
Near-infrared emissive Er(iii) and Yb(iii) molecular nanomagnets in metal–organic chains functionalized by octacyanidometallates(iv)†
 a
Jakub J.
Zakrzewski,
a
Shin-ichi
Ohkoshi,
b
Szymon
Chorazy
*ab
and
Barbara
Sieklucka
*a
a
Jakub J.
Zakrzewski,
a
Shin-ichi
Ohkoshi,
b
Szymon
Chorazy
*ab
and
Barbara
Sieklucka
*a
Abstract
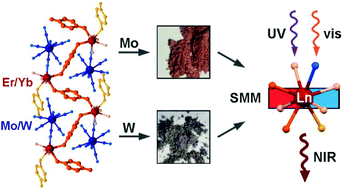
The self-assembly of lanthanide(3+) ions with pyrazine N,N′-dioxide (pzdo) and bis(triphenylphosphine)iminium (PPN+) salts of diamagnetic octacyanidometallates of MoIV and WIV results in one-dimensional (PPN)[LnIII(pzdo)2(MeOH)0.3(H2O)3.7][MIV(CN)8]·7.7H2O·2MeOH (Ln = Er and Yb; M = Mo and W) coordination networks. They are constructed of zigzag metal–organic {LnIII(μ-pzdo)}n chains with terminal [MIV(CN)8]4− metalloligands attached to the LnIII centres. Both Er- and Yb-containing frameworks exhibit field-induced slow magnetic relaxation originating from the field-dependent equilibrium between quantum tunnelling of magnetization and the direct process along with the temperature-dependent Raman and Orbach relaxation processes. Transition metal substitution on [MIV(CN)8]4− sites modifies the intrinsic magnetic anisotropy of lanthanides, as depicted by the subtle change of thermal energy barriers of Orbach relaxation in Er-based systems, while it significantly affects Raman relaxation due to the modulated phonon mode scheme. All reported compounds exhibit strong visible light absorption due to a series of electronic transitions of pzdo ligands and [M(CN)8]4− ions and the appearance of low energy anion–π charge transfer band involving pzdo and octacyanidometallates. These charge transfer states were utilized in achieving the sensitized near-infrared photoluminescence of ErIII and YbIII centers; thus, the energy transfer process is strongly dependent on the nature of the metal centre in the [MIV(CN)8]4− ion. The W(IV)-pzdo system is a good sensitizer for ErIII, while the Mo(IV)-pzdo unit is better for YbIII emission which can be rationalized in terms of the positions of energy levels of their respective donor states. Therefore, we report NIR emissive ErIII and YbIII single-molecule magnets with tunable magnetic and optical properties, uncovering the crucial role of octacyanidometallates and their anion–π interactions with pzdo ligands.
 Please wait while we load your content...
Please wait while we load your content...

