
Rare earth double perovskites: a fertile soil in the field of perovskite oxides
Xiaoyun
Chen
a,
Jun
Xu
 *a,
Yueshan
Xu
a,
Feng
Luo
b and
Yaping
Du
*a
*a,
Yueshan
Xu
a,
Feng
Luo
b and
Yaping
Du
*a
aTianjin Key Lab for Rare Earth Materials and Applications, Center for Rare Earth and Inorganic Functional Materials, School of Materials Science and Engineering & National Institute for Advanced Materials, Nankai University, Tianjin 300350, P.R. China. E-mail: junxu@nankai.edu.cn; ypdu@nankai.edu.cn
bIMDEA Nanoscience, Faraday 9, Ciudad Universitaria de Cantoblanco, 28049 Madrid, Spain
First published on 8th July 2019
Abstract
Tuning the composition of perovskite oxides results in double perovskites with cation ordering. One of the most important families of double perovskites is A2B′B′′O6. With great structural variability, A2B′B′′O6 are promising candidates for a broad range of applications such as magnetic, optical, electrical, and catalytic applications. Rare earth elements, as the ensemble of fifteen lanthanide elements as well as yttrium and scandium, represent an important part of the periodic table due to their unique 4f electrons. The incorporation of rare earth elements into double perovskites may open great opportunities to novel properties and applications. A significant number of rare earth A2B′B′′O6 double perovskites have been reported. However, there is no systematic overview of these materials to date. In this review, we summarize the compositions, syntheses, and applications of rare earth A2B′B′′O6 double perovskites. We hope that it will encourage more researchers to enter this emerging area.
Introduction
The perovskite oxide in a composition of ABO3 was first discovered and named by Rose in the Ural Mountains in 1839.1,2 The ideal ABO3 perovskites belong to a simple cubic structure, in which the A position is occupied by a lanthanide or alkaline earth ion having a large radius and the B position is generally a transition metal ion.1 In this structure, B and O ions constitute a 6-coordinate BO6 octahedron, while the eight BO6 octahedra are distributed over the eight vertices of the cube by the shared O ions and the A ions with 12 oxygen coordination are in the centre of the cube. A variety of metal ions can form ABO3-type compounds with a stable structure and a flexible composition. ABO3 perovskites have been extensively used as ferroelectrics/piezoelectrics (e.g., BaTiO3) as well as efficient and inexpensive catalysts (e.g., LaMnO3),3,4 In addition, this unique structure can exhibit some other interesting characteristics for novel applications such as ferromagnetic/antiferromagnetic conductors (e.g., BaFeO3/SrFeO3),5,6 electromagnetic wave adsorbents (e.g., LaNiO3),7 and photocatalysts (e.g., SrTiO3, KTaO3, NaTaO3, KNbO3, and NaNbO3).8 Crystallization of compounds in perovskite structures is therefore one of the hot spots in solid chemical and physical research.1,2Modulation of the properties of perovskite oxides is often achieved by partial cation substitution, occurring at different degrees at both A and B sites. When the substitution degree at the B site increases to 50%, an ordering of two types of B cations may be present, forming double perovskites in a composition of AB′1/2B′′1/2O3, or A2B′B′′O6.9 Other types of double perovskites also exist, for example, AB′1/3B′′2/3O3.10 However, to narrow down the scope of this review, we will only discuss rare earth A2B′B′′O6 double perovskites, in which rare earth cations can occupy either A or B sites.
Since Steward and Rooksby discovered the first A2B′B′′O6 double perovskite (Ba2CaWO6) in 1951, the study of double perovskites has flourished.11 In principle, this kind of perovskite-like oxide can provide higher structural variability and richer physical–chemical connotations, leaving great opportunities for theoretical modelling, synthetic efforts, property characterization, and applications.9 Due to the different electronic configurations and ionic radii of B′ and B′′ ions, it is possible to explore various properties, such as optical activity, electrical conductivity and magnetic character, by changing their elemental combinations and the coupling between different ions. The B cations of A2B′B′′O6 double perovskites can be arranged perfectly in three ways (Fig. 1). First and the most common circumstance is that B′ and B′′ cations are completely isolated and alternately present at three dimensions, also referred to as the elpasolite structure. The second is that B′ and B′′ cations form a stacked layered structure. The last is an alternating columnar structure of B′ and B′′ cations. However, it is noteworthy that the perfect ordering of B cations in A2B′B′′O6 double perovskites is rare and the ordering degree of B′ and B′′ cations is a fundamental parameter that influences many physical and chemical properties. A typical example is Pb2InNbO6 flux-grown crystals: the crystals with a low ordering degree are ferroelectrics, the crystals with a high ordering degree are antiferroelectrics, whereas the crystals with an intermediate ordering degree undergo a transition from the paraelectric to antiferroelectric and finally to the ferroelectric phase.12 The magnetoresistance behaviour of Sr2FeMoO6 is also dependent on the ordering degree of Fe3+ and Mo5+ ions, in which the ordered samples showed a unique sharp low-field response, probably due to the half-metallic ferromagnetism.13
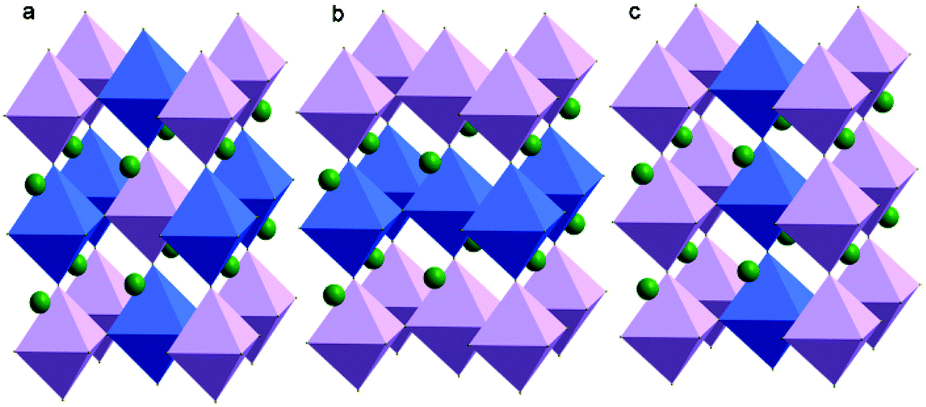 | ||
| Fig. 1 Different B-site cation orderings found in A2B′B′′O6 perovskites: (a) rock-salt, (b) layered and (c) columnar order. | ||
Rare-earth elements (RE) consist of fifteen lanthanide elements, with unique electron configurations of [Xe] 4fn−15d0−16s2 (n = 1–15), as well as the group-IIIB elements Yttrium (Y) and Scandium (Sc). Rare earth materials often exhibit promising magnetic, optical, electrical, and catalytic properties owing to the presence of rare earth elements.14–16 The incorporation of rare earth elements into A2B′B′′O6 double perovskites has therefore attracted considerable attention since their discovery. Despite their importance, to the best of our knowledge, there is no comprehensive overview of this impressive area. They were typically included in more general reviews of perovskite-type oxides.9 Herein, we summarized the compositions, syntheses, and applications of rare earth A2B′B′′O6 double perovskites. We will present the current status of research in this area, discuss the limitation of existing work, and shed light on future studies. We hope that this review will encourage more researchers to enter this emerging area.
Compositions
Under common conditions, the introduction of different cations into the A2B′B′′O6 double perovskite structure may cause lattice distortions, reducing the structural stability. The structural stability of ABO3 perovskites is usually evaluated by the Goldschmidt tolerance factor t.17 In the case of double perovskites, rB is the average of two atomic radii of B′ and B′′. The effective radii of the A and B ions and the oxygen ions rA, rB and rO satisfy the following relationship:
In this formula, t is the Goldschmit tolerance factor and stable perovskites usually satisfy 0.78 < t ≤ 1.05. If the t value exceeds this range, it would belong to another structure type. When t is 1, it is an ideal perovskite structure. When t is less than 1, the effective radius of the A-site ion is smaller than the B-site radius, which means the regular BO6 octahedron is inclined to an orthogonal structure.18 When the B-site ionic radius is so small that t is greater than 1, B ions of the BO6 octahedron are displaced and the crystal structure is distorted into a tetragonal, rhombic or orthogonal structure based on the displacement direction. We calculated the t values of various rare earth A2B′B′′O6 double perovskites and found that they are consistent with the results observed in ABO3 perovskites (Fig. 2).
 | ||
| Fig. 2 Tolerance factor t of some rare earth A2B′B′′O6 perovskites. | ||
To summarize the composition of known rare earth A2B′B′′O6 double perovskites, we first show a periodic table covering all elements present in rare earth A2B′B′′O6 double perovskites (Fig. 3), and then present the detailed compositions in Table 1.19–106 We also predict some unknown rare earth A2B′B′′O6 phases such as La2VTiO6, Pb2ScVO6, La2NiGaO6, Ba2LaTcO6, and La2NiInO6 with the Goldschmidt tolerance factors of 0.95, 0.96, 0.97, 0.96, and 0.90, respectively.
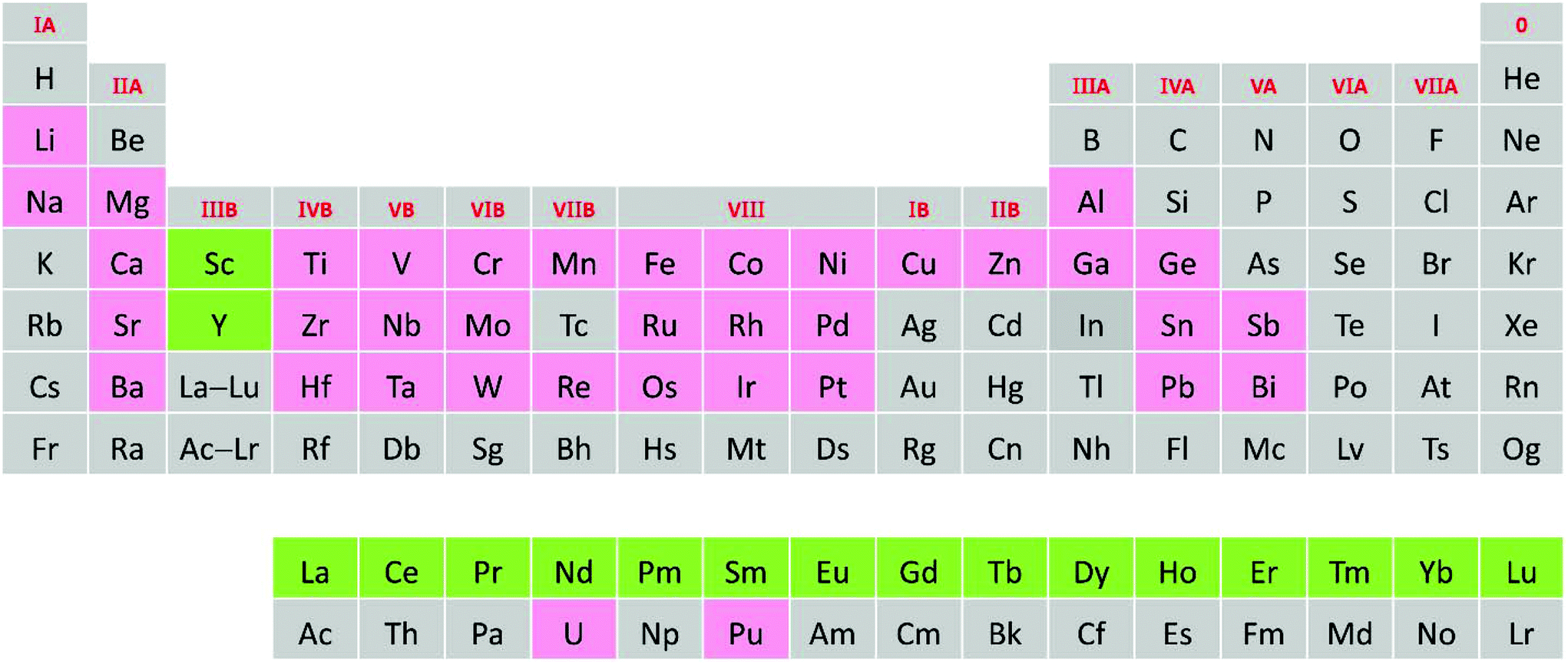 | ||
| Fig. 3 A periodic table illustrating the elements present in rare earth A2B′B′′O6 double perovskites. The rare earth elements are labelled in green while the other metals are labelled in red. The oxygen is not labelled for clarity. | ||
| A | B′ | B′′ |
|---|---|---|
| La | Li, Na | Ir, Mo, Nb, Os, Re, Ru, Sb, V, Al, Ru |
| Mg, Ca | Ge, Hf, Ir, Mn, Pd, Pt, Rh, Ru, Sn, Ti, Zr, Ir, Ti | |
| Zn | Hf, Ir, Mn, Pt, Rh, Ru, Ti | |
| Cu | Ir, Rh, Ru, Sn, Zr, Mn, Ti | |
| Mn | Ir, Rh, Ru, Ti, V | |
| Fe, Co, Ni | Fe, Ir, Rh, Ru, Ti, Pt Mn, Ir, Mn, Rh, Ru, Ti, V, Co, Fe, Ge, Ir, Mn, Pt, Rh, Ru, Sn, Ti, V, Zr, Al | |
| Cr | Co, Fe, Mn, Rh | |
| Sc | Mn | |
| Ga | Co, Fe, Mn, Ni, Ru | |
| Eu, Gd, Nd, Pr, Sm | Li | Ir, Os, Ru |
| Eu, Gd, Nd, Pr, Sm | Na | Ir, Pt, Sn, Ti |
| Eu, Gd, Nd, Pr, Sm | Mn | Rh, Ti |
| Y, Dy, Nd, Eu, Gd, Ho, Nd, Sm, Tb, Tm | Cr, Cu, Ga | Mn |
| Nd, Pr, Sm, Eu, Gd | Cu | Sn, Ti, Ir |
| Dy, Er, Gd, Ho, Nd, Eu, Lu, Pr, Sc, Sm, Tb, Tm, Y, Nd | Ni | Mn, Ru, Pt |
| Dy, Eu, Gd, Ho, Nd, Pr, Sm, Tb | Co | Pt, Ru |
| Eu, Gd, Nd, Pr, Sm | Zn | Pt, Ti |
| Mg | Dy | Bi |
| Ca | Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu, Sc | Nb, Ta |
| Pr, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu, Sc | Sb | |
| Gd | Ru | |
| Sc | Re | |
| Sr | Dy, Er, Eu, Gd, Ho, Lu, Sc, Sm, Tb, Tm, Yb, Y | Ir |
| Dy, Er, Eu, Gd, Ho, Lu, Nd, Pr, Sc, Sm, Tb, Tm, Yb, Y | Sb, Bi | |
| Dy, Sm, Er, Eu, Gd, Ho, Lu, Tb, Nd, Pr, Sc, Tm, Yb, Y | Nb, Ta | |
| Dy, Er, Eu, Gd Ho Yb Y | Mo | |
| Dy, Er, Gd, Ho, Sc, Tb, Tm, Yb, Y | Re | |
| Dy, Eu, Ho, Lu, Sm, Tb, Tm, Yb, Sc, Y | Ru, Os | |
| Dy, Er, Eu, Gd, Ho, Lu, Nd, Sm, Yb | U | |
| Er | Eu | |
| Ce(IV) | Ir, Hf, Ta, Zr | |
| Ba | La, Ce, Pr, Nd, Pr, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu, Sc, Y | V, Nb, Ta |
| La, Ce, Pr, Nd, Pr, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu, Sc, Y | Ir | |
| La, Pr, Nd, Sm, Eu, Gd, Dy, Ho, Er, Tm, Yb, Lu, Tb, Y | Ru, Os | |
| La, Nd, Pr, Sm, Eu, Gd, Tb, Dy, Ho, Er, Tm, Yb, Lu, Sc, Y | Sb, Bi | |
| Pr, Nd, Sm, Eu, Gd, Dy, | Mo, W | |
| La, Nd, Sm, Eu, Gd, Tb, Dy, Ho, Er, Dy, Yb, Lu, Sc, Y | Re | |
| Pb | Dy, Tm, Lu, Sc, Yb, | Nb, Ta |
| Sc | Sb | |
| Eu(II) | La, Eu, Dy, Ho, Er, Tm, Yb, Lu | Ta |
Syntheses
Solid phase synthesis
Most A2B′B′′O6 double perovskites are synthesized via the solid phase synthesis. According to the reactant stoichiometry, well mixed and ground reactants are heated at high temperature to form solids with new phases. In this process, the solid powder undergoes physical and chemical reactions at a temperature lower than the melting point, enabling the thermal diffusion of ions or molecules. This reaction has several advantages, among which the most significant one is that no solvent is needed during the reaction. Solid additives may be included in the starting materials to reduce the reaction temperature. Solid phase synthesis also has several disadvantages: the product is often obtained with impurities; the reaction time is typically long due to the relatively slow diffusion rate in the solid state; the reaction temperature is usually high (>1000 °C); the solid products usually have a non-uniform particle size and poor chemical homogeneity, and so on. Such problems can be alleviated by grinding the solids repeatedly when the calcination temperature and time are gradually increased. For instance, the double perovskites A2ScSbO6 (A = Ca, Sr) were synthesized by a conventional solid phase synthesis method according to the chemical reaction formula 4ACO3 + Sc2O3 + Sb2O5 = 2A2ScSbO6 + 4CO2 (A = Ca, Sr).96 The stoichiometric mixture was first ground in an agate mortar with acetone, then placed in an alumina crucible, and heated in air for 12 h at 600 °C, 12 h at 1000 °C, 24 h at 1200 °C and 72 h at 1500 °C, and cooled to room temperature before raising the heating temperature.High pressure synthesis
It is well known that the phase diagram of matters can alter under high pressure. Azuma and co-workers found that RE2CuSnO6 (RE = Pr, Nd, and Sm) and La2CuZrO6 have been stabilized in the same structure of La2CuSnO6 at high pressures of 6–8 GPa, possessing alternating layers of Cu2+ and Sn4+ (or Zr4+) cations.52 The synthesis was performed between 1000 and 1200 °C. In general, RE2CuSnO6 consists of three types of layers: CuO2, SnO2, and REO layers. The replacement of La3+ with smaller RE3+ cations induces a greater mismatch between the CuO2 and REO layers and a more serious buckling of the CuO2 layers, consistent with the decreasing stability of RE2CuSnO6 phases (La2CuSnO6: synthesized under ambient pressure; Pr2CuSnO6 and Nd2CuSnO6: synthesized under 6 GPa and at 1000 °C; Sm2CuSnO6: synthesized with impurities under 8 GPa and at 1200 °C). In the case of La2CuZrO6, the replacement of Sn4+ with smaller Zr4+ also induces a greater mismatch between the CuO2 and ZrO2 layers as well as the more buckled CuO2 layers. This new phase cannot be obtained under conventional synthetic conditions.Flux synthesis
High-quality single crystals of rare earth double perovskites can grow in molten hydroxide fluxes. Developed by zur Loye's group, molten hydroxides such as NaOH are excellent solvents for the crystallization of both simple and complex rare earth oxides.25 The acid–base properties of hydroxide fluxes, tuned by the water content of the melt, are essential for the dissolution of rare earth oxides. In general, rare earth oxides are only soluble in the so-called acidic “wet” melts in the presence of water. For instance, the single crystals of RELiIrO6 (RE = La, Pr, Nd, Sm and Eu) were grown in a mixed LiOH/KOH flux typically containing 15 wt% of water.26 The flux synthesis is especially useful in the preparation of rare earth double perovskite containing transition metals in unusually high oxidation states (e.g., Ir4+, Ir5+). The large single crystals obtained in this approach (Fig. 4) can enable thorough structure determinations and property characterization.20,25,26,30 | ||
| Fig. 4 A scanning electron microscopy image of a Sm2MgIrO6 single crystal. Reproduced from ref. 30 with permission from Elsevier, copyright 2010. | ||
Hydrothermal synthesis
As a mild synthetic method, the hydrothermal approach allows the co-condensation of different metal cations to obtain complex oxides such as perovskites. However, the large difference of the ionic radii of B′ and B′′ cations is a challenge for hydrothermal synthesis. Nevertheless, optimization of reaction conditions in hydrothermal synthesis indeed enables the synthesis of many rare earth double perovskites. An example is the synthesis of Ba2YSbO6.92 The influences of the temperature, KOH concentration, and B-cation source on the formation of Ba2YSbO6 were investigated (Table 2). The optimal reaction conditions are determined to be 240–260 °C, 10 mol L−1 KOH, and 7 days, respectively. In contrast, a conventional solid phase synthesis of Ba2YSbO6 requires heating mixtures of BaO2, Y2O3, and Sb2O3 at 800, 1000, 1200, and 1300 °C with each thermal treatment lasting 1 day.100| B-cation source | KOH concentration | Reaction temperature (°C) | Products |
|---|---|---|---|
| Y2O3:Sb2O3 | 4 | 240 | BaSb2O6 + Y(OH)3 |
| Y2O3:Sb2O3 | 6 | 240 | Ba2YSbO6 + BaSb2O6 + Y(OH)3 |
| Y2O3:Sb2O3 | 10 | 240 | Ba2YSbO6 |
| Y2O3:Sb2O3 | 14 | 240 | Ba2YSbO6 |
| Y(NO3)3:SbCl5 | 10 | 240 | Ba2YSbO6 + BaSb2O6 + YSbO3 |
| Y(NO3)3:C4H4O6SbK·0.5H2O | 10 | 240 | Ba2YSbO6 + BaSb2O6 + Y(OH)3 |
| Y2O3:Sb2O5 | 10 | 240 | Ba2YSbO6 + BaSb2O6 + Y(OH)3 |
| Y2O3:Sb2O3 | 10 | 160 | Ba2YSbO6 + amorphous |
| Y2O3:Sb2O3 | 10 | 200 | Ba2YSbO6 |
The second example is the one-step mild hydrothermal synthesis of RE2NiMnO6 (RE = Pr, Sm and Ho), which are typically obtained through a solid state reaction between oxides, nitrates and/or carbonate salts.39 The compounds can be obtained by mixing the aqueous solution of all precursors without any pre-treatment. However, the degree of alkalinity of the solution plays a key role: the products were not formed when the alkalinity is less than 6 mol L−1. The increased solubility and enhanced activity of the hydroxide complexes are responsible for the observed differences. In addition, the double perovskites can only form in the presence of KOH. The authors replaced KOH with NaOH, NH3·H2O, and several alkaline solvents and did not find good results. Therefore, they believe that KOH not only maintained the solution alkalinity but also acted as a mineralizer in this reaction. The crystal growth is also sensitive to the reaction temperature and time. With a low reaction temperature (<220 °C) and insufficient reaction time (<12 h), the double perovskites were not present.
Sol–gel synthesis
The preparation of sol–gel technology involves three major steps: preparation of a sol, formation of a wet gel, drying and calcination of the gel. According to the types of precursors, the sol is typically converted into a gel via two approaches: the first is the hydrolysis or hydration of metal salts or metal oxides in water, forming colloidal particles and transforming into a gel. The second is the hydrolysis condensation of metal alkoxides. Owing to the shorter diffusion path and lower diffusion barrier in a well-mixed heterometallic gel, it often requires significantly lower calcination temperature for the formation of rare earth double perovskites.A common route of sol–gel synthesis is the citrate acid-aid process, utilizing the facile formation of various citrate–metal complexes. A study employing both solid phase synthesis and a citrate acid-aided process of La2MMnO6 (M = Co, Ni, and Cu) revealed a milder synthetic condition for the latter method: calcination of the gel for 2 h at 150 °C, 4 h at 400 °C, 4 h at 800 °C, instead of 10 h at 1000 °C.107 Other complexing agents such as glucose and ethylenediamine can also be used.108,109
Microwave sintering
The microwave sintering approach is a modified version of the sol–gel synthesis in which the gel is calcined using the microwave energy rather than the conventional heating. The microwave sintering of complex oxides therefore provides several advantages over the conventional sintering, including rapid heating, improved production rate, enhanced densification, grain growth inhibition, lower energy consumption, and so on.Recently, La2CoMnO6 nanoparticles were prepared by a chemical co-precipitation sol–gel process and subsequently calcined at 900 °C per 10 min using microwave sintering.110 The products have a mean particle size of around 60 nm. Compared to the La2CoMnO6 nanoparticles obtained in other experiments, the nanoparticle size is significantly more uniform, demonstrating the advantage of microwave sintering.
Polymer deposition method
Fabrication of rare earth double perovskite epitaxial films with a low thickness, high crystallinity, and well-defined surface morphology remains a technical challenge to date. The conventional approach is using physical methods such as pulsed laser deposition (PLD) and the sputtering technique. In addition to the strict deposition conditions and high-cost equipment, such physical methods are restricted by coating on a relatively small area. Wet chemical solution depositions such as sol–gel synthesis are simple and more cost-effective. However, the resulting films suffer from the low crack-free thickness and the difficulty in controlling the stoichiometry due to differences in reactivity and solubility among the metals. In 2004, Jia and co-workers developed a novel polymer deposition method to grow metal–oxide films in large areas at low cost.111 As a surfactant assisted sol–gel method, the polyethyleneimine (PEI) present in solution controls the viscosity and binds metal ions, resulting in a relatively homogeneous mixing of metal ions in the gel and the formation of uniform metal–organic films, providing a facile chemical approach to both simple and complex crack-free epitaxial metal oxide films.The polymer deposition method was later used to grow a RE2NiMnO6 (RE = La, Pr, Nd, Sm, Y) epitaxial thin film on the (001) face of LaAlO3 (Fig. 5).112 The synthesis involves a dip-coating method. The aqueous solution containing PEI and EDTA (Ethylene Diamine Tetraacetic Acid) was slowly added into the solution of mixed metal nitrates by using a dropper. The resulting solution was concentrated with stirring in an oil bath of 60 °C. The dip-coating of the stable precursor solution on (001) of the LaAlO3 substrate creates coated polymeric films. After calcination, single phase RE2NiMnO6 epitaxial films were prepared with a typical thickness of about 35.6 nm (Fig. 5).
 | ||
| Fig. 5 The typical cross-sectional area of the La2NiMnO6 thin-film coated at 5000 rpm. Adapted from ref. 112 with permission from Elsevier, copyright 2015. | ||
Sol–gel auto-combustion
Conventional solid phase synthesis of rare earth double perovskites often requires calcination at very high temperature (>1000 °C) with intermediate grinding (e.g., Ba2DySbO6).94 In 2009, Wariar and co-workers proposed a novel single-step auto ignition combustion process for Ba2DySbO6.93 The authors used nitric acid as the oxidizing agent, citric acid as the complexing agent, and ammonium hydroxide as fuel. Sb2O3, which is insoluble in most of the mineral acids, was dissolved in boiling tartaric acid. The solution containing the complex precursor mixture was kept at a pH value of around 7 and then heated at around 250 °C in a ventilated fume hood. The boiling solution was eventually converted into a foam and finally ignited itself, forming powder products. The as-synthesized powder was phase pure and obtained as nanocrystals (20–50 nm). It is worth mentioning that single phase Ba2DySbO6 is typically obtained using solid phase synthesis at a temperature of 1400 °C in air for 48 h with intermediate grindings.94 Using the same method, Palamaru and co-workers later reported the compound Ca2DySbO6, which has never been obtained employing the other synthetic methods.99Applications
Magnetic applications
The cation ordering in rare earth double perovskites usually induces a certain degree of coupling or super-exchange interactions between cations with non-zero spins (or unpaired electrons), resulting in bulk magnetic ordering such as ferromagnetism and antiferromagnetism. More importantly, when a strong coupling between magnetism and other physical phenomena is present, the properties of rare earth double perovskites such as spins, electric charges, and dielectric, thermal and optical properties can be tuned by magnetic and/or electric fields. Since ferromagnetism and antiferromagnetism are common in rare earth materials, we will not mention more details in this review. In contrast, La2NiMnO6 and related materials are used as examples of single-material platforms with multiple functions.Spintronics, as the short name of spin electronics, is an emerging class of devices which combine both electronics and magnetics via the manipulation of electron spins. It can offer great potential for non-conventional functionalities which are impossible with current semiconductor devices, such as quantum computation. It has been predicted that ambient-temperature ferromagnetic semiconductors can become a new generation of spintronic materials. However, the Curie temperature (i.e., ferromagnetic to paramagnetic transition temperature) of most ferromagnetic semiconductors and insulators is significantly lower than room temperature. The rare earth double perovskite La2NiMnO6 is a rare exception (transition at ∼7 °C). In 2005, Subramanian and co-workers reported both magnetocapacitance and magnetoresistance in La2NiMnO6.113 The authors measured the change of the dielectric and resistance response of this material close to the Curie temperature in the presence and absence of external magnetic field. The sudden decrease of the dielectric constant occurs at −53 °C for zero field, while it is at 7 °C for a 0.1 T field and 22 °C for a 1 T field. At −73 °C, there is a 3% decrease in a 1 T field (Fig. 6). Later, Sarma and co-workers found that partially disordered La2NiMnO6 exhibits colossal magnetodielectricity with negligible magnetoresistance.114 In contrast, a colossal magnetoresistance of around 80% was observed at −268 °C in a 80 kOe field in a polycrystalline sample of La2CoMnO6.115 When La is replaced by Y, the results from density-functional calculations predict that the YNiMnO6 compound has an antiferromagnetic ground state.116 In this case, the E*-type ordering breaks inversion symmetry and generates a ferroelectric polarization. An external electric field can therefore be used to tune the transition, allowing electrical control of the magnetization.
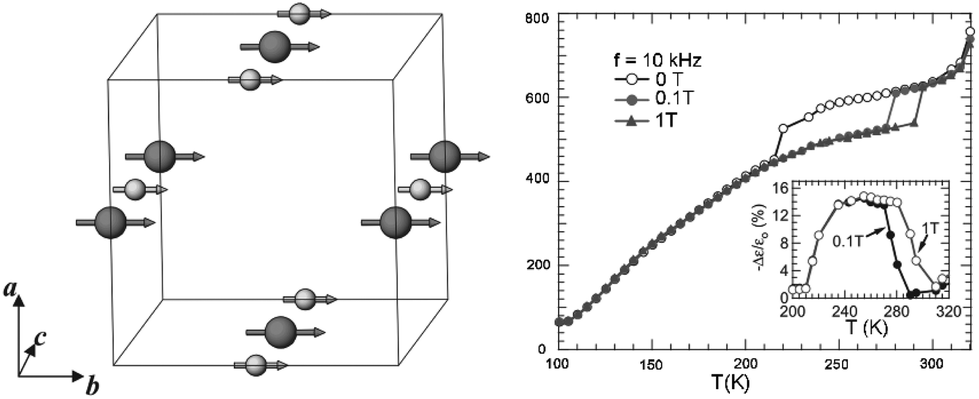 | ||
| Fig. 6 Left: Magnetic structures of La2NiMnO6, in which large spheres are Mn4+ and small spheres are Ni2+, respectively. Magnetic moments are aligned along the b-axis. Right: Temperature dependence of the dielectric constant at 10 kHz for 0, 0.1 and 1 T applied fields. The inset shows the magnetocapacitance effect (−Δε/ε0) with 0.1 and 1 T applied fields. Adapted from ref. 113 with permission from John Wiley and Sons, copyright 2005. | ||
Magnetic refrigeration based on the magnetocaloric effect (MCE) is becoming a promising technique, in which the adiabatic temperature changes due to a change in the magnetic entropy (ΔSm) in an external magnetic field (ΔB). As an eco-friendly alternative and a smarter cooling technology, magnetic refrigeration not only avoids the use of coolants that cause the greenhouse effect and Ozonosphere hole, but also has a theoretical Carnot-Cycle efficiency nearly six times higher than any other process. Several groups have explored the magnetocaloric properties of the RE2NiMnO6 family.117–120 The relatively high values of ΔSm (e.g., 5.2 J kg−1 K−1 for Dy2NiMnO6) and relative cooling power (e.g., 385.1 J kg−1 for Dy2NiMnO6) suggest that they can be used as potential magnetic refrigerants working in wide temperature ranges (Fig. 7).121
 | ||
| Fig. 7 (a) Isothermal magnetization curves in the vicinity of Curie temperature for Dy2NiMnO6. (b) Magnetic entropy change curves as a function of temperature for Dy2NiMnO6 for different magnetic field changes. Adapted from ref. 121 with permission from Elsevier, copyright 2018. | ||
Magnetic nanoparticles have attracted considerable attention in biological and medical areas such as bioseparation, drugs and gene delivery, and others. Tang, Chen and co-workers reported a coprecipitation synthesis of La2NiMnO6 nanoparticles with a crystal size of about 34 to 40 nm.122 These nanoparticles exhibit good adsorbing abilities for the protein bovine serum albumin, with the highest adsorption capacity of 219.6 mg g−1 when the nanoparticles were pre-annealed at 850 °C. When materials exist as nanoparticles, a new physical phenomenon may be observed. Recently, Ravi and others observed a Curie temperature of over 27 °C for the nanoscale La2NiMnO6, which is significantly higher than that of the bulk La2NiMnO6 (∼7 °C).123 The authors thought that oxygen-induced defects may be responsible for such unexpected magnetic properties.
Optical applications
One of the most important applications of rare earth compounds is the photoluminescent material. The local crystal symmetry of cations in A2B′B′′O6 double perovskites, which is critical for luminescence, can be tuned facilely by varying the compositions. For example, the Eu3+-doped Gd2MgTiO6 material utilizes the high excitation efficiency of the charge transfer band of Ti–O as well as the doping of second rare earth ions.124 A weak magnetic dipole transition (5D0–7F1, 590 nm) peak and a strong electric dipole transition (5D0–7F2, 616 nm) peak are present in the emission spectrum, with a maximum red (616 nm) to orange (590 nm) ratio at 15% Eu doping. The 30% Eu doped material exhibits CIE chromaticity coordinates (x = 0.6654 and y = 0.3342) very close to the standard of NTSC (x = 0.670 and y = 0.330). As an almost pure red phosphor, this material may find broad applications for white LEDs.The phosphors with near-infrared (NIR) luminescence have emerged as a new generation of photoluminescent materials. In particular, the NIR emission is in the biologically transparent window (650–950 nm) and thus avoids the self-fluorescence from tissues, resulting in a higher signal-to-noise ratio for fluorescence imaging. These materials can also down-convert the unusable ultraviolet photons to the lower energy phonons that solar cells can adsorb, increasing the overall photovoltaic efficiency. 6-Coordinate Mn4+ with a 3d3 configuration can exhibit intense deep-red and NIR luminescence, sensitive to the crystal field strength. Recently, Qiu and co-workers synthesized Mn4+-doped La2MgGeO6, in which Mg2+ is employed to separate Mn4+ ions and Ge4+ is chosen to offer traps for the capture of radiative energy.125 The products exhibit a strong emission peak between 670 nm and 720 nm with a maximum at 708 nm (Fig. 8a). The luminescence persists after the excitation source is turned off, which can be enhanced by the incorporation of Al3+ ions (Fig. 8b and c). This work demonstrates a new NIR long persistence material.
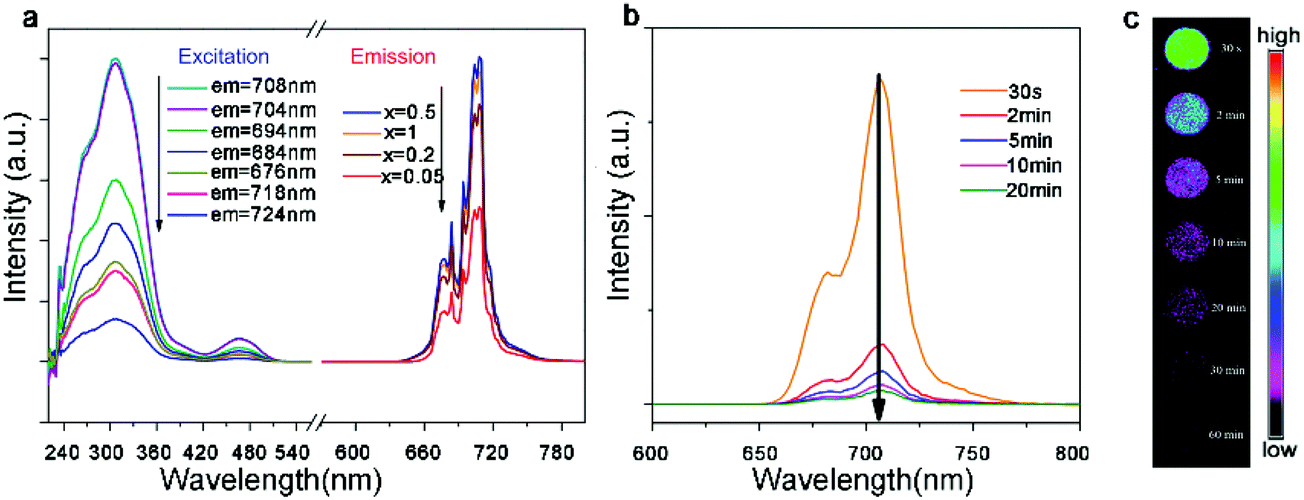 | ||
| Fig. 8 (a) Excitation and emission spectra of La2MgGeO6:Mn4+ samples at room temperature. The emission spectra were excited at 308 nm and excitation spectra were monitored at 676, 684, 694, 704, 708, 718, and 724 nm, respectively. (b) Emission spectra of La2MgGeO6:Mn4+ at different time intervals (30 seconds–20 minutes) after removal of the excitation source. (c) Emission imaging of La2MgGeO6:Mn4+ samples at different time intervals (30 seconds to 60 minutes). The samples were pre-irradiated by a UV lamp for 5 minutes. Adapted from ref. 125 with permission from John Wiley and Sons, copyright 2018. | ||
The new generation of solar cells based on lead halide perovskites have attracted tremendous attention in the last decade.126 However, the low stability of halide perovskites and the use of toxic lead are the major barrier for their practical applications.127 Therefore, it is of critical importance to explore lead free stable perovskites that are suitable for photovoltaic applications. Perovskite oxides are often very stable under ambient conditions but many of them have wide band gaps and extremely poor conductivity.128 As mentioned earlier, the family of RE2NiMnO6 is ferromagnetic semiconductors. In 2017, Sinha and co-workers reported the photovoltaic performance of RE2NiMnO6 (RE = La, Eu, Dy, Lu).129 The measured band gaps are between 1.08 (RE = La) and 1.19 (RE = Dy) eV, which are very close to that of Si (around 1.1 eV) (Fig. 9). It is also striking that these materials also have a long carrier life-time of ∼0.1 ms. The overall photovoltaic performances of RE2NiMnO6-based solar cells are better than those of the ones fabricated with other oxides. Therefore, it may open a new avenue for fabricating cost-effective, green, and commercially successful solar cells.
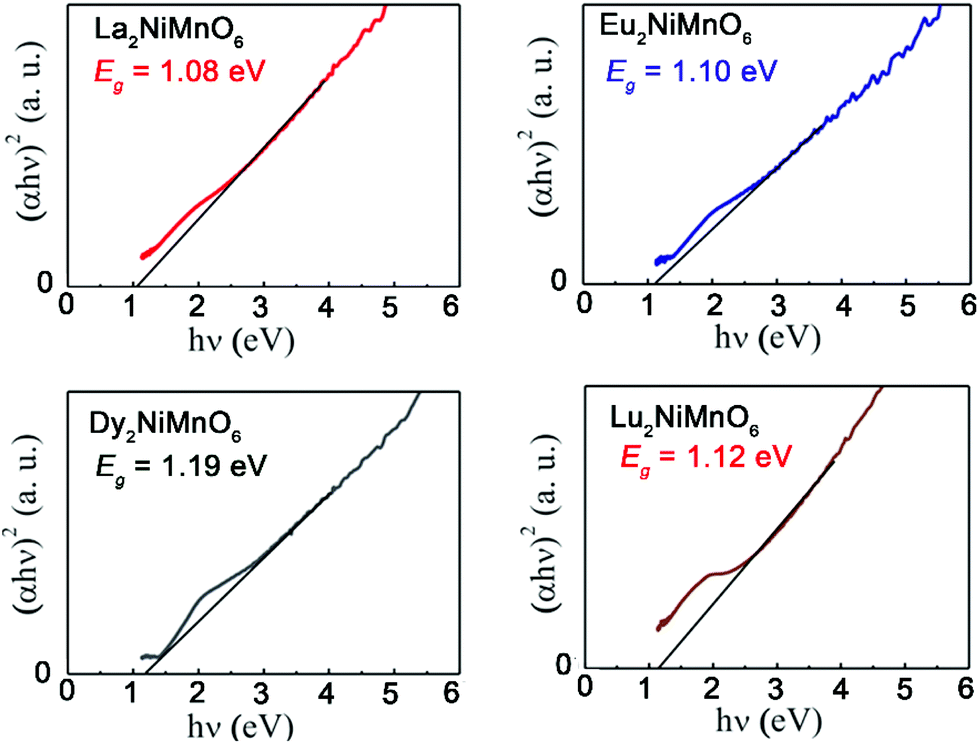 | ||
| Fig. 9 Tauc plot for La2NiMnO6, Eu2NiMnO6, Dy2NiMnO6 and Lu2NiMnO6. Adapted from ref. 129 with permission from Elsevier, copyright 2017. | ||
Electrical applications
Perovskite oxides such as PbTiO3 have been widely used in daily life due to their excellent dielectric, ferroelectric, piezoelectric, electro-optic, and pyro-electric properties. Double perovskites including Pb2ScTaO6 can exhibit comparable or even better performances than PbTiO3. The capacity of facile tuning of both compositions and cation ordering degrees in Pb2ScTaO6 makes it one of the most extensively studied ferroelectrics. However, it also makes the investigation of local structures and phase transitions challenging. The observations and conclusions reported in different groups are often inconsistent. Very recently, Peters and co-workers studied the highly ordered Pb2ScTaO6via dielectric spectroscopy and in situ high-dynamic range electron diffraction.130 The authors were able to measure the degree of ordering as 76% in both local (via TEM) and macroscopic (via XRD) scales (Fig. 10a–c). Further analyses on low temperature electron diffraction patterns revealed the existence of two antiferroelectric displacements in the polar structure (Fig. 10d and e). An unknown phase transition occurs at around −63 °C upon cooling, while it is not present on heating. A possible explanation is that a metastable state was created in a high density of domain walls when freezing the dynamic polar nanodomains.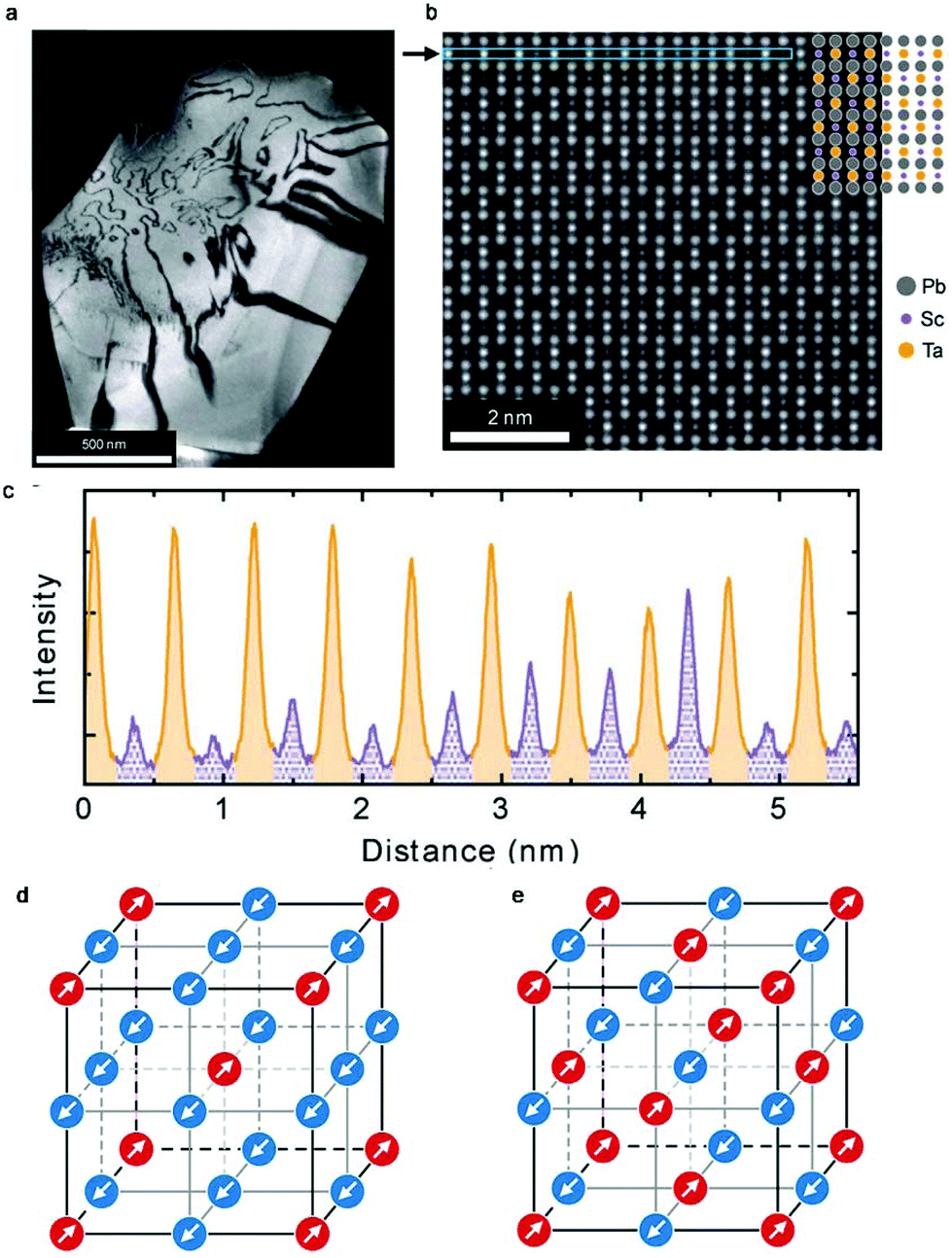 | ||
| Fig. 10 (a) Dark-field g = 111 image of a single grain of Pb2ScTaO6. Bright and dark regions denote ordered regions and antiphase boundaries, respectively. (b) Atomic-resolution annular dark-field image from (a) with strong ordering. (c) An intensity line profile from the region indicated by an arrow in (b). Columns that are expected to contain Sc and Ta are labelled purple (dotted shading) and orange (solid), respectively. (d) Correlated [111] columnar (↑↑) displacement scheme. (e) Anticorrelated [111] (↑↓) displacement scheme. Arrows and colours indicate displacements along the [111] direction. Adapted from ref. 130 with permission from John Wiley and Sons, copyright 2019. | ||
Although the conductivity of many rare earth double perovskites is poor, some are semiconductors. Sr2ErNbO6 has significant frequency dispersion in its dielectric properties. At 100 Hz, the conductivity increases from 1.38 × 10−6 to 2.34 × 10−4 S m−1 with the temperature increased from 30 to 380 °C.131 Further analyses indicate that the conduction mechanism is due to ion hopping. Sr2CeSbO6 is another semiconducting rare earth double perovskite material with the conductivity at 100 Hz changing from 2 × 10−7 to 1.97 × 10−5 S m−1 with the temperature increased from 30 to 430 °C.132 In contrast, the conduction mechanism for this material is due to electron hopping. Similarly, the conductivity of Ca2CeNbO6 varies from 2.79 × 10−7 to 3.5 × 10−5 S m−1 with the temperature increased from 100 to 450 °C.60
Catalytic applications
Catalytic combustion is a promising candidate of conventional flame combustion to achieve higher energy conversion efficiency and reduce emissions of pollutants such as NOx. Catalytic combustion is also safer due to lower combustion temperature and possibility of reacting in a concentration range outside of flammability limits. Simple perovskite oxides (ABO3) have been demonstrated as suitable catalysts with high thermal stability and activity.133 In recent years, rare earth double perovskites have been reported as better catalysts than simple perovskites. For example, Hu and co-workers compared the catalytic performance of La2CuNiO6 with LaNiO3 and LaCuO3 for methane combustion.58 The results indicate that the catalytic activity of La2CuNiO6 was much higher than that of either LaNiO3 or the LaNiO3/LaCuO3 mixture. The 90% conversion temperature of La2CuNiO6 decreased by 63.2 °C than that of LaNiO3. In a later report, the catalytic combustion of propane on La2MMnO6 (M = Co, Ni, and Cu) was measured.107 Infrared spectroscopic data uncovered that the M–O bond strength decreases with cation sizes in the order of Cu > Ni > Co, resulting in more labile O species. The catalyst La2CoMnO6 has the highest activity with 90% conversion at ∼500 °C. It is also found that La2CoMnO6 and La2CuMnO6 are excellent catalysts for the removal of volatile organic compounds.134 100% conversion can be achieved at 150, 200, 300 and 350 °C for isopropyl alcohol, ethanol, toluene, and ethylene, respectively. The catalysts maintain high activity for toluene removal at a high gas hourly space velocity of 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 and are resistive to CO2 and water vapor in gas stream (Fig. 11).
000 h−1 and are resistive to CO2 and water vapor in gas stream (Fig. 11).
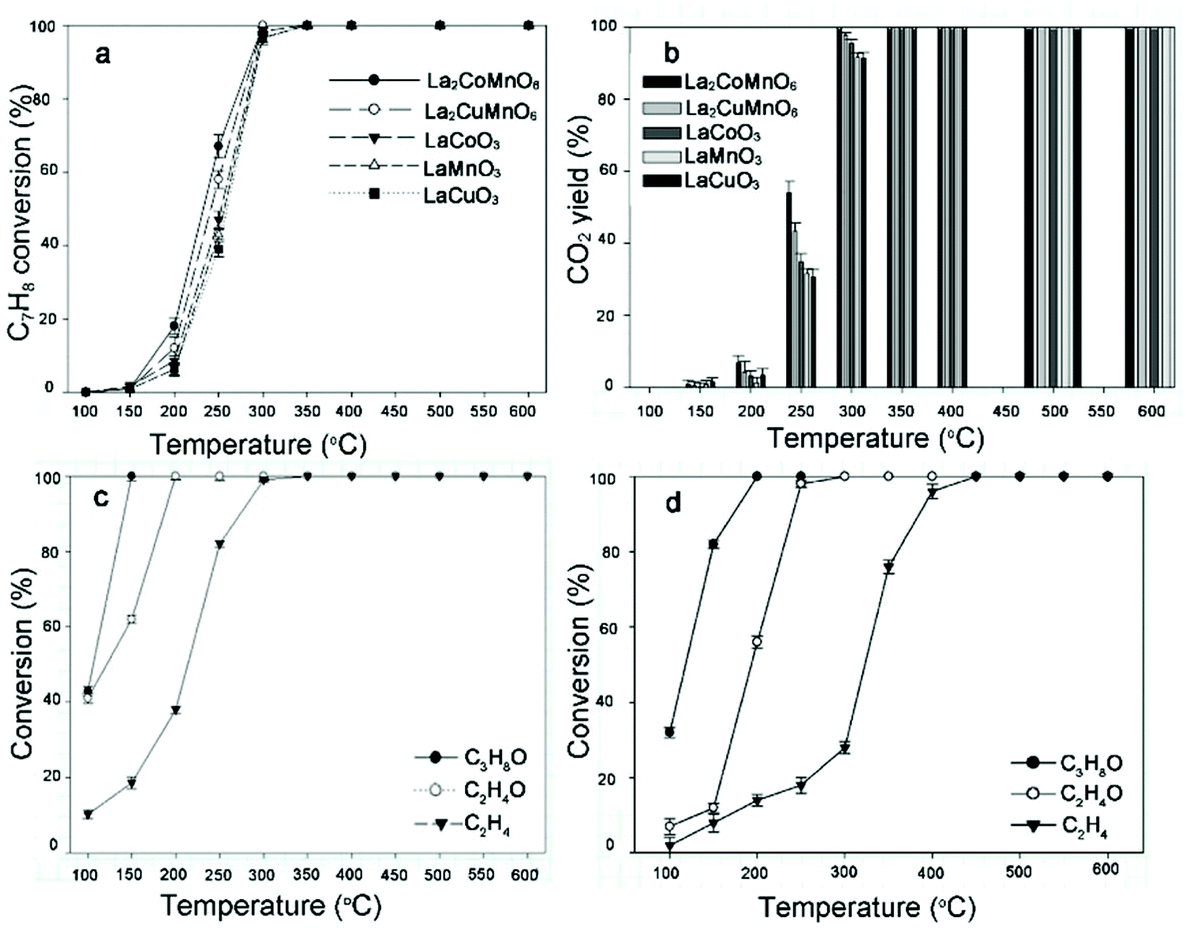 | ||
| Fig. 11 Removal efficiency (a) and CO2 yield (b) of toluene (C7H8) achieved on La2CoMnO6, La2CuMnO6, LaCoO3, LaMnO3, and LaCuO3 as a function of temperature, respectively. Reaction conditions: 300 ppm C7H8 and gas hourly space velocity (GHSV) 30000 h−1. Removal efficiency of isopropyl alcohol (C3H8O), ethanol (C2H6O), and ethylene (C2H4) onLa2CoMnO6 (c) and La2CuMnO6 (d) as a function of temperature. Reaction conditions: 300 ppm C3H8O, 300 ppm C2H4O, 300 ppm C2H4, and GHSV 30000 h−1. Adapted from ref. 134 with permission from Elsevier, copyright 2018. | ||
Microwave induced catalytic oxidation is a new technique in the removal of organic pollutants in wastewater owing to its selective heating ability, higher heating rates and improved energy efficiency. Under microwave irradiation, conducting materials such as activated carbon can adsorb electromagnetic wave energy, creating “hot spots” around the loaded catalysts on the surface. Recently, a La2NiMnO6–cellulose carbon xerogel composite was prepared by a one-step sol–gel approach.135 The catalytic activity measurements found that this composite exhibits higher activities than pure La2NiMnO6 as a result of better dispersion of La2NiMnO6 within a hierarchical porous structure as well as increased active sites for adsorption. Moreover, the composite shows good recyclability and reusability.
Rare earth double perovskites also find applications in the rising field of photocatalysis. Hu and co-workers compared the photocatalytic activities of LaFeO3 and La2FeTiO6 in p-chlorophenol degradation under visible light and found that the latter has much higher activities: after 5 h irradiation, the degradation degree is 62.1% for La2FeTiO6, compared to 49.0% for LaFeO3.109 A possible explanation is that the UV absorption spectrum of La2FeTiO6 is wider. In another report, photocatalytic degradation activities of Ba2REBiO6 (RE = La, Ce, Nd, Sm, Eu, Gd, Dy) were found on methylene blue (MB) and gaseous 2-propanol (IPA) under visible light irradiation.136 The photocatalytic activities of this compound family are generally high. Ba2CeBiO6 is the best candidate for MB degradation. In the case of IPA degradation, a dependence of photocatalytic activity on the lattice parameters was observed, which could be due to a band-gap narrowing induced by the lanthanide contraction, or from the shift of the RE 4f states. It is worth mentioning that the photocatalytic activities of Ba2REBiO6 are all higher than that of BaBiO3. In a later report, the valence states of rare earth ions and band gaps in Ba2REBiO6 were studied.137 Magnetic susceptibility data indicate that the valences of rare earth ions are trivalent except Ce and Pr (tetravalent). The (111) reflection peak in powder XRD patterns, evidence for the RE3+–O–Bi5+ ordering in the double-perovskite structure, was present in all compounds except Ba2CeBiO6. The weak (111) reflection peak of Ba2PrBiO6 verifies the presence of Pr3+. The band gaps were measured from ∼0.7 eV for Ba2CeBiO6, ∼1.1 eV for Ba2PrBiO6, to 1.4–1.7 eV for the other compounds. The high photocatalytic activities of Ba2CeBiO6 and Ba2PrBiO6 reported previously were likely due to the tetravalent state of Ce and Pr.
Another promising area of catalytic applications of rare earth double perovskites is the electrocatalysis. For instance, Ba2REMoO6 (RE = Pr and Nd) were employed as the anode material for solid oxide fuel cells (SOFCs).138 These compounds have good morphologic stability at high temperature and chemical compatibility with the oxide electrolyte. Although the conductivities of Ba2PrMoO6 and Ba2NdMoO6 are as high as 348.5 and 146 S m−1 under humidified CH4 at 800 °C, they are better electrocatalysts for the hydrogen oxidation reaction than the methane oxidation reaction due to the blockage of the methane diffusion path by the deposited carbon. Very recently, a graphene–La2CuNiO6–ZnSe composite was fabricated to enhance the current efficiency of dye-sensitized solar cells (DSSCs) as a counter electrode.139 The conversion efficiency of DSSCs can increase to 11.05%, which is one of the highest efficiencies reported for ternary oxide-based graphene DSSCs. The composite exhibits excellent catalytic activity towards the I3−/I− redox couple and provides a fast diffusion pathway for the electrolyte.
Summary and outlook
This review is the first comprehensive overview on rare earth A2B′B′′O6 double perovskite oxides, focused on their compositions, syntheses and applications. This type of functional material has great structural variability: metals from every group of the periodic table can form A2B′B′′O6 double perovskites with rare earth elements. There have been hundreds of rare earth A2B′B′′O6 double perovskite phases reported to date but it still leaves considerable opportunities for the exploration of new phases. The classical solid phase synthesis remains a universal, robust, and successful approach to obtain rare earth double perovskites. New synthetic routes including high pressure synthesis, flux synthesis, hydrothermal synthesis, sol–gel synthesis, microwave sintering, polymer deposition method, and sol–gel auto-combustion are also useful. The resulting products have great potential in but not limited to various applications such as magnetic, optical, electrical, and catalytic materials. In particular, the novel functionality of rare earth double perovskites including magnetoresistance, magnetocapacitance, multiferroics, white light and long afterglow luminescence, photovoltaic properties, photocatalytic or electrocatalytic activities has attracted tremendous attention in recent years.Despite the importance of rare earth A2B′B′′O6 double perovskites, more effort is required to bring them from laboratory research to applications. The great structural variability of these materials is a double-edged sword that opens tremendous opportunities but also makes the control of compositions and cation ordering, the study of structure–property relationships, and the fabrication of devices quite challenging. In addition, a significant portion of the studies on rare earth double perovskites was conducted by solid physicists which are not well known to chemists. We therefore raise a few issues in the investigations of rare earth double perovskites that might inspire future researchers: firstly, it is desirable to explore new A2B′B′′O6 phases although a large number of materials have been reported; secondly, it is essential to search new synthetic routes or develop current approaches to obtain target products with low cost, tunable composition, and controllable morphology; thirdly, it is of fundamental importance to uncover the various structural information that plays key roles in the properties, such as cation ordering and coupling, phase transition, defects, and dopants; finally, it is critical to broaden their current applications, for example, the majority of rare earth double perovskites are made for their magnetic properties but they might be promising candidates for catalysts or photovoltaic materials. We also hope that this review could attract more chemists to enter this impressive area.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support from the China National Funds for Excellent Young Scientists (21522106), the National Key R&D Program of China (2017YFA0208000), the Open Funds (RERU2019001) of the State Key Laboratory of Rare Earth Resource Utilization, and 111 Project (B18030) from China is gratefully acknowledged.Notes and references
- A. R. Chakhmouradian and P. M. Woodward, Phys. Chem. Miner., 2014, 41, 387–391 CAS.
- J. P. Attfield, P. Lightfoot and R. E. Morris, Dalton Trans., 2015, 44, 10541–10542 CAS , and the Perovskites themed issue.
- M. Acosta, N. Novak, V. Rojas, S. Patel, R. Vaish, J. Koruza, G. A. J. Rossetti and J. Rödel, Appl. Phys. Rev., 2017, 4, 041305 Search PubMed.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546 CAS.
- N. Hayashi, T. Yamamoto, H. Kageyama, M. Nishi, Y. Watanabe, T. Kawakami, Y. Matsushita, A. Fujimori and M. Takano, Angew. Chem., Int. Ed., 2011, 50, 12547–12550 CAS.
- J. B. MacChesney, R. C. Sherwood and J. F. Potter, J. Chem. Phys., 1965, 43, 1907–1913 CAS.
- J. J. Jiang, D. Li, S. J. Li, Z. H. Wang, Y. Wang, J. He, W. Liu and Z. D. Zhang, RSC Adv., 2015, 5, 14584–14591 CAS.
- E. Grabowska, Appl. Catal., B, 2016, 186, 97–126 CAS.
- S. Vasala and M. Karppinen, Prog. Solid State Chem., 2015, 43, 1–36 CAS.
- F. Li, M. J. Cabral, B. Xu, Z. Cheng, E. C. Dickey, J. M. LeBeau, J. Wang, J. Luo, S. Taylor, W. Hackenberger, L. Bellaiche, Z. Xu, L.-Q. Chen, T. R. Shrout and S. Zhang, Science, 2019, 364, 264–268 CAS.
- E. G. Steward and H. P. Rooksby, Acta Crystallogr., 1951, 4, 503–507 CAS.
- A. A. Bokov, I. P. Raevskii, O. I. Prokopalo, E. G. Fesenko and V. G. Smotrakov, Ferroelectrics, 1984, 54, 241–244 Search PubMed.
- D. D. Sarma, E. V. Sampathkumaran, S. Ray, R. Nagarajan, S. Majumdar, A. Kumar, G. Nalini and T. N. Guru Row, Solid State Commun., 2000, 114, 465–468 CAS.
- W. Feng, L.-D. Sun, Y.-W. Zhang and C.-H. Yan, Coord. Chem. Rev., 2010, 254, 1038–1053 CAS.
- H. Dong, S.-R. Du, X.-Y. Zheng, G.-M. Lyu, L.-D. Sun, L.-D. Li, P.-Z. Zhang, C. Zhang and C.-H. Yan, Chem. Rev., 2015, 115, 10725–10815 CAS.
- J. Xu, X. Chen, Y. Xu, Y. Du and C. Yan, Adv. Mater., 2019 DOI:10.1002/adma.201806461.
- V. M. Goldschmidt, Naturwissenschaften, 1926, 14, 477–485 CAS.
- C. Li, K. C. K. Soh and P. Wu, J. Alloys Compd., 2004, 372, 40–48 CAS.
- T. Aharen, J. E. Greedan, C. A. Bridges, A. A. Aczel, J. Rodriguez, G. MacDougall, G. M. Luke, V. K. Michaelis, S. Kroeker, C. R. Wiebe, H. Zhou and L. M. D. Cranswick, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 064436 Search PubMed.
- W. R. Gemmill, M. D. Smith and H.-C. zur Loye, J. Solid State Chem., 2006, 179, 1750–1756 CAS.
- A. A. Aczel, D. E. Bugaris, J. Yeon, C. de la Cruz, H. C. zur Loye and S. E. Nagler, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 014413 Search PubMed.
- A. A. Aczel, D. E. Bugaris, L. Li, J. Q. Yan, C. de la Cruz, H. C. zur Loye and S. E. Nagler, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 014435 Search PubMed.
- M. J. Davis, S. J. Mugavero, K. I. Glab, M. D. Smith and H.-C. zur Loye, Solid State Sci., 2004, 6, 413–417 CAS.
- W. R. Gemmill, M. D. Smith, R. Prozorov and H.-C. zur Loye, Inorg. Chem., 2005, 44, 2639–2646 CAS.
- W. R. Gemmill, M. D. Smith and H.-C. zur Loye, J. Solid State Chem., 2004, 177, 3560–3567 CAS.
- S. J. Mugavero, M. D. Smith and H.-C. zur Loye, J. Solid State Chem., 2005, 178, 200–206 CAS.
- D. A. Landínez Téllez, D. Martínez Buitrago, R. Cardona C, E. W. Barrera and J. Roa-Rojas, J. Mol. Struct., 2014, 1067, 205–209 Search PubMed.
- P. Cai, L. Qin, C. Chen, J. Wang, S. Bi, S. I. Kim, Y. Huang and H. J. Seo, Inorg. Chem., 2018, 57, 3073–3081 CAS.
- M. Swaffer, P. R. Slater, R. K. B. Gover, T. Matsumura, R. Kanno and T. Kamiyama, Chem. Commun., 2002, 1776–1777 CAS.
- S. J. I. Mugavero, A. H. Fox, M. D. Smith and H.-C. zur Loye, J. Solid State Chem., 2010, 183, 465–470 CAS.
- T. Ferreira, G. Morrison, J. Yeon and H.-C. zur Loye, Cryst. Growth Des., 2016, 16, 2795–2803 CAS.
- K. Ouchetto, F. Archaimbault, J. Choisnet and M. Et-Tabirou, Mater. Chem. Phys., 1997, 51, 117–124 CAS.
- D. Yang, P. Zhao, S. Huang, T. Yang and D. Huo, Results Phys., 2019, 12, 344–348 Search PubMed.
- D. K. Mahato, M. Rudra and T. P. Sinha, J. Alloys Compd., 2016, 689, 617–624 CAS.
- E. Rodríguez, M. L. López, J. Campo, M. L. Veiga and C. Pico, J. Mater. Chem., 2002, 12, 2798–2802 Search PubMed.
- R. E. A. Utami, D. Triyono and H. Laysandra, AIP Conf. Proc., 2017, 1862, 030028 Search PubMed.
- K. Asai, K. Fujiyoshi, N. Nishimori, Y. Satoh, Y. Kobayashi and M. Mizoguchi, J. Phys. Soc. Jpn., 1998, 67, 4218–4228 CAS.
- R. J. Booth, R. Fillman, H. Whitaker, A. Nag, R. M. Tiwari, K. V. Ramanujachary, J. Gopalakrishnan and S. E. Lofland, Mater. Res. Bull., 2009, 44, 1559–1564 CAS.
- G. Zhang, G. Li, F. Liao, Y. Fu, M. Xiong and J. Lin, J. Cryst. Growth, 2011, 327, 262–266 CAS.
- N. Nishimori, K. Asai and M. Mizoguchi, J. Phys. Soc. Jpn., 1995, 64, 1326–1333 CAS.
- W. Z. Yang, X. Q. Liu, H. J. Zhao and X. M. Chen, J. Magn. Magn. Mater., 2014, 371, 52–59 CAS.
- B. Orayech, I. Urcelay-Olabarria, G. A. López, O. Fabelo, A. Faik and J. M. Igartua, Dalton Trans., 2015, 44, 13867–13880 CAS.
- M. K. Kim, J. Y. Moon, H. Y. Choi, S. H. Oh, N. Lee and Y. J. Choi, J. Phys.: Condens. Matter, 2015, 27, 426002 CAS.
- Y. Wu, Z. Yu and S. Liu, J. Solid State Chem., 1994, 112, 157–160 CAS.
- S. D. Bhame, V. L. J. Joly and P. A. Joy, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 054426 Search PubMed.
- T. Kawano, J. Takahashi, T. Yamada and H. Yamane, J. Ceram. Soc. Jpn., 2007, 115, 792–796 CAS.
- X. Ding, B. Gao, E. Krenkel, C. Dawson, J. C. Eckert, S.-W. Cheong and V. Zapf, Phys. Rev. B: Condens. Matter Mater. Phys., 2019, 99, 014438 Search PubMed.
- A. E. Smith, A. W. Sleight and M. A. Subramanian, Mater. Res. Bull., 2010, 45, 460–463 CAS.
- C. Schinzer, J. Phys. Chem. Solids, 2000, 61, 1543–1551 CAS.
- C. Schinzer and G. Demazeau, J. Mater. Sci., 1999, 34, 251–256 CAS.
- C. Schinzer, J. Alloys Compd., 1999, 288, 65–75 CAS.
- M. Azuma, S. Kaimori and M. Takano, Chem. Mater., 1998, 10, 3124–3130 CAS.
- R. Ubic, Y. Hu and I. Abrahams, Acta Crystallogr., Sect. B: Struct. Sci., 2006, 62, 521–529 Search PubMed.
- N. Das, M. A. Nath, G. S. Thakur, M. Thirumal and A. K. Ganguli, J. Solid State Chem., 2015, 229, 97–102 CAS.
- P. Gómez-Romero, M. R. Palacín, N. Casañ, A. Fuertes and B. Martínez, Solid State Ionics, 1993, 63–65, 603–608 Search PubMed.
- N. Floros, J. T. Rijssenbeek, A. B. Martinson and K. R. Poeppelmeier, Solid State Sci., 2002, 4, 1495–1498 CAS.
- Z. Yang, W. Wong-Ng, J. A. Kaduk, M. Jang and G. Liu, J. Solid State Chem., 2009, 182, 1142–1148 CAS.
- R. Hu, R. Ding, J. Chen, J. Hu and Y. Zhang, Catal. Commun., 2012, 21, 38–41 CAS.
- R. Hu, Y. Bai, H. Du, H. Zhang, Y. Du, J. Zhang and Q. Zhou, J. Rare Earths, 2015, 33, 1284–1292 CAS.
- C. Bharti and T. P. Sinha, Phys. B, 2011, 406, 1827–1832 CAS.
- Q. Zhou, P. Blanchard, B. J. Kennedy, E. Reynolds, Z. Zhang, W. Miiller, J. B. Aitken, M. Avdeev, L.-Y. Jang and J. A. Kimpton, Chem. Mater., 2012, 24, 2978–2986 CAS.
- K. Henmi, Y. Hinatsu and N. M. Masaki, J. Solid State Chem., 1999, 148, 353–360 CAS.
- A. Dutta, S. Saha, P. Kumari, T. P. Sinha and S. Shannigrahi, J. Solid State Chem., 2015, 229, 296–302 CAS.
- B. Ghosh, A. Dutta, S. Shannigrahi and T. P. Sinha, J. Alloys Compd., 2015, 648, 111–115 CAS.
- Y. Doi and Y. Hinatsu, J. Phys.: Condens. Matter, 2001, 13, 4191–4202 CAS.
- B. J. Kennedy, P. J. Saines, Y. Kubota, C. Minakata, H. Hano, K. Kato and M. Takata, Mater. Res. Bull., 2007, 42, 1875–1880 CAS.
- A. Dutta, T. P. Sinha and D. Das, J. Magn. Magn. Mater., 2014, 360, 211–216 CAS.
- B. Ghosh, S. Halder and T. P. Sinha, J. Am. Ceram. Soc., 2014, 97, 2564–2572 CAS.
- K. Z. Baba-Kishi, P. M. Woodward and K. Knight, Ferroelectrics, 2001, 261, 21–26 CAS.
- A. C. McLaughlin, Solid State Commun., 2006, 137, 354–357 CAS.
- E. J. Cussen, D. R. Lynham and J. Rogers, Chem. Mater., 2006, 18, 2855–2866 CAS.
- R. M. Pinacca, S. A. Larrégola, C. A. López, J. C. Pedregosa, V. Pomjakushin, R. D. Sánchez and J. A. Alonso, Mater. Res. Bull., 2015, 66, 192–199 CAS.
- Y. Sasaki, Y. Doi and Y. Hinatsu, J. Mater. Chem., 2002, 12, 2361–2366 CAS.
- Y. Hinatsu, Y. Izumiyama, Y. Doi, A. Alemi, M. Wakeshima, A. Nakamura and Y. Morii, J. Solid State Chem., 2004, 177, 38–44 CAS.
- Y. Izumiyama, Y. Doi, M. Wakeshima, Y. Hinatsu, Y. Shimojo and Y. Morii, J. Phys.: Condens. Matter, 2001, 13, 1303–1313 CAS.
- Y. Izumiyama, Y. Doi, M. Wakeshima, Y. Hinatsu, K. Oikawa, Y. Shimojo and Y. Morii, J. Mater. Chem., 2000, 10, 2364–2367 CAS.
- Y. Doi, Y. Hinatsu, A. Nakamura, Y. Ishii and Y. Morii, J. Mater. Chem., 2003, 13, 1758–1763 CAS.
- P. D. Battle, J. B. Goodenough and R. Price, J. Solid State Chem., 1983, 46, 234–244 CAS.
- K. Ouchetto, F. Archaimbault, J. Choisnet and M. Et-Tabirou, J. Mater. Sci. Lett., 2000, 19, 371–373 Search PubMed.
- P. D. Battle, C. W. Jones and F. Studer, J. Solid State Chem., 1991, 90, 302–312 CAS.
- C. Sakai, Y. Doi and Y. Hinatsu, J. Alloys Compd., 2006, 408–412, 608–612 CAS.
- Y. Doi and Y. Hinatsu, J. Phys.: Condens. Matter, 1999, 11, 4813–4820 CAS.
- N. G. Parkinson, P. D. Hatton, J. A. K. Howard, C. Ritter, R. M. Ibberson and M.-K. Wu, J. Phys.: Condens. Matter, 2004, 16, 7611–7624 CAS.
- Y. Doi, Y. Hinatsu, K.-I. Oikawa, Y. Shimojo and Y. Morii, J. Mater. Chem., 2000, 10, 797–800 CAS.
- R. Sáez-Puche, E. Climent-Pascual, R. Ruiz-Bustos, M. A. Alario-Franco and M. T. Fernández-Díaz, Prog. Solid State Chem., 2007, 35, 211–219 Search PubMed.
- M. Wakeshima, D. Harada and Y. Hinatsu, J. Mater. Chem., 2000, 10, 419–422 CAS.
- M. Wakeshima, D. Harada, Y. Hinatsu and N. Masaki, J. Solid State Chem., 1999, 147, 618–623 CAS.
- Y. Hinatsu, Y. Doi and M. Wakeshima, J. Solid State Chem., 2013, 206, 300–307 CAS.
- P. J. Saines, B. J. Kennedy, M. M. Elcombe, H. H. Harris, L.-Y. Jang and Z. Zhang, J. Solid State Chem., 2008, 181, 2941–2952 CAS.
- A. Dutta, P. K. Mukhopadhyay, T. P. Sinha, D. Das and S. Shannigrahi, Solid State Sci., 2016, 58, 64–69 CAS.
- S. Otsuka and Y. Hinatsu, J. Solid State Chem., 2015, 227, 132–141 CAS.
- L. Wu, X. Mei and W. Zheng, Mater. Lett., 2006, 60, 2326–2330 CAS.
- C. Vijayakumar, H. Padma Kumar, V. T. Kavitha, S. Solomon, J. K. Thomas, P. R. S. Wariar and J. Koshy, J. Alloys Compd., 2009, 475, 778–781 CAS.
- H. Karunadasa, Q. Huang, B. G. Ueland, P. Schiffer and R. J. Cava, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 8097–8102 CAS.
- W. Wong-Ng, J. A. Kaduk, M. Luong and Q. Huang, Powder Diffr., 2014, 29, 371–378 CAS.
- A. Faik, D. Orobengoa, E. Iturbe-Zabalo and J. M. Igartua, J. Solid State Chem., 2012, 192, 273–283 CAS.
- A. Faik, I. Urcelay, E. Iturbe-Zabalo and J. M. Igartua, J. Mol. Struct., 2010, 977, 137–144 CAS.
- S. A. Larrégola, J. A. Alonso, D. Sheptyakov, M. Algueró, A. Muñoz, V. Pomjakushin and J. C. Pedregosa, Inorg. Chem., 2011, 50, 5545–5557 Search PubMed.
- S. Feraru, P. Samoila, A. I. Borhan, M. Ignat, A. R. Iordan and M. N. Palamaru, Mater. Charact., 2013, 84, 112–119 CAS.
- J. A. Alonso, C. Cascales, P. García Casado and I. Rasines, J. Solid State Chem., 1997, 128, 247–250 CAS.
- R. Phatak, A. K. Yadav, N. Pathak, C. L. Prajapat, U. M. Kasar, M. R. Singh, S. N. Jha, D. Bhattacharyya, A. Das and S. K. Sali, J. Alloys Compd., 2017, 708, 1168–1177 CAS.
- S. K. Awasthi, D. M. Chackruburtty and V. K. Tondon, J. Inorg. Nucl. Chem., 1968, 30, 819–821 CAS.
- Y. Misawa, Y. Doi and Y. Hinatsu, J. Solid State Chem., 2011, 184, 1478–1483 CAS.
- Y. S. Reddy, Y. Markandeya, B. A. Rao and G. Bhikshamaiah, J. Mater. Sci.: Mater. Electron., 2018, 29, 2966–2973 CAS.
- S. Kanungo, K. Mogare, B. Yan, M. Reehuis, A. Hoser, C. Felser and M. Jansen, Phys. Rev. B: Condens. Matter Mater. Phys., 2016, 93, 245148 Search PubMed.
- M. R. Filip and F. Giustino, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5397 CAS.
- J. E. Tasca, A. E. Lavat and M. G. González, J. Asian Ceram. Soc., 2017, 5, 235–241 Search PubMed.
- J. Li, R. Hu, J. Zhang, W. Meng, Y. Du, Y. Si and Z. Zhang, Fuel, 2016, 178, 148–154 CAS.
- R. Hu, C. Li, X. Wang, Y. Sun, H. Jia, H. Su and Y. Zhang, Catal. Commun., 2012, 29, 35–39 CAS.
- M. Penchal Reddy, X. B. Zhou, L. Jing and Q. Huang, Mater. Lett., 2014, 132, 55–58 CAS.
- Q. X. Jia, T. M. McCleskey, A. K. Burrell, Y. Lin, G. E. Collis, H. Wang, A. D. Q. Li and S. R. Foltyn, Nat. Mater., 2004, 3, 529–532 CAS.
- C. Xie, L. Shi, S. Zhou, J. Zhao, H. Liu, Y. Li and D. Yao, Surf. Coat. Technol., 2015, 277, 222–226 CAS.
- N. S. Rogado, J. Li, A. W. Sleight and M. A. Subramanian, Adv. Mater., 2005, 17, 2225–2227 CAS.
- D. Choudhury, P. Mandal, R. Mathieu, A. Hazarika, S. Rajan, A. Sundaresan, U. V. Waghmare, R. Knut, O. Karis, P. Nordblad and D. D. Sarma, Phys. Rev. Lett., 2012, 108, 127201 CAS.
- R. N. Mahato, K. Sethupathi and V. Sankaranarayanan, J. Appl. Phys., 2010, 107, 09D714 Search PubMed.
- S. Kumar, G. Giovannetti, J. van den Brink and S. Picozzi, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 134429 Search PubMed.
- Y. Jia, Q. Wang, Y. Qi and L. Li, J. Alloys Compd., 2017, 726, 1132–1137 CAS.
- X. Luo, Y. P. Sun, B. Wang, X. B. Zhu, W. H. Song, Z. R. Yang and J. M. Dai, Solid State Commun., 2009, 149, 810–813 CAS.
- T. Chakraborty, H. Nhalil, R. Yadav, A. A. Wagh and S. Elizabeth, J. Magn. Magn. Mater., 2017, 428, 59–63 CAS.
- M. Balli, P. Fournier, S. Jandl and M. M. Gospodinov, J. Appl. Phys., 2014, 115, 173904 Search PubMed.
- L. Su, X.-Q. Zhang, Q.-Y. Dong, Y.-J. Ke, K.-Y. Hou, C.-S. Liu and Z.-H. Cheng, J. Alloys Compd., 2018, 746, 594–600 CAS.
- Z.-Y. Wu, C.-B. Ma, X.-G. Tang, R. Li, Q.-X. Liu and B.-T. Chen, Nanoscale Res. Lett., 2013, 8, 207 Search PubMed.
- S. Ravi and C. Senthilkumar, Mater. Lett., 2016, 164, 124–126 CAS.
- N. Ding, Q. Liu, L. Wang, L. Zhang, Y. Feng and Q. Zhang, J. Mater. Sci.: Mater. Electron., 2018, 29, 4122–4127 CAS.
- X. Zhang, J. Nie, S. Liu, Y. Li and J. Qiu, J. Am. Ceram. Soc., 2018, 101, 1576–1584 CAS.
- B. Saparov and D. B. Mitzi, Chem. Rev., 2016, 116, 4558–4596 CAS.
- C. C. Boyd, R. Cheacharoen, T. Leijtens and M. D. McGehee, Chem. Rev., 2019, 119, 3418–3451 CAS.
- W.-J. Yin, B. Weng, J. Ge, Q. Sun, Z. Li and Y. Yan, Energy Environ. Sci., 2019, 12, 442–462 CAS.
- M. Sariful Sheikh, D. Ghosh, A. Dutta, S. Bhattacharyya and T. P. Sinha, Mater. Sci. Eng., B, 2017, 226, 10–17 CAS.
- J. J. P. Peters, A. M. Sanchez, D. Walker, R. Whatmore and R. Beanland, Adv. Mater., 2019, 31, 1806498 Search PubMed.
- R. Mukherjee, S. Chanda, C. Bharti, P. Sahu and T. P. Sinha, Phys. B, 2013, 422, 78–82 CAS.
- C. Bharti and T. P. Sinha, Solid State Sci., 2010, 12, 498–502 CAS.
- N. Labhasetwar, G. Saravanan, S. Kumar Megarajan, N. Manwar, R. Khobragade, P. Doggali and F. Grasset, Sci. Technol. Adv. Mater., 2015, 16, 036002 Search PubMed.
- K. L. Pan, G. T. Pan, S. Chong and M. B. Chang, J. Environ. Sci., 2018, 69, 205–216 Search PubMed.
- Y. Wang, Y. Xiong, J. Wang and X. Zhang, Catal. Commun., 2017, 90, 14–18 CAS.
- T. Hatakeyama, S. Takeda, F. Ishikawa, A. Ohmura, A. Nakayama, Y. Yamada, A. Matsushita and J. Yea, J. Ceram. Soc. Jpn., 2010, 118, 91–95 CAS.
- A. Matsushita, T. Nakane, T. Naka, H. Isago, Y. Yamada and Y. Yamada, Jpn. J. Appl. Phys., 2012, 51, 121802 Search PubMed.
- M. K. Rath and K.-T. Lee, J. Alloys Compd., 2018, 737, 152–159 CAS.
- W.-C. Oh, K. Y. Cho, C. H. Jung and Y. Areerob, Photochem. Photobiol. Sci., 2019, 18, 1389–1397 CAS.
| This journal is © the Partner Organisations 2019 |