
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvatochromic dual luminescence of Eu–Au dyads decorated with chromophore phosphines†
Andrey
Belyaev
 a,
Sofia O.
Slavova
b,
Igor V.
Solovyev
c,
Vladimir V.
Sizov
c,
Janne
Jänis
a,
Elena V.
Grachova
*c and
Igor O.
Koshevoy
*a
a,
Sofia O.
Slavova
b,
Igor V.
Solovyev
c,
Vladimir V.
Sizov
c,
Janne
Jänis
a,
Elena V.
Grachova
*c and
Igor O.
Koshevoy
*a
aDepartment of Chemistry, University of Eastern Finland, Joensuu, 80101, Finland. E-mail: igor.koshevoy@uef.fi
bInstitute of General and Inorganic Chemistry, Bulgarian Academy of Sciences, Sofia, 1113, Bulgaria
cInstitute of Chemistry, St. Petersburg State University, Universitetskii pr. 26, 198504, St. Petersburg, Russia. E-mail: e.grachova@spbu.ru
First published on 1st November 2019
Abstract
Phosphine ligands, containing chromophore substituents –π-spacer–NPh2 (π-spacer = biphenyl, L1; naphthalene-ethynylphenyl, L2; and ethynyl-(phenylethynyl)anthracene, L3), were used to generate the corresponding gold(I) alkynyl complexes Au1–Au3 (alkynyl = ethynylphenyl-2,2′-bipyridine, epbpy). These compounds demonstrate intense fluorescence, which originates from the bipolar donor–π-acceptor systems of coordinated phosphines with negligible contribution from the epbpy fragment. Due to the charge transfer characteristic of the excited state, Au1–Au3 reveal significant emission solvatochromism (particularly discernible for Au2, 428 nm in cyclohexane → 580 nm in acetonitrile). The bipyridine moiety of Au1–Au3 was utilized for binding these metalloligands to the {Eu(tta)3} red emitter (tta = 3-thenoyltrifluoroacetonate) to give a family of novel dyads Au1Eu–Au3Eu. Incomplete energy transfer from L1–L3 to lanthanide ions, which are primarily sensitized by diketonate ligands, leads to dual luminescence. Analogous to the parent complexes Au1–Au3, the fluorescence component of the dyads is highly sensitive to the solvent polarity that provides great opportunities for color tuning, including white light generation.
Introduction
The construction of tunable multicolor photoluminescent compounds and single molecule-based materials, particularly those capable of efficient white-light emission, continues to be an appealing research topic. From practical viewpoint, such species with panchromatic luminescence could significantly advance the fabrication of organic optoelectronic devices (white light emitting diodes and electrochemical cells), and they are highly desired as a gain medium for wide-spectra lasers in the development of new sensing techniques and photoswitches.1 For example, in order to achieve white light emission, a chromophore molecule should generate two complementing bands (e.g. blue and orange) or produce a combination of pure red, green and blue colors. The implementation of multicolor emission from a single molecule or a single component material remains a substantial challenge for molecular design and engineering.Recent progress achieved for panchromatic pure organic and metal–organic luminophores illustrates that multi-band single-molecular emission can be realized via different physical phenomena. Among them, it is possible to identify excited-state intramolecular proton transfer,2 aggregate formation, and conformational change to induce simultaneous locally excited and charge-transfer luminescence3 including the combination of monomer and excimer emission,4 stimulation of dual fluorescence and phosphorescence.5 Nevertheless, these systems often suffer from a lack of control and tuning of their optical properties. An extensively utilized alternative approach implies the binding of two or more individual emitters in one molecular entity through a spacer, thus forming dyads or multi-chromophore assemblies. The reported single-molecule assemblies of this sort represent diverse combinations of constituting components, which may comprise covalently linked organic dyes,6 d-block chromophores connected to organic fluorophores,7 and homo- or heteronuclear d–d,8 d–f9 and f–f10 dyads and triads. In this regard, rare earth metal ions (e.g. Eu(III), Tb(III), Gd(III), Dy(III), and Sm(III)) distinctly stand out in the design of versatile supramolecular aggregates showing multicolor luminescence.9a,c,e Pure colors and very sharp emission bands, long lifetimes and large Stokes shifts of Ln(III) f → f transitions are beneficial characteristics for responsive molecular materials, i.e. chemosensors,10b,11 molecular thermometers,12 biological probes and imaging agents.9c,13 Although the f → f transitions are parity-forbidden by the Laporte rule,14 the excited states of lanthanides can be populated via the energy transfer from, e.g. the adjacent d-metal chromophore.15 This concept was successfully used by De Cola et al. in 2005 who described the d–f heterometallic assembly with panchromatic emission, where the sensitization of the Eu(III) center by means of a partial energy transfer from the blue emitting cyclometalated Ir(III) fragment to the red Eu(III) emitter generates white light with a quantum yield of 7% in solution.16 Later, the utilization of various d-chromophores (Pt(II),17 Ir(III),12d,18 Os(II),19 Ru(II),19,20 Re(I),17b,21etc.) as antennae for lanthanide energy acceptors has rapidly expanded the family of d–f dyads. Analysis of their photophysical behavior indicates that d → f excitation transfer might occur through the conjugated system or by means of the Förster and Dexter mechanisms; some back donation to the d-containing fragment cannot be excluded.
Ultimately, incomplete energy transfer from the organic chromophore to the f-center has been exploited to produce dual or multiple emissions from single molecules.22 Different signals, originating from the f element and the fluorescent motif of these luminophores, can demonstrate distinct responses to the environmental changes that have been applied for sensing purposes.22b,23 A delicate balance between the lanthanide emission and fluorescence in the visible region was demonstrated by Duan et al. in 2009 via rational tailoring of the Eu(III) diketonate unit to the coumarin–rhodamine 6G fluorescent moiety. The resulting dye demonstrates excitation-dependent luminescence in solution and in the film with an overall quantum yield of nearly white emission reaching 2.5%.24 Nonetheless, during the last decade, there has been a limited number of subsequent reports on multicolor luminescence from lanthanide complexes decorated with ancillary fluorophore antenna,25 especially those capable of dynamic emission changes.26
Herein, we describe a facile strategy to prepare dual luminescent compounds by incorporating the beta-diketonate Eu(III) red emissive fragment into the bipyridine-functionalized alkynyl gold(I)–phosphine complexes, which serve as fluorescent metalloligands. For this purpose, we employ phosphines containing extended π-spacers decorated with the diphenylamine donor group. Their coordination to the Au(I) ion converts the PR3 moiety into an electron acceptor and results in a bipolar D–π-A architecture of the pendant chromophore. This feature, in turn, induces an intramolecular charge transfer in the excited state, which is highly sensitive to the polarity of the medium and provides a remarkable tunability of the emission color of the novel dyads.
Results and discussion
Synthesis and characterization
The design of the phosphine building blocks relies on the Ph2P–π-spacer–NPh2 configuration, where the length of the π-spacer is expected to vary the energy of the emission by influencing the photoinduced intramolecular charge transfer (ICT). This process can be conventionally activated in the bipolar phosphine ligands upon the oxidation of the phosphorus atom or its bonding to the Au(I) center.27 For this purpose, we have prepared compounds L128–L3 in good yields from the bromo–π-spacer–NPh2 (π-spacer = biphenyl,29 naphthalene-ethynylphenyl30) and ethynylanthracenyl-ethynyl-diphenylaniline precursors (Scheme S1†). Metalloligands Au1–Au3 were obtained by the depolymerisation of [Au(epbpy)]n (epbpy = 5-(4-ethynylphenyl)-2,2′-bipyridine)31 with phosphines L1–L3 under oxygen-free conditions, which is a common synthetic approach to alkynyl gold(I)–phosphine complexes.5e,32 On the other hand, the binding of the Eu(tta)3 (tta = 3-thenoyltrifluoroacetonate) luminescent motif to the diimine functions occurs under mild conditions12d,25d,e and was successfully utilized to convert gold complexes Au1–Au3 into dyads Au1Eu–Au3Eu, which were isolated in moderately good yields (Scheme 1; for details, see the ESI†). Compounds Au1–Au3 and Au1Eu–Au3Eu are nearly colorless (bearing L1), yellow (L2) and red (L3) powders which are stable in the solid state and in solution under ambient conditions, while phosphines L1–L3 are easily oxidized in air when dissolved. Metalloligands and Au–Eu dyads are readily soluble in organic solvents such as dichloromethane, acetone, chloroform and toluene, sparingly to moderately in diethylether, acetonitrile and cyclohexane, and are practically insoluble in protic media (methanol, ethanol, and water).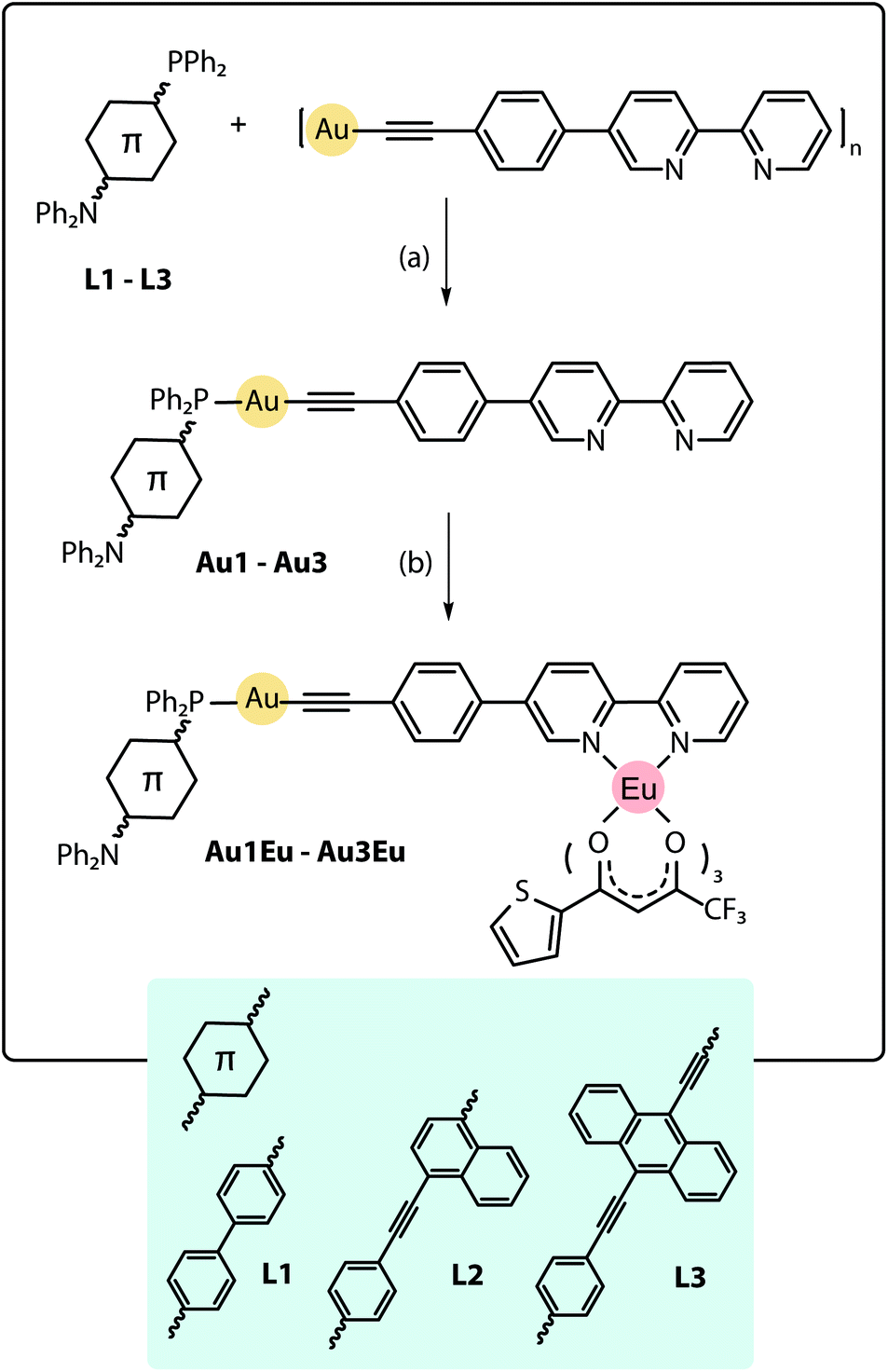 | ||
| Scheme 1 Synthesis of the Au–Eu phosphine-based dyads Au1Eu–Au3Eu, (a) dichloromethane, 1 h, 298 K, yields 86–90%; (b) Eu(tta)3·2H2O, dichloromethane, 1 h, 298 K, yields 72–83%. | ||
The 31P{1H} NMR spectra of Au1–Au3 exhibit singlet peaks each in the range from 41.7 to 16.5 ppm, which are significantly shifted to the low field region compared to the free phosphines L1–L3 (δ = −6.2 to −32.8 ppm) and therefore confirm the coordination of –PPh2 groups to the Au(I) centers. Upon binding to the Eu atoms, the phosphorus NMR signals for dyads Au1Eu–Au3Eu virtually do not change positions with respect to the predecessor metalloligands Au1–Au3.
The 1H NMR spectroscopic patterns for Au1–Au3 (Fig. S1†) are completely compatible with the proposed molecular structures (Scheme 1). The corresponding spectra of the bimetallic compounds reveal a clear effect of introducing the paramagnetic Eu(III) ion into the bipyridine moiety, which is manifested by the visible broadening of the signals and a large low field shift of the resonances (δ = 14.8 and 14.4 ppm) corresponding to the diimine protons nearest to the N atoms (Fig. S2†). Furthermore, these spectra reaffirm the presence of three tta ligands around the Eu center.
Mass spectrometric measurements were performed using ESI+ and atmospheric pressure photoionization (APPI) techniques. For gold complexes, the dominating signals correspond to the protonated molecular ions at m/z 958.26 (Au1), 1032.28 (Au2) and 1106.29 (Au3) (Fig. S3).† In the case of Au–Eu successors, however, only the complicated mixtures of defragmentation species were observed.
The solid state FTIR spectra of Au1Eu–Au3Eu demonstrate intense absorption bands at ∼1600 cm−1, which are attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of the coordinated thenoyltrifluoroacetone (Fig. S4†).25d,e Additionally, the spectrum of Au1Eu exhibits a weak band at ∼2114 cm−1 assigned to the C
O stretching vibrations of the coordinated thenoyltrifluoroacetone (Fig. S4†).25d,e Additionally, the spectrum of Au1Eu exhibits a weak band at ∼2114 cm−1 assigned to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C vibration. Not surprisingly, Au2Eu and Au3Eu compounds also demonstrate distinct stretching frequencies between 2197 and 2114 cm−1, which agree with the presence of several types of acetylene groups in these molecules.
C vibration. Not surprisingly, Au2Eu and Au3Eu compounds also demonstrate distinct stretching frequencies between 2197 and 2114 cm−1, which agree with the presence of several types of acetylene groups in these molecules.
Since the experimental data about the structures of these objects were not obtained, theoretical geometry optimization for all complexes has been carried out (Fig. 1 and S5,† selected bond lengths are listed in Table S1†). The LC-PBE functional with long-range correction33 was chosen because it most accurately reproduces such geometric parameters as the lengths of the Ln–O and Ln–N bonds.34 In general, the coordination of the Eu(tta)3 fragment to the bpy motif has a negligible effect on the stereochemistry of Au(I) environment and of the remote phosphine ligand. The predicted structural parameters for the gold complexes and heterometallic dyads match well with previously reported results of the crystallographic analysis of congener compounds.5e,18a,32b,34
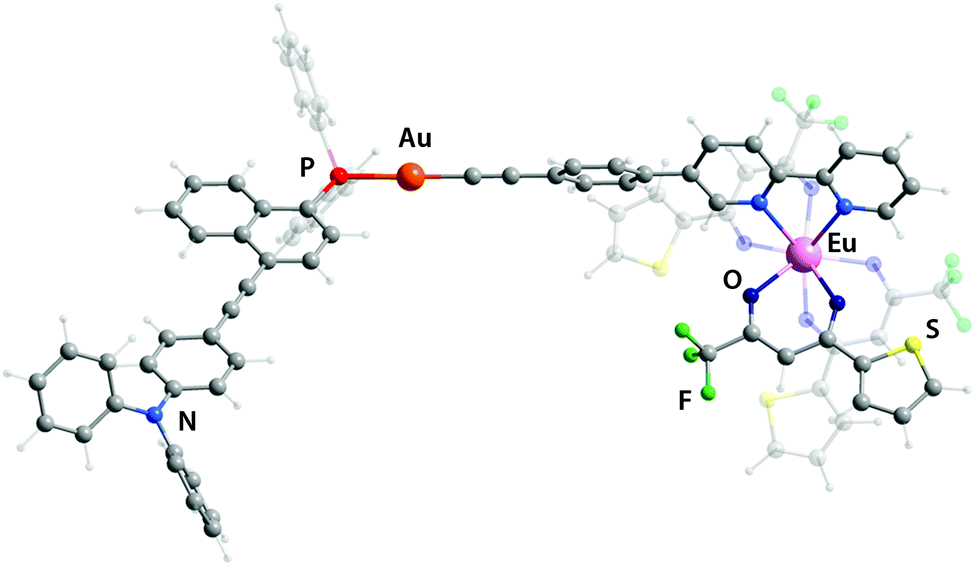 | ||
| Fig. 1 Minimum energy structures of complex Au2Eu (the DFT LC-PBE method). | ||
Photophysical properties
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 M−1 cm−1 for Au1, 67700 M−1 cm−1 for Au2 and 60200 M−1 cm−1 for Au3), attributed to the intraligand π → π* transitions localized on the epbpy fragment.31 This assignment is supported by quantum chemical calculations, according to which the lowest energy excitation S0 → S1 for Au1–Au3 occurs within the phosphine chromophores and shows a gradual growth of the wavelength from 307 to 441 nm, while the transitions S0 → S2 (Au1, Au2) and S0 → S4 (Au3) mainly involve the epbpy aromatic system (Fig. S7 and S8†). Although the predicted absorption energies are overestimated, the general tendencies correlate well with the experimental results.
700 M−1 cm−1 for Au1, 67700 M−1 cm−1 for Au2 and 60200 M−1 cm−1 for Au3), attributed to the intraligand π → π* transitions localized on the epbpy fragment.31 This assignment is supported by quantum chemical calculations, according to which the lowest energy excitation S0 → S1 for Au1–Au3 occurs within the phosphine chromophores and shows a gradual growth of the wavelength from 307 to 441 nm, while the transitions S0 → S2 (Au1, Au2) and S0 → S4 (Au3) mainly involve the epbpy aromatic system (Fig. S7 and S8†). Although the predicted absorption energies are overestimated, the general tendencies correlate well with the experimental results.
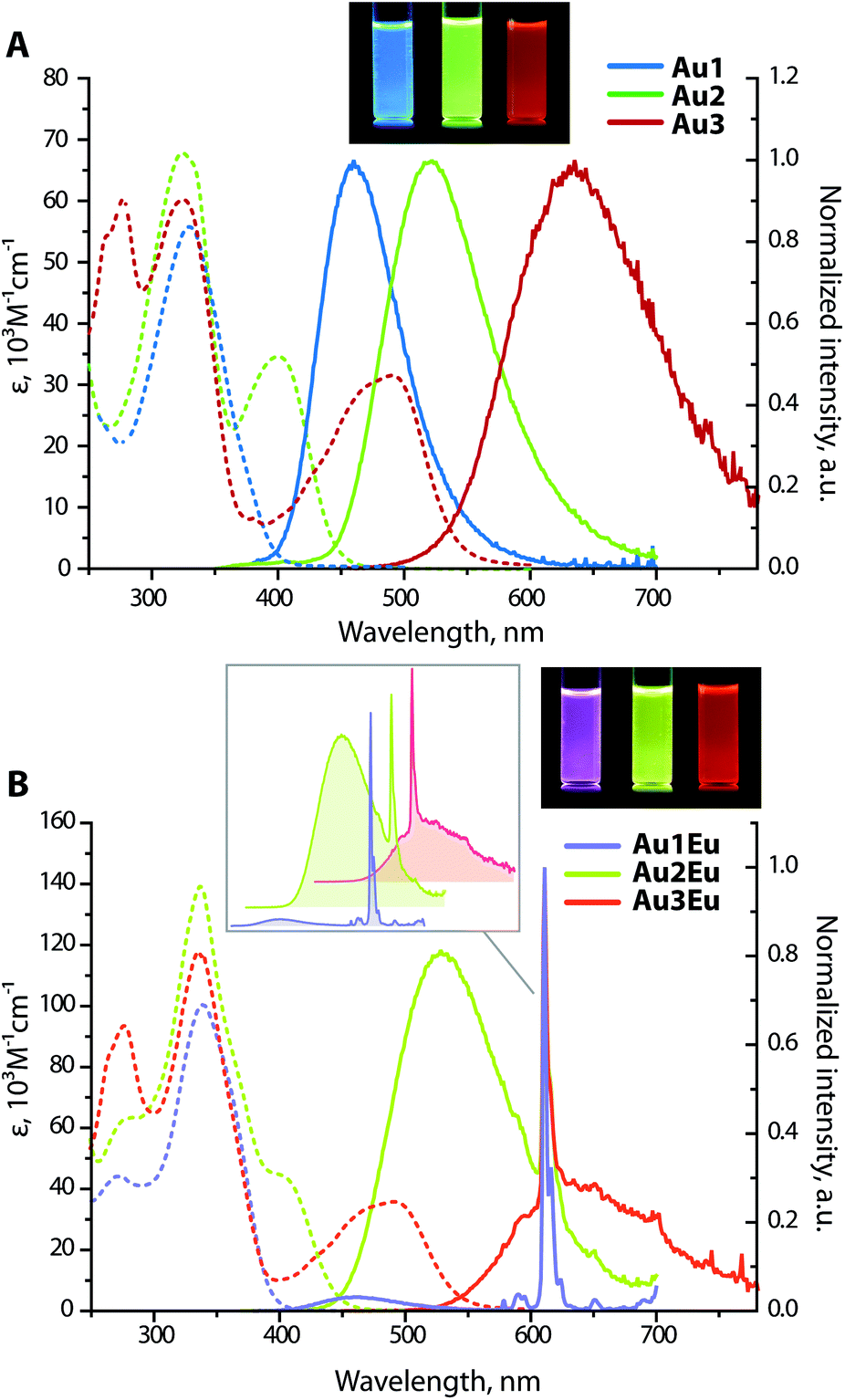 | ||
| Fig. 2 UV-vis absorption (dashed lines) and normalized emission (solid lines) spectra of metalloligands Au1–Au3 (A), and dyads Au1Eu–Au3Eu (B), normalized at 611 nm, the inset exhibits the stacking of the emission spectra (DCE, 298 K, λexc = 360 nm, c = 1 × 10−5 M); the photographs show the corresponding solutions under UV light (a broad band mercury lamp). | ||
|
λ
absa, nm (ε, 10−3 M−1 cm−1) |
λ
ema,b, nm |
Δνc, cm−1 | τ , ns | τ , μs |
Φ
emf |
|
|---|---|---|---|---|---|---|
| a In 1,2-dichloroethane (DCE), 1 × 10−5 M solution. b λ exc = 360 nm. c The difference between the maxima of absorption and fluorescence bands. d Lifetime monitored at the fluorescence band. e Lifetime monitored at 611 nm (5D0–7F2 transition). f Overall quantum yield (quantum yield of the fluorescence band), relative to coumarin 102 dye (Φ = 0.76) in ethanol. | ||||||
| Au1 | 330 (55.7) | 460 | 8560 | 1.8 | — | 0.30 |
| Au2 | 324 (67.9), 400 (34.6) | 525 | 5950 | 1.2 (70), 3.5 (30) | — | 0.31 |
| Au3 | 264 (54.3), 277 (60.2), 325 (60.7), 495 (31.5) | 635 | 4620 | 4.0 | — | 0.29 |
| Au1Eu | 271 (44.1), 339 (100.3) | 460, 580, 592, 611, 654, 700 | 7760 | 2.2 | 280.0 | 0.15(0.04) |
| Au2Eu | 280 (63.2), 337 (139.1), 404 (44.0) | 525, 611 | 5700 | 1.7 | 434.0 | 0.21 |
| Au3Eu | 276 (93.6), 336 (117.8), 495 (35.8) | 611, 635, 705, | 4620 | 4.1 | 404.1 | 0.16 |
Not surprisingly, optical properties of dyads Au1Eu–Au3Eu largely resemble those of gold-containing metalloligands (Fig. 2). The positions and intensities of the LE CT bands remain virtually unaffected upon the coordination of Au1–Au3 to the europium(III) ion. The bands maximized around 335–340 nm, which are nearly the same for all three bimetallic complexes, reveal significantly enhanced intensities (ε are in the order of 105 M−1 cm−1, see Table 1). Such substantial growth of molar absorptivities corresponds to a combination of energetically close π → π* transitions of the phenylene-bipyridine and diketonate (tta) ligands, as suggested by the computational studies (Fig. 3, S8 and S9†), and additionally proves the presence of Eu(tta)3 in the resulting dyads.
 | ||
| Fig. 3 Natural transition orbitals involved in the lowest energy singlet electronic excitation for complex Au2Eu. | ||
Chromophores Au1–Au3 exhibit strong emission under 360 nm excitation in DCE solutions with similar quantum yields around 0.30, while in the solid state, the luminescence at room temperature is completely suppressed apparently due to the aggregation quenching effect. The excited state lifetimes for Au1 (τ = 1.8 ns) and Au3 (τ = 4.0 ns) were obtained by a monoexponential fit of the decay kinetics; for Au2, a biexponential treatment gave two components with τ1 = 1.8 ns (70%) and τ2 = 3.5 ns (30%), and an average value τav of 2.4 ns. Short lifetimes of several ns and insensitivity of the emission intensity to molecular oxygen indicate prompt fluorescence. The broad and structureless emission profiles feature a steady increase in the peak wavelength Au1 (460 nm) < Au2 (525 nm) < Au3 (635 nm). Furthermore, large Stokes shifts for Au1–Au3, found in the range of 4620–8560 cm−1, are typical of the D–π-A architectures and confirm the intramolecular charge transfer characteristic of the lowest excited state. In the case of Au1, the emission energy is comparable to those of the (Ph2N-biphenyl-)3P → E congeners (E = O, 448 nm; S, 450 nm; Se, 450 nm; AuC6F5, 457 nm in dichloromethane).27d With respect to the phosphonium derivatives of ligands L1 and L230 containing stronger electron accepting cationic motifs, the emission and absorption bands of complexes Au1 and Au2 are found at considerably higher energies. The quantum efficiency for Au1 (Φem = 0.30 in DCE) is substantially lower than those for its oxide analogues (0.8 in ethyl acetate for L1O and 0.95 in CH2Cl2 for (Ph2N-biphenyl-)3PO).27b,d One possible nonradiative process might be a partial population of the triplet state T1via relatively slow intersystem crossing (S1 → T1) due to a heavy atom effect induced by a gold ion.5e,35 The absence of phosphorescence under ambient conditions can be explained by weak spin–orbit coupling in the excited state, insufficient for promoting the T1 → S0 radiative relaxation.
Intense phosphine-localized fluorescence of metalloligands Au1–Au3, which is virtually independent on the bipyridine function, is anticipated to achieve panchromatic emission upon the incorporation of the Eu(III) ion, known for its intense red luminescence. As shown in Fig. 2B, dyad Au1Eu exhibits two emission components. The minor HE signal centered at 460 nm matches the fluorescence band of the parent complex Au1, and the major set of LE peaks with maxima at 580, 592, 611, 654, and 700 nm correspond to the characteristic intraconfigurational 5D0 → 7Fn (n = 0–4) electronic 4f–4f transitions of the Eu(III) ion; the hypersensitive transition 5D0 → 7F2 dominates over other bands. The ratio of the integral intensities for 5D0 → 7F2 and 5D0 → 7F1 transition (I2/I1) equals 12 that indicates the low symmetry of the Eu(III) local environment. The overall quantum efficiency for Au1Eu amounts to 0.15, where fluorescence contributes only 0.04 and the rest 0.11 of the intensity appear from the lanthanide. An appreciable decrease in the singlet HE emission for Au1Eu compared to the parent Au1 (Φem = 0.30) suggests the partial energy transfer from the metalloligand to the Eu(III) center.
This process is supported by the TDDFT calculated energy diagram (Fig. 4 and S8†). The singlet levels of the tta, epbpy (32894 cm−1) and phosphine L1 (32467 cm−1) components in Au1Eu have very close energies and therefore are expected to be simultaneously populated upon photoexcitation and thermal equilibration. The lowest triplet state of the dyad is localized on the diketonate ligand (∼20500 cm−1) meaning that the ET is also possible from the higher lying triplet levels of epbpy (∼22500 cm−1) and L1 (∼23700 cm−1) to the 5D0 state of Eu(III) (∼17500 cm−1).36 Direct sensitization of the Eu(III) ion by means of the epbpy ligand is confirmed by the emergence of the lanthanide emission upon treating Au1–Au3 fluorophores with Eu(NO3)3 (Fig. S10).†
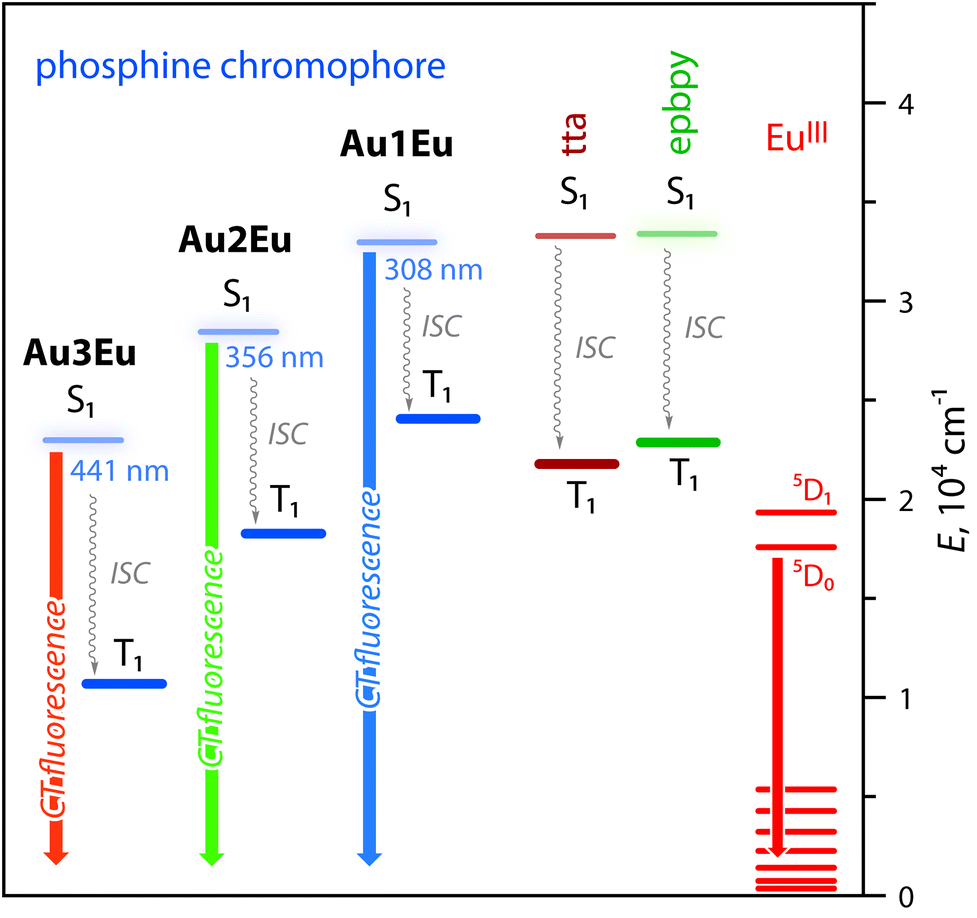 | ||
| Fig. 4 Excited state diagrams for complexes Au1Eu–Au3Eu predicted by TDDFT analysis. The energies of tta and epbpy ligands are virtually the same for all dyads (see the complete diagram in Fig. S8†). | ||
The luminescence spectra of Au2Eu and Au3Eu exhibit two-component profiles dominated by fluorescence bands at 525 and 635 nm, respectively, accompanied by the sharp emission peak of the europium ion positioned at 611 nm. The predicted lowest energy singlet and triplet excited states for the gold complexes and the dyads correspond to phosphine ligands (Fig. 4 and S8†) that prevent the ET from L2 and L3 to the Eu-coordinated bpy and tta moieties. The total quantum yields for Au2Eu (Φem = 0.21) and Au3Eu (Φem = 0.16) also decrease with respect to their gold precursors Au2 and Au3. The lower lying charge transfer triplets (∼18000 cm−1 for Au2Eu and ∼10530 cm−1 for Au3Eu) might allow back transfer of the excitation energy from 5D0(Eu(III)) to dark 3CT(L2/L3) states that presumably accounts for the decrease in the intensity of both lanthanide and phosphine-centered singlet emissions.
The emission lifetimes for Au1Eu–Au3Eu, determined by the monoexponential fit of the fluorescence decays, correlate with the corresponding values for metalloligands Au1–Au3. The luminescence for Au1Eu–Au3Eu monitored at 611 nm produced rather long lifetimes of 280.0, 434.0 and 404.1 μs, respectively, which are typical of the radiative relaxation of the Eu(III) ion.37
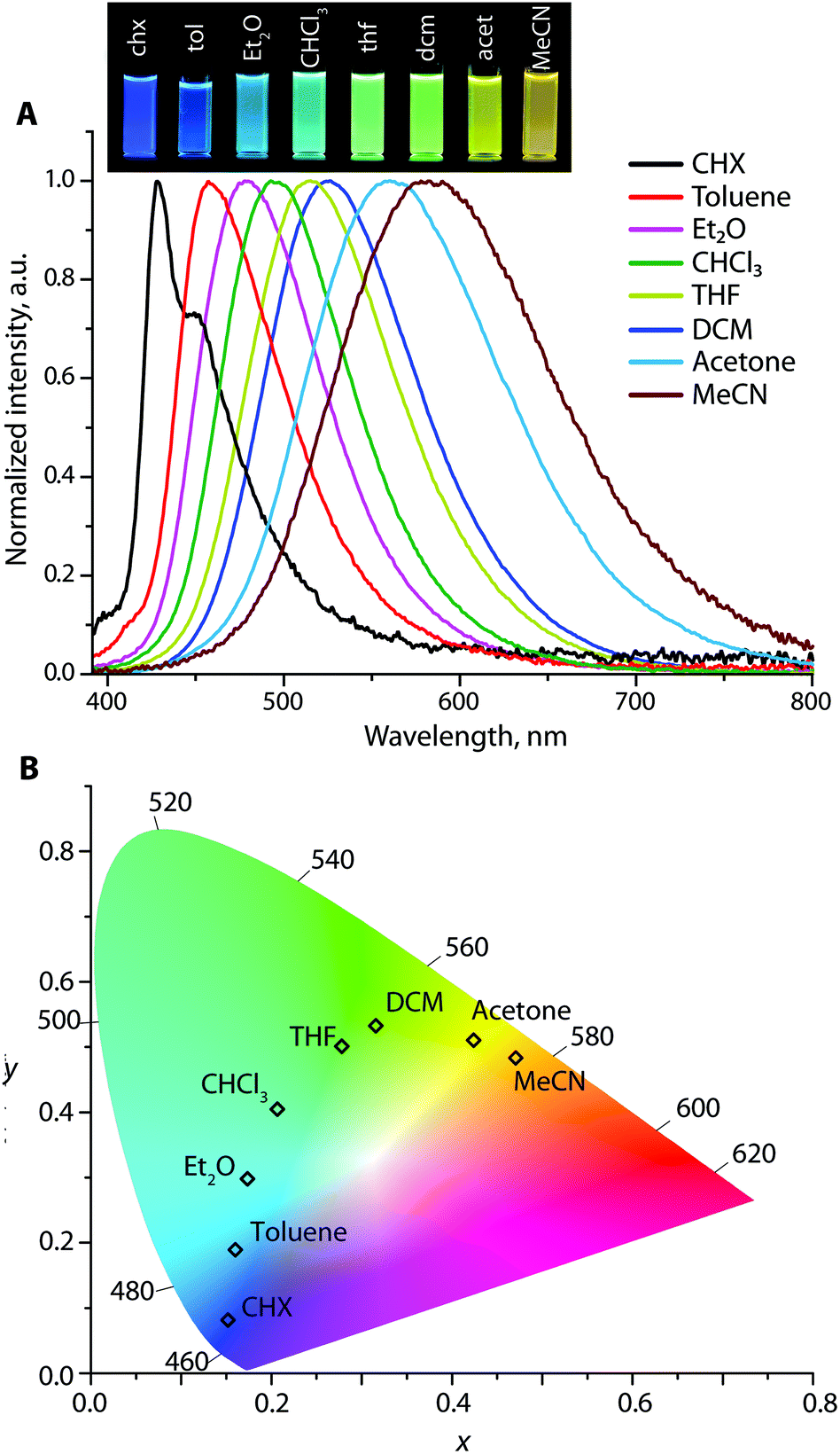 | ||
| Fig. 5 (A) The emission spectra (λexc = 365 nm, normalized on the fluorescence band) and (B) CIE 1931 coordinates of Au2 in different solvents (the photograph shows the corresponding solutions under a broad band mercury lamp). | ||
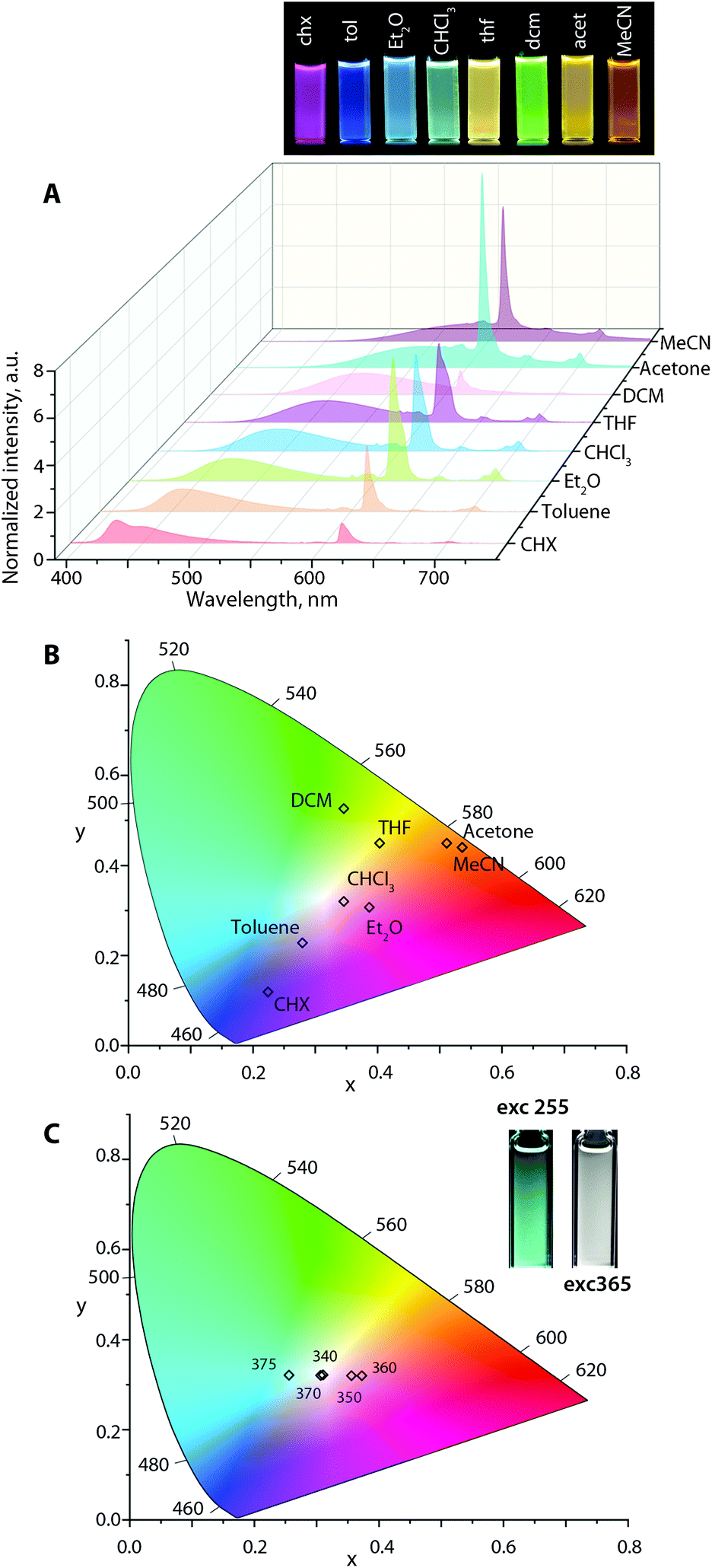 | ||
| Fig. 6 (A) The emission spectra (λexc = 365 nm, normalized on the fluorescence band; the photograph shows the corresponding solutions under a broad band mercury lamp). (B) CIE 1931 coordinates for Au2Eu in different solvents (λexc = 365 nm). (C) CIE 1931 coordinates for Au2Eu in chloroform at different excitation wavelengths, and the inset shows the visual appearance of the solutions under 255 and 365 nm excitation. | ||
| CHX | Toluene | Et2O | CHCl3 | THF | DCM | Acetone | MeCN | |
|---|---|---|---|---|---|---|---|---|
| a Measured at 0.15 O.D. at the excitation wavelength. b Fluorescence band, λexc = 365 nm. c (x, y) coordinates calculated from emission spectra. d Only Eu-emission was observed. | ||||||||
| Au1Eu | ||||||||
|
λ
absa |
342 | 339 | 337 | 336 | 339 | 338 | 334 | 335 |
|
λ
ema,b |
403, 423 | 425 | 435 | 445 | 450 | 460 | 475 | 490 |
| CIEc | 0.34, 0.13 | 0.56, 0.26 | 0.27, 0.12 | 0.30, 0.16 | 0.29, 0.18 | 0.52, 0.28 | 0.35, 0.28 | 0.52, 0.33 |
| Au2Eu | ||||||||
|
λ
absa |
334, 405 | 336, 402 | 330, 393 | 321, 402 | 334, 396 | 322, 401 | 393 | 328, 392 |
|
λ
ema,b |
428, 450 | 460 | 480 | 495 | 515 | 525 | 560 | 580 |
| CIEc | 0.22, 0.11 | 0.28, 0.23 | 0.39, 0.31 | 0.35, 0.32 | 0.40, 0.45 | 0.34, 0.52 | 0.51, 0.45 | 0.53, 0.44 |
| Au3Eu | ||||||||
|
λ
absa |
336, 494 | 337, 498 | 333, 486 | 334, 492 | 336, 488 | 336, 494 | 332, 484 | 333, 483 |
|
λ
ema,b |
520, 555 | 550 | 572 | 590 | —d | 630 | —d | —d |
| CIEc | 0.37, 0.58 | 0.46, 0.53 | 0.58, 0.41 | 0.59, 0.39 | 0.64, 0.36 | 0.61, 0.38 | 0.66, 0.33 | 0.67, 0.33 |
Increasing the solvent polarity from cyclohexane to acetonitrile leads to an insignificant, but detectable, hypsochromic shift for 7–13 nm (296–820 cm−1) of the CT band in the absorption spectra of metalloligands Au1–Au3 (Fig. S11†). Small negative solvatochromism can be tentatively explained by the solvent–molecule interaction that leads to a certain stabilization of the ground state S0 in a medium of higher polarity. Analysis of the composition of NTOs involved in the LE electronic transitions supports these observations. Considering complex Au3 as an example, one can see that both the NTO and NTO* orbitals are predominantly localized on diphenylaniline and diethynyl–anthracene fragments (Fig. S7†). For the solvent with the lowest polarity (cyclohexane), their contributions to the NTO are 22% and 76%. This ratio changes with increasing the solvent polarity, reaching 30%:69% for acetonitrile. Unlike the NTO orbital, NTO* appears to be unaffected by the properties of the solvent.
In nonpolar cyclohexane, the emission spectra of the Au1–Au3 appear as roughly structured bands with vibronic progressions of ca. 1140–1210 cm−1, which are attributed to the relaxation of locally excited (LE) states (Fig. 5, S12 and S13†). For example, the signal of Au1 with maxima at 403 (0 → 0 transition) and 423 nm (Δν = 1170 cm−1) correlates with the fluorescence profile of N,N-diphenyl-biphenyl amine38 but is red-shifted for ∼50 nm due to the presence of the electron-accepting Ph2P → Au group. In more polar solvents, the emission bands for Au1–Au3 become broad and featureless, and gradually move to a low energy region due to the prevailing charge transfer characteristic of the radiative relaxation. For instance, Au2 in cyclohexane produces nearly pure deep blue emission (λem = 428 and 450 nm) with the Commission Internationale de l'Eclairage (CIE) coordinates (0.15, 0.08), green in dichloromethane λem = 525 nm (CIE 0.31, 0.53), and orange λem = 580 nm (0.47, 0.48) in acetonitrile, revealing a prominent red shift of 152 nm (4719 cm−1) (Fig. 5). However, the diversity of colors for Au1 and Au3 is less pronounced; their CIE coordinates vary from violet-blue to green (0.15, 0.02) → (0.20, 0.34) for Au1, and from green-yellow to red (0.33, 0.61) → (0.48, 0.40) for Au3 (Fig. S12 and S13†). The Stokes shifts for Au1–Au3 vary from 1177–5126 cm−1 in cyclohexane to larger values of 5689–10173 cm−1 in polar acetonitrile that points to a significant stabilization of the ICT excited state.
The solvent-dependent behavior of Au1–Au3 was examined using the Lippert–Mataga approach (see Fig. S14† and the description of the procedure in the Experimental section),39 which treats the Stokes shift of a CT fluorophore as a linear function of the solvent polarity. The slopes of these plots for metalloligands Au1–Au3 provide the changes in the dipole moments (Δμ = μes − μgs), which occur upon excitation, and amount to ca. 24, 33 and 37 D, respectively; these values are typical of neutral donor–acceptor molecules.
The absorption spectra of dyads Au1Eu–Au3Eu slightly differ from those of metalloligands Au1–Au3. The main intense peak centered at ∼330 nm is attributed to the π → π* tta-centered transitions, which are almost insensitive to the solvent polarity, while the CT bands follow the same trend as for Au1–Au3, and show a small hypsochromic shift of ca. 11–13 nm upon polarity increase (Fig. S11†).
Analogous to Au1–Au3, the emission profiles for Au1Eu–Au3Eu (Fig. 6, S15 and S16†) exhibit a bathochromic shift in the broad fluorescence band in polar media. In contrast, the position of sharp 5D0 → 7Fn (n = 0–4) Eu(III) transitions maximized at 611 nm remains unaffected (although the intensities vary) that results in a large diversity of the luminescence colors according to the calculated CIE coordinates. For instance, at the excitation wavelength of 365 nm, Au2Eu in CHCl3 and Et2O exhibits nearly white emission with coordinates (0.35, 0.32) and (0.39, 0.31). In more polar MeCN and acetone solutions, the color of all dyads appears in the reddish-orange zone of the chromaticity diagram (for Au2Eu CIE 0.51, 0.45 and 0.53, 0.44, respectively; Fig. 6) due to a significant quenching of the fluorescence band and the domination of the lanthanide luminescence. The emissions of Au1Eu and Au3Eu dyads correspond to different regions of the color space. The color of Au3Eu steadily changes from green-yellow (cyclohexane) to red (acetonitrile), but Au1Eu shows irregular alterations from magenta (diethyl ether) to orange (acetonitrile) and red (toluene) (Fig. S15 and S16).† In addition to the solvatochromic effect, the excitation energy distinctly affects the intensity ratio of the fluorescence and lanthanide emission bands, as illustrated by the response of the Au2Eu complex in CHCl3 solution to the excitation wavelengths, ranging from 280 nm to 400 nm (Fig. S17†). Notably, using the radiation from 320 to 375 nm allowed influencing primarily the x coordinate (0.26, 0.32 → 0.37, 0.32), and, particularly with 340 and 370 nm excitation, to achieve the CIE values (0.31, 0.32), which are close to pure white color. When the excitation shifted to lower energy (390–400 nm), only the fluorescence band was observed for Au2Eu, which confirms the absence of energy transfer from the intraphosphine 1CT state to the europium ion.
Conclusions
The chromophore-containing phosphines L1–L3, based on diphenylamine-functionalized conjugated systems, produce highly fluorescent species Au1–Au3 upon coordination to the gold(I)-alkynyl fragments. Their photophysical properties are determined by the intraligand (L1–L3) electronic transitions that are confirmed by the DFT computational analysis. Due to the intrinsic donor–acceptor nature of the metal-bound phosphine motifs, complexes Au1–Au3 undergo photoinduced intramolecular charge transfer, which results in pronounced luminescence solvatochromism (428 nm in cyclohexane → 580 nm in acetonitrile for Au2). Furthermore, the incorporation of the chelating bipyridine unit into the Au-connected alkynyl offers a facile opportunity to utilize compounds Au1–Au3 as metalloligands. As a proof of concept, these gold(I) precursors were combined with the {Eu(tta)3} moiety to give a family of dyads Au1Eu–Au3Eu. Efficient sensitization of the lanthanide emission by diketonates (tta) and partial excitation energy transfer from intra-phosphine 1CT states, particularly suppressed for L2 and L3, lead to the dual luminescence combined with fluorescence and europium-centered transitions. The dynamic features of Au1–Au3 emission behavior, governed by the properties of the medium, are retained in bimetallic assemblies Au1Eu–Au3Eu. This, together with the excitation-dependent ratio of the phosphine and lanthanide emissions, offers rich opportunities for tuning the luminescence color. The described complexes represent the first examples of merging the remote charge-transfer highly solvatochromic fluorophores with red-emitting Eu centers and demonstrate a convenient approach in the design of dual emissive complexes with widely variable luminescence characteristics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors appreciate the financial support from the Academy of Finland (grant 317903, I. O. K.) and the Russian Science Foundation (grant 16-13-10064, photophysical and computational investigations, E. V. G., V. V. S., and S. O. S.). The work was carried out using the core facilities of St Petersburg State University Research Park (Centre for Magnetic Resonance, Centre for Optical and Laser Materials Research, Computer Centre).References
- (a) I. D. W. Samuel and G. A. Turnbull, Chem. Rev., 2007, 107, 1272–1295 CrossRef CAS; (b) A. J. C. Kuehne and M. C. Gather, Chem. Rev., 2016, 116, 12823–12864 CrossRef CAS PubMed; (c) G. M. Farinola and R. Ragni, Chem. Soc. Rev., 2011, 40, 3467–3482 RSC; (d) M.-S. Wang and G.-C. Guo, Chem. Commun., 2016, 52, 13194–13204 RSC; (e) D. Kim and S. Y. Park, Adv. Opt. Mater., 2018, 6, 1800678 CrossRef; (f) S.-C. Lee, J. Heo, H. C. Woo, J.-A. Lee, Y. H. Seo, C.-L. Lee, S. Kim and O. P. Kwon, Chem. – Eur. J., 2018, 24, 13706–13718 CrossRef CAS; (g) D. Li, J. Wang and X. Ma, Adv. Opt. Mater., 2018, 6, 1800273 CrossRef.
- (a) V. S. Padalkar and S. Seki, Chem. Soc. Rev., 2016, 45, 169–202 RSC; (b) S. Park, J. E. Kwon, S. H. Kim, J. Seo, K. Chung, S.-Y. Park, D.-J. Jang, B. M. Medina, J. Gierschner and S. Y. Park, J. Am. Chem. Soc., 2009, 131, 14043–14049 CrossRef CAS; (c) K.-C. Tang, M.-J. Chang, T.-Y. Lin, H.-A. Pan, T.-C. Fang, K.-Y. Chen, W.-Y. Hung, Y.-H. Hsu and P.-T. Chou, J. Am. Chem. Soc., 2011, 133, 17738–17745 CrossRef CAS; (d) B. Li, J. Lan, D. Wu and J. You, Angew. Chem., Int. Ed., 2015, 54, 14008–14012 CrossRef CAS; (e) S. Tsuchiya, K.-I. Sakai, K. Kawano, Y. Nakane, T. Kikuchi and T. Akutagawa, Chem. – Eur. J., 2018, 24, 5868–5875 CrossRef CAS; (f) B. Li, L. Zhou, H. Cheng, Q. Huang, J. Lan, L. Zhou and J. You, Chem. Sci., 2018, 9, 1213–1220 RSC.
- (a) X.-H. Jin, C. Chen, C.-X. Ren, L.-X. Cai and J. Zhang, Chem. Commun., 2014, 50, 15878–15881 RSC; (b) Z. Xie, C. Chen, S. Xu, J. Li, Y. Zhang, S. Liu, J. Xu and Z. Chi, Angew. Chem., Int. Ed., 2015, 54, 7181–7184 CrossRef CAS; (c) D. Tu, P. Leong, S. Guo, H. Yan, C. Lu and Q. Zhao, Angew. Chem., Int. Ed., 2017, 56, 11370–11374 CrossRef CAS; (d) X. Feng, C. Qi, H.-T. Feng, Z. Zhao, H. H. Y. Sung, I. D. Williams, R. T. K. Kwok, J. W. Y. Lam, A. Qin and B. Z. Tang, Chem. Sci., 2018, 9, 5679–5687 RSC; (e) D. Li, W. Hu, J. Wang, Q. Zhang, X.-M. Cao, X. Ma and H. Tian, Chem. Sci., 2018, 9, 5709–5715 RSC.
- (a) J. Kalinowski, M. Cocchi, D. Virgili, V. Fattori and J. A. G. Williams, Adv. Mater., 2007, 19, 4000–4005 CrossRef CAS; (b) A. Sakai, M. Tanaka, E. Ohta, Y. Yoshimoto, K. Mizuno and H. Ikeda, Tetrahedron Lett., 2012, 53, 4138–4141 CrossRef CAS; (c) Q.-Y. Yang and J.-M. Lehn, Angew. Chem., Int. Ed., 2014, 53, 4572–4577 CrossRef CAS.
- (a) C.-C. Hsu, C.-C. Lin, P.-T. Chou, C.-H. Lai, C.-W. Hsu, C.-H. Lin and Y. Chi, J. Am. Chem. Soc., 2012, 134, 7715–7724 CrossRef CAS; (b) F. Geist, A. Jackel and R. F. Winter, Dalton Trans., 2015, 44, 3974–3987 RSC; (c) Z. Mao, Z. Yang, Y. Mu, Y. Zhang, Y.-F. Wang, Z. Chi, C.-C. Lo, S. Liu, A. Lien and J. Xu, Angew. Chem., Int. Ed., 2015, 54, 6270–6273 CrossRef CAS; (d) P. Irmler and R. F. Winter, Dalton Trans., 2016, 45, 10420–10434 RSC; (e) I. Kondrasenko, K.-Y. Chung, Y.-T. Chen, J. Koivistoinen, E. V. Grachova, A. J. Karttunen, P.-T. Chou and I. O. Koshevoy, J. Phys. Chem. C, 2016, 120, 12196–12206 CrossRef CAS; (f) H. Wu, C. Hang, X. Li, L. Yin, M. Zhu, J. Zhang, Y. Zhou, H. Ågren, Q. Zhang and L. Zhu, Chem. Commun., 2017, 53, 2661–2664 RSC; (g) C. Zhou, S. Zhang, Y. Gao, H. Liu, T. Shan, X. Liang, B. Yang and Y. Ma, Adv. Funct. Mater., 2018, 28, 1802407 CrossRef; (h) K. Tabata, T. Yamada, H. Kita and Y. Yamamoto, Adv. Funct. Mater., 2019, 29, 1805824 CrossRef.
- (a) R. Kawagoe, I. Takashima, S. Uchinomiya and A. Ojida, Chem. Sci., 2017, 8, 1134–1140 RSC; (b) Y. Okorochenkova, M. Porubský, S. Benická and J. Hlaváč, Chem. Commun., 2018, 54, 7589–7592 RSC.
- (a) J. E. Yarnell, J. C. Deaton, C. E. McCusker and F. N. Castellano, Inorg. Chem., 2011, 50, 7820–7830 CrossRef CAS; (b) T. Yoshihara, Y. Yamaguchi, M. Hosaka, T. Takeuchi and S. Tobita, Angew. Chem., Int. Ed., 2012, 51, 4148–4151 CrossRef CAS; (c) Q. Zhao, X. Zhou, T. Cao, K. Y. Zhang, L. Yang, S. Liu, H. Liang, H. Yang, F. Li and W. Huang, Chem. Sci., 2015, 6, 1825–1831 RSC; (d) S. K. Gupta, A. Haridas and J. Choudhury, Chem. – Eur. J., 2017, 23, 4770–4773 CrossRef CAS.
- (a) S. Van Wallendael and D. P. Rillema, J. Chem. Soc., Chem. Commun., 1990, 1081–1082 RSC; (b) E. C. Glazer, D. Magde and Y. Tor, J. Am. Chem. Soc., 2005, 127, 4190–4192 CrossRef CAS; (c) Z. Chen, K. Y. Zhang, X. Tong, Y. Liu, C. Hu, S. Liu, Q. Yu, Q. Zhao and W. Huang, Adv. Funct. Mater., 2016, 26, 4386–4396 CrossRef CAS; (d) K. S. Kisel, A. S. Melnikov, E. V. Grachova, P. Hirva, S. P. Tunik and I. O. Koshevoy, Chem. – Eur. J., 2017, 23, 11301–11311 CrossRef CAS; (e) Z. Hao, F. Meng, P. Wang, Y. Wang, H. Tan, Y. Pei, S. Su and Y. Liu, Dalton Trans., 2017, 46, 16257–16268 RSC; (f) F. Liu, J. Wen, S.-S. Chen and S. Sun, Chem. Commun., 2018, 54, 1371–1374 RSC.
- (a) J.-C. G. Bünzli and C. Piguet, Chem. Soc. Rev., 2005, 34, 1048–1077 RSC; (b) M. D. Ward, Coord. Chem. Rev., 2010, 254, 2634–2642 CrossRef CAS; (c) S. V. Eliseeva and J.-C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC; (d) H.-B. Xu, J.-G. Deng and B. Kang, RSC Adv., 2013, 3, 11367–11384 RSC; (e) M. Pan, W.-M. Liao, S.-Y. Yin, S.-S. Sun and C.-Y. Su, Chem. Rev., 2018, 118, 8889–8935 CrossRef CAS.
- (a) T. J. Sørensen, M. Tropiano, O. A. Blackburn, J. A. Tilney, A. M. Kenwright and S. Faulkner, Chem. Commun., 2013, 49, 783–785 RSC; (b) T. J. Sørensen, A. M. Kenwright and S. Faulkner, Chem. Sci., 2015, 6, 2054–2059 RSC; (c) O. Kotova, S. Comby, C. Lincheneau and T. Gunnlaugsson, Chem. Sci., 2017, 8, 3419–3426 RSC.
- (a) M. Tropiano and S. Faulkner, Chem. Commun., 2014, 50, 4696–4698 RSC; (b) S. Shuvaev, M. Starck and D. Parker, Chem. – Eur. J., 2017, 23, 9974–9989 CrossRef CAS; (c) S.-Y. Huang and V. C. Pierre, Chem. Commun., 2018, 54, 9210–9213 RSC; (d) G. Bao, S. Zha, Z. Liu, Y.-H. Fung, C.-F. Chan, H. Li, P.-H. Chu, D. Jin, P. A. Tanner and K.-L. Wong, Inorg. Chem., 2018, 57, 120–128 CrossRef CAS.
- (a) H. Peng, M. I. J. Stich, J. Yu, L.-N. Sun, L. H. Fischer and O. S. Wolfbeis, Adv. Mater., 2010, 22, 716–719 CrossRef CAS; (b) K. Yanagisawa, Y. Kitagawa, T. Nakanishi, T. Seki, K. Fushimi, H. Ito and Y. Hasegawa, Chem. – Eur. J., 2018, 24, 1956–1961 CrossRef CAS; (c) X.-Q. Guo, L.-P. Zhou, L.-X. Cai and Q.-F. Sun, Chem. – Eur. J., 2018, 24, 6936–6940 CrossRef CAS; (d) H. Zhang, J. Jiang, P. Gao, T. Yang, K. Y. Zhang, Z. Chen, S. Liu, W. Huang and Q. Zhao, ACS Appl. Mater. Interfaces, 2018, 10, 17542–17550 CrossRef CAS.
- (a) A. J. Amoroso and S. J. A. Pope, Chem. Soc. Rev., 2015, 44, 4723–4742 RSC; (b) J.-C. G. Bünzli, J. Lumin., 2016, 170, 866–878 CrossRef; (c) K. Y. Zhang, Q. Yu, H. Wei, S. Liu, Q. Zhao and W. Huang, Chem. Rev., 2018, 118, 1770–1839 CrossRef CAS.
- O. Laporte and W. F. Meggers, J. Opt. Soc. Am., 1925, 11, 459–463 CrossRef CAS.
- M. D. Ward, Coord. Chem. Rev., 2007, 251, 1663–1677 CrossRef CAS.
- P. Coppo, M. Duati, V. N. Kozhevnikov, J. W. Hofstraat and L. De Cola, Angew. Chem., Int. Ed., 2005, 44, 1806–1810 CrossRef CAS.
- (a) T. K. Ronson, T. Lazarides, H. Adams, S. J. A. Pope, D. Sykes, S. Faulkner, S. J. Coles, M. B. Hursthouse, W. Clegg, R. W. Harrington and M. D. Ward, Chem. – Eur. J., 2006, 12, 9299–9313 CrossRef CAS; (b) F. Kennedy, N. M. Shavaleev, T. Koullourou, Z. R. Bell, J. C. Jeffery, S. Faulkner and M. D. Ward, Dalton Trans., 2007, 1492–1499 RSC; (c) H.-B. Xu, J. Ni, K.-J. Chen, L.-Y. Zhang and Z.-N. Chen, Organometallics, 2008, 27, 5665–5671 CrossRef CAS; (d) O. J. Stacey, A. J. Amoroso, J. A. Platts, P. N. Horton, S. J. Coles, D. Lloyd, C. F. Williams, A. J. Hayes, J. J. Dunsford and S. J. A. Pope, Chem. Commun., 2015, 51, 12305–12308 RSC.
- (a) D. Sykes, I. S. Tidmarsh, A. Barbieri, I. V. Sazanovich, J. A. Weinstein and M. D. Ward, Inorg. Chem., 2011, 50, 11323–11339 CrossRef CAS; (b) A. Baschieri, S. Muzzioli, E. Matteucci, S. Stagni, M. Massi and L. Sambri, Dalton Trans., 2015, 44, 37–40 RSC; (c) A. Jana, B. J. Crowston, J. R. Shewring, L. K. McKenzie, H. E. Bryant, S. W. Botchway, A. D. Ward, A. J. Amoroso, E. Baggaley and M. D. Ward, Inorg. Chem., 2016, 55, 5623–5633 CrossRef CAS PubMed.
- T. Lazarides, N. M. Tart, D. Sykes, S. Faulkner, A. Barbieri and M. D. Ward, Dalton Trans., 2009, 3971–3979, 10.1039/B901560D.
- K. Sénéchal-David, S. J. A. Pope, S. Quinn, S. Faulkner and T. Gunnlaugsson, Inorg. Chem., 2006, 45, 10040–10042 CrossRef.
- L. R. Hill, O. A. Blackburn, M. W. Jones, M. Tropiano, T. J. Sørensen and S. Faulkner, Dalton Trans., 2013, 42, 16255–16258 RSC.
- (a) S. J. A. Pope, Polyhedron, 2007, 26, 4818–4824 CrossRef CAS; (b) M. Andrews, J. E. Jones, L. P. Harding and S. J. A. Pope, Chem. Commun., 2011, 47, 206–208 RSC.
- (a) J. Wang, H. Chen, F. Ru, Z. Zhang, X. Mao, D. Shan, J. Chen and X. Lu, Chem. – Eur. J., 2018, 24, 3499–3505 CrossRef CAS; (b) M. Rong, X. Yang, L. Huang, S. Chi, Y. Zhou, Y. Shen, B. Chen, X. Deng and Z.-Q. Liu, ACS Appl. Mater. Interfaces, 2019, 11, 2336–2343 CrossRef CAS.
- G. He, D. Guo, C. He, X. Zhang, X. Zhao and C. Duan, Angew. Chem., Int. Ed., 2009, 48, 6132–6135 CrossRef CAS.
- (a) G.-L. Law, K.-L. Wong, H.-L. Tam, K.-W. Cheah and W.-T. Wong, Inorg. Chem., 2009, 48, 10492–10494 CrossRef CAS; (b) A. H. Shelton, I. V. Sazanovich, J. A. Weinstein and M. D. Ward, Chem. Commun., 2012, 48, 2749–2751 RSC; (c) J. Zhang, H. Li, P. Chen, W. Sun, T. Gao and P. Yan, J. Mater. Chem. C, 2015, 3, 1799–1806 RSC; (d) K. Singh, R. Boddula and S. Vaidyanathan, Inorg. Chem., 2017, 56, 9376–9390 CrossRef CAS; (e) R. Boddula, K. Singh, S. Giri and S. Vaidyanathan, Inorg. Chem., 2017, 56, 10127–10130 CrossRef CAS.
- H.-B. Xu, P.-C. Jiao, B. Kang, J.-G. Deng and Y. Zhang, Sci. Rep., 2013, 3, 2199 CrossRef.
- (a) P.-A. Bouit, A. Escande, R. Szűcs, D. Szieberth, C. Lescop, L. Nyulászi, M. Hissler and R. Réau, J. Am. Chem. Soc., 2012, 134, 6524–6527 CrossRef CAS; (b) X.-K. Liu, C.-J. Zheng, M.-F. Lo, J. Xiao, C.-S. Lee, M.-K. Fung and X.-H. Zhang, Chem. Commun., 2014, 50, 2027–2029 RSC; (c) E. Yamaguchi, C. Wang, A. Fukazawa, M. Taki, Y. Sato, T. Sasaki, M. Ueda, N. Sasaki, T. Higashiyama and S. Yamaguchi, Angew. Chem., Int. Ed., 2015, 54, 4539–4543 CrossRef CAS; (d) I. Kondrasenko, Z.-H. Tsai, K.-Y. Chung, Y.-T. Chen, Y. Y. Ershova, A. Doménech-Carbó, W.-Y. Hung, P.-T. Chou, A. J. Karttunen and I. O. Koshevoy, ACS Appl. Mater. Interfaces, 2016, 8, 10968–10976 CrossRef CAS.
- X. Yang, X. Yan, H. Guo, B. Liu, J. Zhao, G. Zhou, Y. Wu, Z. Wu and W.-Y. Wong, Dyes Pigm., 2017, 143, 151–164 CrossRef CAS.
- Z. H. Li, M. S. Wong, Y. Tao and M. D'Iorio, J. Org. Chem., 2004, 69, 921–927 CrossRef CAS.
- A. Belyaev, Y.-H. Cheng, Z.-Y. Liu, A. J. Karttunen, P.-T. Chou and I. O. Koshevoy, Angew. Chem., Int. Ed., 2019, 58, 13456–13465 CrossRef CAS.
- I. V. Solovjov, A. Kondinski, K. Y. Monakhov, I. O. Koshevoy and E. V. Grachova, Inorg. Chem. Front., 2018, 5, 160–171 RSC.
- (a) G. E. Coates and C. Parkin, J. Chem. Soc., 1962, 3220–3226 RSC; (b) V. W.-W. Yam, S. W.-K. Choi and K.-K. Cheung, Organometallics, 1996, 15, 1734–1739 CrossRef CAS.
- (a) J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed; (b) H. Iikura, T. Tsuneda, T. Yanai and K. Hirao, J. Chem. Phys., 2001, 115, 3540–3544 CrossRef CAS.
- V. Khistiaeva, A. S. Melnikov, S. Slavova, V. Sizov, G. L. Starova, I. O. Koshevoy and E. V. Grachova, Inorg. Chem. Front., 2018, 5, 3015–3027 RSC.
- Y.-C. Chang, K.-C. Tang, H.-A. Pan, S.-H. Liu, I. O. Koshevoy, A. J. Karttunen, W.-Y. Hung, M.-H. Cheng and P.-T. Chou, J. Phys. Chem. C, 2013, 117, 9623–9632 CrossRef CAS.
- W. T. Carnall, G. L. Goodman, K. Rajnak and R. S. Rana, J. Chem. Phys., 1989, 90, 3443–3457 CrossRef CAS.
- H. Xu, K. Yin and W. Huang, ChemPhysChem, 2008, 9, 1752–1760 CrossRef CAS.
- G. Chen, W. Li, T. Zhou, Q. Peng, D. Zhai, H. Li, W. Z. Yuan, Y. Zhang and B. Z. Tang, Adv. Mater., 2015, 27, 4496–4501 CrossRef CAS.
- (a) M. Maus, W. Rettig, D. Bonafoux and R. Lapouyade, J. Phys. Chem. A, 1999, 103, 3388–3401 CrossRef CAS; (b) E. Sakuda, Y. Ando, A. Ito and N. Kitamura, J. Phys. Chem. A, 2010, 114, 9144–9150 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic, other experimental and computational details. See DOI: 10.1039/c9qi01015g |
| This journal is © the Partner Organisations 2020 |