
Substituent effects on the fluorescent spin-crossover Fe(ii) complexes of rhodamine 6G hydrazones†

Abstract
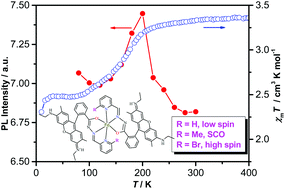
Two new complexes were constructed via the coordination of fluorophores with Fe(II) ions to study the effects of ligand substituents on fluorescent-spin crossover (SCO) materials. Single-crystal structural determination suggests that the central iron(II) of complexes 1–2 adopted an N4O2 coordination configuration, chelated by two ligands L1 or L2. The π⋯π stacking interaction between the π-conjugated xanthene groups of the adjacent ligands for complex 1 is apparently stronger than that for complex 2. Magnetic susceptibility measurements show that complex 1 is high spin, while complex 2 is diamagnetic at room temperature. Interestingly, the desolvated form 1-d of complex 1 exhibits SCO behavior (Tc = 175 K) without hysteresis. Significantly, complex 1-d displays the correlation between the spin crossover and the fluorescence.
 Please wait while we load your content...
Please wait while we load your content...

