
Solvent switching smart metal–organic framework as a catalyst of reduction and condensation†
Farzaneh
Rouhani
,
Behnam
Gharib
and
Ali
Morsali
 *
*
Department of Chemistry, Faculty of Sciences, Tarbiat Modares University, P.O. Box 14115-175, Tehran, Iran. E-mail: morsali_a@modares.ac.ir
First published on 16th July 2019
Abstract
The organization of a Zn-based metal–organic framework (MOF) as the first solvent switching catalyst has been achieved via in situ ligand incorporation. The catalytic mechanism of TMU-60 (Zn (OBA) (L*)) can be changed by a special solvent which led to reversible changes in its structure. The switching process leads to the production of two completely different products from the same raw material. The local reversible dynamic conversion of the ligand functionality from amine to imine on impact of chloroform led to a noticeable hydrogen generation/selective aldehyde reduction to the corresponding alcohol in the absence of a reductant. The catalyst recovery process was easily and quickly performed by immersing the framework in DMF. 1H NMR spectroscopy, IR spectroscopy and the results of GC and GC mass confirm the switching process. However, in the presence of acetonitrile, a different catalytic mechanism, i.e., the aldol condensation reaction, occurred by TMU-60 in a short period at ambient temperature. This study represents a simple approach for the construction of a switchable MOF, and these smart systems may provide a unique MOF platform for many applications, particularly as catalysts, in the future.
1. Introduction
Stimuli-responsive materials that are capable of altering their structure or properties in response to external stimuli, such as temperature,1 spin crossover,2 light,3 redox,4 and pH5 and in limited cases, solvent molecules,6 have recently captured considerable attention on account of their important role in constructing functional materials.7 In general, the term “molecular switch” is referred to a reversible and controllable transformation process between two or more stable molecular states created by an external stimulus.8–10 These transformations sometimes lead to different properties.11,12 A catalyst is key for the effective and efficient conversion of available building blocks for high-value molecules and materials.13,14 Previously, chemists have focused on the invention of new catalysts and optimization of their performance.15–17 Recently, inspired by nature, chemists have started to turn their attention to the development of catalysts whose activity can be switched by an external stimulus in different chemical processes. Potential applications include using the states of multiple switchable catalysts to control the sequences of transformations, thus producing different products from a pool of building blocks according to the order and type of stimuli applied.18 The use of switchable catalysts does not have a long history but has shown some interesting results. In 2010, Sohtome and Nagasawa described a Mannich-type reaction based on an enantiodivergent organocatalyst using a conformationally flexible guanidine/bisthiourea catalyst.19 When the reaction was performed in nonpolar solvents, such as m-xylene, excellent yields and enantiocontrol (99% yield, 97% ee) in favour of the (S)-enantiomer were achieved. In contrast, when the reaction was performed using polar aprotic solvents, such as acetonitrile, the (R)-enantiomer was formed with similar yield and enantioselectivity. In another impressive example, Melchiorre et al. described the diastereodivergent Michael addition of alkyl thiols to α,β-unsaturated ketones catalyzed by a cinchona alkaloid derivative in which the sense of diastereoselectivity could be reversed by the choice of the solvent and the acid additive that was employed.20 The switching process strongly depends on the hydrogen-bonding ability of the acid-anion. Solvent-dependent stereoselectivity of other catalytic systems have also been reported.21–25 The use of MOFs, a new class of crystalline and porous materials, particularly as a switchable catalyst, have advantages such as the possibility of computational studies of structural states, high surface area, permanent porosity and chemical and thermal stability, which make solvent switching MOFs a dream heterogeneous catalyst.26 Moreover, because of sufficient space in the pores, MOFs overcome the possible problems caused by incomplete conversion and steric hindrance due to intramolecular interactions.27,28 Also, the suitable functional groups of MOFs, which were achieved via in situ or post-synthesis methods, can affect the quality of molecular switch in the pores. Among the three general methods for the preparation of switchable MOFs, the incorporation of the switchable molecules into MOF pores,3,29,30 functionalization of the linkers as switchable groups,31,32 and the direct use of switching ligands for synthesizing MOFs,1,33,34 we chose the third method which is proper for catalytic applications.Despite fairly extensive studies on MOFs,35–38 since 2009, when the first switchable MOF was designed and synthesized, very limited studies (fewer than 50) were documented in the area, and only one study has been performed on solvent switching MOFs.6 To date, no reports have been documented on changing the catalytic mechanism using solvents. Tunable active sites of stimuli-responsive MOFs may serve as enzyme-like catalysts that are capable of catalyzing specific reactions.
The selective reduction of aldehydes is one of the most important goals of organic chemistry. Two common methods for the reduction of aldehydes to their corresponding alcohols are known. One is the use of a hydrogen source and a precious metal (such as a complex containing ruthenium)39,40 and another is the Meerwein–Ponndorf–Verley (MPV) reduction method.41,42 However, the replacement of rare, toxic and precious metals by cheap and non-toxic compounds as catalysts for the hydrogenation reaction is an important and attractive target.43,44 Although the MPV method uses inexpensive metals for the reduction process, it requires the presence of a sacrificial alcohol and high temperature. Despite the effective performance, this method does not selectively act between aldehyde and ketone. It should be noted that many of the catalysts can still work at high temperatures and require the high pressure of hydrogen gas.45
Herein, for the first time, we succeeded in designing and synthesizing a solvent switching MOF that demonstrated hydrogen generation/selective aldehyde reduction in one solvent and self-condensation of aldehydes in another solvent. The use of this unique structure as a catalyst offers tremendous properties, such as infinite catalytic cycling, selective product related to the used solvent, no need for hydrogen sources or sacrificial alcohols for the reduction process, good performance at ambient temperature, and simple separation of reaction mixtures. The tetrahydropyrazine-functionalized pillared MOF [Zn(OBA)(L*)]·DMF (TMU-60) was synthesized using Zn(NO3)2·4H2O and 4,4′-oxybis(benzoic acid) (H2OBA) and 5,6-di(pyridin-4-yl)-1,2,3,4-tetrahydropyrazine ligand. L* was synthesized by the in situ C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C coupling reaction of N1,N2-bis(pyridin-4-ylmethylene)ethane-1,2-diamine (L). The obtained TMU-60 is a functional solvent switching catalyst for the reduction and self-condensation of aldehydes. In the presence of chloroform as a co-solvent, the dynamic conversion of L* into L** (dihydropyrazine moieties) led to the production of hydrogen and the selective reduction of the aldehyde to the related alcohol at ambient temperature without the need for hydrogen. The hydrogen production is related to the reversible dynamic conversion of tetrahydropyrazine to dihydropyrazine moieties. However, TMU-60 in the presence of non-oxidizing solvents such as acetonitrile acts as an efficient basic catalyst for the self-condensation reaction of aldehydes.
C coupling reaction of N1,N2-bis(pyridin-4-ylmethylene)ethane-1,2-diamine (L). The obtained TMU-60 is a functional solvent switching catalyst for the reduction and self-condensation of aldehydes. In the presence of chloroform as a co-solvent, the dynamic conversion of L* into L** (dihydropyrazine moieties) led to the production of hydrogen and the selective reduction of the aldehyde to the related alcohol at ambient temperature without the need for hydrogen. The hydrogen production is related to the reversible dynamic conversion of tetrahydropyrazine to dihydropyrazine moieties. However, TMU-60 in the presence of non-oxidizing solvents such as acetonitrile acts as an efficient basic catalyst for the self-condensation reaction of aldehydes.
As far as we know, this is the first report of a solvent switching MOF which plays the catalytic role with two separate catalytic mechanisms in the presence of two different solvents. It could be a new step in the development of solvent switching MOFs. Research in this field can open new horizons in science and technology.
2. Experimental
2.1. Synthesis of [Zn(OBA) (L*)]·DMF (TMU-60)
A mixture of Zn(NO3)2·4H2O, 4,4′-oxybis(benzoic acid) (H2OBA) and N1,N2-bis (pyridin-4-ylmethylene) ethane-1,2-diamine (L) in DMF was heated at 60 °C for 5 min to dissolve all components. The resulting yellow solution was transferred to a Teflon autoclave and heated to 120 °C for 72 h. The red crystals of TMU-60 were separated via filtration and washed once with DMF and three times with cold ethanol to remove the unreacted materials, followed by drying in air. Yield: ∼20%. IR (KBr, cm−1) for TMU-60: IR (KBr, cm−1): 461.99(m), 656.61(m), 775.74(m), 837.65(m), 1016.40(w), 1075.05(w), 1145.28(m), 1225.62(s), 1375.15(s), 1494.46(m), 1575.20(m), 1606.10(s), 1674.15(s), 3245.75(m). Elemental analysis (%) calculated: C 59.96, H 3.56, N 6.66. Found: C 60.0, H 3.5, N 7.1. Prior to any catalytic reaction, solvent exchange was used to remove the solvents that fill the pores. For this purpose, the crystals were immersed in dichloromethane for two days, and the solvent was refreshed every 12 h. Subsequently, they were separated by filtration and placed under heat at 120 °C for 24 h. The ligand synthesis method and explanations of raw materials and the apparatus are given in the ESI.†2.2. Synthesis of 5,6-di(pyridin-4-yl)-1,2,3,4-tetrahydropyrazine (L*) ligand
L ligand (0.5 mmol) was dissolved in 7 ml of DMF for 5 min to obtain a clear yellow-colored solution. After the complete dissolution of the ligand, 0.05 g of sodium cyanide was added to the solution and heated to 120 °C for 20 min. Changes in the color of the solution from yellow to red indicated the completion of the reaction. The reaction solvent was evaporated after 20 min at 80 °C, and the remaining sediment was washed with ethanol to remove the unreacted material and stored for further analysis. Mp: 70.2 °C. IR (KBr, cm−1): 3301(s), 3032(m), 2843(s), 1667(m), 1595(s), 1406(s), 1318(w), 1218(w), 1095(w), 992 (w), 823(m), 586(m). 1H NMR (500 MHz, DMSO): δ 10.3 (d, 2H, –NH), 8.3 (d, 4 H, ArH), 7.61 (d, 4 H, Ar–H) and 3.9 (s, 4 H, HC-C). Elemental analysis (%) calculated: C 67.27, H 6.59, N 26.15. Found: C 67.2, H 6.63, N 26.15. The mass and 1H NMR spectrum of L and L* ligand are presented in ESI (Fig. S1–S3†).2.3. General procedure for catalytic reactions
After activation, the catalyst (6 mol% based on the amount of ligand present in the structure relative to the aldehyde) was placed in a round bottom tube. Then, 0.5 mmol of aldehyde and 2 ml of solvent were added to the container and stirred at 5 °C, 15 °C, 25 °C and 35 °C for different times. In addition, 25 μl of benzene was added to all reactions as an internal standard. In the case of the reduction reaction, 5% chloroform (v/v) was added to the reaction solvent. The conversion was measured by the GC with the HP-5 column and the Agilent software and in some cases by the GC-mass. After the desired time, the catalysts were separated from the reaction mixture by centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 3 min, and then 0.5 μl of the supernatant liquid was immediately injected into the apparatus. After completion of the reaction time and separation of the catalyst from the mixture, the reaction solution was exposed to ambient temperature for 24 h to evaporate the solvent and solid products were then analyzed via1H NMR spectroscopy. The separated catalyst was washed several times with a fresh reaction solvent and dried at ambient temperature for use in subsequent experiments.
000 rpm for 3 min, and then 0.5 μl of the supernatant liquid was immediately injected into the apparatus. After completion of the reaction time and separation of the catalyst from the mixture, the reaction solution was exposed to ambient temperature for 24 h to evaporate the solvent and solid products were then analyzed via1H NMR spectroscopy. The separated catalyst was washed several times with a fresh reaction solvent and dried at ambient temperature for use in subsequent experiments.
3. Results and discussion
As described in the Experimental section, TMU-60 was synthesized using the solvothermal method by heating the mixture of Zn(NO3)2·4H2O, H2OBA and N1,N2-bis (pyridine-4-ylmethylene) ethane-1,2-diamine (L) in DMF at 120 °C for 2 days. Suitable X-ray quality red crystals were obtained with the [Zn(OBA)(L*)]·DMF structure. L* = 5,6-dipyridin-4-yl-1,2,3,4-tetrahydropyrazine ligand was formed in situ from the L ligand by aldimine CC coupling and cyclization reaction.35 As shown in Scheme 1, the L* ligand acts as the intermediate between L and L**. The proposed mechanism for the cyclization reaction of L is presented in Scheme S1.† The mechanism shows that in the presence of an oxidizing agent the reaction proceeds and dihydropyrazine species are formed, but due to lack of oxygen in the reaction vessel, L* is formed instead of L**.
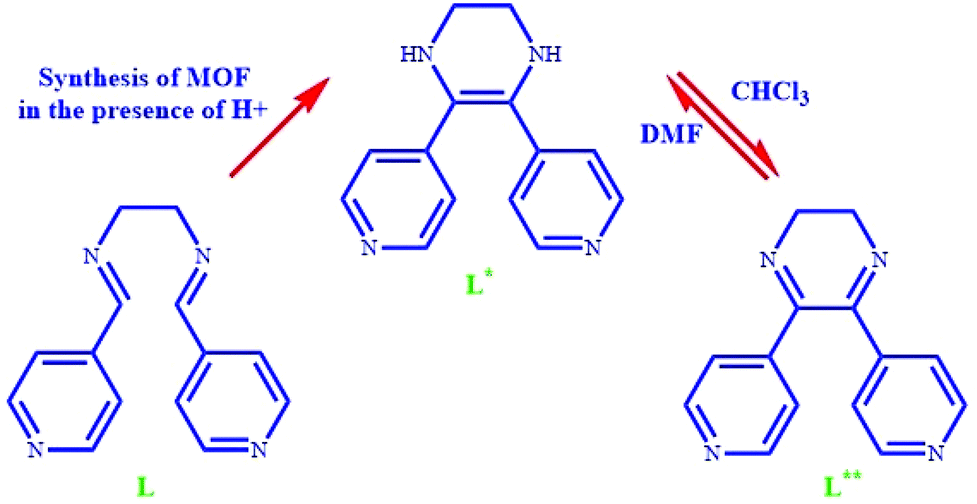 | ||
| Scheme 1 Schematic of N1,N2-bis(pyridin-4-ylmethylene)ethane-1,2-diamine (L), 5,6-di(pyridin-4-yl)-1,2,3,4-tetrahydropyrazine (L*) and 5,6-di(pyridin-4-yl)-2,3-dihydropyrazine (L**). | ||
The 3D structure of the TMU-60 crystallizes in the Ibca space group, and is built up from Zn2(COO)4 paddle-wheel secondary building units that are connected together by OBA ligands. Twofold interpenetrated structure of [Zn(oba)(L*)]·DMF (TMU-60) was obtained by connecting the 2D sheets, which were extended in three dimensions through L* ligands. TMU-60 is based on a binuclear Zn(II) unit (Fig. 1A) and shows 1D pores running along the crystallographic a-axis (Fig. 1).
 | ||
| Fig. 1 Representation of 2D sheets formed by the interaction of a paddle-wheel Zn2(CH3COO)4 secondary building unit (A) and H2OBA ligand (B). The presence of L* pillar leads to the expansion of the structure in 3D (D and E). Color code: C, gray; O, red; N, blue; H: yellow; Zn: green. (E) The topology of TMU-60 in the direction of a (right) and c (left) axes. | ||
The average distances of Zn–O and Zn–N in the structure are 2.040 and 2.026 Å, respectively35 (CCDC number: 1862166†). In order to study the porosity of TMU-60, nitrogen gas adsorption was performed at 77 K and 1 bar. The results show that the Brunauer–Emmett–Teller (BET) surface area is 171.522 m2 g−1 and can adsorb 65 cm3 g−1 N2 (Fig. S4†) and the pore distribution is shown in Fig. S5.† As shown in Fig. S6,† the thermal analysis of the TMU-60 framework reflects the loss of a DMF molecule located in the pore in the temperature range of 100–250 °C (calculated 11.51%, found 11%). Therefore, the structure is stable up to around 270 °C and the release of the ligand at 280 °C leads to the destruction of the framework. To ensure the thermal stability of the framework, the structure was placed at 250 °C for 12 h. The XRD and BET patterns of the heat-treated framework remain completely intact, which confirm the thermal stability of the structure until 250 °C (Fig. S6†).
Based on previous in situ synthesis experiments of the ligand-containing secondary amine37 and the possibility of reversible structural transformation of these types of frameworks in special solvents,6 the TMU-60 framework was synthesized to achieve a smart catalyst that responded to environmental conditions.
The ligands containing primary amine are suitable catalytic species for synthesis and post-synthesis;46–48 but due to their high tendency to metal ions, they are often occupied by the metal building units. One of the best methods to introduce the secondary amine with good catalytic properties is the in situ synthesis. According to our studies, the L ligand, which contains secondary amine, can be simply synthesized via in situ method. As noted above, L* is the intermediate of L and L**, and in the presence of an oxidant, it can be turned to L**.35,37 In order to study the possible structural changes in solvents, TMU-60 was immersed in conventional organic solvents at room temperature, as shown in Fig. 2. As it is easily recognizable, TMU-60 shows a significant red to brown color change in chloroform (less than 5 min); however, in other solvents, even after immersion for 12 h, it remains completely unchanged. The IR spectrum of TMU-60 after immersion in some solvents is shown in Fig. S7,† and it has the same pattern as that of the TMU-60 powder before immersion in any solvent. So, it can be concluded that other solvents could not oxidize the L* ligand. It should be noted that some minor changes in the peak at 1677 cm−1, which are related to trapped DMF solvent, are dependent on the solvent ability to remove DMF from the pores in the structure. It is worth noting that, despite the color change, structure crystallinity remains entirely unchanged. Both structures (L* and L**) are V-shaped; however, probably with the formation of the imine, the conjugation of the structure is more complete and the color of the structure becomes darker. Also, the N2 adsorption–desorption diagram of O-TMU-60 is shown in Fig. S8.†
 | ||
| Fig. 2 (A) The color change of TMU-60 in the presence of different VOCs. (B) Significant color change of TMU-60 in chloroform after ultrasonication for 1 min. The powder color of TMU-60(C) and O-TMU-60(D) after immersion for 5 min in chloroform. | ||
The XRD pattern of the immersed crystal for 2 h in chloroform remains intact (Fig. 3A). The IR spectra of TMU-60 and O-TMU-60 are shown in Fig. 3B. NH stretching and bending peaks at 3245 and 1575 cm−1 are related to the secondary amine in the L* ligand of TMU-60 and the absence of the peaks in O-TMU-60, respectively, which confirm the conversion of the ligand to O-TMU-60. Also, 1H NMR spectra shown in Fig. 3C demonstrates the amine removal and the formation of the imine.
 | ||
| Fig. 3 (A) Intact XRD pattern of TMU-60 after immersion in chloroform for 2 h, (B) IR spectrum of TMU-60 before and after of activation and oxidation and (C) 1H NMR spectrum of TMU-60 and O-TMU-60. | ||
Chloroform is a common organic solvent, and in some cases, it acts as an oxidizing agent and reduces to dichloromethane.49–51 The solvent was injected into the GC-mass apparatus to ensure that no other oxidizing agent is available in the solvent (such as peroxide, which may have formed over time in the vessel). In addition, in the absence of any reactant, after immersion for 2 h of TMU-60 in chloroform, the solvent was separated from the structure by centrifugation and injected into the GC apparatus. The results show that both chloroform and dichloromethane are present in the reaction medium. Due to the process of dichloromethane synthesis and its price compared to chloroform, this simple transformation is very important. Accordingly, we believe that chloroform can, in addition to reduction, oxidize the tetrahydropyrazine in L* to dihydropyrazine by a reversible method through a dynamic transformation that occurs at the same time as the color changes. Dichloromethane observation in the reaction environment and the color change of the structure is consistent with the proposed mechanism in Scheme 2.
 | ||
| Scheme 2 Proposed mechanism for the oxidation of L* to L** in the adjacent of chloroform. | ||
On the other hand, since the dihydropyrazine in L** is electron deficient, it can simultaneously accept electrons and two hydrogen atoms, and then convert to tetrahydropyrazine.52,53 The color change from brown to red and in the IR and 1H NMR spectra of O-TMU-60 after washing with DMF indicates the change and the conversion of L** to L*. This conversion does not reduce the crystallinity of the structure. Thus, TMU-60 oxidizes easily to O-TMU-60 and O-TMU-60 reduces to TMU-60. After five conversion cycles, the XRD pattern remains unchanged (Fig. S6†). The above results show that due to the conversion of L* to L** and thus the release of two hydrogen atoms, TMU-60 can be easily used as a reducing agent.
To investigate the effect of TMU-60 as a reduction catalyst, the reaction was studied using acetaldehyde as a reactant in DMF/chloroform. As shown in entry 1 (Table 1), due to the conversion of TMU-60 to O-TMU-60 in the presence of chloroform, hydrogen was released in the reaction medium and O-TMU-60 as a catalyst facilitated the reduction of aldehyde to alcohol. Also, due to the presence of DMF in the reaction vessel, the catalyst recovery process to the effective state occurred in the reaction solution, and the presence of a small amount of the catalyst was sufficient for the aldehyde reduction reaction. This cycle can be repeatedly continued only with the addition of a reactant (aldehyde) without the need for external catalyst recovery. The increase in the reaction temperature up to 35 °C speeds up the reaction rate to an insignificant amount. Therefore, 6 mol% of catalyst, time duration of 2 h and ambient temperature were selected as optimal conditions. To investigate the TMU-60 reducing selectivity, the reaction was performed in 90% DMF and 10% chloroform with various reactants, such as acetone, acrylaldehyde, 2-butene and methyl vinyl ketone. The results of entries 7–10 indicate that there is no reaction and confirm that TMU-60 acts selectively toward the aldehyde. For further examination, we extended the reaction range to other aldehydes in the abovementioned conditions. For this purpose, the reaction was performed in the presence of TMU-60 as a source of hydrogen and catalyst on various reactants, such as formaldehyde, butyraldehyde, benzaldehyde, and cinnamaldehyde. The GC curves of the reduction of acetaldehyde, butyraldehyde, and cinnamaldehyde are shown in Fig. S9–12.† As expected, in the reaction of cinnamaldehyde, carbonyl is reduced and the CC bond remains intact. The completion of the reduction process in less than 2 h and at room temperature indicates the selectivity and effectiveness of the catalyst. The proposed mechanism for the reduction of aldehyde in the presence of TMU-60 is shown in Scheme 3.
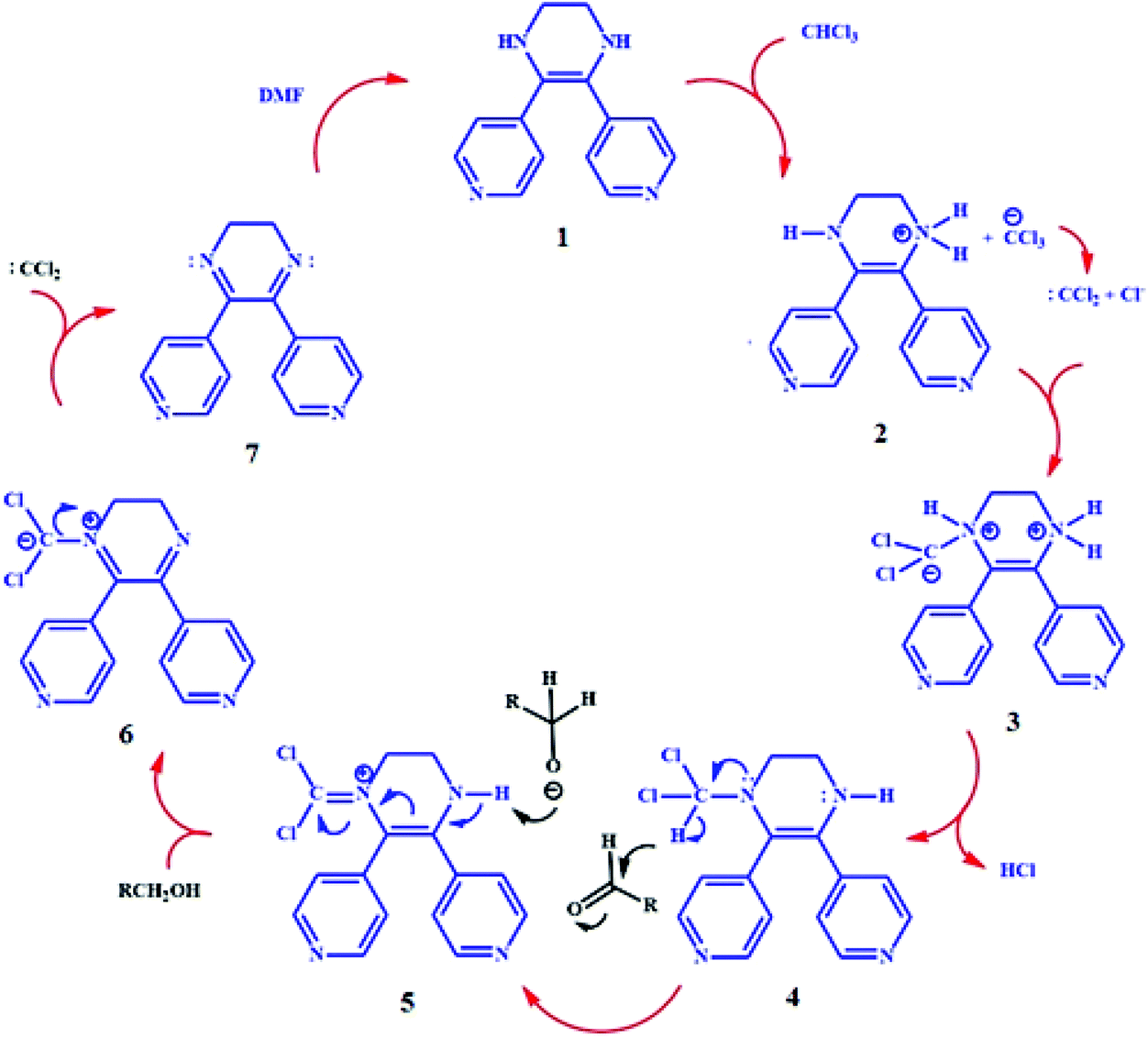 | ||
| Scheme 3 Proposed mechanism for the reduction of aldehyde in the presence of TMU-60. | ||
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Reactant | Solvent | Mol% cat | Time (h) | T (°C) | Yield (%) | |
| Reduction (selectivity number) | Condensation | ||||||
| a Allyl alcohol. b Phenylallyl alcohol. | |||||||
| 1 | Acetaldehyde | CHCl3/DMF | 4 | 2 | 25 | 63 (90) | 7 |
| 2 | Acetaldehyde | CHCl3/DMF | 4 | 4 | 25 | 70 (91) | 7 |
| 3 | Acetaldehyde | CHCl3/DMF | 4 | 24 | 25 | 76 (89) | 9 |
| 4 | Acetaldehyde | CHCl3/DMF | 6 | 2 | 25 | 70 (94) | 4 |
| 5 | Acetaldehyde | CHCl3/DMF | 8 | 2 | 25 | 73 (91) | 4 |
| 6 | Acetaldehyde | CHCl3/DMF | 6 | 2 | 35 | 72 (85) | 9 |
| 7 | Acetone | CHCl3/DMF | 6 | 4 | 25 | 3 | 5 |
| 8 | Acrylaldehyde | CHCl3/DMF | 6 | 4 | 25 | 61a (99) | — |
| 9 | 2-Butene | CHCl3/DMF | 6 | 4 | 25 | 1.1 | — |
| 10 | Methyl vinyl ketone | CHCl3/DMF | 6 | 4 | 25 | 3.3 | — |
| 11 | Formaldehyde | CHCl3/DMF | 6 | 2 | 25 | 79 (95) | 3 |
| 12 | Butyraldehyde | CHCl3/DMF | 6 | 2 | 25 | 69 (92) | 5 |
| 13 | Benzaldehyde | CHCl3/DMF | 6 | 2 | 25 | 70 (95) | 2 |
| 14 | Cinnamaldehyde | CHCl3/DMF | 6 | 2 | 25 | 54b (99) | — |
| 15 | Furfural | CHCl3/DMF | 6 | 2 | 25 | 65 (99) | — |
| 16 | 3-Methyl-2-butenal | CHCl3/DMF | 6 | 2 | 25 | 56 (81) | 11 |
| 17 | Acrolein | CHCl3/DMF | 6 | 2 | 25 | 67 (91) | 6 |
| 18 | 4-Cl-Benzaldehyde | CHCl3/DMF | 6 | 2 | 25 | 60 (97) | — |
| 19 | 4-OH-Benzaldehyde | CHCl3/DMF | 6 | 2 | 25 | 73 (96) | — |
| 20 | 4-NO2-Benzaldehyde | CHCl3/DMF | 6 | 2 | 25 | 45 (97) | — |

As shown in Scheme 3, dichlorocarbene is the byproduct of stage 6 of the catalytic cycle. This byproduct can advance the reaction cycle from step 3 and therefore reduce the need for chloroform as an oxidant. Since the conversion of O-TMU-60 to TMU-60 needs DMF, different ratios of these two solvents were investigated as the reaction solvent and adding the 5% v/v of chloroform to DMF was obtained as an optimal value (Table S2†). On the other hand, the catalytic performance of TMU-60 in comparison with other aldehyde reduction catalysis is listed in Table S3.†
Also, due to the structural features of TMU-60, the presence of the secondary amine in the framework and the proper orientation toward the pores, the ability of the framework to catalyze the aldehyde condensation reaction is well predictable.
To study the catalytic efficiency of TMU-60, the reaction was performed using acetaldehyde as a conventional organic solvent. The results presented in Table 2 clearly show that the solvent type strongly affects the catalyst selection path. In the presence of conventional organic solvents other than chloroform, the structural transformation of TMU-60 does not occur in its oxidized form and hydrogen is not released. Based on the proposed mechanism in Scheme 4, the catalyst chooses the alkaline path for aldehyde condensation. In addition to acetaldehyde, other aldehydes were studied as reactive agents. As the temperature decreased, the condensation product formed with a higher proportion. Time lapse led to the removal of water from 3-hydroxybutanal and the formation of crotonaldehyde. By prolonging the reaction time to more than 24 h, higher molecular weight products were obtained from the repeated aldol condensation mechanism. The 1H NMR spectra of the products of acetaldehyde and butyraldehyde condensation are given in Fig. S13 and S14.† Moreover, the mass spectra obtained from the GC mass analysis of the above-mentioned products are shown in Fig. S15.† The GC curves for the condensation reaction of acetaldehyde and butyraldehyde are presented in Fig. S16 and S17,† respectively.
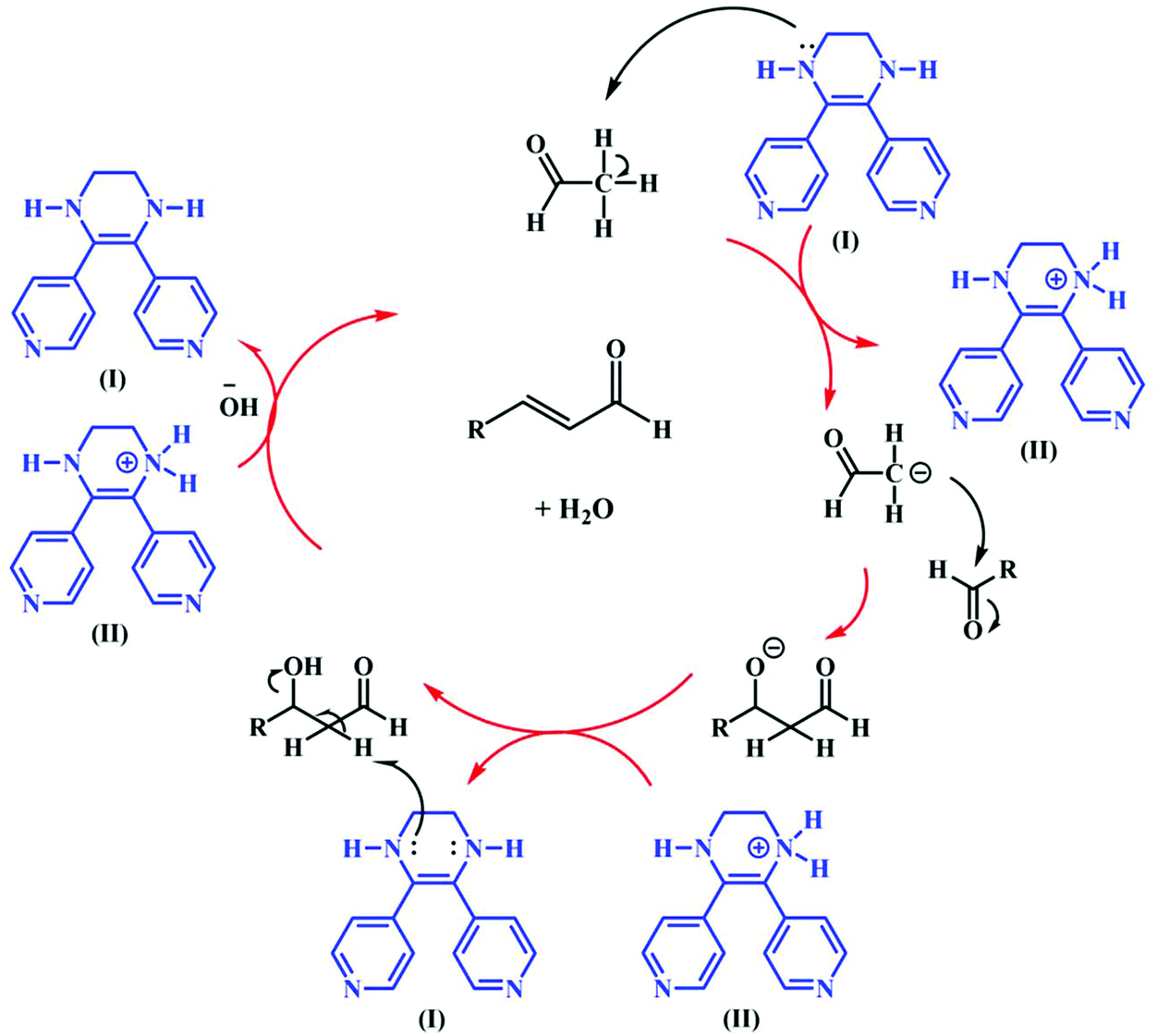 | ||
| Scheme 4 Proposed mechanism for the self-condensation of aldehyde in the presence of TMU-60 as a catalyst and acetonitrile. | ||
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Reactant | Solvent | Mol% cat | Time (h) | T (°C) | Yield (%) | |
| Reduction | Condensation (selectivity number) | ||||||
| 1 | Acetaldehyde | Acetonitrile | 6 | 2 | 25 | 60 (85) | |
| 1 | Acetaldehyde | Acetonitrile | 6 | 4 | 25 | — | 73 (75) |
| 2 | Acetaldehyde | Acetonitrile | 6 | 4 | 15 | — | 76.0 (90) |
| 3 | Acetaldehyde | Acetonitrile | 6 | 4 | 5 | — | 85.1 (95) |
| 4 | Acetaldehyde | DCM | 6 | 4 | 25 | — | 19.6 (65) |
| 5 | Acetaldehyde | DMF | 6 | 4 | 25 | — | 5.0 |
| 6 | Acetaldehyde | Toluene | 6 | 4 | 25 | — | 10.3 |
| 7 | Acetaldehyde | Hexane | 6 | 4 | 25 | — | 4.7 |
| 8 | Acetaldehyde | Acetonitrile | — | 12 | 25 | — | 3.0 |
| 9 | Butyraldehyde | Acetonitrile | 6 | 2 | 25 | — | 47.0 (85) |
| 10 | Butyraldehyde | Acetonitrile | 6 | 4 | 25 | 52.4 (77) | |
| 11 | Butyraldehyde | DCM | 6 | 4 | 25 | — | 16.1 |
| 12 | Butyraldehyde | DMF | 6 | 4 | 25 | — | — |
| 13 | Butyraldehyde | Toluene | 6 | 4 | 25 | — | 7.3 |
| 14 | Butyraldehyde | Hexane | 6 | 4 | 25 | — | 4.5 |
| 15 | Acetaldehyde | CHCl3/DMF | 5 mg L* | 2 | 25 | 61 (95) | — |
| 16 | Acetaldehyde | Acetonitrile | 5 mg L* | 2 | 25 | — | 57.2 (67) |
| 17 | Acetaldehyde | Acetonitrile | 5 mg L** | 2 | 25 | — | — |
| 18 | Cinnamaldehyde | Acetonitrile | 6 | 4 | 25 | 4 | 60 (85) |
| 19 | 3-Methyl-2-butenal | Acetonitrile | 6 | 4 | 25 | 5 | 56 (81) |
| 20 | Acrolein | Acetonitrile | 6 | 4 | 25 | — | 63 (85) |
| 21 | Acetone | Acetonitrile | 6 | 4 | 25 | — | 4 |
| 22 | Furfural | Acetonitrile | 6 | 4 | 25 | — | — |
| 23 | Benzaldehyde | Acetonitrile | 6 | 4 | 25 | 3 | — |
| 24 | Propionaldehyde | Acetonitrile | 6 | 4 | 25 | 5 | 65 (80) |

In the control reaction, under optimized conditions and in the presence of acetaldehyde in various solvents but without catalyst, no conversion of acetaldehyde to alcohol or crotonaldehyde was observed in 48 h. In order to ensure that the catalytic process performed due to the role of the ligand, L* and L** were separately used as catalysts. The result of L* is comparable to the catalytic activity of TMU-60, which indicates that the active portion of the TMU-60 is the L* ligand. The other possibility is that Zn plays a role as a catalyst for the reaction. Given that the entire of coordination sphere of the Zn(II) is occupied by the TMU-60 constructor ligands, the only possible case is the partial degradation of the structure and the release of metal ions in the reaction medium. The intact XRD pattern of the structure even after five cycles confirms the significant stability of the framework (Fig. 4A). However, ICP studies on the reaction solution after 4 h indicate a very small amount (2 ppm) of Zn, which confirms the stability of the structure and the absence of free Zn in the reaction vessel as a catalyst.
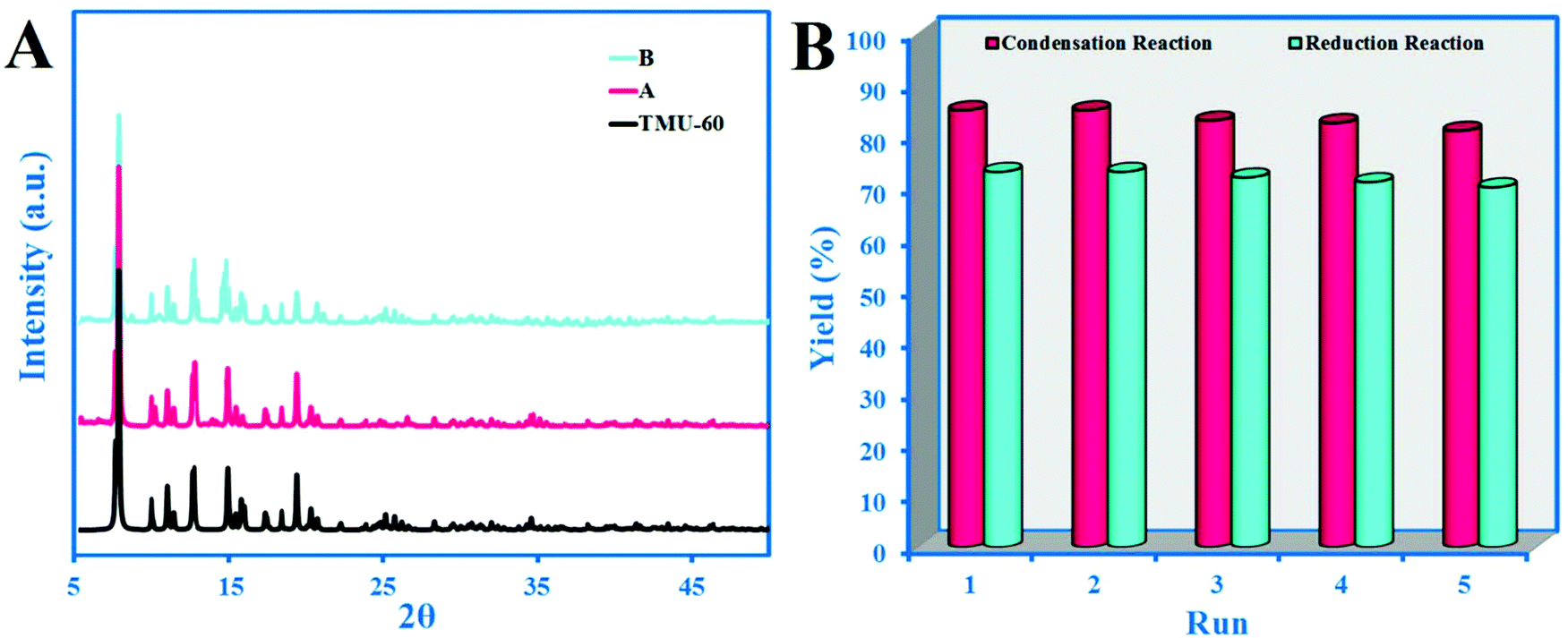 | ||
| Fig. 4 (A) XRD pattern of TMU-60 after five cycle condensation (red) and reduction reaction (blue) and (B) catalytic performance of the TMU-60 with repeated cycles. | ||
It should be noted that the use of TMU-60 instead of L* facilitates the separation of the catalyst from the reaction mixture. In addition, MOF pores act like a small vessel for the reaction. As a result, the reaction components become closer together and eventually the reaction rate increases.
3.1. Reuse of the catalyst
In order to study the reusing possibility of TMU-60 as a catalyst, after a condensation reaction in acetonitrile, the catalyst was separated by filtration and washed with fresh solvent. After five repeated cycles, the reaction efficiency dropped slightly (Fig. 4), indicating the stability and reusability of the catalyst. Fig. 4A shows the intact XRD pattern of the TMU-60 framework after five cycles. However, in the aldehyde reduction reaction, due to the presence of a solvent mixture, the catalyst returns to the active form in the reaction vessel and there is no need for the recovery process. The results presented in Fig. 4B show that the framework retains its performance even after five cycles. The intact XRD pattern of the framework after five cycles in the aldol condensation and reduction reaction confirms the significant stability of the framework.4. Conclusion and outlook
For the first time, a solvent switching 3D metal–organic framework (TMU-60) with [Zn(OBA)(L*)·DMF] structure was designed and used as a smart catalyst that selects two separate catalytic mechanisms only by the impact of the catalytic amount of a special solvent. The framework with a ligand containing tetrahydropyrazine, which can be converted reversibly to dihydropyrazine in chloroform by releasing hydrogen and converting from amine to imine form, can convert aldehydes to the corresponding alcohol selectively and quickly. However, in the presence of non-oxidizing solvents such as acetonitrile, the framework act as an alkaline catalyst and the condensation reaction of aldehyde is performed at room temperature. Despite the significant progress of MOFs over the past two decades, few studies on switching MOF have been documented. Therefore, this field can be extended for the use of porous materials, which could be a new horizon in the development and use of MOFs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support of this investigation by Tarbiat Modares University is gratefully acknowledged. Also, we appreciate Dr Ali Ramazani and Dr Hamed Moghanni for their helpful comments.References
- N. Ogihara, N. Ohba and Y. Kishida, On/off switchable electronic conduction in intercalated metal-organic frameworks, Sci. Adv., 2017, 3(8), e1603103 CrossRef PubMed.
- K. Tarafder, S. Kanungo, P. M. Oppeneer and T. Saha-Dasgupta, Pressure and temperature control of spin-switchable metal-organic coordination polymers from ab initio calculations, Phys. Rev. Lett., 2012, 109(7), 077203 CrossRef CAS PubMed.
- A. Knebel, L. Sundermann, A. Mohmeyer, I. Strauß, S. Friebe, P. Behrens and J. r. Caro, Azobenzene Guest Molecules as Light-Switchable CO2 Valves in an Ultrathin UiO-67 Membrane, Chem. Mater., 2017, 29(7), 3111–3117 CrossRef CAS.
- P. R. McGonigal, P. Deria, I. Hod, P. Z. Moghadam, A.-J. Avestro, N. E. Horwitz, I. C. Gibbs-Hall, A. K. Blackburn, D. Chen and Y. Y. Botros, Electrochemically addressable trisradical rotaxanes organized within a metal–organic framework, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(36), 11161–11168 CrossRef CAS PubMed.
- S. Rodríguez-Hermida, M. Y. Tsang, C. Vignatti, K. C. Stylianou, V. Guillerm, J. Pérez-Carvajal, F. Teixidor, C. Viñas, D. Choquesillo-Lazarte and C. Verdugo-Escamilla, Switchable Surface Hydrophobicity–Hydrophilicity of a Metal–Organic Framework, Angew. Chem., Int. Ed., 2016, 55(52), 16049–16053 CrossRef PubMed.
- S. A. A. Razavi, M. Y. Masoomi and A. Morsali, Stimuli-Responsive Metal–Organic Framework (MOF) with Chemo-Switchable Properties for Colorimetric Detection of CHCl3, Chem. – Eur. J., 2017, 23(51), 12559–12564 CrossRef CAS PubMed.
- X. Meng, B. Gui, D. Yuan, M. Zeller and C. Wang, Mechanized azobenzene-functionalized zirconium metal-organic framework for on-command cargo release, Sci. Adv., 2016, 2(8), e1600480 CrossRef PubMed.
- S. Castellanos, F. Kapteijn and J. Gascon, Photoswitchable metal organic frameworks: turn on the lights and close the windows, CrystEngComm, 2016, 18(22), 4006–4012 RSC.
- M. D. Manrique-Juarez, S. Rat, L. Salmon, G. Molnar, C. M. Quintero, L. Nicu, H. J. Shepherd and A. Bousseksou, Switchable molecule-based materials for micro-and nanoscale actuating applications: Achievements and prospects, Coord. Chem. Rev., 2016, 308, 395–408 CrossRef CAS.
- O. Drath and C. Boskovic, Switchable cobalt coordination polymers: Spin crossover and valence tautomerism, Coord. Chem. Rev., 2018, 375, 256–266 CrossRef CAS.
- A. Halder and D. Ghoshal, Structure and properties of dynamic metal–organic frameworks: a brief accounts of crystalline-to-crystalline and crystalline-to-amorphous transformations, CrystEngComm, 2018, 20(10), 1322–1345 RSC.
- S. Horike, S. Shimomura and S. Kitagawa, Soft porous crystals, Nat. Chem., 2009, 1(9), 695 CrossRef CAS PubMed.
- Y. Li, X. Zhang, P. Xu, Z. Jiang and J. Sun, The design of a novel and resistant Zn (PZDC)(ATZ) MOF catalyst for the chemical fixation of CO2 under solvent-free conditions, Inorg. Chem. Front., 2019, 6(1), 317–325 RSC.
- J. Lan, M. Liu, X. Lu, X. Zhang and J. Sun, Novel 3D nitrogen-rich metal organic framework for highly efficient CO2 adsorption and catalytic conversion to cyclic carbonates under ambient temperature, ACS Sustainable Chem. Eng., 2018, 6(7), 8727–8735 CrossRef CAS.
- S. Abedi and A. Morsali, Ordered mesoporous metal–organic frameworks incorporated with amorphous TiO2 as photocatalyst for selective aerobic oxidation in sunlight irradiation, ACS Catal., 2014, 4(5), 1398–1403 CrossRef CAS.
- A. Askarinejad, M. Bagherzadeh and A. Morsali, Catalytic performance of Mn3O4 and Co3O4 nanocrystals prepared by sonochemical method in epoxidation of styrene and cyclooctene, Appl. Surf. Sci., 2010, 256(22), 6678–6682 CrossRef CAS.
- M. Y. Masoomi, S. Beheshti and A. Morsali, Mechanosynthesis of new azine-functionalized Zn(II) metal–organic frameworks for improved catalytic performance, J. Mater. Chem. A, 2014, 2(40), 16863–16866 RSC.
- V. Blanco, D. A. Leigh and V. Marcos, Artificial switchable catalysts, Chem. Soc. Rev., 2015, 44(15), 5341–5370 RSC.
- Y. Sohtome, S. Tanaka, K. Takada, T. Yamaguchi and K. Nagasawa, Solvent-Dependent Enantiodivergent Mannich-Type Reaction: Utilizing a Conformationally Flexible Guanidine/Bisthiourea Organocatalyst, Angew. Chem., Int. Ed., 2010, 49(48), 9254–9257 CrossRef CAS PubMed.
- X. Tian, C. Cassani, Y. Liu, A. Moran, A. Urakawa, P. Galzerano, E. Arceo and P. Melchiorre, Diastereodivergent asymmetric sulfa-Michael additions of α-branched enones using a single chiral organic catalyst, J. Am. Chem. Soc., 2011, 133(44), 17934–17941 CrossRef CAS PubMed.
- S. Arseniyadis, A. Valleix, A. Wagner and C. Mioskowski, Kinetic resolution of amines: a highly enantioselective and chemoselective acetylating agent with a unique solvent-induced reversal of stereoselectivity, Angew. Chem., 2004, 116(25), 3376–3379 CrossRef.
- A. B. Northrup and D. W. MacMillan, Two-step synthesis of carbohydrates by selective aldol reactions, Science, 2004, 305(5691), 1752–1755 CrossRef CAS PubMed.
- T. Yamamoto, T. Yamada, Y. Nagata and M. Suginome, High-molecular-weight polyquinoxaline-based helically chiral phosphine (PQXphos) as chirality-switchable, reusable, and highly enantioselective monodentate ligand in catalytic asymmetric hydrosilylation of styrenes, J. Am. Chem. Soc., 2010, 132(23), 7899–7901 CrossRef CAS PubMed.
- M. Messerer and H. Wennemers, Reversing the enantioselectivity of a peptidic catalyst by changing the solvent, Synlett, 2011,(04), 499–502 CAS.
- R. J. Chew, X. R. Li, Y. Li, S. A. Pullarkat and P. H. Leung, Pd-Catalyzed Enantiodivergent and Regiospecific phospha-Michael Addition of Diphenylphosphine to 4-oxo-Enamides: Efficient Access to Chiral Phosphinocarboxamides and Their Analogues, Chem. – Eur. J., 2015, 21(12), 4800–4804 CrossRef CAS PubMed.
- Z. Wang, L. Heinke, J. Jelic, M. Cakici, M. Dommaschk, R. J. Maurer, H. Oberhofer, S. Grosjean, R. Herges and S. Bräse, Photoswitching in nanoporous, crystalline solids: an experimental and theoretical study for azobenzene linkers incorporated in MOFs, Phys. Chem. Chem. Phys., 2015, 17(22), 14582–14587 RSC.
- N. Fuentes, A. Martín-Lasanta, L. Á. de Cienfuegos, M. Ribagorda, A. Parra and J. M. Cuerva, Organic-based molecular switches for molecular electronics, Nanoscale, 2011, 3(10), 4003–4014 RSC.
- B. Gui, X. Meng, Y. Chen, J. Tian, G. Liu, C. Shen, M. Zeller, D. Yuan and C. Wang, Reversible tuning hydroquinone/quinone reaction in metal–organic framework: Immobilized molecular switches in solid state, Chem. Mater., 2015, 27(18), 6426–6431 CrossRef CAS.
- D. G. D. Patel, I. M. Walton, J. M. Cox, C. J. Gleason, D. R. Butzer and J. B. Benedict, Photoresponsive porous materials: the design and synthesis of photochromic diarylethene-based linkers and a metal–organic framework, Chem. Commun., 2014, 50(20), 2653–2656 RSC.
- J. Park, Q. Jiang, D. Feng and H. C. Zhou, Controlled generation of singlet oxygen in living cells with tunable ratios of the photochromic switch in metal–organic frameworks, Angew. Chem., 2016, 128(25), 7304–7309 CrossRef.
- J. W. Brown, B. L. Henderson, M. D. Kiesz, A. C. Whalley, W. Morris, S. Grunder, H. Deng, H. Furukawa, J. I. Zink and J. F. Stoddart, Photophysical pore control in an azobenzene-containing metal–organic framework, Chem. Sci., 2013, 4(7), 2858–2864 RSC.
- K. Müller, A. Knebel, F. Zhao, D. Bléger, J. Caro and L. Heinke, Switching Thin Films of Azobenzene-Containing Metal–Organic Frameworks with Visible Light, Chem. – Eur. J., 2017, 23(23), 5434–5438 CrossRef PubMed.
- L. Pan, G. Liu, H. Li, S. Meng, L. Han, J. Shang, B. Chen, A. E. Platero-Prats, W. Lu and X. Zou, A Resistance-Switchable and Ferroelectric Metal–Organic Framework, J. Am. Chem. Soc., 2014, 136(50), 17477–17483 CrossRef CAS PubMed.
- C. A. Fernandez, P. C. Martin, T. Schaef, M. E. Bowden, P. K. Thallapally, L. Dang, W. Xu, X. Chen and B. P. McGrail, An electrically switchable metal-organic framework, Sci. Rep., 2014, 4, 6114 CrossRef CAS PubMed.
- F. Rouhani and A. Morsali, Fast and Selective Heavy Metal Removal by a Novel Metal-Organic Framework Designed with In-Situ Ligand Building Block Fabrication Bearing Free Nitrogen, Chem. – Eur. J., 2018, 24(21), 5529–5537 CrossRef CAS PubMed.
- F. Rouhani and A. Morsali, Highly effective Brønsted base/lewis acid cooperative catalysis: a new Cd metal–organic framework for the synthesis of Hantzsch 1, 4-DHPs at ambient temperature, New J. Chem., 2017, 41(24), 15475–15484 RSC.
- F. Rouhani, A. Morsali and P. Retailleau, Simple One-Pot Preparation of a Rapid Response AIE Fluorescent Metal-Organic Framework, ACS Appl. Mater. Interfaces, 2018, 10(42), 36259–36266 CrossRef CAS PubMed.
- F. Rouhani, F. Rafizadeh-Masuleh and A. Morsali, Highly Electro-Conductive Metal-Organic Framework; Tunable by Metal Ion Sorption Quantity, J. Am. Chem. Soc., 2019, 141(28), 11173–11182 CrossRef CAS PubMed.
- N. Gorgas, B. Stöger, L. F. Veiros and K. Kirchner, Highly efficient and selective hydrogenation of aldehydes: a well-defined Fe(II) catalyst exhibits noble-metal activity, ACS Catal., 2016, 6(4), 2664–2672 CrossRef CAS PubMed.
- L. Bonomo, L. Kermorvan and P. Dupau, Ruthenium-Catalyzed Highly Chemoselective Hydrogenation of Aldehydes, ChemCatChem, 2015, 7(6), 907–910 CrossRef CAS.
- E. Plessers, G. Fu, C. Y. X. Tan, D. E. De Vos and M. B. Roeffaers, Zr-based MOF-808 as Meerwein–Ponndorf–Verley reduction catalyst for challenging carbonyl compounds, Catalysts, 2016, 6(7), 104 CrossRef.
- P. J. Larson, J. L. Cheney, A. D. French, D. M. Klein, B. J. Wylie and A. F. Cozzolino, Anchored Aluminum Catalyzed Meerwein–Ponndorf–Verley Reduction at the Metal Nodes of Robust MOFs, Inorg. Chem., 2018, 57(12), 6825–6832 CrossRef CAS PubMed.
- R. M. Bullock, Catalysis without precious metals, John Wiley & Sons, 2011 Search PubMed.
- S. Rösler, J. Obenauf and R. Kempe, A Highly Active and Easily Accessible Cobalt Catalyst for Selective Hydrogenation of C–O Bonds, J. Am. Chem. Soc., 2015, 137(25), 7998–8001 CrossRef PubMed.
- G. Wienhoefer, F. A. Westerhaus, K. Junge, R. Ludwig and M. Beller, A Molecularly Defined Iron-Catalyst for the Selective Hydrogenation of α, β-Unsaturated Aldehydes, Chem. – Eur. J., 2013, 19(24), 7701–7707 CrossRef CAS PubMed.
- Z. Yin, S. Wan, J. Yang, M. Kurmoo and M.-H. Zeng, Recent advances in post-synthetic modification of metal–organic frameworks: New types and tandem reactions, Coord. Chem. Rev., 2019, 378, 500–512 CrossRef CAS.
- Y. Luan, Y. Qi, H. Gao, R. S. Andriamitantsoa, N. Zheng and G. Wang, A general post-synthetic modification approach of amino-tagged metal–organic frameworks to access efficient catalysts for the Knoevenagel condensation reaction, J. Mater. Chem. A, 2015, 3(33), 17320–17331 RSC.
- S. Hu, J. Yan, X. Huang, L. Guo, Z. Lin, F. Luo, B. Qiu, K.-Y. Wong and G. Chen, A sensing platform for hypoxanthine detection based on amino-functionalized metal organic framework nanosheet with peroxidase mimic and fluorescence properties, Sens. Actuators, B, 2018, 267, 312–319 CrossRef CAS.
- A. Grostern, M. Duhamel, S. Dworatzek and E. A. Edwards, Chloroform respiration to dichloromethane by a Dehalobacter population, Environ. Microbiol., 2010, 12(4), 1053–1060 CrossRef CAS PubMed.
- A. J. Włodarczyk, P. P. Romańczyk, T. Lubera and S. S. Kurek, Electrocatalytic activity of bisalkoxide molybdenum nitrosyl complexes in the reduction of chloroform, Electrochem. Commun., 2008, 10(12), 1856–1859 CrossRef.
- J. Feng and T.-T. Lim, Pathways and kinetics of carbon tetrachloride and chloroform reductions by nano-scale Fe and Fe/Ni particles: comparison with commercial micro-scale Fe and Zn, Chemosphere, 2005, 59(9), 1267–1277 CrossRef CAS PubMed.
- Y. Zhao, Y. Li, Z. Qin, R. Jiang, H. Liu and Y. Li, Selective and colorimetric fluoride anion chemosensor based on s-tetrazines, Dalton Trans., 2012, 41(43), 13338–13342 RSC.
- B. L. Schottel, H. T. Chifotides and K. R. Dunbar, Anion-π interactions, Chem. Soc. Rev., 2008, 37(1), 68–83 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1862166. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qi00714h |
| This journal is © the Partner Organisations 2019 |