
A theoretical investigation into the first-row transition metal–O2 adducts†
Xiao-Xi
Li
a,
Kyung-Bin
Cho
 *ab and
Wonwoo
Nam
*a
*ab and
Wonwoo
Nam
*a
aDepartment of Chemistry and Nano Science, Ewha Womans University, Seoul 03760, Korea. E-mail: wwnam@ewha.ac.kr
bDepartment of Chemistry, Chonbuk National University, Jeonju 54896, Korea. E-mail: workforkyung@jbnu.ac.kr
First published on 18th June 2019
Abstract
An extensive investigation into various M–O2 species (M = CrI, MnI, FeI, CoI, NiI, CuI) has been conducted using a Density Functional Theory (DFT) approach, generating MI–O2, MII–superoxo or MIII–peroxo species. Two different ligands, 12-TMC and 14-TMC, are used to gauge the effects of the ligand ring-size. In general, theory reproduces the experimental results (where available) well enough to give confidence in the calculations. In addition to the usual calculated features of the individual metal complexes, a statistical analysis has been done by comparing the M–O2 species across the periodical system. It is found that the O2 binding energy diminishes with higher metal atomic number, while an end-on structure becomes gradually favored. Also, multi-spin state reactivity becomes more likely for metals above Fe. The spin density on O2 (and with it the formal oxidation state of the metal) is more dependent on the prevailing spin state of the compound rather than the metal type per se, and the higher flexibility of the larger 14-TMC ring has also been verified. The theoretical methods used are also evaluated regarding their accuracy.
Introduction
In biochemical systems, evolution has taken advantage of the fact that air (and with it, O2) is easily accessible.1 Not only is the O–O bond energy rich which can be utilized to drive the reaction, but the oxygen atoms can be attached to substrates to make them solvable in in vivo systems. The O–O bond break is frequently catalysed by metal centres; thus, an initial metal–dioxygen species is expected to form.2 Depending on the external electron and proton donor sources, one could expect the presence of one or more type(s) of such intermediates, e.g. metal–superoxo, –peroxo and –hydroperoxo species.3–5 It is evident that the initial oxidation state and type of the metal is also a key factor, as for instance both metal(II)- and metal(III)–peroxo species could be plausibly formed.6,7 In addition to the fact that these species may be en route to form high-valent metal-oxo species8 (which are usually the prime candidates to be the oxygenating species9,10), there are suggestions that these species can act as the oxygenating species themselves as well.11 Thus, the literature is full of studies on both synthetic and biologically occurring metal–dioxygen species, in order to understand the inherent features of these species.12–16Our current study is an attempt to contribute to this field by comparing the effects and features of different metals to each other, in an otherwise identical setting. The last condition however, is not easy to achieve. From already published studies, it is sometimes difficult to compare similar species side-by-side due to differences in methods, ligands, metals, solvents etc. used in experiments. This heterogeneity of the studies is probably necessary, as the individual species may not be stable for studies in different experimental conditions. However, much of this problem can be circumvented in theoretical studies, where the stability is not an issue. Even though theoretical methods also have methodological variations between the studies, at least over-the-board homogeneity is possible. The added advantage is that species not yet experimentally isolated could also be studied and compared. Thus, we opted for a theoretical approach in our current study.
In this study, we have calculated [(L)MO2]+ species (Fig. 1), where M is one of the six transition metals (Cr, Mn, Fe, Co, Ni and Cu) with an oxidation state of +1 and O2 is a neutral species. Hence, any re-distribution of electrons within the complex could result in MII–superoxo or MIII–peroxo species depending on the precise circumstances. The ligands (L) of both 12-TMC (1,4,7,10-tetramethyl-1,4,7,10-tetraazacyclo-dodecane) and 14-TMC (1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane) were used, enabling us to compare the effects of the ligand ring size. The particular choice of the formal metal(III)-peroxide and TMC ligands for this study is due to the existence of several experimental results for this set of compounds.17 The existing experimental studies are not a complete set, but enough to allow us to assess the accuracy of our calculations. Therefore, the present work is also a study in how well theoretical methods can match up to and predict experiments. For each of the metal species, we have calculated all the probable spin states, without the O2 adduct as well as with it in both side-on and end-on fashions. The calculated structures presented here include 97 gas-phase optimized structures and 105 solvent-phase optimized ones. We will denote each species as 1M for [(12-TMC)MO2]+ species and 2M for [(14-TMC)MO2]+ species (M = Cr, Mn, Fe, Co, Ni or Cu).
 | ||
| Fig. 1 The models used in this study. [(12-TMC)MO2]+ (upper row) and [(14-TMC)MO2]+ (lower row) with O2 binding in either a side-on (left column) or an end-on (right column) fashion. M = Cr, Mn, Fe, Co, Ni or Cu. H-Atoms were omitted for clarity. | ||
Methods
Computational details
Calculations were performed using density functional theory (DFT)18 as implemented in ORCA19 package. Two functionals, B3LYP20–24 and BP86,21,25,26 were used in this study to estimate the energy accuracy of hybrid vs. pure functionals for these systems. The basis set used was CP(PPP)27,28 for the metal, and Def2-TZVPP29 for the rest. The optimizations were done at the BP86/[CP(PPP)+Def2-TZVPP]/GAS level (where GAS denotes gas-phase), whereby a single-point frequency calculation was carried out at the same level. This also enabled us to calculate the free energy (ΔG) at this level. Also, a single-point B3LYP//BP86/[CP(PPP)+Def2-TZVPP]/GAS calculation was done on that geometry for energy comparisons. The effect of the functional for the geometry optimization itself was investigated only in few cases where it could possibly matter (as detailed in relevant sections). The effects of solvent (acetonitrile) were dealt with separately as a new optimization at the BP86/[CP(PPP)+Def2-TZVPP]/COSMO level, with the solvent being included as a dielectric medium using the COSMO30 scheme as implemented in ORCA. A subsequent single-point B3LYP/[CP(PPP)+Def2-TZVPP]/COSMO calculation was done for the BP86/[CP(PPP)+Def2-TZVPP]/COSMO optimized geometry as well. No frequency calculation has been carried out due to questions of the validity of such calculation on the solvent optimized structures.31 Dispersion calculations, where it is suspected to matter, was done by the DFT-D3 program to correct the energies.32 To test its effect on the geometries as well, the representative species (vide infra) were reoptimized at the BP86-D level. The RMSDs were found to be on average at 0.07 Å for 1M and 0.05 Å for 2M (see ESI, Table S9†) These values are much smaller than the RMSDs found when comparing the BP86 structures (without dispersion) to the X-ray structures (Table 1); hence the dispersion effect on geometry optimizations were deemed to be of little consequence to our study and not used. To test the sensitivity of the calculations to basis sets, the entire set of calculations were also conducted with CP(PPP) + TZVP28,33 basis set, without no significant bearing for the current results (data not shown). The data quoted in the text are taken from the BP86/[CP(PPP)+Def2-TZVPP]/COSMO calculations, unless explicitly stated otherwise. A more in-depth look at the electronic structures was done with Natural Bond Orbital (NBO) 6.0 program.34 All the data can be found in the ESI.†| Metal | 12-TMC | 14-TMC | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| End-on or Side-on | Bond lengths M–O(avg)/O–O (Å) | RMSDb core/all (Å) | Spin S | End-on or Side-on | Bond lengths M–O(avg)/O–O (Å) | RMSDb core/all (Å) | Spin S | |||||||
| DFT | EXP | DFT(solv) | EXP | DFT | EXP | DFT | EXP | DFT(solv) | EXP | DFT | EXP | |||
| a The theoretical values presented in this table correspond to the lowest energy spin states. In case there are ambiguities in the theoretical results, the spin state matching experiments are chosen. For the theoretical distances, solvent optimized geometries are given. Values for all spin states are given in ESI.† b Root mean square deviation of the atomic positions between the calculated and the X-ray structures. Core is defined as consisting of the metal, O and N atoms. c The results are ambiguous depending on different functionals as well as the solution phase and/or free energy corrections, that can be chosen to match the experimental values. d EXAFS value. e The distance to the bonding oxygen only, in the spin state matching the DFT column. | ||||||||||||||
| Cr | S | 1.88/1.47 | 3/2 | S | 1.89/1.45 | 3/2 | ||||||||
| Mn37,47 | S | S | 1.86/1.44 | 1.85/1.41 | 0.34/0.98 | 2 | 2 | S | S | 1.86/1.43 | 1.88/1.40 | 0.04/0.13 | 2 | 2 |
| Fe36,49 | S | 1.94/1.44 | ?c | S | S | 1.95/1.43 | 1.91/1.46 | 0.05/0.13 | ?c | 5/2 | ||||
| Co38,39,65 | S | S | 1.88/1.43 | 1.87/1.44 | 0.03/0.36 | 0 | 0 | S | S | 1.87/1.38 | 1.88d/— | N/A | ?c | 1 |
| Ni35,40 | S | S | 1.88/1.40 | 1.89/1.39 | 0.03/0.11 | ?c | 1/2 | E | E | 1.93e/1.31 | 1.98d/— | N/A | 3/2 | 1/2 |
| Cu | E | 2.42/1.30 | 1 | E | 2.06/1.29 | 1 | ||||||||
Models used
When it comes to geometry, TMC is notorious regarding possible variations in the structure. For instance, there may or may not be an axial (solvent) ligand bound to the opposite side of the molecular oxygen. Also, the methyl groups of TMC can be oriented all syn or trans to the bound oxygen, or a mix thereof. The ring structure symmetry is also subject to variations: given a particular C–C bond in the ligand, its corresponding bond on the other side could be either parallel or crossing to it. A full investigation into all these and more possible geometries in combination with all possible spin states would require some resources, with little gain in our understanding of the core metal system. Instead, we have opted to start our calculations from the known X-ray structure for NiIII–peroxo species for the 12-TMC ligand (1Ni)35 and FeIII–peroxo species for the 14-TMC ligand (2Fe).36 In these structures, the molecular oxygen is bound syn to the methyl groups without any other axial ligands. Also in 2Fe, for a given C–C bond, the counterpart at the opposite side of the ring is not parallel but crossing. Using the same ligand conformation, we only replace the metal atom. While the ligand conformation may or may not differ for other metal species (not all the species have X-ray structures solved), this gives us the advantage to be able to compare the different metal species with a consistent constant influence from the ligand.Results
Correlation to experiments
Before discussing new conclusions drawn from the calculations, it is necessary to assess the reliability of the calculations. This is done by comparing two experimentally available criteria: geometry and ground spin state.Having decided on the ligand conformation (see Methods section), the most obvious geometric feature is whether the dioxygen molecule (O2) is bound in an end-on or a side-on fashion. There are seven cases in total, where the binding mode of O2 to the metal is experimentally known: a side-on mode for 1M (M = Mn, Co, Ni) and 2M (M = Mn, Fe, Co), and an end-on mode for 2Ni (Table 1). The calculations are in good agreement with all these seven cases, regardless of the DFT functional or solvent phase used. Moreover, the calculations give the same unanimous answers to the potential structures of the remaining unknown structures. Consequently, the predictive value of the calculations for the binding geometries is high, and should reflect the preferred O2 binding mode correctly for the unknown structures as well.
To date, there are only five X-ray structures available among the investigated species. For the 1Ni species, the Ni–O and O–O bond lengths differ by less than 0.01 Å between the calculated and X-ray structures (Table 1). Overlaying only the core section (defined as the metal, O and N atoms), the root-mean square deviation (RMSD) of the atom positions is 0.03 Å, while overlaying the whole atoms results in a RMSD of 0.11 Å. Thus, the calculation seems to be able to reproduce the X-ray structure quite excellently. The RMSD is only marginally larger for 1Co species (0.36 Å), but this is mostly due to the outlying part of the ligand as the core has the same RMSD as in the Ni case (0.03 Å). Similarly, the calculations on 2Fe and 2Mn species show good agreement with experiments as well (Table 1). For the 1Mn species, the agreement is a little bit worse, with a RMSD of 0.34 Å at the core. In this case, the discrepancy indeed comes from the different C–C bond configurations of the TMC ligand; our use of the 12-TMC ligand configuration from Ni differs slightly from what is experimentally obtained in the Mn case. However, we have shown in our earlier work that a small RMSD of 0.03 Å could be obtained if we were to calculate the 12-TMC configuration corresponding to what has been obtained experimentally for the Mn case.37 As our stated goal is to compare the change of the metal but not the ligand, we keep the 12-TMC structure as calculated in this study, noting that the experimental and theoretical Mn–O and O–O distance differences are still within 0.03 Å (Table 1).
Regarding the spin states, there are six cases where the ground states are experimentally determined. Theory reproduces the experiments unambiguously only in two cases: 2Mn and 2Fe (Table 1). If the “right” functional or energy treatment is chosen, theory can match the experiments in three other cases: 1Co, 1Ni, and 2Co. For 1Co, the electronic energies of both functionals, with or without solvent, predict an S = 0 ground state, in agreement with experiments. Using Gibb's free energy, S = 0 is indeed the ground state at T = 10 K (at which the experiments were done), but not at 298 K. For the 1Ni case, BP86 prefers the S = 1/2 state, while B3LYP prefers S = 3/2 instead. For 2Co, BP86 prefers S = 0, while B3LYP prefers S = 2. There are conflicting reports about the preferred spin state of this species,38,39 but the latest NMR spectra taken at 233 K points to an S = 1 state for this species.39 Indeed, inclusion of thermal effects at 233 K (i.e. obtaining Gibb's free energy) corrects the BP86 energy to an S = 1 preference (see ESI, Table S1†). Only in one case do the current DFT methodologies not match the experimentally determined spin state at all: 2Ni. The erroneous results on Ni from DFT calculations have been observed before,40–43 and is discussed in the relevant section below.
The overall conclusion drawn from the above described comparisons of geometry and spin state between the calculated and experimental structures indicates that the accuracy can be fairly good, if some special attention is given to certain cases.
Orbitals
We begin this section by presenting the orbitals involved and the principle of orbital mixings that defines all the species in this study. We define the z-axis as along the normal of the ligand plane (in the case of end-on, this would be along the M–O bond) and the x-axis in the direction of the O–O bond, projected perpendicular to the z-axis (Fig. 2). The five atomic 3d-orbitals on the metal are usually defined as dxy, dxz, dyz, dz2 and dx2−y2. As the last orbital mixes with the ligand nitrogen px/y orbitals in a head-to-head fashion, we denote the generated antibonding molecular orbital as σ*z2−y2. On the peroxide side, each of the oxygen atoms contains three p orbitals that can mix with the corresponding p orbitals on the other oxygen atom to form the six classical O2 bonding π- and anti-bonding π*-orbitals. Of these, only the valence orbitals named π*y and π*z are of interest in our study. These π*y/z orbitals mix with the metal d orbitals in different fashion depending on the structure, and understanding this mixing enables us to correctly describe the electronic configurations of the metal–peroxo species. | ||
| Fig. 2 The five metal valence orbitals (dxy, dxz, dyz, dz2 and σ*x2–y2) combine with two O2 valence orbitals (π*y and π*z) to create a seven valence orbital system of an M–O2 complex. (a) Side-on system where the dxy orbital on the metal combines with the π*y orbital on O2 to form δ and δ* orbitals. Similarly, the dxz orbital combines with the π*z orbital on O2 to form π and π* orbitals. The actual electron occupation shown is for the S = 5/2 side-on FeI + O2 species. (b) End-on system where the dyz orbital combines with the π*y orbital to form π and π* orbitals. The dz2 orbital combines with the π*z orbital to form δ and δ* orbitals. The electron occupation shown is for the S = 1 end-on CuI + O2 species. | ||
For the side-on bound general M–O2 species, the five d orbitals of the metal are in Fig. 2a shown to overlap symmetrically with the O2 π*-orbitals. The lowest lying d orbital, dxy, mixes with π*y to form two new orbitals, a bonding δ-orbital and an anti-bonding δ*-orbital. Similarly, dxz is found to mix with π*z resulting in a low-energy bonding (π) and a high-energy anti-bonding (π*) orbitals as well. Although dyz shows some signs of mixing with the bonding πy-orbital of the oxygen, this mixing is relatively small and not significant for our discussion below; hence, we regard this as a pure dyz orbital. In the absence of an axial ligand, dz2 does not really mix with other orbitals, and we retain its designation as dz2. Thus, the seven resulting orbitals of the side-on mode species are π, δ, δ*, dyz, dz2, σ*x2–y2 and π*. Hence, while the metals are usually described to have “five d orbitals” in colloquial language, strictly speaking this may not be the case outside of the isolated metal atom. In our case, molecular interaction creates seven molecular orbitals, which have metal atomic d orbital components. However, usually (and fortunately) only the five valence orbitals are energetically high enough to matter in reactions, thereby supporting the “five d orbitals” notion.
For the end-on species (Fig. 2b), the orbital mixing occurs slightly different. As expected, the O2 π* orbitals now overlap the metal orbitals asymmetrically, with only the proximal oxygen atom part interacting with the metal. π*y now mixes heavily with dyz to form the bonding and anti-bonding pair of orbitals (π and π*). The π*z orbital on the other hand now overlaps with the dz2 orbital in a head-to-head fashion to create σ/σ* orbital. The seven generated orbitals here are thus denoted as σ, dxy, π, dxz, σ*x2–y2, π* and σ*.
Depending on the exact nature of the metal and spin state, the degree of mixing can vary and other alternative descriptions of the involved orbitals are possible. However, we found that significant deviations from the above descriptions only occur in case of high energy spin configurations, where there are some room for alternative orbital mixings. The above description was general and systematic enough for us to apply it to our systems of interest, which are the ground states of each species. Henceforth, we will use the above defined orbital terminology. Table 2 shows the energetically lowest valence orbital occupations found in this study using the above described orbital nomenclature.
| Side-on | π | δ | δ* | d yz | d z 2 | σ*x2–y2 | π* |
|---|---|---|---|---|---|---|---|
| a According to B3LYP, there is a β-electron in this orbital. b According to BP86, there is a β-electron in this orbital. | |||||||
| 1 Cr, 2Cr | ↑↓ | ↑↓ | ↑ | ↑ | ↑ | ||
| 1 Mn, 2Mn | ↑↓ | ↑↓ | ↑ | ↑ | ↑ | ↑ | |
| 1 Fe, 2Fe | ↑↓ | ↑↓ | ↑ | ↑ | ↑ | ↑ | ↑ |
| 1Co | ↑↓ | ↑↓ | ↑↓ | ↑↓ | ↑↓ | ||
| 2 Co | ↑↓ | ↑↓ | ↑(↓)a | ↑↓ | ↑(↓)b | ↑ | |
| 1Ni | ↑↓ | ↑↓ | ↑↓ | ↑↓ | ↑↓ | ↑ | |
| End-on | σ | d xy | π | d xz | σ*x2–y2 | π* | σ* |
| 2 Ni | ↑↓ | ↑↓ | ↑↓ | ↑↓ | ↑ | ↑ | ↑ |
| 1 Cu, 2Cu | ↑↓ | ↑↓ | ↑↓ | ↑↓ | ↑↓ | ↑ | ↑ |
However, an alternate view is available for the end-on 12-TMC species. In the 14-TMC case, the metal–N ligands form a fairly planar plane, which is not the case for 12-TMC. Due to the small ring size, the “plane” is heavily distorted in the 12-TMC case. In fact, it is so distorted that one of the ligand nitrogen atoms forms almost a linear N–M–O direction, which can serve as the x-axis. Together with a perpendicular M–N direction, we can define a new “plane”. Looking at the five d orbitals of the metals, they fit remarkably well to this coordinate system, where the metal orbitals simply switch designations (σ*x2–y2 → σ*z2, dxy → dyz, etc.) due to the change of the coordinate axis (x → z, y → y, z → −x). However, in the interest of comparability to the 14-TMC species, we approximate the heavily distorted 12-TMC plane as planar, and designate the orbitals with the same nomenclature as close as possible to the 14-TMC orbitals.
Individual metal–O2 species
In this section, we detail the individual results obtained for all the metal–O2 species separately. A statistical analysis over all the species will be presented afterwards based on the designated representative species in each of the subsections below. All the energy values for all the species and computational protocols are presented in the ESI, Table S1.† | ||
| Fig. 3 Energy difference between the ground state and the second lowest state, without changing end-on/side-on geometry for (a) 12-TMC and (b) 14-TMC ligand. | ||
The O2 moiety shows almost zero in Mulliken spin density distribution, a fact that curiously enough occurs in many of the other MO2 species studied here as well (Table 3). A closer look at the orbitals shows that the relevant electrons are located in the δ, δ* and π orbitals, which are partially delocalized on the O2 moiety (Fig. 2). Examining δ(β), one finds that this orbital is of significantly less mixed character than δ(α). δ(β) is centred more on O2 rather than Cr, hence it's β-electron spin density is roughly 1 on the O2 part and 0 on the Cr part. This cancels out the combined 1 of α-spin from both δ(α) and δ*(α) orbitals; i.e. the contribution of spin density on the O2 moiety from both δ and δ* orbitals is zero. The π orbital on the other hand is more evenly delocalized between Cr and O2. Therefore, it contributes partially with its α (0.5) and β (0.4) electrons to the electron count, but largely cancel each other out in terms of spin.
| Total electron count | Oxidation state assignment based on | |||||||
|---|---|---|---|---|---|---|---|---|
| Metal | O2 | Total electron count | Valence orbital analysis | |||||
| α | β | Δα–β | α | β | Δα–β | |||
| a Round-off error implicates this S = 1 species to appear to be in an S = 1/2 state. Adding spin from the ligand to Cu spin count will imply CuII. b Round-off error implicates this S = 1/2 or 3/2 species to appear to be in an S = 1 state. Adding spin from the ligand to Ni spin count will imply NiII. | ||||||||
| 1 Cr | 12.93 | 10.06 | 2.86 | 8.55 | 8.39 | 0.16 | 4CrI + 1O2 | 4[CrIII–peroxo] |
| 1 Mn | 13.64 | 9.89 | 3.75 | 8.51 | 8.44 | 0.07 | 5MnI + 1O2 | 5[MnIII–peroxo] |
| 1 Fe | 14.09 | 10.32 | 3.78 | 8.95 | 8.07 | 0.88 | 5FeII + 2O2˙− | 6[FeII–superoxo] |
| 1 Co | 12.96 | 12.96 | 0.00 | 8.50 | 8.50 | 0.00 | 1CoI + 1O2 | 1[CoIII–peroxo] |
| 1 Ni | 13.73 | 12.97 | 0.76 | 8.41 | 8.50 | –0.09 | 2NiI + 1O2 | 2[NiIII–peroxo] |
| 1 Cu | 14.09 | 13.70 | 0.39 | 8.96 | 7.59 | 1.37 | 1CuI + 2O2˙−a | 3[CuII–superoxo] |
| 2 Cr | 12.92 | 10.02 | 2.90 | 8.53 | 8.41 | 0.12 | 4CrI + 1O2 | 4[CrIII–peroxo] |
| 2 Mn | 13.65 | 9.86 | 3.79 | 8.48 | 8.46 | 0.03 | 5MnI + 1O2 | 5[MnIII–peroxo] |
| 2 Fe | 14.09 | 10.33 | 3.76 | 8.95 | 8.05 | 0.91 | 5FeII + 2O2˙− | 6[FeII–superoxo] |
| 2 Co | 13.66 | 12.06 | 1.59 | 8.48 | 8.30 | 0.18 | 3CoI + 1O2 | 3[CoIII–peroxo] |
| 2 Ni | 14.05 | 12.71 | 1.34 | 8.96 | 7.66 | 1.30 | 2NiI + 2O2˙−b | 4[NiII –superoxo] |
| 2 Cu | 14.08 | 13.71 | 0.37 | 8.96 | 7.58 | 1.38 | 1CuI + 2O2˙−a | 3[CuII–superoxo] |
To investigate the electron distribution in more detail, we have performed NBO calculations, where the default Lewis structure determined by the program was used on all the representative species in this study. NBO analysis on 1Cr and 2Cr yielded a Mulliken natural population of 13 α-electrons and 10 β-electrons on the Cr atom (Table 3). One arrives therefore to the conclusion that the Cr atom has 23 electrons and 3 in spin density, i.e. an S = 3/2 CrI species. At the same time, the O2 moiety has 8.5 α-electrons and 8.4 β-electrons. This would imply almost 17 electrons on O2 (i.e. superoxo), but as the fractional 0.5 α- and 0.4 β-electrons almost cancel each other out in spin density distribution, these decimals may be best treated here as round-off errors. This would imply 16 electrons on O2, and the resulting designation should be a singlet O2 species. Taken together, the spatial distribution of the electrons is thus more consistent with a 4CrI–1O2 designation (the multiplicity number M = 2S + 1 is used as the left superscript), which would not be compatible with the conventional oxidation state designation of these kinds of species. Given that there are some error margins to consider when dealing with Mulliken analysis, and in order to confirm the existing spectroscopic conventions, a better designation would be S = 3/2 CrIII–peroxo (see Discussion section). This species would produce the same spin density distribution and the same number of electrons in the valence orbitals as 4CrI–1O2.
Assuming the S = 5/2 ground state, Table 3 shows that the Fe structures have one radical on the O2 moiety and four on Fe, seemingly implicating a FeII–superoxo. This stems from the single occupations of the π* and σ* orbitals, which spin-polarizes the O2 moiety to its superoxo character. As mentioned above, the obtained bond lengths indicate that the calculated geometries match experiments (Table 1) and therefore, the calculation results should warrant a serious consideration. However, a FeII–superoxo structure is not (yet) supported experimentally. Based on the Raman spectroscopy and X-ray structure data, this structure has been determined to be a FeIII–peroxo species.36,49 As mentioned above, the X-ray geometries are well reproduced by the current calculations, while the Raman vibrations are reasonably close (BP86 gas-phase Fe–O/O–O stretching frequencies: 431/957 cm−1, experimental values: 487/825 cm−1).36 Noteworthy is that if S = 3/2 is assumed in 2Fe, B3LYP gives that the δ* orbital is doubly occupied (while BP86 reveals that dz2 is doubly occupied instead). The configuration in this case is more akin to FeIII–peroxo, in a similar pattern to the other MO2 structures. Hence, depending on the spin state, the O2 peroxo/superoxo character fluctuates.
For 2Co, BP86 gas- and solvent-phase calculations favour a side-on S = 0 state, while B3LYP yields an S = 2 spin state. The BP86 preference for S = 0 was found in earlier calculations as well.39 Again, the temperature effect seems to matter here as well, as the calculated BP86 free energies (at 233 K) yield an S = 1 preference. Indeed, the most recent NMR experiments at 233 K have determined that this structure is in an S = 1 state.39 Examining our computationally obtained S = 1 structure, we find that it exhibits a “half side-on/end-on” structure, where one Co–O bond is longer than the other (1.80 Å/1.94 Å, average 1.87 Å). This is somewhat in contradiction to earlier calculations, where symmetric Co–O2 distances were obtained, despite a very similar calculation setup (albeit a slightly larger basis sets was applied here).39 However, the average Co–O (1.87 Å) and O–O distances are similar to earlier theoretical results,39 and match the EXAFS Co–O bond distance of 1.88 Å.39 We therefore assign the half side-on/end-on S = 1 structure as the representative state for 2Co. The earlier calculations assigned this structure as a CoIII–peroxo structure based on the Mulliken spin density distribution analysis for the S = 1 spin state,39 which is about the same as in the current study. An interesting note however is that the electronic structure is different between the functionals: BP86 finds a β-electron in the dz2 orbital, while B3LYP finds this electron in the δ* orbital. The former electron occupation makes the structure slightly ferromagnetically coupled between Co and O2, while the latter spin distribution makes the structure slightly anti-ferromagnetically coupled (see ESI, Tables S2 vs. S3†).
For 2Ni, the present and previous40,41 DFT calculations uniformly prefer the S = 3/2 state over S = 1/2, irrespective of functional, solvent or thermal additions.53 Noteworthy is that even ab initio CASSCF calculations yielded an S = 3/2 state for this structure.42 While an end-on species is experimentally supported by EXAFS,35,40 EPR suggest that this species is in an S = 1/2 state.40 The reason for this last discrepancy to theoretical results is not known. Both spin states exhibit calculated O–O bond vibrations close to the experimentally obtained values to be of no help in identifying the ground state. Also, in three out of five calculation protocols, the end-on S = 1/2 structure is not even the second favoured structure. In fact, the B3LYP side-on S = 3/2 structure is slightly preferred over even the end-on S = 3/2 structure, let alone the S = 1/2 species (optimizing the geometries with B3LYP did not change this fact). The only support the calculations give to this being an S = 1/2 species is the calculated Ni–O bond distance. This bond was determined by EXAFS to be 1.98 Å,40 which is in fact close to the calculated S = 1/2 value (1.96 Å, Table S4 in ESI†). However, the calculated S = 3/2 value is also close (1.93 Å) and therefore also plausible. In terms of spin density distribution, there is 1.2 α-spin density on Ni and 0.5 β-spin density on O2 in the end-on S = 1/2 state. Rounding off both these values strictly to 1 introduces a round off error, as it will yield an S = 1 2NiIII–2O2˙− species. There is, however, partial significant spin on the 14-TMC ligand (0.3). If we reason that this spin also belongs to Ni as much of it comes from the singly occupied σ*x2−y2 orbital, the round offs yield a 3NiII–2O2˙− species. Thus, the spin distribution seems to imply a borderline case between anti-ferromagnetic coupled 3NiII–2O2˙− and 2NiIII–1O22−. In the S = 3/2 state, the spin density distribution is higher on both Ni (1.3) and O2 (1.3), which can also be rounded off to 3NiII–2O2˙− with addition of 0.4 spin from the ligand to Ni. Both spin states could thus fit into the description of “NiII–superoxo” as described by XAS.40
Since the spin state determination was done by EPR comparison to known metal–superoxo S = 3/2 species (and not to S = 3/2 Ni species specifically as this has not been reported before, see footnote 16 of ref. 40), it leaves some room for possible experimental errors. We chose in this publication to designate 2Ni as an end-on S = 3/2 species since the theoretical values overwhelmingly support this structure, but a final reconciliation between theory and experiment in this issue is pending.
Both 1Cu and 2Cu yield similar results from the calculations. The preferred spin state is S = 1, as was earlier found in a DFT study on 2Cu with a Cl axial ligand.62 The preferred structures obtained here are the end-on species. Hence, these are our representative structures for both 1Cu and 2Cu. The O–O bond distances are short, 1.30 Å for 1Cu and 1.29 Å for 2Cu. The spin density distribution is ambiguous 1.4 on the O2 moiety and, in conjunction with the short O–O bond length, would probably be designated as a superoxo species (i.e. CuII–superoxo). The NBO calculation reveals 0.3 spin on Cu, which can be raised to 0.6 by adding spin from the ligand to it, being closer to the expected 1 for an S = 1/2 CuII species. However, given the weak Cu–O bond strength (under 20 kcal mol−1 depending on the calculation protocol) and the long Cu–O bond lengths of 1.96 Å and 2.06 Å for 1Cu and 2Cu, respectively, a borderline case between 2CuII–2O2˙− and a weakly interacting 1CuI–3O2 species seems to be plausible in this case.
Statistical analysis of the MO2 structures
In the below section, we aim to utilize our extensive data on the individual MO2 structures to gain some insights into the trends in metal variations.As Fig. 3 shows, there is a considerable energy gap (around 20 kcal mol−1) for 1Cr and 2Cr to switch its spin state. Therefore, any multi-spin state reactivity here can be ruled out. The massive preference for the ground spin state is clear for any of the computational protocol given; indicating that the spin state preference is very reliable. For 1Mn and 2Mn, the energy gap is in the 15 kcal mol−1 range that varies depending on the methods and ligand. This makes multi-spin state reactivity not easy, but still within the theoretically possible range and cannot be ignored. For all the other metals, the gap is typically within 5 kcal mol−1 and thus spin transitions are real and possible, which must be considered in any studies of these species.
 | ||
| Fig. 4 The M–O2 binding energies for (a) 12-TMC and (b) 14-TMC ligand. | ||
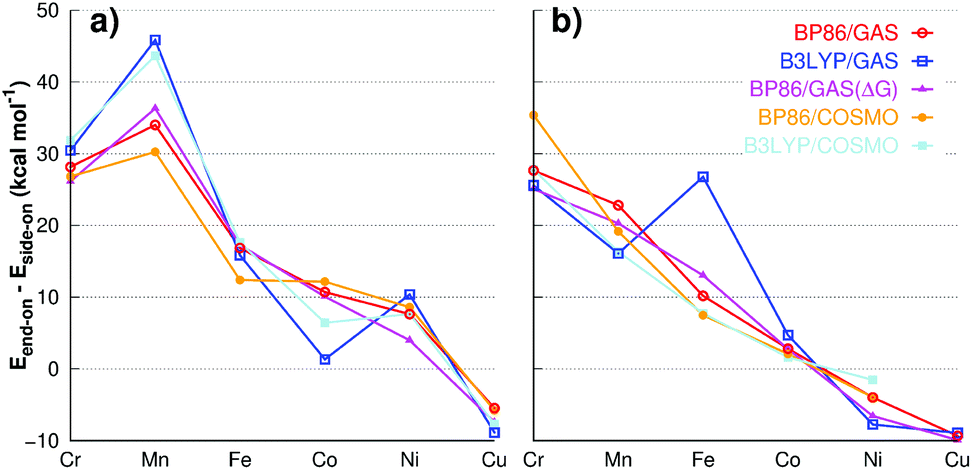 | ||
| Fig. 5 Energy differences (Eend-on − Eside-on) for various M–O2 complexes with (a) 12-TMC and (b) 14-TMC ligand. | ||
 | ||
| Fig. 6 Spin density distribution on the O2 moiety for various M–O2 complexes with (a) 12-TMC and (b) 14-TMC ligand. The spin state chosen for each of the metal structure is corresponding to the representative state for each species, as discussed in the text. | ||
Discussion
Ring size effect
In term of differences between 12-TMC and 14-TMC ligands, the general trends on spin state stability, O2 binding energies and spin density distribution are similar (Fig. 3, 4 and 6). Looking at the average M–N bond lengths, it is clear that these bonds in 2M are longer than those in 1M, due to the larger ring size (Fig. 7a). As a result, this probably causes the σ*x2−y2 orbital energy to be lower and closer to the dz2 orbital in 2M compared to the corresponding case in 1M. This may be the cause to why 1Co prefers to avoid occupying the σ*x2−y2 orbital by switching to an S = 0 state (Table 2), while the more accessible σ*x2−y2 orbital in 2Co is occupied to form an S = 1 ground state. | ||
| Fig. 7 (a) The average M–N distance, showing that the 14-TMC ligand affords larger M–N distances and (b) the smallest N–M–N angle, showing that 14-TMC fluctuates more and therefore is more flexible. | ||
The flexibility of the ligand binding can also be illustrated with the (smallest) N–M–N angle, where the two N atoms oppose each other, and therefore indicate the planarity of the M–(4 × N) plane. In 2M, this angle deviates from 125° to 149° within the representative species (a span of 24°, Fig. 7b). In 1M however, this angle is markedly more constrained, ranging from 105° to 114° (a span of 9°). The larger ring size thus generates more flexibility. The smaller N–M–N angle also means that the O2 binding site is wider and more accessible to O2. This can be seen from the O2 binding energies, where the binding is few kcal mol−1 stronger for 1M than 2M (Fig. 4). The calculations thus corroborate experimental results,35,65 that explain the preference of a side-on binding mode for 1Nivs. an end-on binding mode for 2Ni as a result of the spatial openness at the O2 binding site.
Metal oxidation states
The availability of the exact electronic structure enables us to investigate the question of whether a certain structure is in fact an MI, MII or MIII state, coupled to O2, superoxo or peroxo moiety, respectively. This is, however, a challenging task from a computational point of view. One might, naturally, assume that the electron count would have something to do with it. A useful case was discussed in some length above for 1Cr and 2Cr, vide supra. Thus, the electron count by NBO generally supports a MMI–1O2 designation in our current study (Table 3). However, as MI–O2 species are clearly not acceptable in light of current conventions, most computational chemists look at the number of electrons in the valence orbitals only, combined with the spin density distribution. In the current study, most of the species were found to have no spin on the O2 part and with the number of electrons in the five highest valence orbitals matching the isolated MIII, consistent with a MIII–peroxo designation. Hence, this is the oxidation state we generally present above (with the exception of Fe and Cu based structures, vide supra). Noteworthy is that the excess radical distribution would be the same for MMIII–peroxo as for MMI–1O. There is also a good agreement with the geometries and spin states obtained in comparison to experiments; implying that the calculations and experiments in fact agree but that the oxidation states interpretations may need some further reconciliations between theoreticians and experimentalists.Comparing with experiments, our calculated oxidation states differ from the experimentally determined ones in the case of Fe. We determine these species to be an FeII–superoxo species because of the high-spin S = 5/2 configuration that is only attainable for Fe, yielding a total spin of just below 1 on the O2 moiety (Table 3). The experiments indicate an FeIII–peroxo species instead based on both the O–O distance and resonance Raman frequencies, which are both reproduced by our calculations. For Cu based structures, no experimental structure exists, but if attainable, we predict it to be a borderline case to a CuI–O2 species.
Computational protocol dependence
In this study, we have deliberately made use of BP86 geometries only as this functional is generally known to compete with (and even excel over) B3LYP when it comes to the quality of geometries, while being faster to compute.66 We have shown here that the obtained BP86 geometries are indeed in agreement with experiments, where available (Table 1). B3LYP is generally thought of as being more accurate in energy evaluations, while BP86 seemed to be a better fit for Ni species specifically.43 This prompted us to consider both energies in our evaluations. We consider both the SCF gas-phase energy and the Gibb's free energy, and we also incorporate acetonitrile solvent effects on both geometries and energies for comparison. There are individual cases where the choice of computational protocol matter (vide supra), but if looking at trends over multiple species (as in Fig. 3–6), the conclusions will most likely be the same. Hence, we do not see any excessive method dependence in our calculations, but there are enough deviations to imply that methodology checks are always warranted in any individual calculation.Conclusions
We have used five different computational protocols (BP86//BP86/GAS ΔE, B3LYP//BP86/GAS ΔE, BP86//BP86/GAS ΔG, BP86//BP86/COSMO ΔE and B3LYP//BP86/COSMO ΔE) to evaluate the characteristics of six MI + O2 compounds (M = Cr, Mn, Fe, Co, Ni and Cu) with two different closely related ligands, 12-TMC and 14-TMC. The geometries, valence orbital electron occupations and oxidation states are either predicted or confirmed (where experiments are available). A statistical comparison over all the metals reveals that as the metal atomic number goes up, (1) O2 binding energy decreases, (2) spin state preference becomes less clear, (3) end-on structure is gradually being stabilized, and becomes preferred at Ni or Cu. In addition, the spin density distributions on the O2 group are seen to be more dependent on the individual spin state rather than the metal atomic number, and experimental observations about 14-TMC having a more flexible ring wrapping than 12-TMC have also been confirmed. Determining the most likely structure and spin state, we find that 1Cr and 2Cr are side-on S = 3/2 CrIII–peroxo species, 1Mn and 2Mn are side-on S = 2 MnIII–peroxo species, 1Fe and 2Fe are side-on S = 5/2 FeII–superoxo species, 1Co is a side-on S = 0 CoIII–peroxo species, 2Co is a half side-on/end-on S = 1 CoIII–peroxo species, 1Ni is a side-on S = 1/2 NiIII–peroxo species, 2Ni is an end-on S = 3/2 NiII–superoxo species, and finally 1Cu and 2Cu are end-on S = 1 CuII–superoxo species.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was gratefully supported by the NRF of Korea through CRI (NRF-2012R1A3A2048842 to W. N.), GRL (NRF-2010-00353 to W. N.), and MSIP (NRF-2013R1A1A2062737 to K.-B. C.). Professors Joan S. Valentine and Jaeheung Cho are acknowledged for ideas and consultations.Notes and references
- Active Oxygen in Biochemistry, ed. J. S. Valentine, C. S. Foote, A. Greenberg and J. F. Liebman, Springer, Boston, MA, 1995 Search PubMed.
- M. Unno, H. Chen, S. Kusama, S. Shaik and M. Ikeda-Saito, J. Am. Chem. Soc., 2007, 129, 13394 CrossRef CAS PubMed.
- N. Kitajima, H. Komatsuzaki, S. Hikichi, M. Osawa and Y. Moro-oka, J. Am. Chem. Soc., 1994, 116, 11596 CrossRef CAS.
- D. P. de Sousa, J. O. Bigelow, J. Sundberg, L. Que, Jr. and C. J. McKenzie, Chem. Commun., 2015, 51, 2802 RSC.
- C. J. Cramer and W. B. Tolman, Acc. Chem. Res., 2007, 40, 601 CrossRef CAS.
- J. Rosenthal and D. G. Nocera, Acc. Chem. Res., 2007, 40, 543 CrossRef CAS PubMed.
- E. E. Chufán, S. C. Puiu and K. D. Karlin, Acc. Chem. Res., 2007, 40, 563 CrossRef PubMed.
- Y.-M. Lee, S. Hong, Y. Morimoto, W. Shin, S. Fukuzumi and W. Nam, J. Am. Chem. Soc., 2010, 132, 10668 CrossRef CAS PubMed.
- S. Shaik, D. Kumar, S. P. de Visser, A. Altun and W. Thiel, Chem. Rev., 2005, 105, 2279 CrossRef CAS PubMed.
- Metal-Oxo and Metal-Peroxo Species in Catalytic Oxidations, ed. B. Meunier, Springer, Berlin Heidelberg, 2000 Search PubMed.
- A. D. Vaz, S. J. Pernecky, G. M. Raner and M. J. Coon, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 4644 CrossRef CAS.
- W. Nam, Acc. Chem. Res., 2007, 40, 465 CrossRef CAS.
- A. J. Jasniewski and L. Que, Jr., Chem. Rev., 2018, 118, 2554 CrossRef CAS PubMed.
- M. Guo, T. Corona, K. Ray and W. Nam, ACS Cent. Sci., 2019, 5, 13 CrossRef CAS.
- W. Nam, Y.-M. Lee and S. Fukuzumi, Acc. Chem. Res., 2018, 51, 2014 CrossRef CAS PubMed.
- K. Ray, F. F. Pfaff, B. Wang and W. Nam, J. Am. Chem. Soc., 2014, 136, 13942 CrossRef CAS PubMed.
- J. Cho, R. Sarangi and W. Nam, Acc. Chem. Res., 2012, 45, 1321 CrossRef CAS PubMed.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133 CrossRef.
- F. Neese, ORCA – an ab initio, Density Functional and Semiempirical program package, Version 3.01, University of Bonn, 2010 Search PubMed.
- The B3LYP/G functional as first implemented in the Gaussian packages was used here.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B, 1986, 33, 8822 CrossRef PubMed.
- J. P. Perdew, Phys. Rev. B, 1986, 34, 7406 CrossRef.
- The ORCA basis set ‘CoreProp’ was used. This basis is based on the TurboMole DZ basis developed by Ahlrichs and coworkers and obtained from the basis set library under ftp://ftp.chemie.uni-karlsruhe.de/pub/basen (now defunct).
- The Ahlrichs (2d2fg,3p2df) polarization functions were obtained from the TurboMole basis set library under ftp://ftp.chemie.uni-karlsruhe.de/pub/basen (now defunct). Sc-Zn: 2p functions from Wachters JCP, 52 (1970), 1033 plus one f-function from the TurboMole library.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- S. Sinnecker, A. Rajendran, A. Klamt, M. Diedenhofen and F. Neese, J. Phys. Chem. A, 2006, 110, 2235 CrossRef CAS PubMed.
- J. Ho, A. Klamt and M. L. Coote, J. Phys. Chem. A, 2010, 114, 13442 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829 CrossRef.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis and F. Weinhold, NBO 6.0, Theoretical Chemistry Institute, University of Wisconsin-Madison, 2013 Search PubMed.
- J. Cho, R. Sarangi, J. Annaraj, S. Y. Kim, M. Kubo, T. Ogura, E. I. Solomon and W. Nam, Nat. Chem., 2009, 1, 568 CrossRef CAS PubMed.
- J. Cho, S. Jeon, S. A. Wilson, L. V. Liu, E. A. Kang, J. J. Braymer, M. H. Lim, B. Hedman, K. O. Hodgson, J. S. Valentine, E. I. Solomon and W. Nam, Nature, 2011, 478, 502 CrossRef CAS PubMed.
- H. Kang, J. Cho, K.-B. Cho, T. Nomura, T. Ogura and W. Nam, Chem. – Eur. J., 2013, 19, 14119 CrossRef CAS PubMed.
- Y. Jo, J. Annaraj, M. S. Seo, Y.-M. Lee, S. Y. Kim, J. Cho and W. Nam, J. Inorg. Biochem., 2008, 102, 2155 CrossRef CAS PubMed.
- R. Sarangi, J. Cho, W. Nam and E. I. Solomon, Inorg. Chem., 2011, 50, 614 CrossRef CAS PubMed.
- M. T. Kieber-Emmons, J. Annaraj, M. S. Seo, K. M. Van Heuvelen, T. Tosha, T. Kitagawa, T. C. Brunold, W. Nam and C. G. Riordan, J. Am. Chem. Soc., 2006, 128, 14230 CrossRef CAS PubMed.
- R. Latifi, L. Tahsini, D. Kumar, G. N. Sastry, W. Nam and S. P. de Visser, Chem. Commun., 2011, 47, 10674 RSC.
- J. Zapata-Rivera, R. Caballol and C. J. Calzado, J. Comput. Chem., 2012, 33, 1407 CrossRef CAS PubMed.
- K.-B. Cho, J. Cho, S. Shaik and W. Nam, J. Phys. Chem. Lett., 2014, 5, 2437 CrossRef CAS PubMed.
- J. Cho, J. Woo and W. Nam, J. Am. Chem. Soc., 2010, 132, 5958 CrossRef CAS PubMed.
- J. Cho, J. Woo and W. Nam, J. Am. Chem. Soc., 2012, 134, 11112 CrossRef CAS PubMed.
- A. Yokoyama, J. E. Han, J. Cho, M. Kubo, T. Ogura, M. A. Siegler, K. D. Karlin and W. Nam, J. Am. Chem. Soc., 2012, 134, 15269 CrossRef CAS PubMed.
- M. S. Seo, J. Y. Kim, J. Annaraj, Y. Kim, Y.-M. Lee, S.-J. Kim, J. Kim and W. Nam, Angew. Chem., Int. Ed., 2007, 46, 377 CrossRef CAS PubMed.
- J. Annaraj, J. Cho, Y.-M. Lee, S. Y. Kim, R. Latifi, S. P. de Visser and W. Nam, Angew. Chem., Int. Ed., 2009, 48, 4150 CrossRef CAS PubMed.
- J. Annaraj, Y. Suh, M. S. Seo, S. O. Kim and W. Nam, Chem. Commun., 2005, 4529 RSC.
- L. M. Blomberg, M. R. A. Blomberg and P. E. M. Siegbahn, J. Inorg. Biochem., 2005, 99, 949 CrossRef CAS PubMed.
- S. Shaik, S. Cohen, Y. Wang, H. Chen, D. Kumar and W. Thiel, Chem. Rev., 2010, 110, 949 CrossRef CAS PubMed.
- K.-B. Cho, H. Chen, D. Janardanan, S. P. de Visser, S. Shaik and W. Nam, Chem. Commun., 2012, 48, 2189 RSC.
- The side-on S = 3/2 species is predicted to be a TS by BP86 frequency calculation, yielding an imaginary frequency of i143.84 cm−1 and higher in energy than the S = 1/2 species. However, both single-point and optimized B3LYP calculations actually predict this structure to be the energetically lowest structure.
- S. Itoh, Curr. Opin. Chem. Biol., 2006, 10, 115 CrossRef CAS PubMed.
- K. Fujisawa, M. Tanaka, Y. Moro-oka and N. Kitajima, J. Am. Chem. Soc., 1994, 116, 12079 CrossRef CAS.
- P. J. Donoghue, A. K. Gupta, D. W. Boyce, C. J. Cramer and W. B. Tolman, J. Am. Chem. Soc., 2010, 132, 15869 CrossRef CAS PubMed.
- J. Y. Lee, R. L. Peterson, K. Ohkubo, I. Garcia-Bosch, R. A. Himes, J. Woertink, C. D. Moore, E. I. Solomon, S. Fukuzumi and K. D. Karlin, J. Am. Chem. Soc., 2014, 136, 9925 CrossRef CAS PubMed.
- S. Kim, J. Y. Lee, R. E. Cowley, J. W. Ginsbach, M. A. Siegler, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2015, 137, 2796 CrossRef CAS PubMed.
- N. W. Aboelella, E. A. Lewis, A. M. Reynolds, W. W. Brennessel, C. J. Cramer and W. B. Tolman, J. Am. Chem. Soc., 2002, 124, 10660 CrossRef CAS PubMed.
- A. M. Reynolds, B. F. Gherman, C. J. Cramer and W. B. Tolman, Inorg. Chem., 2005, 44, 6989 CrossRef CAS PubMed.
- B. F. Gherman and C. J. Cramer, Inorg. Chem., 2004, 43, 7281 CrossRef CAS PubMed.
- A. Ansari, P. Jayapal and G. Rajaraman, Angew. Chem., Int. Ed., 2015, 54, 564 CAS.
- M. Swart and M. Costas, Spin States in Biochemistry and Inorganic Chemistry, John Wiley & Sons, Ltd, Oxford, UK, 1st edn, 2015 Search PubMed.
- W.-Y. Tcho, B. Wang, Y.-M. Lee, K.-B. Cho, J. Shearer and W. Nam, Dalton Trans., 2016, 45, 14511 RSC.
- J. Cho, R. Sarangi, H. Y. Kang, J. Y. Lee, M. Kubo, T. Ogura, E. I. Solomon and W. Nam, J. Am. Chem. Soc., 2010, 132, 16977 CrossRef CAS PubMed.
- M. Bühl and H. Kabrede, J. Chem. Theory Comput., 2006, 2, 1282 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Energies, NBO results, Mulliken spin density distributions, geometries and coordinates. See DOI: 10.1039/c9qi00407f |
| This journal is © the Partner Organisations 2019 |