. J. Toal and W. C. Trogler, J. Mater. Chem., 2006, 16, 2871–2883 RSC.
- Y. Xia, L. Song and C. Zhu, Anal. Chem., 2011, 83, 1401–1407 CrossRef CAS.
- K. Zhang, H. Zhou, Q. Mei, S. Wang, G. Guan, R. Liu, J. Zhang and Z. Zhang, J. Am. Chem. Soc., 2011, 133, 8424–8427 CrossRef CAS.
- S. Zhang, L. Ding, F. Lü, T. Liu and Y. Fang, Spectrochim. Acta, Part A, 2012, 97, 31–37 CrossRef CAS.
- I. S. Kovalev, O. S. Taniya, N. V. Slovesnova, G. A. Kim, S. Santra, G. V. Zyryanov, D. S. Kopchuk, A. Majee, V. N. Charushin and O. N. Chupakhin, Chem. – Asian J., 2016, 11, 758–781 CrossRef.
- T. Liu, L. Ding, K. Zhao, W. Wang and Y. Fang, J. Mater. Chem., 2012, 22, 1069–1077 RSC.
- E. V. Verbitskiy, A. A. Baranova, K. I. Lugovik, M. Z. Shafikov, K. O. Khokhlov, E. M. Cheprakova, G. L. Rusinov, O. N. Chupakhin and V. N. Charushin, Anal. Bioanal. Chem., 2016, 408, 4093–4101 CrossRef CAS.
- L. Fan, Y. Hu, X. Wang, L. Zhang, F. Li, D. Han, Z. Li, Q. Zhang, Z. Wang and L. Niu, Talanta, 2012, 101, 192–197 CrossRef CAS.
- W. J. Peveler, A. Roldan, N. Hollingsworth, M. J. Porter and I. P. Parkin, ACS Nano, 2015, 10, 1139–1146 CrossRef.
- R. Liang, S. Xu, D. Yan, W. Shi, R. Tian, H. Yan, M. Wei, D. G. Evans and X. Duan, Adv. Funct. Mater., 2012, 22, 4940–4948 CrossRef CAS.
- Y. W. Lin, W. L. Tseng and H. T. Chang, Adv. Mater., 2006, 18, 1381–1386 CrossRef CAS.
- W. K. Bae, J. Kwak, J. Lim, D. Lee, M. K. Nam, K. Char, C. Lee and S. Lee, Nano Lett., 2010, 10, 2368–2373 CrossRef CAS.
- R. J. Spear, W. P. Norris and R. W. Read, Tetrahedron Lett., 1983, 24, 1555–1558 CrossRef CAS.
- A. A. Antipov and G. B. Sukhorukov, Adv. Colloid Interface Sci., 2004, 111, 49–61 CrossRef CAS.
- L. Krasemann and B. Tieke, Langmuir, 2000, 12, 187–193 Search PubMed.
- B. Qin, H. Chen, H. Liang, L. Fu, X. Liu, X. Qiu, S. Liu, R. Song and Z. Tang, J. Am. Chem. Soc., 2010, 132, 2886–2888 CrossRef CAS.
- F. Fant, A. De Sloovere and K. Matthijsen, Environ. Pollut., 2001, 111, 503–507 CrossRef CAS.
- D. T. Mcquade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537–2574 CrossRef CAS.
- M. L. Chabinyc, D. T. Chiu, J. C. McDonald, A. D. Stroock, J. F. Christian, A. M. Karger and G. M. Whitesides, Anal. Chem., 2001, 73, 4491–4498 CrossRef CAS.
- A. J. Demello, Nature, 2006, 442, 394–402 CrossRef CAS.
- S. Haeberle and R. Zengerle, Lab Chip, 2007, 7, 1094–1110 RSC.
- P. E. Sheehan and L. J. Whitman, Nano Lett., 2005, 5, 803–807 CrossRef CAS.
- V. Srinivasan, V. K. Pamula and R. B. Fair, Lab Chip, 2004, 4, 310–315 RSC.
- S. F. Xu, H. Z. Lu, J. H. Li, X. L. Song, A. X. Wang, L. X. Chen and S. B. Han, ACS Appl. Mater. Interfaces, 2013, 5, 8146–8154 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S11 and Table S1. See DOI: 10.1039/c8qm00430g |
| This journal is © the Partner Organisations 2019 |