
Photodetection of DNA mismatches by dissymmetric Ru(II) acridine based complexes†
Martin
Gillard
 a,
Baptiste
Laramée-Milette
b,
Quentin
Deraedt
a,
Garry S.
Hanan
b,
Fredérique
Loiseau
c,
Jérôme
Dejeu
c,
Eric
Defrancq
*c,
Benjamin
Elias
*a and
Lionel
Marcélis
*a
a,
Baptiste
Laramée-Milette
b,
Quentin
Deraedt
a,
Garry S.
Hanan
b,
Fredérique
Loiseau
c,
Jérôme
Dejeu
c,
Eric
Defrancq
*c,
Benjamin
Elias
*a and
Lionel
Marcélis
*a
aInstitute of Condensed Matter and Nanosciences (IMCN), Molecular Chemistry, Materials and Catalysis (MOST), Université catholique de Louvain (UCLouvain), Place Louis Pasteur 1, bte L4.01.02, B-1348 Louvain-la-Neuve, Belgium. E-mail: benjamin.elias@uclouvain.be; lionel.marcelis@uclouvain.be; Fax: +3210474168; Tel: +3210473014
bDepartement de Chimie, Université de Montréal, 2900 Boulevard Edouard-Montpetit, Montréal, Québec H3T 1J4, Canada
cDépartement de Chimie Moléculaire, Université Grenoble-Alpes (UGA), UMR CNRS 5250, CS 40700, 38058 Grenoble, France. E-mail: eric.defrancq@univ-grenoble-alpes.fr
First published on 22nd May 2019
Abstract
The early detection of DNA mutations such as DNA mismatches is of major interest. Indeed, the accumulation of mismatches into the genome arises from deficiencies of the cellular mismatch repair machinery that is often associated with several types of cancers being resistant to classic chemotherapeutics. In this context, ruthenium(II) compounds bearing a planar extended ligand appear to be excellent candidates as DNA photoprobes since they exhibit high affinity for DNA as well as tuneable luminescence properties. Herein, we report on the synthesis of a novel dissymmetric acridine based Ru(II) complex, [Ru(bpy)2napp]2+, along with the study of its ability to photodetect DNA mismatches. We also investigated the origin of the ability of the complex to photodetect mismatches via CD-melting assays and bio-layer interferometry. Interestingly, this behaviour may be attributed to a better protection of the excited state of the complex from non-radiative deexcitation sources (e.g., collisions with the solvent, oxygen photosensitization, etc.) when intercalated into well-matched compared to mismatched DNA.
Introduction
Cancer is a major health concern as it accounted for 18.1 million new cases and 9.6 million deaths worldwide in 2018.1 Even though considerable progress has been made during the last few decades, the current cancer treatments and diagnosis show limitations such as a poor differentiation between cancer and healthy cells, leading to undesired side effects.2 This has stimulated the design of drugs able to specifically target a biological signature of cancer.3,4 In this context, the early detection of mismatch repair (MMR) deficient cancers is of high interest as these cancer cells exhibit a well differentiated genetic material due to the accumulation of DNA mismatches.5,6 Being highly prevalent in many cancer types that are cast under the generic name of Lynch syndrome,7–9 the development of new drugs to diagnose and to treat these cancers is of major issue as they often exhibit resistance to conventional treatments.9,10In the chemical space, metallotherapeutics and more precisely ruthenium(II) compounds appear to be attractive candidates for theranostic applications with some complexes attaining advanced stages of clinical trials.11–18 In particular, Ru(II) complexes bearing a planar extended ligand showed a strong affinity for DNA19 and proved to be very sensitive to their microenvironment once intercalated into the double helix. The reference of the field is the [Ru(bpy)2dppz]2+ (bpy = 2,2′-bipyridine; dppz = dipyrido [3,2-a:2′,3′-c]phenazine) complex that has been widely studied as a DNA “light switch”. Indeed, under aqueous conditions, the luminescence of the complex is totally quenched, but recovers in the presence of DNA.20 This behaviour was explained by a better protection of the complex from non-radiative deexcitation sources (mainly the solvent and oxygen) once intercalated into the double helix.19–21
Aiming to take advantage of the extreme sensitivity of [Ru(bpy)2dppz]2+ to its microenvironment for detecting DNA mismatches, some analogues have been recently designed and they showed different luminescence properties in the presence of mismatches relative to totally well-matched DNA.22–24 As a general rule, it has been shown that structural modifications of the complexes by the use of either (i) a dissymmetrical planar extended ligand24–26 or (ii) bulkier ancillary ligands27 lead to an enhancement of the mismatch recognition ability. This has been associated with a higher binding affinity towards the mismatch sites resulting from their weaker thermodynamic stability.28,29
In this context, our group has been investigating the potential of dissymmetric acridine based complexes to differentiate DNA mismatches from well-matched DNA. The Ru(II) complexes [Ru(phen)2dpac]2+1 (dpac = dipyrido[3,2-a:2′,3′-c]acridine) and [Ru(bpy)2npp]2+2 (npp = naphtho[1,2-b]pyrido[3,2-f][1,7]phenanthroline) were reported to differentiate mismatches based on their luminescence properties.26,30 In this paper, we describe the straightforward synthesis of a new dissymmetric complex [Ru(bpy)2napp]2+3 (napp = naphtho[2,1-b]pyrido[3,2-f][1,7]phenanthroline), along with the study of its photophysics in the absence and in the presence of well- and mismatched DNA (Fig. 1).
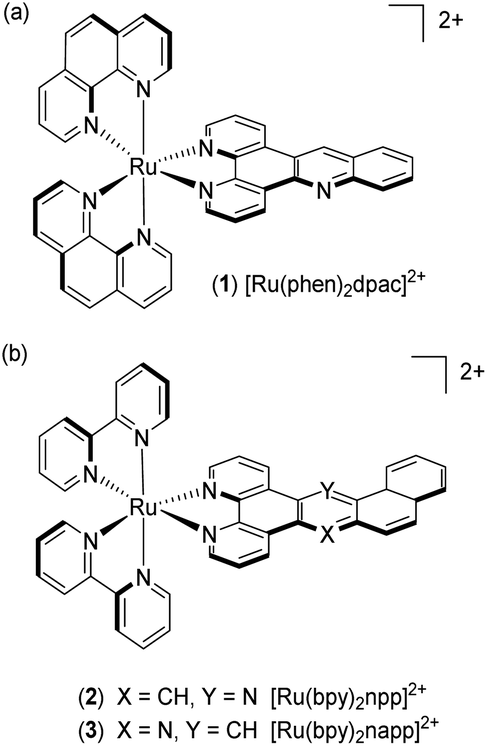 | ||
| Fig. 1 Structure of complexes [Ru(phen)2dpac]2+1, [Ru(bpy)2npp]2+2 and [Ru(bpy)2napp]2+3. | ||
Results
Synthesis procedures
The synthesis strategy of the napp ligand follows the general scheme reported by our group.30 The key step consists in the formation of the acridine core via a Tröger's base modified reaction involving 5-amino-phenanthroline and amino-alcohol 7. The preparation of amino-alcohol 7 was achieved in four steps via the isatin pathway starting from commercially available β-bromonaphthalene (Scheme 1). Copper(I) catalysed the coupling of β-bromonaphthalene with aqueous ammonia yielding β-aminonaphthalene 4 (82% yield).31 The isatin derivative 5 was regioselectively formed in 78% yield by the addition of di-ethyl-mesoxalate in acetic acid and subsequent air oxidation under basic conditions.32 Next, anthranilic ester 6 was obtained in 36% yield after treatment with hydrogen peroxide in the presence of sodium methoxide followed by a decarboxylation step under acidic conditions. Lastly, reduction by LiAlH4 furnished amino-alcohol 7 (90% yield). The condensation of 7 with 5-amino-phenanthroline afforded the napp ligand in 71% yield. The structure of napp was confirmed by NMR spectroscopy, and HRMS analysis (see the ESI†).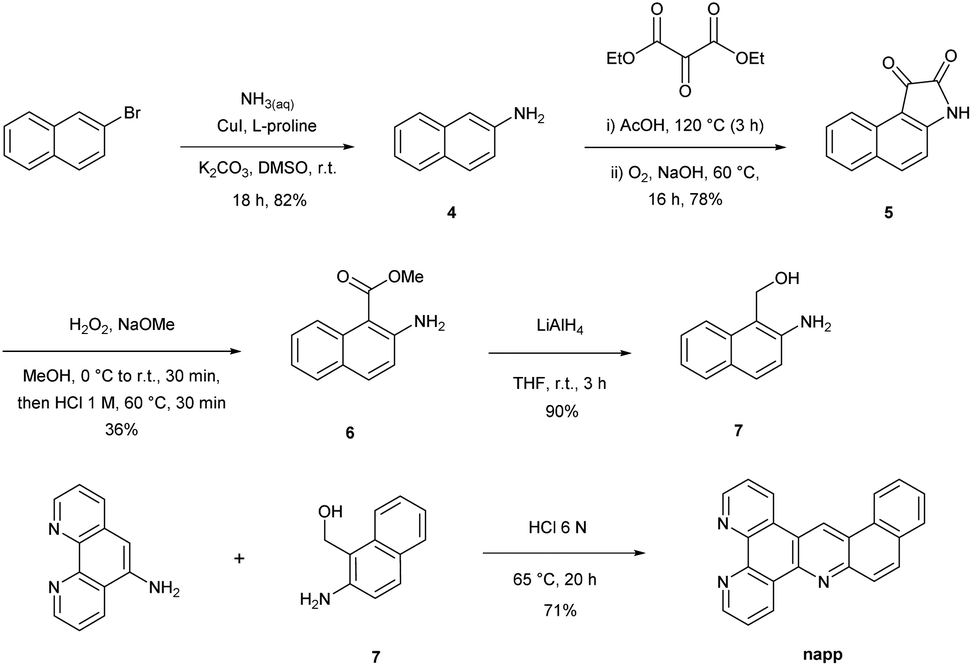 | ||
| Scheme 1 Synthesis of the napp ligand. | ||
[Ru(bpy)2napp]2+ complex 3 was synthesized by the direct chelation of the napp ligand onto a [Ru(bpy)2Cl2] precursor. Complex 3 was isolated as an orange solid and characterized by 1H-NMR spectroscopy, HRMS (see the ESI†) and X-ray crystallography (Fig. 2). 1H-NMR spectroscopy shows unambiguously the absence of symmetry, which induces the non-equivalence of (i) the protons of napp and (ii) of the bipyridine moieties. X-ray diffraction analysis was fully consistent with NMR spectroscopy data and confirmed the structure of complex 3 in which the ruthenium(II) ion resides within an octahedral geometry. It also showed the enhanced stabilization of the packing induced by parallel displaced π–π stacking interactions between two complexes.
 | ||
| Fig. 2 X-ray crystal structure of 3. Displacement ellipsoids of non-hydrogen atoms are drawn at the 50% probability level. Hydrogen atoms were omitted for clarity. | ||
Electrochemistry
The electrochemical data were obtained by cyclic voltammetry in dry deoxygenated CH3CN or N,N-dimethylformamide (Table 1 and ESI†). Not surprisingly, the oxidation potential of 3 is close to that of the Ru2+/Ru3+ oxidation of [Ru(bpy)3]2+ (1.34 V vs. Ag/AgCl) as already observed for complexes 1 and 2.26 Three reversible reduction waves were detected within the investigated potential window with the less energetic reduction occurring at ca. −1.24 V vs. Ag/AgCl, which is ascribed to the addition of an electron onto the planar extended ligand. This hypothesis is supported by the fact that phenazine containing complexes, e.g. [Ru(bpy)2dppz]2+ (termed Ru-DPPZ), display an anodically shifted reduction (≈−1.0 V vs. Ag/AgCl) compared with acridine complexes probably due to the presence of the additional nitrogen atom which further stabilizes the added electron. Altogether, the electrochemical data demonstrated that the HOMO of complexes 1–3 is likely localized on the metal centre while the LUMO is localized on the planar extended ligand.| Complex | E 1/2 ox [V vs. Ag/AgCl] | E 1/2 red [V vs. Ag/AgCl] | ||
|---|---|---|---|---|
| a Measured in dry acetonitrile. b Measured in dry N,N-dimethylformamide. The electrochemical data for complexes [Ru(bpy)3]2+, [Ru(bpy)2dppz]2+, [Ru(phen)2dpac]2+ and [Ru(bpy)2npp]2+ are from references.21,26,30 | ||||
| [Ru(bpy)3]2+ | 1.34 | −1.28 | −1.47 | −1.71 |
| [Ru(bpy)2dppz]2+ | 1.29 | −0.97 | −1.39 | −1.62 |
| [Ru(phen)2dpac]2+1 | 1.35 | −1.22 | −1.35 | −1.66 |
| [Ru(bpy)2npp]2+2 | 1.39 | −1.23 | −1.38 | −1.60 |
| [Ru(bpy)2napp]2+3 | 1.35 | −1.24 | −1.38 | −1.65 |
Light absorption data
Light absorption data were recorded at ambient temperature in water and in acetonitrile under an air atmosphere (Table 2). The absorption spectra show typical shapes for Ru(II) complexes bearing polypyridyl ligands as described for [Ru(bpy)3]2+ (Fig. 3).21 Indeed, the comparison of the data obtained for 1–3 with the literature allows us to assign the strong absorption bands (ε ≈ 105 M−1cm−1) in the UV region to ligand-centred (LC) transitions and the broad absorption bands at ca. 450 nm (ε ≈ 104 M−1cm−1) to metal-to-ligand charge-transfer (MLCT). In the case of Ru-DPPZ, the absorption of a photon leads to the formation of a charge-separated state in which an electron is transferred from ruthenium(II) towards the phen or phenazine part of the dppz ligand.33 In the case of complex 3, a small bathochromic shift (Δλ = 8 nm) of the MLCT transition at 456 nm compared to 1 is noticeable as already reported for 2. This is ascribed to the increased conjugation provided by the additional phenyl of the “elbow-shaped” ligands.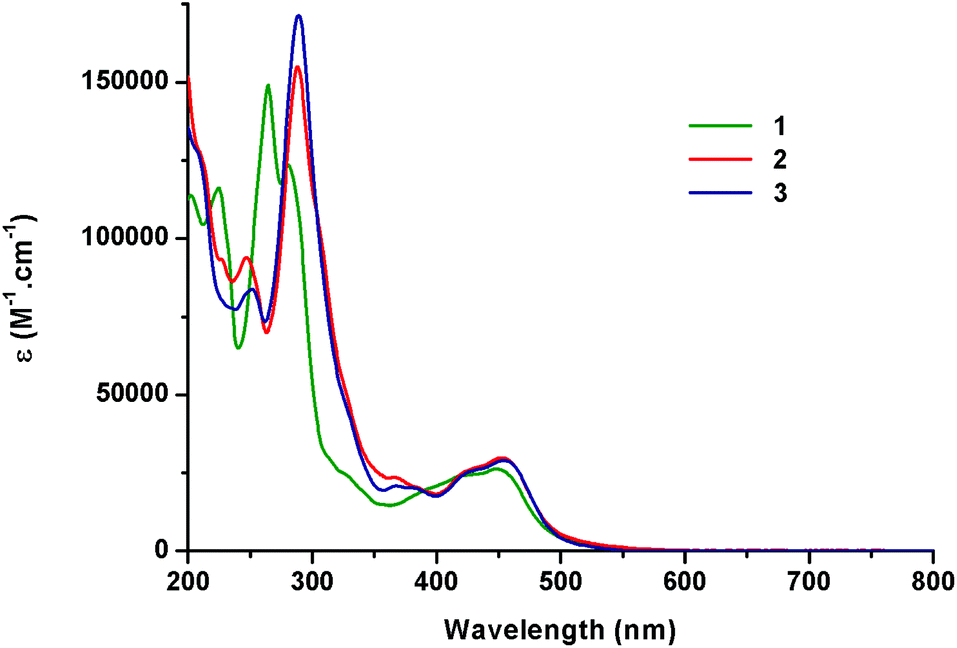 | ||
| Fig. 3 Absorption spectra of complexes 1–3 in acetonitrile under air. | ||
| Complex | Absorbance λmax [nm] (ε [104 M−1cm−1])a | |
|---|---|---|
| CH3CN | H2O | |
| a Measurements were performed with 1.10−5 mol L−1 solutions of the complex at room temperature. Extinction coefficients are reported in brackets. Absorption bands in the visible region (ε ≈ 104 M−1 cm−1 around λ ≈ 400–450 nm) are attributed to Metal-to-Ligand Charge-Transfer (MLCT) transitions. The absorption data for complexes [Ru(bpy)3]2+, [Ru(bpy)2dppz]2+, [Ru(phen)2dpac]2+ and [Ru(bpy)2npp]2+ are from references.21,26,30 | ||
| [Ru(bpy)3]2+ | 250 (2.51), 285 (8.71), 323 (sh), 345 (sh), 452 (1.45) | 250, 286, 322, 345, 451 |
| [Ru(bpy)2dppz]2+ | 255 (4.18), 284 (9.36), 352 (sh), 357 (1.56), 366 (1.55), 448 (1.57) | 255, 283, 352, 357, 365, 448 |
| [Ru(phen)2dpac]2+1 | 224 (15.1), 264 (16.3), 280, (11.6), 448 (2.62) | 224, 264 (16.2), 280, 448 (2.62) |
| [Ru(bpy)2npp]2+2 | 210 (11.1), 248 (9.21) 287 (15.6), 457 (2.94) | 210, 248, 287 (15.6), 457 (2.93) |
| [Ru(bpy)2napp]2+3 | 246 (3.31), 289 (6.82), 456 (1.24) | 210, 243, 286 (6.83), 456 (1.24) |
Computational studies
To better understand the photophysics of complex 3 along with the influence of the formation of the hydrogen bond with the non-chelating nitrogen of napp at the excited state, the ground- and excited-state electronic structures of the complex and its protonated form in water were investigated by means of DFT/time-dependent (TD-DFT) calculations. In accordance with electrochemical data, the calculations of the electronic structures of complex 3 and its protonated form in water locate the HOMO on the metal centre (see the ESI†). Concerning the LUMO of 3 in water, the calculations show a distribution between the planar extended ligand (66% contribution) and the ancillary ligands (34% contribution). Conversely, the LUMO of the protonated form of complex 3 appears to be fully localized on the protonated napp ligand. The experimental absorption spectra match well with the calculated transitions, which allows us to attribute the different transitions by using natural transition orbitals (NTOs).34 The less energetic transitions that occur between the HOMO and the LUMO of the complex seem to be 1MLCT-type transitions that stand between the metal centred HOMO and the napp- (major) and bpy-centred (minor) LUMO, LUMO+1 and LUMO+3. Concerning the protonated complex, the less energetic transition appears to be a HOMO–LUMO transition occurring between the metal-centred HOMO and the fully napp centred LUMO. Regarding the calculations obtained for 3 and its protonated form in water, it is likely that the formation of the hydrogen bond with the non-chelating nitrogen of napp has a strong influence on the photophysics of the complex. Indeed, the excited electron is mainly located on the planar extended ligand, which can be protonated. Therefore, in polar protic solvents such as water, the microenvironment of the napp ligand should play a major role in the photophysics of the complex (vide infra).Light emission data
Emission, quantum yield and lifetime measurements of 3 recorded in water and in acetonitrile suggest similar photophysical properties compared to 1 and 2 (Table 3). Indeed, the three complexes exhibit typical characteristics of 3MLCT-type transitions as they possess (i) a broad unstructured emission subject to the strong solvatochromic effect and (ii) a relatively long (≈microsecond) lifetime of the excited state. This 3MLCT-type transition is promoted by the substantial spin orbit coupling of Ru(II). In addition, the charge-separated state character is confirmed by (i) less energetic emissions in water than in acetonitrile resulting from a better stabilization of the dipolar excited state in a more polar solvent (ii) the hypsochromic shift of the emission band at 77 K (λmax = 567–577 nm) and (iii) the large kr value (>104 s−1) (Table 3). A decrease in the luminescence intensity in air-equilibrated solvents compared to nitrogen-purged solutions is observed for all the complexes which indicates the photosensitization of 3O2 by the excited complexes (data not shown).| Complex | Emission λmaxa,b[nm] | Emission λmaxb at 77 K [nm] | Φ em , | τ em [ns]c | k r [103 s−1] | ||||
|---|---|---|---|---|---|---|---|---|---|
| CH3CN | H2O | EtOH/MeOH 4/1 | CH3CN | H2O | CH3CN | H2O | CH3CN | H2O | |
| a Measurements were made with solutions 1 × 10−5 mol L−1 in complex under air. b λ exc = 450 nm. c Measurements were made with 1 × 10−5 mol L−1 solutions of the complex under argon. d Measurements relative to [Ru(bpy)3]2+ in the nitrogen purged aqueous solution (Φem = 0.063) and in nitrogen purged acetonitrile (Φem = 0.094).35 e No luminescence was observed in H2O. The photophysical data for complexes [Ru(bpy)3]2+, [Ru(phen)3]2+, [Ru(bpy)2dppz]2+, [Ru(phen)2dpac]2+ and [Ru(bpy)2npp]2+ are from references.20,23,36–38 | |||||||||
| [Ru(bpy)3]2+ | 604 | 604 | 578 | 0.062 | 0.042 | 855 | 630 | 77 | 69 |
| [Ru(phen)3]2+ | 604 | 606 | 564 | 0.028 | 0.072 | 460 | 920 | 61 | 75 |
| [Ru(bpy)2dppz]2+ | 610 | —e | 581 | 0.031 | —e | 692 | —e | 45 | —e |
| [Ru(phen)2dpac]2+1 | 597 | 604 | 567 | 0.038 | 0.066 | 710 | 999 | 54 | 66 |
| [Ru(bpy)2npp]2+2 | 604 | 606 | 577 | 0.083 | 0.092 | 702 | 750 | 118 | 123 |
| [Ru(bpy)2napp]2+3 | 601 | 606 | 576 | 0.072 | 0.12 | 812 | 841 | 89 | 143 |
The chemical modification of the acridine core via the addition of an aromatic ring results in a bathochromic shift of the emission maxima both in CH3CN and H2O. This means that the presence of the additional phenyl ring in complexes 2 and 3 leads to a better stabilization of the excited state compared to complex 1. Interestingly, we also observed longer excited state lifetimes and increased kr values of our complexes in water than in acetonitrile. It was also observed that the luminescence lifetimes of complexes 1–3 are inversely related to the hydrophobic and steric hindrance around their acridine core nitrogen (2 < 3 < 1).
Photophysics in the presence of DNA
As shown in Fig. 4, the luminescence of complex 3 was significantly enhanced in the presence of increasing concentrations of DNA, in agreement with the previous studies on complexes 1 and 2.26,30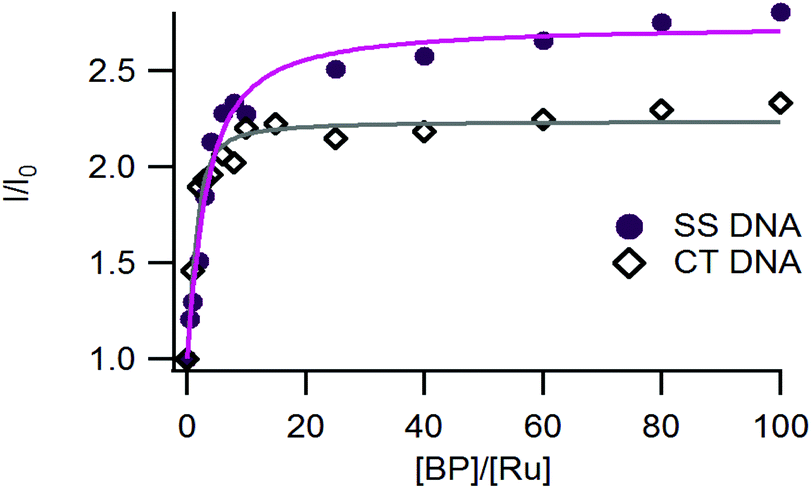 | ||
| Fig. 4 Relative luminescence intensity (I/I0) of 3 as a function of the relative DNA base pair concentration ([BP]/[Ru]) with I0, the luminescence of the complex in the absence of DNA (SS-DNA salmon sperm DNA full circles and Calf Thymus-DNA (empty diamonds)). Measurements were performed by using 10 μM of the complex in 50 mM Tris-HCl buffer, with 50 mM NaCl at pH 7.4 under ambient air conditions. Irradiation at 450 nm. The fitted curves were obtained by using a modified McGhee-von Hippel equation and drawn in purple for SS-DNA and in grey for CT-DNA. | ||
The affinity constants of complexes 1–3 for DNA have been estimated using a modified McGhee-von Hippel model that fits the titration curves (Table 4).39 Complex 3 exhibits a micromolar range affinity for DNA which is in agreement with our previous studies on complexes 1 and 2. Since our complexes are structurally close to Ru-DPPZ and possess high affinities for DNA, we assume that they also intercalate into DNA.
| Complex | SS-DNA | CT-DNA | ||
|---|---|---|---|---|
| K D (μM) | I/I0 max | K D (μM) | I/I0 max | |
| a Binding constants were obtained by using a McGhee-von Hippel type equation; the binding site was fixed to two base pairs per complex (best fit). Errors estimated to 5%. | ||||
| [Ru(phen)2dpac]2+1 | 0.55 | 2.6 | 2.4 | 2.8 |
| [Ru(bpy)2npp]2+2 | 1.3 | 2.7 | 12 | 2.4 |
| [Ru(bpy)2napp]2+3 | 1.2 | 2.7 | 10 | 2.2 |
Photophysics in the presence of mismatched DNA
The ability of complex 3 to recognize mismatched DNA base pairs from well-matched DNA was investigated by using a series of short hairpin-shaped DNA models. These hairpin DNA models contain (or not) a single mismatch base pair near the centre of the duplex (see the sequences in Fig. 5).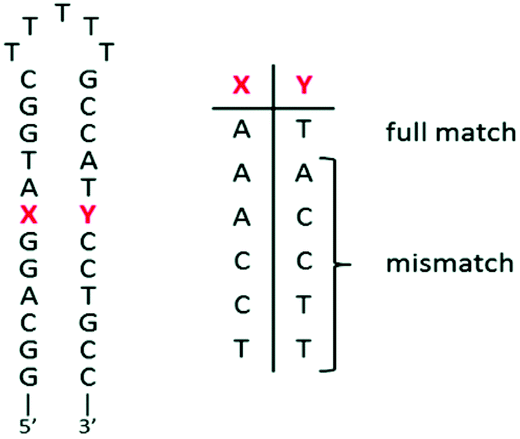 | ||
| Fig. 5 Schematic representation of the short DNA hairpins used for titrations. | ||
A 2-fold decrease of luminescence intensity has been shown for complex 1 in the presence of mismatched DNA versus the well-matched sequence. Nevertheless, complex 1 is unable to differentiate the different types of mismatched hairpins.30 As mentioned previously, the elbow-shaped structure of 2 and 3 would be of interest to obtain such specificity for mismatches. Indeed, complex 2, bearing the elbow on the non-chelating nitrogen side proved to better discriminate the different mismatches.26
Interestingly, complex 3, which bears the elbow at the opposite side of the non-chelating nitrogen, showed better differentiation capabilities. Indeed, complex 3 displayed higher luminescence intensity decreases from well-matched DNA to mismatched DNA to reach a 3-fold luminescence intensity diminution for the TT mismatched hairpin (Fig. 6). Furthermore, complex 3 was found to be able to discriminate the different mismatches with a 2-fold luminescence intensity difference between AC and TT mismatched sequences (see Fig. S22 in the ESI†).
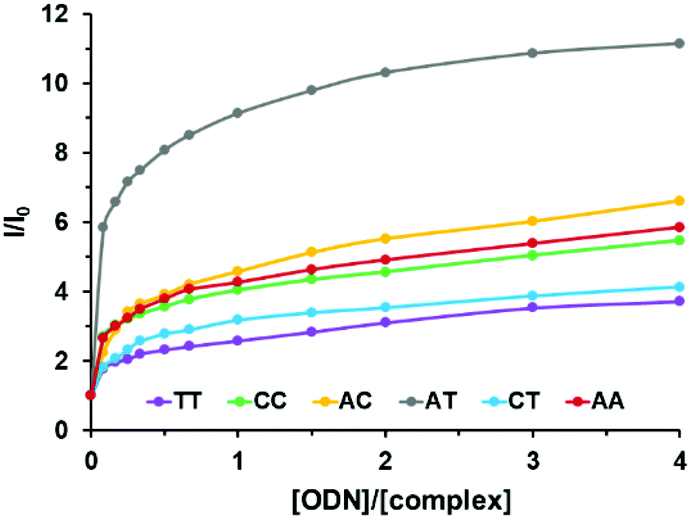 | ||
| Fig. 6 Relative luminescence intensity (I/I0) of 3 as a function of the relative hairpin oligonucleotide concentration ([ODN]/[Ru]) with I0, the luminescence of the complex in the absence of DNA (λexc = 450 nm). Measurements were performed using 1 μM of the complex in Tris-HCl buffer 5 mM, NaCl 1 mM, pH 7.5 under ambient air conditions. Lines are used as guides for the eye and do not correspond to a fitting process. | ||
To better understand the origin of the difference of the luminescence intensity of complex 3 between well-matched and mismatched DNA, binding affinity studies were performed by using circular dichroism (CD) melting assays and bio-layer interferometry (BLI).
Binding affinity studies
CD melting assays were implemented to assess a potential difference of affinity of complex 3 for well-matched versus TT-mismatched DNA sequences. The latter was chosen because it showed the largest difference in luminescence intensity with the AT well-matched sequence. The measurements were carried out under the same conditions as for steady state luminescence titrations (Tris-HCl buffer 5 mM, NaCl 1 mM, pH 7.5). The correct folding of well-matched and TT-mismatched oligonucleotides into the double helix structure was confirmed by the presence of a negative peak at 248 nm and a positive peak at 278 nm (see Fig. S23 in the ESI†).40 Upon the addition of complex 3 no change in the CD spectrum was observed suggesting that the complex did not induce structural modifications in the double helix structure of the hairpins. After the addition of one equivalent to either the well-matched or TT-mismatched hairpin, we observed an increase of the melting temperatures by 5.0 °C and 4.6 °C respectively, which indicates a stabilization of the double helix as a result of the intercalation of the complex into the duplex (Table 5 and Fig. S24†). However, it does not suggest a significant difference of affinity of complex 3 for any of the two hairpins.| XY | T m (°C) (±0.5) | ΔTm (°C) (±1) | |
|---|---|---|---|
| Melting temperatures were measured using 2.5 μM of the complex and hairpin in Tris-HCl buffer 5 mM, NaCl 1 mM, pH 7.5 under ambient air conditions.a Measurements performed for the hairpins alone.b Measurements performed in the presence of one equivalent of complex 3. | |||
| AT | 62.4a | 67.4b | 5.0 |
| TT | 54.3a | 58.9b | 4.6 |
To further determine the affinities of complex 3 for the different hairpins, we next performed the bio-layer interferometry analysis. This technique allows the determination of the association and dissociation kinetic constants between the compound of interest and its target, which directly gives access to the affinity constant (such as surface plasmon resonance). BLI has been used to study biomolecular interactions between large biomolecules, such as protein–membrane interactions and more recently for the interactions of small molecules with G-quadruplex DNA.17,41 The six different hairpin sequences were tested under the same conditions as that of steady state luminescence titrations and CD melting assays (Tris-HCl buffer 5 mM, NaCl 1 mM, pH 7.5). As an example, BLI sensorgrams recorded for the interaction of complex 3 with the fully well-matched hairpin and the TT-mismatch containing hairpin are displayed in Fig. 7. Complex 3 exhibited affinity constants close to the micromolar for each hairpin (0.90 to 3.0 μM). Thus no noticeable difference of affinity for a particular hairpin could be revealed (Table 6).
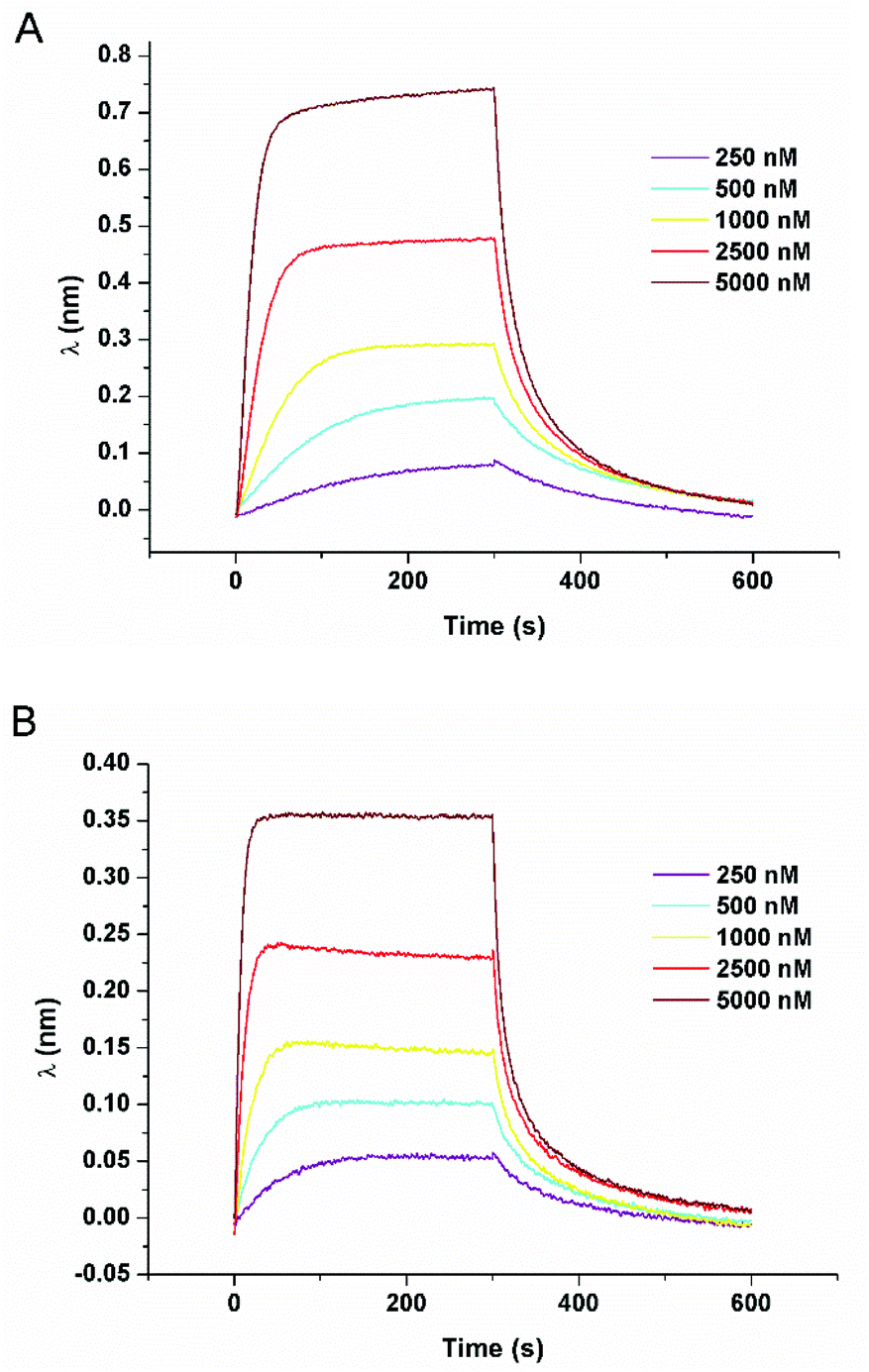 | ||
| Fig. 7 BLI response curves for the association and dissociation of 3 in the presence of well-matched hairpin (A) and the TT-mismatch containing hairpin (B). Measurements are performed using 250 to 5000 nM of the complex in Tris-HCl buffer 5 mM, NaCl 1 mM, pH 7.5 under ambient air conditions. | ||
| XY | k on (104 M−1 s−1) | k off (10−2 s−1) | K D (μM) |
|---|---|---|---|
| Equilibrium dissociation constants were deduced from the kinetic rate constants. Measurements were performed using a concentration range from 0.5 μM to 20 μM of the complex in Tris-HCl buffer 5 mM, NaCl 1 mM, pH 7.5 under ambient air conditions. Errors are estimated to 10%. See Fig. S25 for all the sensorgrams. | |||
| AT | 0.846 | 2.45 | 2.90 |
| AA | 1.52 | 1.92 | 1.26 |
| AC | 0.933 | 2.78 | 2.98 |
| CC | 2.70 | 5.50 | 2.04 |
| CT | 3.77 | 3.39 | 0.90 |
| TT | 2.58 | 2.85 | 1.11 |
Discussion
Computational studies and electrochemical and photophysical data for complexes 1–3 allow us to study their photophysics to better understand the photophysical behaviour of the complexes alone and in the presence of DNA.Photophysical scheme for complexes 1–3 alone
By comparing the photophysical data of acridine-based complexes 1–3 with Ru-DPPZ, we suggest that these systems follow a different photophysical scheme than phenazine based complexes. Indeed, it is well accepted that Ru-DPPZ and its derivatives are less luminescent in water than in acetonitrile since the formation of two hydrogen bonds between water and the two phenazine core nitrogens leads to a non-emissive 3MLCT “dark state”.20 In the case of acridine-based complexes, we observe the opposite trend as complexes 1–3 display longer luminescence lifetimes, increased kr values and less energetic emission wavelengths in water with respect to acetonitrile (Table 3). As already reported for the [Ru(phen)3]2+ complex,42 we assume that acridine-based complexes display a lower luminescence excited state in a more polar solvent (e.g. water vs. acetonitrile), which renders the non-emissive thermally populated 3MC excited state more difficult to reach. Higher stabilisation of the luminescent excited state in H2O may also be promoted by H-bonding with the nitrogen of the acridine core.When comparing the photophysical data of complexes 1–3, we notice that luminescence lifetimes in H2O appear to be correlated to the relative steric and hydrophobic hindrance around the acridine core nitrogen (2 < 3 < 1), which is not the case in the non-protic CH3CN. Indeed, the more the non-chelating nitrogen is hindered, the less it can form H-bond and the shorter is the luminescence lifetime of the complex in water (Table 3).
Based on these observations, the computational studies, and the electrochemical and photophysical data, we propose a new photophysical scheme that is based on the schemes established for Ru-DPPZ and [Ru(phen)3]2+ (Scheme 2).37,42 As for Ru-DPPZ, light absorption followed by inter-system crossing (ISC) populates a 3MLCT excited state centred on the bpy/phen moiety surrounding the metal centre (state A). This state then quickly converts to another more stable 3MLCT in which the electron is localized solely on the acridine part of the planar extended ligand (state B). Depending on the solvent and on the acridine nitrogen accessibility, a lower mono-hydrogen bonded 3MLCT (state C) can be reached, which is in thermal equilibrium with state B. Finally, a non-emissive metal centred 3MC excited state may also be populated either from state B or C.
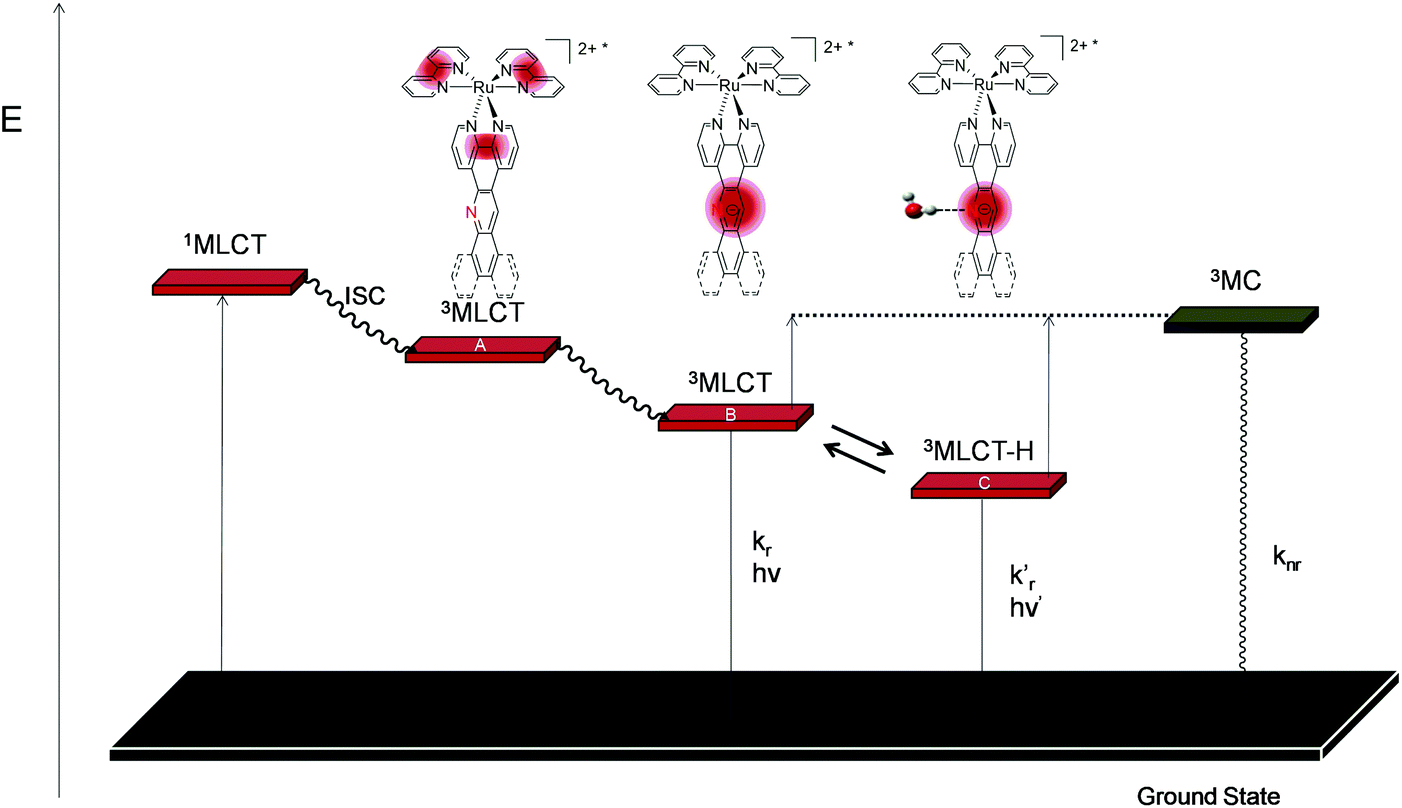 | ||
| Scheme 2 Simplified photophysical scheme for Ru(II) polypyridyl metal complexes bearing an acridine based planar extended ligand. | ||
Photophysical behaviour in the presence of DNA and mismatch containing DNA
As depicted above, the luminescence of complexes 1–3 is strongly influenced by the presence of DNA and is differently altered in the presence of mismatch containing DNA. Indeed, as already reported for other Ru(II) photoprobes, a strong enhancement of the luminescence of complex 3 in the presence of DNA was observed. This can be attributed to a protection of the complexes induced by the intercalation into the double helix that prevent the excited state from non-radiative deactivation such as collisions with the solvent and oxygen photosensitization.Besides, the fact that complexes 1–3 exhibit a significant difference of luminescence intensity between well-matched and mismatched DNA may either be explained by a difference of the affinity of the complexes for mismatched DNA and/or it may arise from the high sensitivity of the complexes to their microenvironment once intercalated into DNA. According to CD melting assays and BLI, it appears that complex 3 exhibits very similar affinities for mismatch containing and totally well-matched hairpins. This means that the ability of complex 3 to photoprobe DNA mismatches does not arise from a difference in binding affinity. As a result, we assume that the origin of the intensified luminescence in the presence of well-matched compared to mismatched DNA is probably due to a better protection of the complex from non-radiative deexcitation sources when intercalated in well-matched DNA. This assumption can be supported by the fact that mismatch containing DNA is known to be less rigid which may provide better access of the solvent and oxygen to the intercalated complex leading to a lower luminescence intensity.
In addition, the fact that complex 3 displays better differentiation capabilities than 1 and 2 indicates (i) the importance of dissymmetry in the mismatch recognition process and (ii) the major role played by the non-chelating nitrogen of the acridine core in the photophysics of our complexes in the presence of DNA.
Conclusions
In this work, we report on the synthesis of a new dissymmetric acridine based ligand, napp, which depicts an interesting dissymmetric shape. The corresponding Ru(II) complex 3 has been fully characterized by 1H-NMR and HRMS and its elbow orientation has been established by X-ray crystallography. The photophysics of 3 has been studied by computational, electrochemical and photophysical means. They suggest that the less energetic electronic transition consists in an MLCT mainly occurring between the metal core and the napp ligand. In addition, our results indicate that a 3MLCT localized on the acridine part of the napp ligand in equilibrium with the hydrogen-bonded form of the complex 3MLCT-H are the two main emissive states. Comparing the photophysical data obtained for 3 with the ones reported for 1 and 2 in acetonitrile and in water allowed us to propose a new photophysical scheme for acridine based Ru(II) complexes. It mainly differs from the most recent scheme described for phenazine based Ru(II) complexes as we propose that the mono-H-bonded complex is more emissive than the non-H-bonded complex due to the higher energy needed to reach the non-emissive 3MC state.Mainly, complex 3 shows interesting mismatched DNA recognition capabilities as the complex appears to be up to three times more luminescent in the presence of the fully well-matched compared to the TT-mismatch containing hairpin. The origin of this difference was investigated by using CD melting assays and bio-layer interferometry analysis. They revealed that the complex exhibits a micromolar affinity for DNA and does not possess a significant difference of affinity for the well-matched hairpin relative to the mismatch containing hairpins. The origin of the luminescence intensity difference may thus be attributed to a better protection of the complex excited state from non-radiative deexcitation sources (e.g., collisions with the solvent, oxygen photosensitization) when intercalated into well-matched DNA compared to mismatched DNA. In addition, the enhancement of the mismatch recognition ability of 3 compared to 1 and 2 suggests that both the “elbow-shape” of the ligand and the hydrophobic hindrance of the non-chelating nitrogen incorporated into the planar extended ligand play an important role in the mismatch photodetection process.
Experimental
Materials and instrumentation
[Ru(bpy)2Cl2] and 5-amino-1,10-phenanthroline were synthesized according to previously described literature protocols.28,43 All solvents and reagents for the synthesis were of reagent grade and were used without any further purification. All solvents for the spectroscopic and electrochemical measurements were of spectroscopic grade. Water was purified with a Millipore Milli-Q system.Calf thymus DNA Type I (CT-DNA) and salmon sperm DNA (SS-DNA) were purchased from Sigma-Aldrich. Hairpin ODNs were purchased from Eurogentech. DNA and ODN concentrations were determined spectroscopically (λ260 nm = 6600 M−1 cm−1/bp for CT-DNA and SS-DNA;44,45λ260 nm = 260![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 for ODN-AT, 264900 M−1 cm−1 for ODN-AA, 253300 M−1 cm−1 for ODN-CC, 259100 M−1 cm−1 for ODN-AC, 254200 M−1 cm−1 for ODN-CT, and 257500 M−1 cm−1 for ODN-TT). The molar extinction coefficients of hairpin oligonucleotides are values calculated based on the base content of each sequence. Biotinylated hairpin ODN's were synthesized on a Controlled Pore Glass solid support by using the phosphoramidite approach with an Applied Biosystems 3400 DNA/RNA Synthesizer (1 μmol scale).
000 M−1 cm−1 for ODN-AT, 264900 M−1 cm−1 for ODN-AA, 253300 M−1 cm−1 for ODN-CC, 259100 M−1 cm−1 for ODN-AC, 254200 M−1 cm−1 for ODN-CT, and 257500 M−1 cm−1 for ODN-TT). The molar extinction coefficients of hairpin oligonucleotides are values calculated based on the base content of each sequence. Biotinylated hairpin ODN's were synthesized on a Controlled Pore Glass solid support by using the phosphoramidite approach with an Applied Biosystems 3400 DNA/RNA Synthesizer (1 μmol scale).
1H NMR experiments were performed in CDCl3, CD3OD or CD3CN on a Bruker AC-300 Avance II (300 MHz) or on a Bruker AM-500 (500 MHz) at 20 °C. The chemical shifts (given in ppm) were measured vs. the residual peak of the solvent as the internal standard. High-resolution mass spectrometry (HRMS) spectra were recorded on a Q-Exactive orbitrap from ThermoFisher using reserpine as the internal standard. Samples were ionized by electrospray ionization (ESI; capillary temperature = 320 °C, vaporizer temperature = 320 °C, sheath gas flow rate = 5 mL min−1).
Cyclic voltammetry was carried out in a one-compartment cell, using a glassy carbon disk working electrode (approximate area = 0.03 cm2), a platinum wire counter electrode, and an Ag/AgCl reference electrode. The potential of the working electrode was controlled by an Autolab PGSTAT 100 potentiostat through a PC interface. The cyclic voltammograms were recorded with a sweep rate of 300 mV s−1, in dried acetonitrile (Sigma-Aldrich, HPLC grade). The concentration of the complexes was 8 × 10−4 mol L−1, with 0.1 mol L−1 tetrabutylammonium perchlorate as the supporting electrolyte. Before each measurement, the samples were purged with nitrogen.
UV-vis absorption spectra were recorded on a Shimadzu UV-1700. Room temperature fluorescence spectra were recorded on a Varian Cary Eclipse instrument. The luminescence intensity at 77 K was recorded on a FluoroLog3 FL3-22 from Jobin Yvon equipped with an 18 V, 450 W xenon short arc lamp and an R928P photomultiplier, using an Oxford Instrument Optistat DN nitrogen cryostat controlled by an Oxford Intelligent Temperature Controller (ITC503S) instrument. Luminescence lifetime measurements were performed after irradiation at λ = 400 nm obtained by the second harmonic of a titanium/sapphire laser (picosecond Tsunami laser spectra physics 3950-M1BB + 39868-03 pulse picker doubler) at a 80 kHz repetition rate. The Fluotime 200 from AMS technologies was used for the decay acquisition. It consists of a GaAs microchannel plate photomultiplier tube (Hamamatsu model R3809U-50) followed by a time-correlated single photon counting system from Picoquant (PicoHarp300). The ultimate time resolution of the system is close to 30 ps. Luminescence decays were analyzed with FLUOFIT software available from Picoquant.
CD-analyses were performed on a Jasco J810 spectro-polarimeter using a 1 cm length quartz cuvette. Prior to CD analysis, the oligonucleotides were annealed by heating the sample at 95 °C for 5 min under the buffer conditions and cooling it overnight to room temperature. Spectra were recorded at 5 °C increments from 25 °C to 90 °C over the wavelength range from 220 to 330 nm. For each temperature, the spectrum was an average of three scans with a 0.5 s response time, a 1 nm data pitch, a 4 nm bandwidth and a 200 nm min−1 scanning speed. For CD melting experiments, the ellipticities of the hairpins were recorded at 248 nm. Melting temperatures were obtained using a Boltzmann-type fit on Origin software. Each curve fit was only accepted with a R value >0.99.
Bio-layer interferometry experiments were performed using sensors coated with streptavidin (SA sensors) purchased from Forte Bio (PALL). Prior to use, they were immersed for 10 minutes in a buffer before functionalization to dissolve the sucrose layer. Then the sensors were dipped for 15 minutes in DNA containing solutions (biotinylated hairpin oligonucleotides) at 100 nM and rinsed in the buffer solution (Tris-HCl buffer 5 mM, NaCl 1 mM, pH 7.5 and 0.5% v/v surfactant P20) for 10 minutes. The functionalized sensors were next dipped in the ruthenium complex containing solution at different concentrations (see the ESI†) for 2 minutes interspersed by a rinsing step in the buffer solution for 4 minutes. Reference sensors without DNA immobilization were used to subtract the non-specific adsorption on the SA layer. The sensorgrams were fit using a 1:1 interaction model. The reported values are the means of representative independent experiments, and the errors provided are standard deviations from the mean. Each experiment was repeated at least two times.
Synthesis procedures and characterization
:1 Cy/EtOAc) to give 2-aminonaphthalene as a white solid (1.66 g, 11.6 mmol, 82%). Rf 0.24 (3:1 Cy/EtOAc); 1H NMR (CDCl3, 300 MHz) δ 7.72–7.63 (2H, m, H7–6), 7.60 (d, J = 8.2 Hz, 1H, H4), 7.59 (d, J = 8.2 Hz, 1H, H3), 7.25–7.19 (m, 1H, H5), 6.98 (s, 1H, H1), 6.94 (1H, dd, J = 8.5, 2.3 Hz, H8), 3.81 (s, broad, 2H, NH2). Data are consistent with the literature values.46
:1 Cy/EtOAc); 1H NMR (CD3CN, 300 MHz) δ 8.89 (1H, s, NH), 8.49 (1H, dd, J = 8.3, 0.9 Hz, H5), 8.17 (1H d, J = 8.6 Hz H4), 7.87 (1H, d, J = 8.2 Hz, H8), 7.67 (1H, ddd, J = 8.3, 7.0, 1.2 Hz, H7), 7.44 (1H, ddd, J = 8.2, 7.0, 1.2 Hz, H6), 7.20 (1H, d, J = 8.6 Hz, H3). Data are consistent with the literature values.48
:4 CH2Cl2/Cy) to afford methyl 2-amino-1-naphthoate as a white solid (390 mg, 0.423 mmol, 36%). Rf 0.28 (6:4 CH2Cl2/Cy); 1H NMR (CDCl3, 300 MHz) δ 8.38 (1H, d, J = 8.8 Hz, H8), 7.55 (2H, m, H4, H5), 7.38 (1H, ddd, J = 8.6, 6.9, 1.5 Hz, H7), 7.15 (1H, ddd, J = 8.0, 6.9, 1.0 Hz, H6), 6.71 (1H, d, J = 8.9 Hz, H3), 5.72 (2H, s, NH2), 3.91 (3H, s, OMe).
:1 CH2Cl2/MeOH) to afford naphtho[2,1-b]pyrido[3,2-f][1,7]phenanthroline (napp) as a beige solid (224 mg, 0.679 mmol, 71%). 1H NMR (CDCl3, 500 MHz) δ 10.09 (1H, s, Hd), 9.61 (1H, dd, Ja–b = 8.1, Ja–c = 1.8 Hz, Ha), 9.29 (1H, dd, Jk–l = 8.3, Jk–m = 1.4 Hz, Hk), 9.14 (1H, dd, Jc–b = 4.3, Jc–a = 1.8 Hz, Hc), 9.10 (1H, dd, Jm–l = 4.3, Jm–k = 1.5 Hz, Hm), 9.04 (1H, d, Je–f = 8.2 Hz, He), 8.12 (1 H, d, Jj–i = 9.1 Hz, Hj), 8.05 (1 H, d, Jj–i = 9.2 Hz, Hj), 8.03 (1 H, d, Jh–g = 7.3 Hz, H), 7.85–7.79 (2 H, m, Hf,g), 7.79–7.74 (2 H, m, Hl,b); HRMS-ESI calculated for C23H14N3 ([M + H]+): m/z 332.11822, found: m/z 332.11816.
:1:0.5 CH3CN/H2O/KNO3sat) to afford [Ru(bpy)2napp]2+3 as an orange solid (28 mg, 0.027 mmol, 66%). The counter-anion exchange from PF6− to Cl− was performed by adding small portions of NBu4Cl to a solution of the complex in acetone. Rf 0.35 (CH3CN/H2O/KNO3sat 10:1:1/2); 1H NMR (CD3CN, 500 MHz) δ (ppm), 10.47 (1H, s, Hd), 9.81 (1H, d, Ja–b = 8.2, Ja–c = 1.2 Hz, Ha), 9.59 (1H, dd, Jc–b = 8.2, Jc–a = 1.2 Hz, Hc), 9.24 (1H, d, Jm–l = 8.2 Hz, Hm), 8.53 (4H, m, H5, H5′, H6, H6′), 8.34 (1H, d, Ji–j = 9.1 Hz, Hi), 8.25 (1H, d, Jj–i = 9.1 Hz, Hj), 8.16 (2H, m, H4, H4′), 8.12 (3H, m, He, Hf, Hg), 8.01 (2H, m, H7, H7′), 7.95 (1H, m, Hl), 7.91–7.84 (5H, m, Hb, H2,H2′, Hm, Hh), 7.73 (1H, d, J9–8 = 5.4 Hz, H9), 7.69 (1H, d, J9′−8′ = 5.6 Hz, H9′), 7.49–7.44 (2H, m, H3, H3′), 7.28–7.21 (m, 2H, H8, H8′); HRMS-ESI calculated for [C43H29N7F6PRu]+: m/z 890.11728, found: m/z 890.11700 and for [C43H29N7Ru]2+: m/z 372.57617, found: m/z 372.57640. The product yield was confirmed by elemental composition analysis and X-ray crystallography.
Conflicts of interest
The authors declare no competing interest.Acknowledgements
M. G. and B. E. gratefully acknowledge the Université catholique de Louvain, the Fonds National pour la Recherche Scientifique (F.R.S.-F.N.R.S.) and the Prix Pierre et Colette Bauchau for financial support. We also thank Dr. Koen Robeyns for scientific and technical support. B. L.-M. and G. S. H. thank the Natural Sciences and Engineering Research Council (NSERC) of Canada for financial support. The NanoBio-ICMG platform (FR 2607) is acknowledged for the technical support. This work was supported by IDEX Grenoble and the Region Auvergne-Rhône-Alpes for financial support. We also thank Oceane Thery and Hugues Bonnet for technical support.References
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, CA-Cancer J. Clin., 2018, 68, 394–424 CrossRef.
- W. Marx, N. Kiss, A. L. McCarthy, D. McKavanagh and L. Isenring, J. Acad. Nutr. Diet., 2016, 116, 819–827 CrossRef PubMed.
- S. A. Dugger, A. Platt and D. B. Goldstein, Nat. Rev. Drug Discovery, 2017, 17, 183 CrossRef PubMed.
- D. M. Hyman, B. S. Taylor and J. Baselga, Cell, 2017, 168, 584–599 CrossRef CAS.
- D. Guillotin and S. A. Martin, Exp. Cell Res., 2014, 329, 110–115 CrossRef CAS PubMed.
- G. Ponti, M. Manfredini, A. Tomasi and G. Pellacani, Gene, 2016, 589, 127–132 CrossRef CAS PubMed.
- C. Rosty, M. D. Walsh, N. M. Lindor, S. N. Thibodeau, E. Mundt, S. Gallinger, M. Aronson, A. Pollett, J. A. Baron, S. Pearson, M. Clendenning, R. J. Walters, B. N. Nagler, W. J. Crawford, J. P. Young, I. Winship, A. K. Win, J. L. Hopper, M. A. Jenkins and D. D. Buchanan, Fam. Cancer, 2014, 13, 573–582 CrossRef CAS PubMed.
- J. M. Helder-Woolderink, E. A. Blok, H. F. A. Vasen, H. Hollema, M. J. Mourits and G. H. De Bock, Eur. J. Cancer, 2016, 55, 65–73 CrossRef CAS.
- P. Boland, M. Yurgelun and R. Boland, CA-Cancer J. Clin., 2018, 68, 217–231 CrossRef PubMed.
- F. Honecker, H. Wermann, F. Mayer, A. J. Gillis, H. Stoop, R. J. van Gurp, K. Oechsle, E. Steyerberg, J. T. Hartmann, W. N. Dinjens, J. W. Oosterhuis, C. Bokemeyer and L. H. Looijenga, J. Clin. Oncol., 2009, 27, 2129–2136 CrossRef CAS PubMed.
- D.-L. Ma, T. Xu, D. S.-H. Chan, B. Y.-W. Man, W.-F. Fong and C.-H. Leung, Nucleic Acids Res., 2011, 39, e67–e67 CrossRef CAS.
- D. S.-H. Chan, H.-Z. He, C.-H. Leung and D.-L. Ma, Nucleic Acids Res., 2013, 41, 4345–4359 CrossRef.
- C. Mari, V. Pierroz, S. Ferrari and G. Gasser, Chem. Sci., 2015, 6, 2660–2686 RSC.
- F. E. Poynton, S. A. Bright, S. Blasco, D. C. Williams, J. M. Kelly and T. Gunnlaugsson, Chem. Soc. Rev., 2017, 46, 7706–7756 RSC.
- S. Thota, D. A. Rodrigues, D. C. Crans and E. J. Barreiro, J. Med. Chem., 2018, 61, 5805–5821 CrossRef CAS PubMed.
- G. Piraux, L. Bar, M. Abraham, T. Lavergne, H. Jamet, J. Dejeu, L. Marcélis, E. Defrancq and B. Elias, Chem. – Eur. J., 2017, 23, 11872–11880 CrossRef CAS PubMed.
- J. Weynand, A. Diman, M. Abraham, L. Marcélis, H. Jamet, A. Decottignies, J. Dejeu, E. Defrancq and B. Elias, Chem. – Eur. J., 2018, 24, 19216–19227 CrossRef CAS PubMed.
- K. Lin, Z.-Z. Zhao, H.-B. Bo, X.-J. Hao and J.-Q. Wang, Front. Pharmacol., 2018, 9, 1–10 CrossRef CAS PubMed.
- H.-K. Liu and P. J. Sadler, Acc. Chem. Res., 2011, 44, 349–359 CrossRef CAS PubMed.
- A. E. Friedman, J. C. Chambron, J. P. Sauvage, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1990, 112, 4960–4962 CrossRef CAS.
- E. Amouyal, A. Homsi, J.-C. Chambron and J.-P. Sauvage, J. Chem. Soc., Dalton Trans., 1990, 1841–1845 RSC.
- A. Granzhan, N. Kotera and M.-P. Teulade-Fichou, Chem. Soc. Rev., 2014, 43, 3630–3665 RSC.
- J. K. Barton, A. N. Boynton and K. M. Boyle, in DNA-targeting Molecules as Therapeutic Agents, The Royal Society of Chemistry, 2018, pp. 367–390 Search PubMed.
- A. N. Boynton, L. Marcélis, A. J. McConnell and J. K. Barton, Inorg. Chem., 2017, 56, 8381–8389 CrossRef CAS PubMed.
- A. J. McConnell, M. H. Lim, E. D. Olmon, H. Song, E. E. Dervan and J. K. Barton, Inorg. Chem., 2012, 51, 12511–12520 CrossRef CAS PubMed.
- Q. Deraedt, L. Marcelis, F. Loiseau and B. Elias, Inorg. Chem. Front., 2017, 4, 91–103 RSC.
- A. N. Boynton, L. Marcélis and J. K. Barton, J. Am. Chem. Soc., 2016, 138, 5020–5023 CrossRef CAS PubMed.
- E. Rüba, J. R. Hart and J. K. Barton, Inorg. Chem., 2004, 43, 4570–4578 CrossRef PubMed.
- M. H. Lim, H. Song, E. D. Olmon, E. E. Dervan and J. K. Barton, Inorg. Chem., 2009, 48, 5392–5397 CrossRef CAS PubMed.
- Q. Deraedt, L. Marcélis, T. Auvray, G. S. Hanan, F. Loiseau and B. Elias, Eur. J. Inorg. Chem., 2016, 2016, 3649–3658 CrossRef CAS.
- L. Jiang, X. Lu, H. Zhang, Y. Jiang and D. Ma, J. Org. Chem., 2009, 74, 4542–4546 CrossRef CAS.
- G. Reissenweber and D. Mangold, Angew. Chem., Int. Ed., 1981, 20, 882–883 CrossRef.
- R. M. Hartshorn and J. K. Barton, J. Am. Chem. Soc., 1992, 114, 5919–5925 CrossRef CAS.
- R. L. Martin, J. Chem. Phys., 2003, 118, 4775–4777 CrossRef CAS.
- A. M. Brouwer, Pure Appl. Chem., 2011, 83, 2213–2228 CAS.
- J. V. Caspar and T. J. Meyer, J. Am. Chem. Soc., 1983, 105, 5583–5590 CrossRef CAS.
- J. Olofsson, B. Önfelt and P. Lincoln, J. Phys. Chem. A, 2004, 108, 4391–4398 CrossRef CAS.
- I. Ortmans, B. Elias, J. M. Kelly, C. Moucheron and A. Kirsch-DeMesmaeker, Dalton Trans., 2004, 668–676 RSC.
- J. D. McGhee and P. H. von Hippel, J. Mol. Biol., 1974, 86, 469–489 CrossRef CAS.
- J. Kypr, I. Kejnovská, D. Renciuk and M. Vorlícková, Nucleic Acids Res., 2009, 37, 1713–1725 CrossRef CAS PubMed.
- J. Wallner, G. Lhota, D. Jeschek, A. Mader and K. Vorauer-Uhl, J. Pharm. Biomed. Anal., 2013, 72, 150–154 CrossRef CAS PubMed.
- A. Boisdenghien, C. Moucheron and A. Kirsch-De Mesmaeker, Inorg. Chem., 2005, 44, 7678–7685 CrossRef CAS PubMed.
- S. Ji, H. Guo, X. Yuan, X. Li, H. Ding, P. Gao, C. Zhao, W. Wu, W. Wu and J. Zhao, Org. Lett., 2010, 12, 2876–2879 CrossRef CAS.
- E. M. Castanheira, M. S. D. Carvalho, A. R. O. Rodrigues, R. C. Calhelha and M.-J. R. Queiroz, Nanoscale Res. Lett., 2011, 6, 379 CrossRef.
- R. B. Dixit, T. S. Patel, S. F. Vanparia, A. P. Kunjadiya, H. R. Keharia and B. C. Dixit, Sci. Pharm., 2011, 79, 293–308 CrossRef CAS PubMed.
- G. D. Vo and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 11049–11061 CrossRef CAS PubMed.
- T. C. Bruice, J. Am. Chem. Soc., 1957, 79, 702–705 CrossRef CAS.
- A. S. Karpenko, M. O. Shibinskaya, N. M. Zholobak, Z. M. Olevinskaya, S. A. Lyakhov, L. A. Litvinova, M. Y. Spivak and S. A. Andronati, Pharm. Chem. J., 2006, 40, 595–602 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1894480. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9qi00133f |
| This journal is © the Partner Organisations 2019 |