
Structural, electrochemical and photophysical behavior of Ru(II) complexes with large bite angle sulfur-bridged terpyridyl ligands†
Christopher M.
Brown
 ,
Nicole E.
Arsenault
,
Trevor N. K.
Cross
,
Duane
Hean
,
Zhen
Xu
and
Michael O.
Wolf
*
,
Nicole E.
Arsenault
,
Trevor N. K.
Cross
,
Duane
Hean
,
Zhen
Xu
and
Michael O.
Wolf
*
Department of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, BC V6T 1Z1, Canada. E-mail: mwolf@chem.ubc.ca
First published on 31st October 2019
Abstract
Ruthenium(II) terpyridyl species typically suffer from a distortion away from the ideal octahedral geometry due to the rigidity of the terpyridine ligand. In this study we report a series of new sulfur-bridged terpyridyl ligands (1a–c) which offer enhanced flexibility due to sulfur spacers linking the pyridine rings, in addition to the ability to tune electronic properties with electron-donating (Me) and electron-withdrawing (CF3) substituents in the 4 and 4′′-positions of the sulfur-bridged ligands. The resulting Ru(II) complexes are detailed herein as both homoleptic (2a–c) and heteroleptic (3a–c) species, with the synthesis, structural characterization, photophysical and redox properties discussed, and the sulfur-bridged ligands allowing geometries closer to that of a “perfect” octahedron. All six species were found to be non-emissive at room temperature (with emissivity increasing on cooling to 77 K), and the electrochemical properties were greatly altered due to the increased bite angle of the sulfur-bridged ligands. The electrochemical and photophysical HOMO–LUMO gap can be easily tuned by altering the substituents in the 4 and 4′′-positions.
Introduction
For more than 80 years, ruthenium-based polypyridyl species have been extensively studied. [Ru(tpy)2]2+ (tpy = 2,2′:6′,2′′-terpyridine)1 and [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine)2 were first synthesized in the 1930s, and the seminal details of their photophysical properties described a few decades later.3,4 Since these reports, a great deal of research has focused on Ru(II)-polypyridine complexes as their photophysical and electrochemical properties lend themselves as ideal candidates for a wide range of applications. These include (but are not limited to): solar energy conversion;5–7 light-emitting devices;8–10 photocatalysis;11 electrocatalysis;12 molecular electronics;13,14 and as bioimaging probes.15In particular, Ru(II) species are excellent analogs for studying artificial photosynthesis; and polynuclear species have been used to mimic the chromophores in the light-harvesting reactions in photosystem II (PSII).16,17 While tris-bidentate Ru(II) species have many advantageous properties for this purpose (e.g. [Ru(bpy)3]2+ possesses a long-lived room temperature excited-state lifetime of ∼860 ns),18 the chirality of these complexes leads to structural issues in large supramolecular assemblies. To combat this, bis-tridentate Ru(II) complexes can be used as the absence of stereoisomers leads to greater control over larger systems.19 Though stereochemical control is improved, excited-state properties are worsened relative to those of tris-bidentate Ru(II) complexes. For example [Ru(tpy)2]2+ has an excited-state lifetime of only 0.25 ns and is practically non-emissive at room temperature.20 These limitations are due to a distortion from the ideal octahedral geometry imposed by the tpy ligands, causing the non-emissive d–d metal-centered triplet state (3MC) to become close in energy to the emissive triplet metal-to-ligand charge transfer (3MLCT) state.21
Judicious ligand design can help overcome these shortcomings, with a number of approaches established to increase the 3MLCT–3MC energy gap, either through stabilizing the 3MLCT state or destabilizing the 3MC state.21,22 Successful strategies include: (a) appending electron-donating substituents to the tpy to destabilize the highest-occupied molecular orbital (HOMO) or appending electron-withdrawing substituents to the tpy ligand to stabilize the lowest-unoccupied molecular orbital (LUMO);9,20,23–26 and (b) expansion of the chelate rings to alleviate the ring strain about the metal center through the addition of a spacer between the pyridyl rings.27–32 Tuning the ring-strain of Ru(II) complexes extends beyond photoluminescence applications, with these metallic species showing uses as molecular photocatalysts and as electrocatalysts.
Our group has recently shown that sulfur-bridged, bidentate, chelating ligands can be used to affect the photophysical properties of transition metal complexes.33–36 One interesting observation from this work is the bridging sulfur affords a greater flexibility about the ligand, and thus N^N bite angles come close to 90°; towards that of a “perfect” octahedron. In light of this, we set about designing a system whereby bridging sulfur atoms are used to alleviate the ring strain in Ru(II)-terpyridine species, with the electronics further tuned by appending electron-donating and electron-withdrawing groups. Thus, in this work we report the synthesis and properties of these new sulfur-bridged terpyridyl analogues (pro-ligands 1a–c) and their resulting Ru(II) complexes as homoleptic (2a–c) and heteroleptic (3a–c) species.
Synthesis and structural characterization
Sulfide-bridged terpyridyl ligands
The sulfide-bridged terpyridyl ligands 2,6-bis(pyridin-2-ylthio)pyridine (TPS, 1a), 2,6-bis((4-methylpyridin-2-yl)thio)pyridine (Me-TPS, 1b) and 2,6-bis((4-(trifluoromethyl)pyridin-2-yl)thio)pyridine (CF3-TPS, 1c) were prepared by the routes shown in Scheme 1. Ligand 1a was synthesized via the reaction of 2,6-dibromopyridine with 2-mercaptopyridine.37 For methyl and trifluoromethyl substituted species (1b and 1c, respectively) higher yields were obtained by first preparing the disulfide precursor38 prior to reaction with 2,6-dibromopyridine. The three ligands were characterized using NMR spectroscopy and high-resolution mass spectrometry (HR-MS).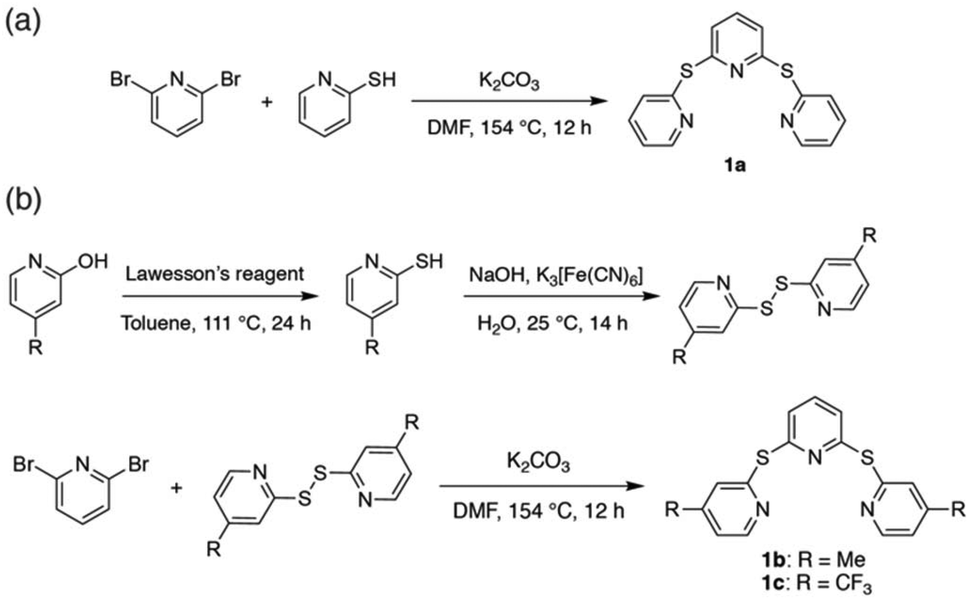 | ||
| Scheme 1 Preparation of sulfide-bridged terpyridyl pro-ligands 1a–c. | ||
Ruthenium(II) complexes
All six ruthenium(II) complexes were synthesized using a microwave reactor as detailed by Johansson et al.39 (Scheme S1†). Homoleptic species [Ru(TPS)2][PF6]2 (2a), [Ru(Me-TPS)2][PF6]2 (2b) and [Ru(CF3-TPS)2][PF6]2 (2c) were prepared by reaction of cis-Ru(DMSO)4Cl2 with the appropriate pro-ligand (1a–c) in ethylene glycol. Heteroleptic complexes [Ru(tpy)(TPS)][PF6]2 (3a), [Ru(tpy)(Me-TPS)][PF6]2 (3b) and [Ru(tpy)(CF3-TPS)][PF6]2 (3c) were prepared by reacting 1 equivalent of the appropriate pro-ligand 1a–c with Ru(tpy)Cl3 in ethylene glycol. The metallic species were structurally characterized by solution NMR spectroscopy, HR-MS and X-ray crystallography. 1H and 13C NMR signals were assigned using 2D NMR experiments (1H–1H COSY, 1H–1H NOESY, 1H–13C HSQC and 1H–13C HMBC) and all six complexes are shown to be octahedral in solution (Fig. S1–24†). Attempts to form Ru(II) complexes with analogous sulfone-bridged (SO2) ligands were unsuccessful, likely due to an instability caused by photo-ejection of the SO2-bridged ligand.35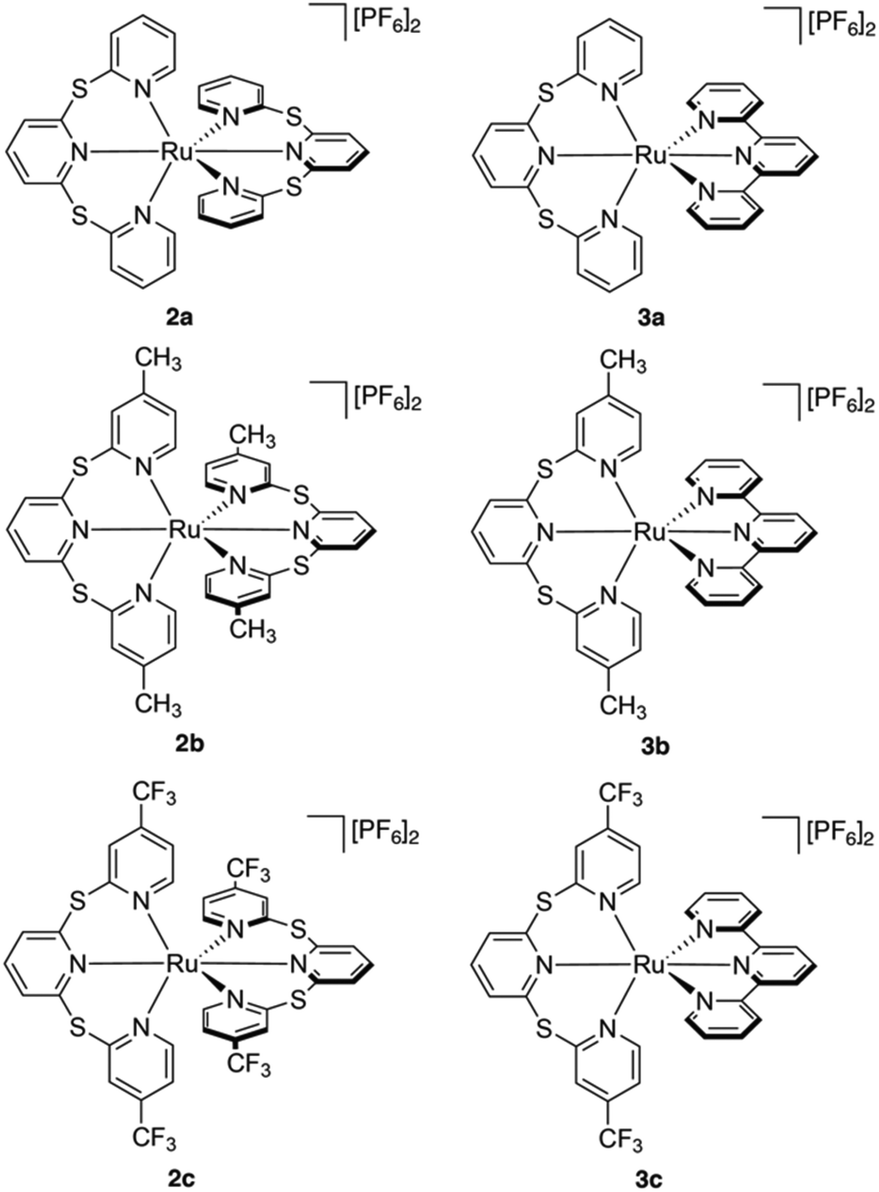
Crystallographic studies
X-ray quality crystals were obtained by slow diffusion of Et2O into acetone and their single crystal structures shown in Fig. 1. Complexes 2a and 3a contain CH3CN solvent molecules from previous crystallization attempts. A summary of the crystal data parameters is included in Table S1.† Complexes 2a–c and 3c crystallize in a monoclinic space group, while complexes 3a and 3b crystallize in the triclinic space group. All six complexes show the two tridentate ligands meridionally coordinated to the ruthenium atom, producing a distorted octahedral geometry. The bridging sulfur atom in the sulfur-bridged tridentate ligands provides flexibility allowing near-octahedral angles in 2a–c and 3a–c (Table S2†). The average trans N–Ru–N angles for sulfur-bridged ligands in the six species are 178.9°, which is in good agreement with literature values for Ru(II) terpyridyl species with –NMe– spacers between the pyridyl rings.30,31,40 The distorted octahedral geometry in the homoleptic species 2a–2c is due to the steric interactions between the two tridentate sulfur-bridged ligands, while the distortion in the heteroleptic complexes 3a–3c is due to the rigidity of the tpy ligand with an average trans N–Ru–Nu bond angle of 158.8°, which is in agreement with other [Ru(tpy)(L)]2+ metal complexes.30,41,42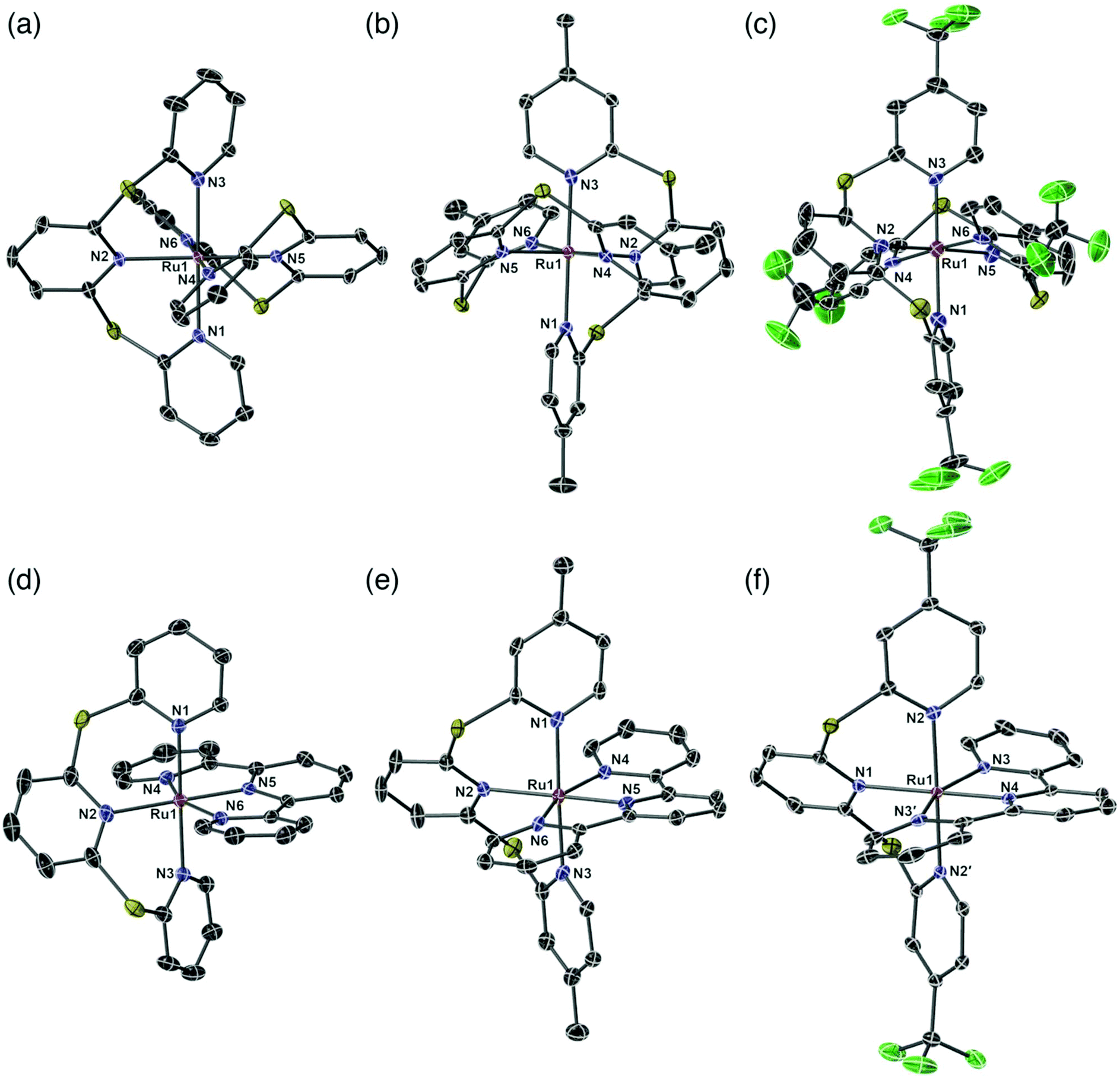 | ||
| Fig. 1 Perspective views of single crystal structures of complexes (a) 2a; (b) 2b; (c) 2c; (d) 3a; (e) 3b and (f) 3c. [PF6]− anions, solvent molecules and hydrogen atoms removed for clarity. Thermal ellipsoids are shown at a 50% level. | ||
The Ru–N bond lengths are consistent throughout the series and are in agreement with similar species (Table S3†).30,31,40,41 The longest average Ru–N bond lengths are seen in 2c, due to the electron withdrawing properties of the fluorinated CF3-TPS (1c) ligands (2c: Ru–N length = 2.101 Å). The shortest average Ru–N bond length is seen in 2b and is due to the electron donating properties of the methylated ligand Me-TPS (1b) (2b: Ru–N length = 2.091 Å). For heteroleptic complexes 3a–3c the longest Ru–N bond lengths are from the central pyridyl core unit in the sulfur-bridged ligands (bond lengths: Ru1–N2 = 2.124 Å; Ru1–N2 = 2.127 Å; and Ru1–N1 = 2.143 Å for 3a, 3b and 3c respectively). The shortest Ru–N bond lengths are from the nitrogen atom of the central pyridyl unit in the tpy ligand (lengths: Ru1–N5 = 1.958 Å; Ru1–N5 = 1.960 Å; and Ru1–N4 = 1.975 Å for 3a, 3b and 3c respectively). All sulfur-bridged ligand units in 2a–c and 3a–c are in a helical arrangement about the Ru metal center (absolute configuration is not determined). Dihedral angles between the planes of the central pyridyl rings trans to each other are 49.43°, 50.88° and 53.51° for 2a–c respectively, and 48.55°, 47.09° and 56.27° for 3a–c respectively.
The O-value (octahedricity value) measures the mean absolute deviation of a set of N–Ru–N angles from the ideal octahedral values, where an O-value = 0 indicates no deviation.43,44 The O-values of 2a–2c and 3a–3c were calculated using eqn (S1)† and compared to homoleptic literature compounds [Ru(bpy)3]2+,45 [Ru(tpy)2]2+,46 [Ru(pbpy)2]2+,47 [Ru(Lket)2]2+ (ref. 40) and [Ru(dgpy)2]3+,32 and heteroleptic literature compounds [Ru(Ph-tpy)(dgpy)]2+,32 [Ru(ddpd)(EtOOC-tpy)]2+ (ref. 31) and [Ru(ttpy)(bpe)]2+ (ref. 48) (Table 1). Chemical structures of the literature species are illustrated in Chart S1.†
| Homoleptic complexes | Octahedricity value (O) | Heteroleptic complexes | Octahedricity value (O) |
|---|---|---|---|
| 2a | 2.96 | 3a | 7.97 |
| 2b | 3.49 | 3b | 7.86 |
| 2c | 2.84 | 3c | 8.1 |
| [Ru(bpy)3]2+ (ref. 45) | 7.12 | [Ru(Ph-tpy)(dgpy)]3+ (ref. 32) | 7.93 |
| [Ru(tpy)2]2+ (ref. 46) | 11.1 | [Ru(ddpd)(EtOOC-tpy)]2+ (ref. 31) | 7.76 |
| [Ru(pbpy)2]2+ (ref. 47) | 7.09 | [Ru(ttpy)(bpe)]2+ (ref. 48) | 9.84 |
| [Ru(Lket)2]2+ (ref. 40) | 0.938 | ||
| [Ru(dgpy)2]2+ (ref. 32) | 5.12 |
The O-values for 2a–2c (2.96, 3.49 and 2.84, respectively) trend closer to the ideal octahedral geometry as the sp3 sulfur linkers provide conformational flexibility when compared to the rigidity of [Ru(tpy)2]2+ (11.1) and [Ru(dgpy)2]2+ (5.12) systems. Of the homoleptic species discussed, [Ru(Lket)2]2+ (0.94) is the closest to the ideal octahedral geometry, suggesting that the 120° angle at the sp2 carbonyl linkers in the expanded tpy system provide the optimal geometry. Despite existing as a homoleptic species, complex [Ru(pbpy)2]2+ (7.09) has a comparable O-value to [Ru(tpy)2]2+ and the heteroleptic compounds discussed herein. This is due to the sp3 carbon linker confining the conformational flexibility.
Heteroleptic species 3a–c contain one tpy ligand and one expanded ligand, therefore the O-values are expected to increase as the complex deviates from an ideal octahedron. Complexes 3a–c (7.97, 7.86 and 8.10, respectively) have O-values comparable to [Ru(ddpd)(EtOOC-tpy)]2+ (7.76) and [Ru(Ph-tpy)(dgpb)]3+ (7.93); these systems all contain sp3 atoms bridging the pyridyl rings of the expanded ligand. These results confirm that by expanding the bite angle of a tpy ligand with sulfur bridges (the TPS system), metal centers with geometries closer to that of a perfect octahedron can be achieved.
Electrochemical data
Electrochemical potentials of the metal complexes were determined by cyclic voltammetry (CV) to obtain insight into the charge transfer absorption events (Fig. 2 and S31–36†). Oxidation and reduction potentials of both homoleptic and heteroleptic complexes are reported in Table 2, along with those of the known complex [Ru(tpy)2]2+.49 The Ru3+/Ru2+ reversible oxidation couple for 2a occurs at 1.01 V, while for 2b this occurs at a slightly lower potential (0.90 V) compared to 2a, and for 2c at a significantly higher potential (1.33 V). This would suggest the HOMO is more destabilized in the more electron donating Me-TPS-coordinated complex 2b, while the HOMO of the electron withdrawing CF3-TPS coordinated complex 2c is more stabilized in energy, when compared to the HOMO of the unsubstituted TPS-coordinated complex 2a. Oxidation potentials for heteroleptic complexes do not experience as large a variation between 3a–c compared to the homoleptic complexes. Complexes 3a and 3b have similar oxidation potentials (0.95 V and 0.96 V respectively), however 3c has a slightly higher oxidation potential (1.10 V) indicating the HOMO is stabilized when only one CF3-TPS ligand is coordinated to ruthenium in a heteroleptic complex.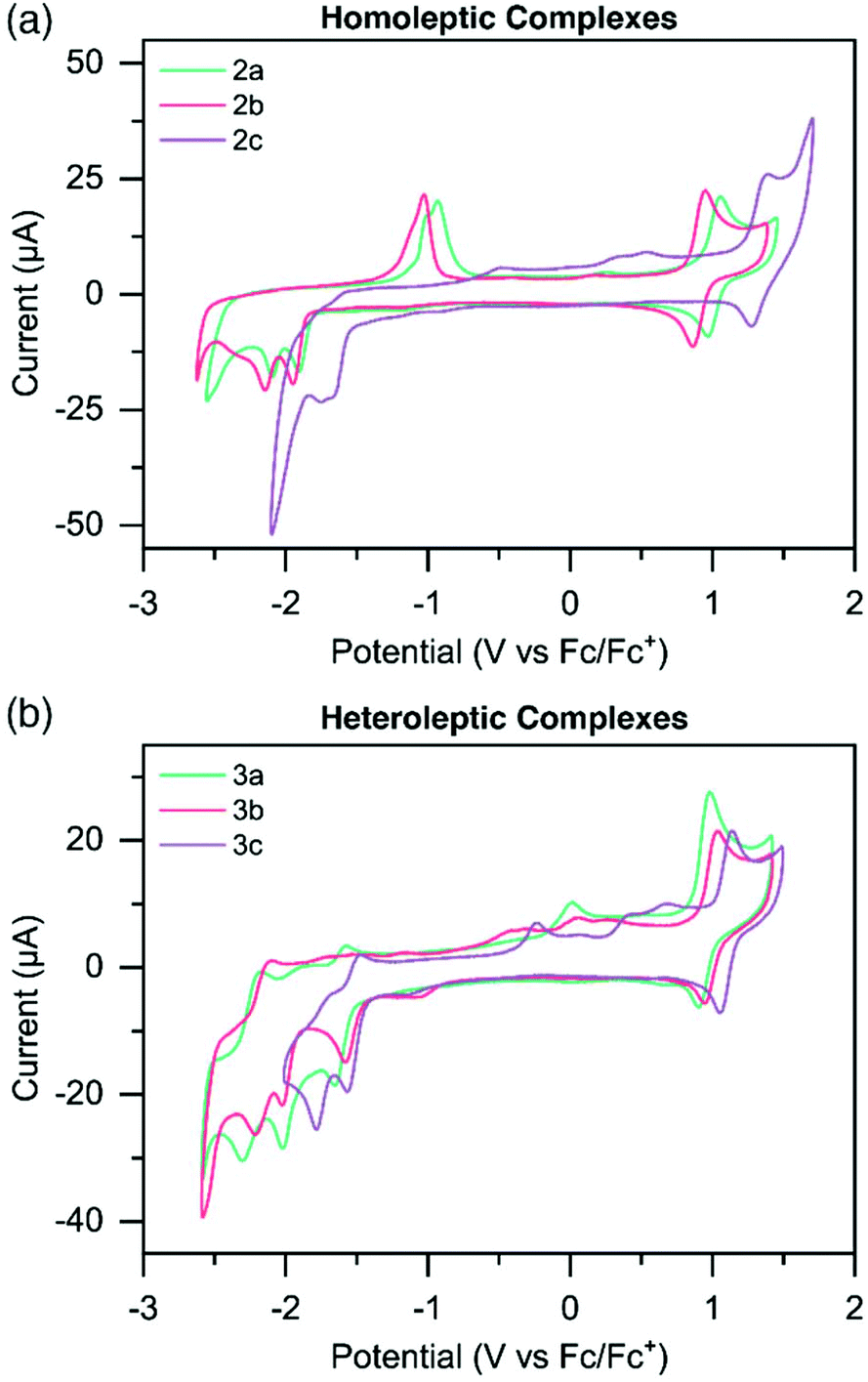 | ||
| Fig. 2 Cyclic voltammograms of (a) homoleptic species 2a–c; and (b) heteroleptic species 3a–c at 1 mM concentrations in dry CH3CN sparged with N2, utilizing (n-Bu4N)PF6 as the electrolyte in 0.1 M concentrations. Sweep rate = 100 mV s−1. Potentials were recorded vs. Fc/Fc+ as an added internal standard. | ||
| E ox [V] [ip,a/ip,c] | E red [V] [ip,c/ip,a] | E g [eV] | ||
|---|---|---|---|---|
| Electrochemical | Optical | |||
| a Measurements were carried out in CH3CN solution at concentrations of 1 mM, with scan rates of 100 mV s−1 and Fc/Fc+ employed as an internal standard. b Irreversible redox event. c Irreversible redox event when cycled through increasingly negative potentials. | ||||
| [Ru(tpy)2]2+ (ref. 49) | 0.92 | −1.62, −1.87 | — | — |
| 2a | 1.01 [1.2] | −1.91b, −2.09b | 2.92 | 2.75 |
| 2b | 0.90 [1.4] | −1.95b, −2.15b | 2.85 | 2.70 |
| 2c | 1.33 [2.7] | −1.66b, −1.75b | 2.99 | 2.88 |
| 3a | 0.95 [2.0] | −1.61c, [1.6] | 2.56 | 2.55 |
| −2.02b, −2.24 | ||||
| 3b | 0.96 [1.6] | −1.57c, [1.2] | 2.53 | 2.53 |
| −2.06b, −2.19 | ||||
| 3c | 1.10 [1.1] | −1.52, −1.73 | 2.62 | 2.60 |
The homoleptic complexes exhibit irreversible reduction peaks at more negative potentials compared to [Ru(tpy)2]2+.49 The first reduction potentials of 2a (−1.91 V) and 2b (−1.95 V) are at significantly more negative potentials compared to [Ru(tpy)2]2+, suggesting the electron donating effects of the TPS and Me-TPS ligands destabilize the LUMO energies of the respective complexes compared to the analogous [Ru(tpy)2]2+ complex. Interestingly, 2a and 2b have similar first reduction potentials suggesting the increased electron donating properties of the Me group in 2b does not significantly affect the LUMO energy compared to the unsubstituted TPS ligand on 2a. Complex 2c has a significantly less negative first reduction potential (−1.66 V) compared to the other homoleptic complexes, suggesting the electron withdrawing CF3 group helps counteract the electron donating effects of the TPS ligand, and a stabilization of the LUMO occurs when compared to 2a. Heteroleptic complexes 3a, 3b and 3c all have reversible reduction potentials at similar potential values to [Ru(tpy)2]2+ (−1.61 V, −1.57 V, −1.52 V). The reversibility and similarity in potential values suggests that for the heteroleptic complexes the LUMO resides on the tpy ligand.
The electrochemical HOMO–LUMO energy gap (Eg) for all complexes was estimated as the difference between the oxidation potential and the first reduction potential. It should be noted that for the homoleptic complexes, reduction potentials are reported for the peak of the irreversible reduction wave, which leads to an inherent error in the calculated Eg and thus slightly higher values.
The second reduction potentials of all complexes were irreversible, estimating that for both homo- and heteroleptic complexes the LUMO+1 resides on the respective sulfur-bridged ligand. When complexes were doubly reduced, new irreversible oxidation waves appeared on the reverse scan and steadily increased in current intensity when complexes were cycled to increasingly negative potentials (Fig. S31 and S32†). This is especially noticeable for complexes 2a and 2b, where significant oxidation peaks appear at ∼ −1.0 V (Fig. 2a). Complex 2c and complexes 3a–c also show similar irreversible oxidation waves, however with less current. These oxidation peaks appear both when cycled first through positive potentials, and when cycled first through negative potentials (Fig. S31–36†).
We propose these growing oxidation waves are from a new intermediate complex forming when the second reduction of the complex occurs. A report by Sarkar and coworkers50 found that with strongly electron donating groups attached to terpyridine ligands coordinated around Ru(II) centers, one of the pyridyl arms can detach from the metal center, allowing for surrounding solvent to coordinate to the Ru, stabilizing the complex. The highly reduced solvated complex is re-oxidized at a higher oxidation potential, which allows for removal of the solvent molecule and re-coordination of the ligand arm to the Ru center, completing the redox cycle.
We propose a similar redox mechanism is likely occurring with complexes reported herein. We postulate that the electron-donating sulfur-bridged ligand is partially unbound from the Ru center at high reduction states (Scheme S2†). The metal center then has a free coordination site which solvent may coordinate to, forming a solvated complex. The highly reduced solvated complex is then re-oxidized at a higher potential, and the complex reverts back to the unsolvated, fully-bound form. For complexes 2a and 2b (which have the strongest electron donating ligands), the growing oxidation peak is significantly larger than in 2c and 3a–c, indicating there is a higher concentration of intermediate solvated complex.
These results suggest that Ru(II) complexes containing sulfur-bridged terpyridyl ligands could be useful for electrocatalysis, whereby reducing these complexes to the second reduction state opens up a coordination site on the metal center, potentially allowing chemical reactions to occur at the ruthenium center.
Photophysical data
Absorption spectroscopy
The UV-vis absorption profiles for pro-ligands 1a–c in DCM solutions are shown in Fig. 3a and Table 3. All three species exhibit features at energies higher than 350 nm, and these are assigned to π–π* transitions on the pyridyl rings. 1a also shows a lower-energy feature centered at 360 nm.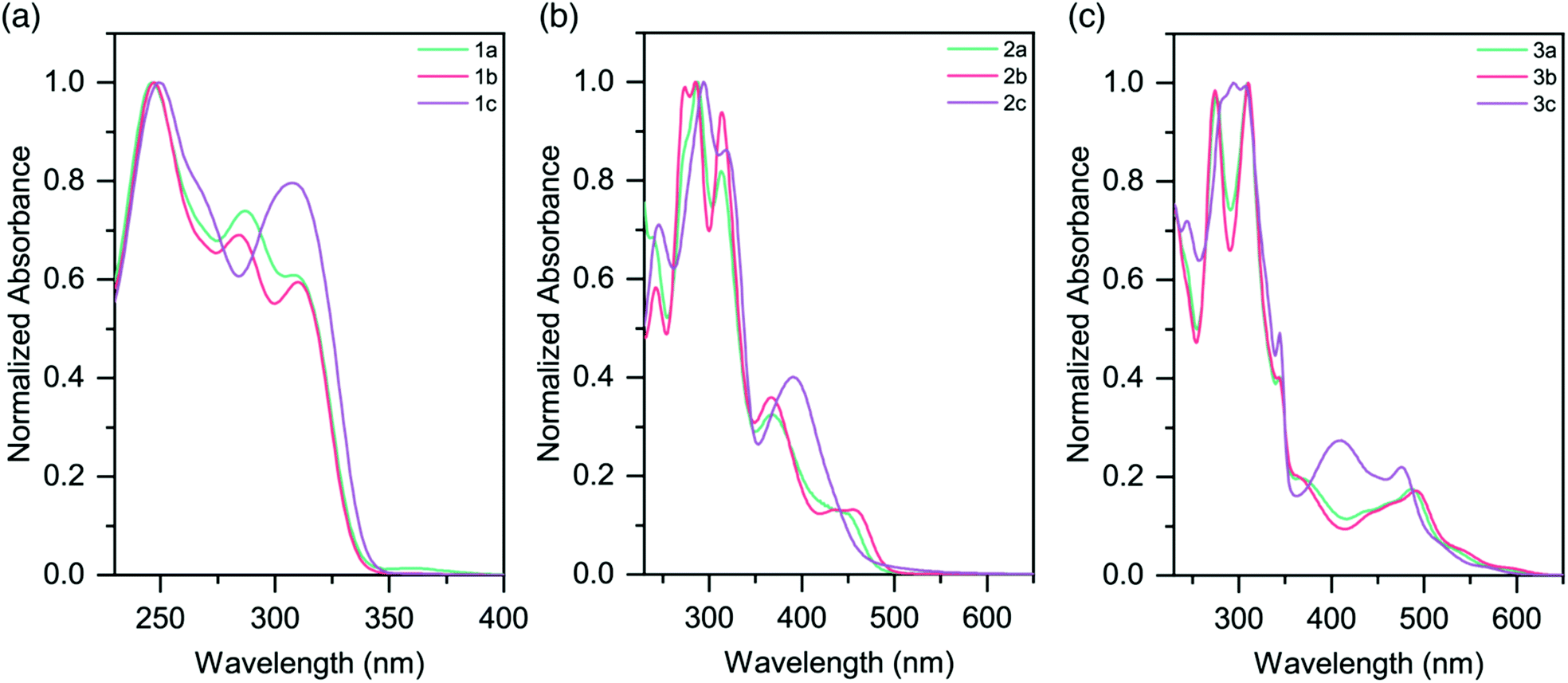 | ||
| Fig. 3 Absorption spectra of (a) pro-ligands 1a–c in DCM solutions; (b) homoleptic complexes 2a–c in CH3CN solutions (c) heteroleptic complexes 3a–c in CH3CN solutions. | ||
| Compound | Absorbance [solution, 298 K] | Emission [77 K] |
|---|---|---|
| λ max [nm] | λ max [nm] | |
a DCM solutions.
b CH3CN solutions.
c Thin films drop-cast from acetone on to quartz substrates.
d Non-emissive at 77 K.
e Frozen glass (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 EtOH:MeOH).
f Conditions not given.
g Frozen glass (propionitrile:butyronitrile 4:5).
h Frozen glass (isobutyronitrile). :1 EtOH:MeOH).
f Conditions not given.
g Frozen glass (propionitrile:butyronitrile 4:5).
h Frozen glass (isobutyronitrile).
|
||
| Pro-ligands | ||
| 1a | 246, 287, 309, 360a | — |
| 1b | 247, 285, 310a | — |
| 1c | 249, 269, 308a | — |
| Homoleptic complexes | ||
| 2a | 369, 452b | 612c |
| 2b | 367, 436, 455b | 615c |
| 2c | 390b | —d |
| [Ru(tpy)2]2+ (ref. 19) | 476b | 598e |
| [Ru(pbpy)2]2+ (ref. 47) | 477b | 610f |
| [Ru(Lket)2]2+ (ref. 40) | 432, 500, 522, 562b | 613g |
| Heteroleptic complexes | ||
| 3a | 437, 460, 486, 532, 590b | 641, 698shc |
| 3b | 443, 466, 491, 545, 600b | 649, 704shc |
| 3c | 411, 476, 521, 572b | 612, 665shc |
| [Ru(ttpy)(bpe)]2+ (ref. 48) | 482b | 650e |
| [Ru(ddpd)(EtOOC-tpy)]2+ (ref. 31) | 473, 517, 582, 630b | 707h |
The absorption spectra for the homoleptic Ru(II) complexes (2a–c) are shown in Fig. 3b. All show intense high energy bands between 230–350 nm, assigned to ligand-centered transitions. Through Time-Dependent Density Functional Theory (TD-DFT) calculations we ascribe lower energy features to 1MLCT absorptions (Fig. S38 and 39†). Complex 2a exhibits a 1MLCT maximum at 452 nm. The peak for the methyl-substituted species 2b is slightly red-shifted relative to this (λmax = 455 nm), while the peak for the CF3-substituted species 2c is substantially blue-shifted (λmax = 390 nm). The cause of these shifts is the electron-donating and electron-withdrawing properties of the substituents, respectively. These trends are in good agreement with the Eg values from the electrochemical data (Table 2). Additionally of note is the overall blue-shift of the 1MLCT band of complexes 2a–c when compared to that of [Ru(tpy)2]2+ (λmax = 474 nm).20
MLCT absorptions are affected by the geometry of the complex, and the observed blue-shift is consistent with the reduction in steric strain about the Ru(II) centers in 2a–c as they approach that of a more-perfect octahedron.31 It is likely that the electron-donating properties of the sulfur atom in the TPS ligands slightly red-shifts the absorptions of the complexes. Analogous homoleptic complexes which contain sp3 moieties (such as N, O, CH2) bridging the pyridyl rings in the expanded ligand system do not exist in the literature, and so direct comparisons are not possible.
Heteroleptic complexes 3a–c also show similar ligand π–π* transitions in the UV region. A greater number of bands are present, presumably due to the mixture of tpy and sulfur-bridged ligands. In comparison to 2a–c, the visible absorptions are bathochromically shifted, with bands tailing out to 650 nm (Fig. 3c and Table 3). These features are in agreement with our electrochemical data, which in conjunction with the lack of tailing character seen in the homoleptic complexes, indicates that these lower energy transitions are due to a 1MLCT to the tpy ligand, as opposed to the sulfur-bridged ligand. The optical band gaps of the complexes are in agreement with the electrochemical Eg calculated, demonstrating that the optical properties of the complexes follow the same trend as the redox properties (Table 2).
Photoluminescence spectroscopy
None of the six Ru(II) complexes displayed luminescence in Ar-sparged CH3CN solutions at room temperature, nor in the solid state at room temperature. This is attributed to deactivation of the 3MLCT state by thermal population of the non-emissive 3MC state. Therefore, to probe the excited state properties, low temperature photoluminescence spectroscopy was employed (Table 3 and Fig. 4, S41†). Solid samples of 2a–c and 3a–c were drop-cast from acetone onto quartz substrates and allowed to air dry, leaving neat thin films of good quality. Complexes 2a and 2b were found to be emissive at 77 K (λmax = 612 and 615 nm, respectively), with each exhibiting a broad featureless emission. The nature of this emission was probed by studying the spin density of the species using Density Functional Theory (DFT). As the compound is excited to the triplet state, the total spin becomes 2 due to the unpaired electron. By studying the spin densities, we get information on how the spin density is distributed, giving a general and qualitative description of the triplet excited state (Table S11†). Calculations assigned the emissive state as a triplet metal-centered (3MC) (Fig. S46†). The slight bathochromic shift observed in 2b is attributed to the electron-donating methyl groups in the 4 and 4′′ positions of the sulfur-bridged ligand. Complex 2c, however, was found to be non-emissive at 77 K. DFT calculations assign the excited state as 3MC (Fig. S46c†). The electron-withdrawing –CF3 groups in 2c greatly stabilize both the HOMO and LUMO levels when compared to 2a and 2b (and [Ru(tpy)2]2+), as noted in the electrochemical properties of the species. This aligns it closer in energy to the 3MC state, whereby a thermally-activated internal conversion can occur (even at 77 K), resulting in the non-emissive character of the complex.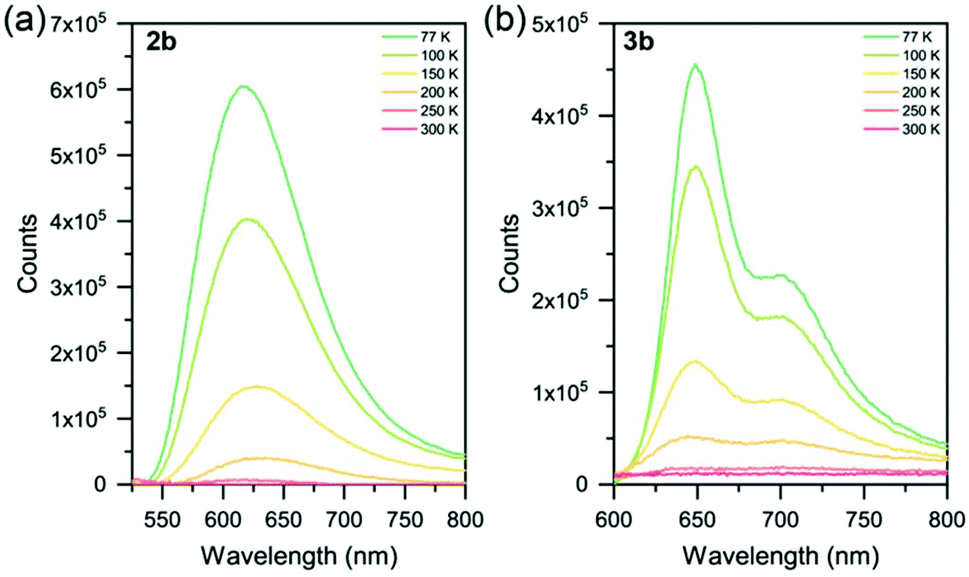 | ||
| Fig. 4 Variable temperature photoluminescence spectra of complexes (a) 2b and (b) 3b from 77–300 K. Solid samples as neat thin-films on quartz substrates. | ||
At 77 K, complexes 2a and 2b display a similar emission to [Ru(p-toyl-tpy)2]2+,29 a homoleptic species with a –CH2– spacer between two pyridyl groups to alleviate ring strain with an emission maximum of 609 nm. Another method of improving octahedricity is the use of more-rigid ligands, such as in [Ru(dqxp)2]2+,51 a homoleptic complex containing 2,6-di(quinoxaline-5-yl)pyridine units. The increased π-conjugation (in addition to the reduced ring strain) leads to emission that is red-shifted relative to complexes 2a and 2b.
Complexes 3a, 3b and 3c were found to be emissive at 77 K (λmax = 641, 649 and 612 nm, respectively) with all three complexes exhibiting structured photoluminescence spectra with a lower energy shoulder. This structure is very similar in nature to that of [Ru(tpy)2]2+ at 77 K (ref. 20 and 52) indicating that the 3MLCT involves the tpy ligand and not the sulfur-bridged ligand, with this assignment corroborated by DFT calculations (though the nature of 3c is a mixture of 3MLCT and 3MC, again caused by the electron-withdrawing -CF3 groups on the sulfur-bridged ligand). Relative shifts in λmax between 3a–c are due to the electron-donating and electron-withdrawing substituents appended, as described for the homoleptic species (2a–c) above.
On comparing homoleptic complexes 2a–c to heteroleptic complexes 3a–c, there is a higher energy gap seen for the homoleptic vs. the heteroleptic species. This is in good agreement with the electrochemical, absorption and DFT data.
Conclusions
In summary, this work details the synthesis and characterization of a series of new sulfur-bridged terpyridyl ligands (1a–c) and their resulting homoleptic [Ru(L)2]2+ (2a–c) and heteroleptic [Ru(tpy)(L)]2+ (3a–c) complexes. The Ru(II) species were structurally characterized with NMR and single crystal XRD techniques, in addition to photophysical, electrochemical and computational data.All six complexes were found to be stable in solution. Single crystal XRD experiments shows that the addition of sulfur spacers between the pyridyl rings affords a greater flexibility that results in reduced ring strain and complex geometries closer to that of a “perfect” octahedron, with average trans N–Ru–N angles calculated at 178.9°. Future functionalization at the 4, 4′ or 4′′-positions could lead to highly-ordered, multi-nodal systems.
All Ru(II) species were found to be non-emissive at room temperature due to deactivation of the triplet metal-to-ligand charge transfer (3MLCT) state by thermal population of the non-emissive triplet metal-centered (3MC) state. 2a and 2b were both emissive as neat thin films at 77 K, with a broad, featureless emission assigned to a 3MC state. 2c however was non-emissive at 77 K due to the domination of a 3MC state. Complexes 3a–c are emissive at 77 K and all three show a structured profile due to a 3MLCT emission from the tpy ligand. Excited state assignments were corroborated with Density Functional Theory (DFT) calculations. Altering the substituents at the 4 and 4′′-positions of the sulfur-bridged ligands with electron-donating or electron-withdrawing groups allows facile tuning of the photophysical and electrochemical HOMO–LUMO gap. Interestingly, complexes 2a and 2b display significant oxidation peaks at ∼–1.0 V. This is attributed to formation of a new intermediate complex when the second reduction of the complex occurs. We propose that the increased flexibility afforded by the bridging sulfur atoms allows one of the pyridyl rings on the sulfur-bridged ligand to become partially unbound from the Ru center at higher reduction states, resulting in the formation of a solvated complex. The redox-triggered changes observed in complexes 2a and 2b could open avenues towards redox switches (such as electrochromic dyes). The facile synthesis of the sulfur-bridged ligands, coupled with improved octahedricity at the metal center, leads to enhanced tuneability of the electronic properties. This will lead to complexes which can be more easily tailored to a range of electrocatalytic reactions.
Experimental section
General considerations
All experiments were conducted under an inert N2 atmosphere using standard Schlenk-line techniques unless otherwise stated. Microwave reactions were performed on a Biotage Initiator 2.3 microwave synthesizer. The following chemicals were purchased and used without further purification: 2-hydroxy-4-methylpyridine and ammonium hexafluorophosphate from Alfa Aesar; 2-hydroxy-4-(trifluoromethyl)pyridine from AK Scientific; 2,6-dibromopyridine, 2-mercaptopyridine and Lawesson's reagent from Sigma Aldrich; 2,2′:6,2′′-terpyridine and ruthenium(III) chloride hydrate from Strem. 2,2′-Bis(4-methylpyridyl)disulfide,38cis-Ru(DMSO)4Cl253 and Ru(tpy)Cl354 were prepared according to the relevant literature procedures.
Spectroscopy
1H NMR and 13C NMR spectra were collected using either a Bruker AV-400 or AV-600 spectrometer and referenced to the residual protonated solvent peak. NMR solvents (from either Cambridge Isotope Laboratories or Aldrich) were used as received. Electrospray ionization mass spectrometry data were obtained using a Bruker Esquire LC ion trap mass spectrometer. All solvents used for photophysical measurements were analytical HPLC grade and used as received from Fisher Scientific. Electronic absorption spectra were recorded on a Varian Cary 5000 UV-vis-near-IR spectrophotometer. Photoluminescence data were collected on a Photon Technology International (PTI) QuantaMaster 50 fluorimeter fitted with an integrating sphere, double excitation monochromator, and utilizing a 75 W Xe arc lamp as the source. Low temperature photoluminescence measurements were conducted using an Oxford Instruments Optistat DN utilizing an ITC601 temperature controller. Solution spectra were collected using 1 cm2 quartz cells (Starna Cells). Solid samples were drop-cast on to quartz slides (Ted Pella Inc.) from acetone, with the solvent allowed to evaporate under ambient conditions, yielding thin films that were uniform by eye.Crystallography
Single crystal X-ray data were collected using a Bruker APEX DUO diffractometer with graphite monochromated Mo Kα radiation (λ = 0.71073 Å) at 100 K. Raw frame data were processed using APEX2.55 The program SAINT+, v. 7.6856 was used to reduce the data and the program SADABS57 was used to make corrections to the empirical absorptions. Space group assignments were made using XPREP on all compounds. In all cases, the structures were solved in the OLEX258 suite of programs by direct methods using SHELXS-9759 and refined using full-matrix least-squares/difference Fourier techniques on F2 using SHELXL-97.60 All non-hydrogen atoms were refined anisotropically. Diagrams and publication material were generated using CrystalMaker.Electrochemistry
Electrochemical experiments were performed using a Pine AFCBP1 bipotentiostat in an airtight three-electrode cell with a glassy carbon working electrode, platinum mesh counter electrode, and silver wire pseudo-reference electrode. Cyclic voltammetry (CV) was performed on 1 mM solutions of the complexes in an electrolyte solution consisting of 0.1 M (n-Bu4N)PF6 (recrystallized 3 times from EtOH) in anhydrous CH3CN sparged with N2 prior to use. CV experiments were executed with sweep rates of 100 mV s−1. Potentials were recorded vs. ferrocene as an added internal standard. Electrochemical band gaps were calculated using the oxidation potential and the first reduction potential.Computational methods
Density functional theory (DFT) calculations were carried out using the Gaussian 16 Rev.B01 suite of programs.61 The B3LYP functional with 6-31G** basis set (for C, H, N, F, and S atoms)62–64 and the LANL2DZ effective-core pseudopotential (for Ru)65 was employed to optimize the geometry of the ground-states (S0) and lowest-energy triplet states (T1) of all complexes with no symmetry restrictions. All the calculations were performed with solvent (CH3CN) included. Solvent effects were considered within the self-consistent reaction field (SCRF) theory using the polarized continuum model (PCM) approach.66,67 Time-dependent DFT (TD-DFT) calculations were also performed to understand the nature of vertical excitation transitions, and were executed at the same level as the DFT.68–70Synthetic details
To a round-bottomed flask was added the obtained 4-(trifluoromethyl)pyridine-2-thiol (1.06 g, 5.91 mmol, 1.0 equiv.), NaOH (0.284 g, 7.09 mmol, 1.2 equiv.) and H2O (50 mL). The solution was stirred for 30 min at room temperature, following which K3[Fe(CN)6] (2.33 g, 7.09 mmol, 1.2 equiv.) in H2O (50 mL) was added. The reaction mixture was stirred for 14 h at room temperature, and the resultant precipitate was filtered, washed with H2O and dried under vacuum to give pure 1,2-bis(4-(trifluoromethyl)pyridin-2-yl)disulfane as an off-white solid (1.01 g, 2.84 mmol, 96%). 1H NMR (400 MHz, CD2Cl2) δ 8.65 (dt, J = 5.1, 0.8 Hz, 2H), 7.82 (dt, J = 1.6, 0.8 Hz, 2H), 7.37 (ddd, J = 5.0, 1.6, 0.7 Hz, 2H). 13C{1H} NMR (101 MHz, CD2Cl2) δ 160.7, 151.4, 140.0 (q, J = 34.3 Hz), 123.1 (q, J = 273.6 Hz), 117.7 (q, J = 3.4 Hz), 116.4 (q, J = 3.8 Hz). 19F{1H} NMR (377 MHz, CD2Cl2) δ −65.0. ESI-MS calcd for C12H7N2F6S2: 356.9955; found 356.9952 [M + H]+.
:5 DCM:MeOH) yielding a yellow oil (1.09 g, 3.67 mmol, 69%). 1H NMR (400 MHz, CD2Cl2) δ 8.49 (ddd, J = 4.8, 2.0, 0.9 Hz, 2H), 7.61–7.50 (m, 3H), 7.43 (dt, J = 8.0, 1.0 Hz, 2H), 7.25 (d, J = 7.8 Hz, 2H), 7.20–7.14 (m, 2H). 13C{1H} NMR (101 MHz, CD2Cl2) δ 158.0, 156.6, 150.6, 138.2, 137.5, 126.8, 123.5, 122.5. ESI-MS calcd for C15H12N3S2: 298.0473; found 298.0471 [M + H]+.
:5 DCM:EtOAc) yielding a white powder (0.254 g, 0.586 mmol, 58%). 1H NMR (400 MHz, CD2Cl2) δ 8.63 (d, J = 5.1 Hz, 2H), 7.68 (d, J = 1.5 Hz, 2H), 7.67–7.64 (m, 1H), 7.42 (d, J = 7.8 Hz, 2H), 7.33 (dd, J = 5.1, 1.6 Hz, 2H). 13C{1H} NMR (101 MHz, CD2Cl2) δ 158.5, 156.0, 151.0, 138.9 (q, J = 34.3 Hz), 138.4, 124.7, 122.7 (q, J = 274.5 Hz) 121.0 (q, J = 3.9 Hz), 117.2 (q, J = 3.6 Hz). 19F{1H} NMR (377 MHz, CD2Cl2) δ −65.1. ESI-MS calcd for C17H10N3F6S2: 434.0221; found 434.0220 [M + H]+.
:5:5 CH3CN:H2O:sat. KNO3 (aq.)), yielding a yellow powder (0.067 g, 0.068 mmol, 80%). 1H NMR (400 MHz, CD3CN) δ 8.02–7.93 (m, 6H), 7.88–7.77 (m, 8H), 7.07 (dt, J = 5.9, 1.3 Hz, 4H), 6.93 (ddd, J = 7.6, 5.9, 1.8 Hz, 4H). 13C{1H} NMR (101 MHz, CD3CN) δ 160.1, 157.8, 157.3, 140.1, 139.8, 131.3, 128.9, 126.8. ESI-MS calcd for C30H22N6F6PS4102Ru: 840.9473; found 840.9474 [M–PF6]+.
:5:5 CH3CN:H2O:sat. KNO3 (aq.)) yielding an orange powder (0.068 g, 0.073 mmol, 65%). 1H NMR (400 MHz, CD3CN) δ 8.58 (ddd, J = 5.6, 1.6, 0.7 Hz, 2H), 8.53 (d, J = 8.1 Hz, 2H), 8.41 (ddd, J = 8.1, 1.4, 0.7 Hz, 2H), 8.30–8.22 (m, 3H), 8.16 (dd, J = 8.5, 6.9 Hz, 1H), 8.06 (td, J = 7.9, 1.5 Hz, 2H), 7.69 (ddd, J = 7.9, 1.7, 0.7 Hz, 2H), 7.62 (td, J = 7.7, 1.6 Hz, 2H), 7.39 (ddd, J = 7.4, 5.6, 1.4 Hz, 2H), 6.88 (ddd, J = 7.5, 6.0, 1.7 Hz, 2H), 6.73 (ddd, J = 6.0, 1.6, 0.7 Hz, 2H). 13C{1H} NMR (101 MHz, CD3CN) δ 159.8, 159.5, 159.2, 159.1, 153.6, 139.9, 139.3, 137.4, 131.2, 129.3, 127.1, 126.1, 125.7, 125.7. ESI-MS calcd for C30H22N6F6PS2102Ru: 777.0033; found 777.0037 [M − PF6]+.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Natural Sciences and Engineering Research Council (NSERC) of Canada for funding.C. M. B. thanks Dr Maria Ezhova for assistance with NMR characterizations. D. H. thanks Dr Brian Patrick for help with refinement of the crystal structures. Z. X. thanks Compute Canada (http://www.computecanada.ca) for the provision of computational resources.Notes and references
- G. Morgan and F. H. Burstall, J. Chem. Soc., 1937, 1649–1655 RSC.
- F. H. Burstall, J. Chem. Soc., 1936, 173 RSC.
- J. P. Paris and W. W. Brandt, J. Am. Chem. Soc., 1959, 81, 5001–5002 CrossRef CAS.
- D. W. Fink and W. E. Ohnesorge, J. Am. Chem. Soc., 1969, 91, 4995–4998 CrossRef CAS.
- K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159–244 CrossRef CAS.
- P. G. Bomben, K. C. D. Robson, B. D. Koivisto and C. P. Berlinguette, Coord. Chem. Rev., 2012, 256, 1438–1450 CrossRef CAS.
- A. Carella, F. Borbone and R. Centore, Front. Chem., 2018, 6, 6624–6653 Search PubMed.
- A. Wu, D. Yoo, J. K. Lee and M. F. Rubner, J. Am. Chem. Soc., 1999, 121, 4883–4891 CrossRef CAS.
- H. J. Bolink, L. Cappelli, E. Coronado and P. Gaviña, Inorg. Chem., 2005, 44, 5966–5968 CrossRef CAS.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS.
- S. Angerani and N. Winssinger, Chem. – Eur. J., 2019, 25, 6661–6672 CrossRef CAS.
- J.-W. Lee and B. N. Popov, J. Solid State Electrochem., 2007, 11, 1355–1364 CrossRef CAS.
- S. Rigaut, J. Perruchon, L. Le Pichon, D. Touchard and P. H. Dixneuf, J. Organomet. Chem., 2003, 37–44 CrossRef.
- A. Benameur, P. Brignou, E. Di Piazza, Y.-M. Hervault, L. Norel and S. Rigaut, New J. Chem., 2011, 35, 2105–2109 RSC.
- F. E. Poynton, S. A. Bright, S. Blasco, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2017, 46, 7706–7756 RSC.
- L. Hammarström, Curr. Opin. Chem. Biol., 2003, 7, 666–673 CrossRef.
- C. W. Cady, R. H. Crabtree and G. W. Brudvig, Coord. Chem. Rev., 2008, 252, 444–455 CrossRef CAS.
- J. K. McCusker, Acc. Chem. Res., 2003, 36, 876–887 CrossRef CAS.
- J. P. Sauvage, J. P. Collin, J. C. Chambron, S. Guillerez, C. Coudret, V. Balzani, F. Barigelletti, L. De Cola and L. Flamigni, Chem. Rev., 1994, 94, 993–1019 CrossRef CAS.
- M. Maestri, N. Armaroli, V. Balzani, E. C. Constable and A. M. W. C. Thompson, Inorg. Chem., 1995, 34, 2759–2767 CrossRef CAS.
- A. K. Pal and G. S. Hanan, Chem. Soc. Rev., 2014, 43, 6114–6184 RSC.
- E. A. Medlycott and G. S. Hanan, Chem. Soc. Rev., 2005, 34, 110–133 RSC.
- M. I. J. Polson, N. J. Taylor and G. S. Hanan, Chem. Commun., 2002, 1356–1357 RSC.
- G. J. E. Davidson, S. J. Loeb, P. Passaniti, S. Silvi and A. Credi, Chem. – Eur. J., 2006, 12, 3233–3242 CrossRef CAS.
- F. S. Han, M. Higuchi and D. G. Kurth, J. Am. Chem. Soc., 2008, 130, 2073–2081 CrossRef CAS.
- J. Fortage, G. Dupeyre, F. Tuyèras, V. Marvaud, P. Ochsenbein, I. Ciofini, M. Hromadová, L. Pospísil, A. Arrigo, E. Trovato, F. Puntoriero, P. P. Lainé and S. Campagna, Inorg. Chem., 2013, 52, 11944–11955 CrossRef CAS.
- A. K. Pal, S. Serroni, N. Zaccheroni, S. Campagna and G. S. Hanan, Chem. Sci., 2014, 5, 4800–4811 RSC.
- L. Hammarström and O. Johansson, Coord. Chem. Rev., 2010, 254, 2546–2559 CrossRef.
- M. Abrahamsson, M. J. Lundqvist, H. Wolpher, O. Johansson, L. Eriksson, J. Bergquist, T. Rasmussen, H.-C. Becker, L. Hammarström, P.-O. Norrby, B. Åkermark and P. Persson, Inorg. Chem., 2008, 47, 3540–3548 CrossRef CAS.
- A. Breivogel, C. Förster and K. Heinze, Inorg. Chem., 2010, 49, 7052–7056 CrossRef CAS.
- A. Breivogel, M. Meister, C. Förster, F. Laquai and K. Heinze, Chem. – Eur. J., 2013, 19, 13745–13760 CrossRef CAS.
- A. K. Pal, N. Zaccheroni, S. Campagna and G. S. Hanan, Chem. Commun., 2014, 50, 6844–6846 Search PubMed.
- C. M. Brown, M. J. Kitt, Z. Xu, D. Hean, M. B. Ezhova and M. O. Wolf, Inorg. Chem., 2017, 56, 15110–15118 CrossRef CAS.
- C. M. Brown, V. Carta and M. O. Wolf, Chem. Mater., 2018, 30, 5786–5795 CrossRef CAS.
- E. Caron, C. M. Brown, D. Hean and M. O. Wolf, Dalton Trans., 2019, 48, 1263–1274 RSC.
- C. M. Brown, C. Li, V. Carta, W. Li, Z. Xu, P. H. F. Stroppa, I. D. W. Samuel, E. Zysman-Colman and M. O. Wolf, Inorg. Chem., 2019, 58, 7156–7168 CrossRef CAS.
- C. Chachaty, G. C. Pappalardo and G. Scarlata, J. Chem. Soc., Perkin Trans. 2, 1976, 1234–1235 RSC.
- O. S. Kanishchev and W. R. Dolbier Jr., Angew. Chem., Int. Ed., 2014, 54, 280–284 Search PubMed.
- M. Jäger, R. J. Kumar, H. Görls, J. Bergquist and O. Johansson, Inorg. Chem., 2009, 48, 3228–3238 CrossRef.
- F. Schramm, V. Meded, H. Fliegl, K. Fink, O. Fuhr, Z. Qu, W. Klopper, S. Finn, T. E. Keyes and M. Ruben, Inorg. Chem., 2009, 48, 5677–5684 CrossRef CAS.
- C. Bhaumik, D. Saha, S. Das and S. Baitalik, Inorg. Chem., 2011, 50, 12586–12600 CrossRef CAS.
- M. W. Cooke, M.-P. Santoni, B. Hasenknopf and G. S. Hanan, Dalton Trans., 2016, 45, 17850–17858 RSC.
- T. Österman, M. Abrahamsson, H. C. Becker, L. Hammarström and P. Persson, J. Phys. Chem. A, 2012, 116, 1041–1050 CrossRef.
- M. J. Lundqvist, PhD thesis, Uppsala University, 2006.
- K. S. Low, J. M. Cole, X. Zhou and N. Yufa, Acta Crystallogr., Sect. B: Struct. Sci., 2012, 68, 137–149 CrossRef CAS.
- K. Lashgari, M. Krttikos, R. Norrestam and T. Norrby, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 64–67 CrossRef.
- H. Wolpher, O. Johansson, M. Abrahamsson, M. Kritikos, L. Sun and B. Åkermark, Inorg. Chem. Commun., 2004, 7, 337–340 CrossRef CAS.
- M. Abrahamsson, H. Wolpher, O. Johansson, J. Larsson, M. Kritikos, L. Eriksson, P. O. Norrby, J. Bergquist, L. Sun, B. Åkermark and L. Hammarström, Inorg. Chem., 2005, 44, 3215–3225 CrossRef CAS.
- M. Beley, J. P. Collin, J. P. Sauvage, H. Sugihara, F. Heisel and A. Miehé, J. Chem. Soc., Dalton Trans., 1991, 46, 3157–3159 RSC.
- J. Klein, A. Stuckmann, S. Sobottka, L. Suntrup, M. van der Meer, P. Hommes, H.-U. Reissig and B. Sarkar, Chem. – Eur. J., 2017, 23, 12314–12325 CrossRef CAS.
- G. A. Parada, L. A. Fredin, M. P. Santoni, M. Jäger, R. Lomoth, L. Hammarström, O. Johansson, P. Persson and S. Ott, Inorg. Chem., 2013, 52, 5128–5137 CrossRef CAS.
- A. Amini, A. Harriman and A. Mayeux, Phys. Chem. Chem. Phys., 2004, 6, 1157–1164 RSC.
- I. P. Evans, A. Spencer and G. Wilkinson, J. Chem. Soc., Dalton Trans., 1973, 204–206 RSC.
- B. P. Sullivan, J. M. Calvert and T. J. Meyer, Inorg. Chem., 1980, 19, 1404–1407 CrossRef CAS.
- Bruker, APEX2 (Version 2), User Manual M86-E01078, Bruker AXS Inc., Madison, Wisconsin, USA, 2006 Search PubMed.
- Bruker, SAINT+ (Version 7.68), Bruker AXS Inc., Madison, Wisconsin, USA, 2009 Search PubMed.
- SADABS, Bruker AXS Inc., 2014 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Peralta Jr., F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian, Inc., Wallingford, CT, 2016.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- S. Miertuš, E. Scrocco and J. T. C. Physics, J. Organomet. Chem., 1981, 55, 117–129 Search PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS.
- C. Jamorski, M. E. Casida and D. R. Salahub, J. Chem. Phys., 1996, 104, 5134–5147 CrossRef CAS.
- M. Petersilka, U. J. Gossmann and E. K. U. Gross, Phys. Rev. Lett., 1996, 76, 1212–1215 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, crystallographic data, electrochemical data, photophysical data and computational calculations. CCDC 1935270–1935275. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qi01009b |
| This journal is © the Partner Organisations 2020 |